
Design, Synthesis and Antiproliferative Evaluation of Bis-Indole Derivatives with a Phenyl Linker: Focus on Autophagy



Abstract
1. Introduction
2. Results and Discussion
2.1. Chemistry
2.2. Biological Activity
2.2.1. MTT Screening Assay
2.2.2. BrdU Cell Proliferation Assay
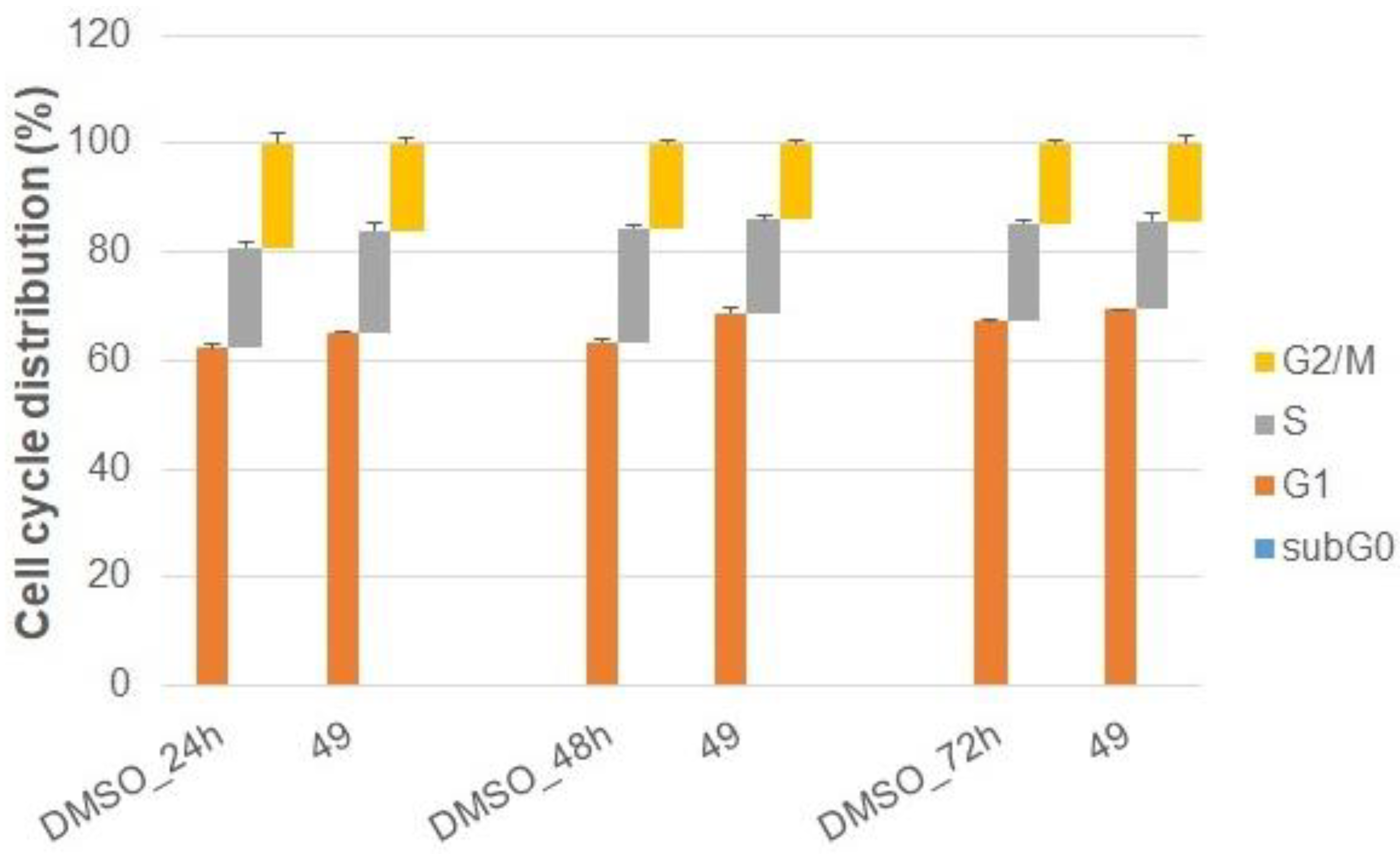
2.2.3. Cell Cycle Analysis
2.2.4. Annexin V/PI Staining
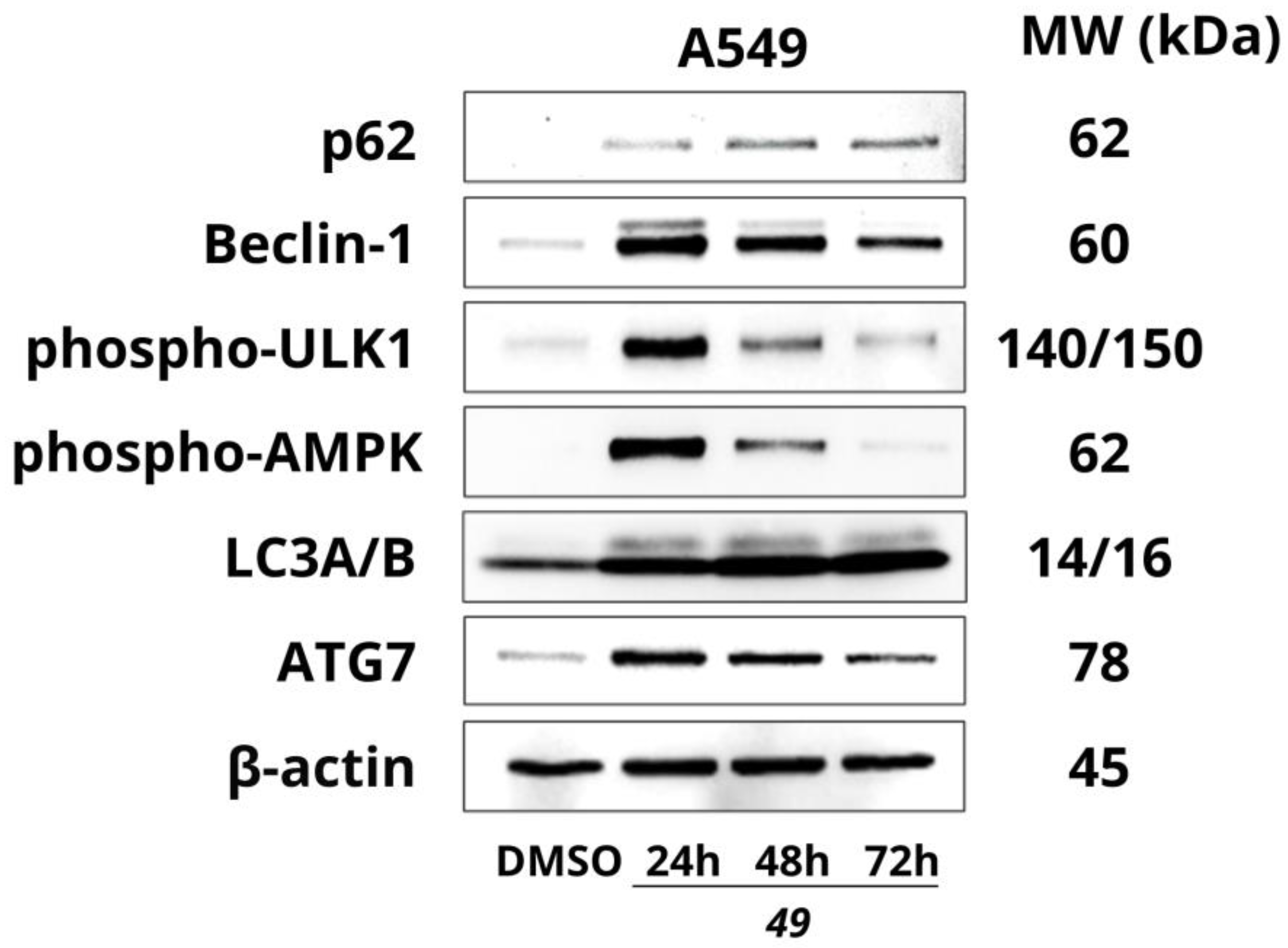
2.2.5. Effect of Compound 49 on the Expression of Autophagy-Related Proteins
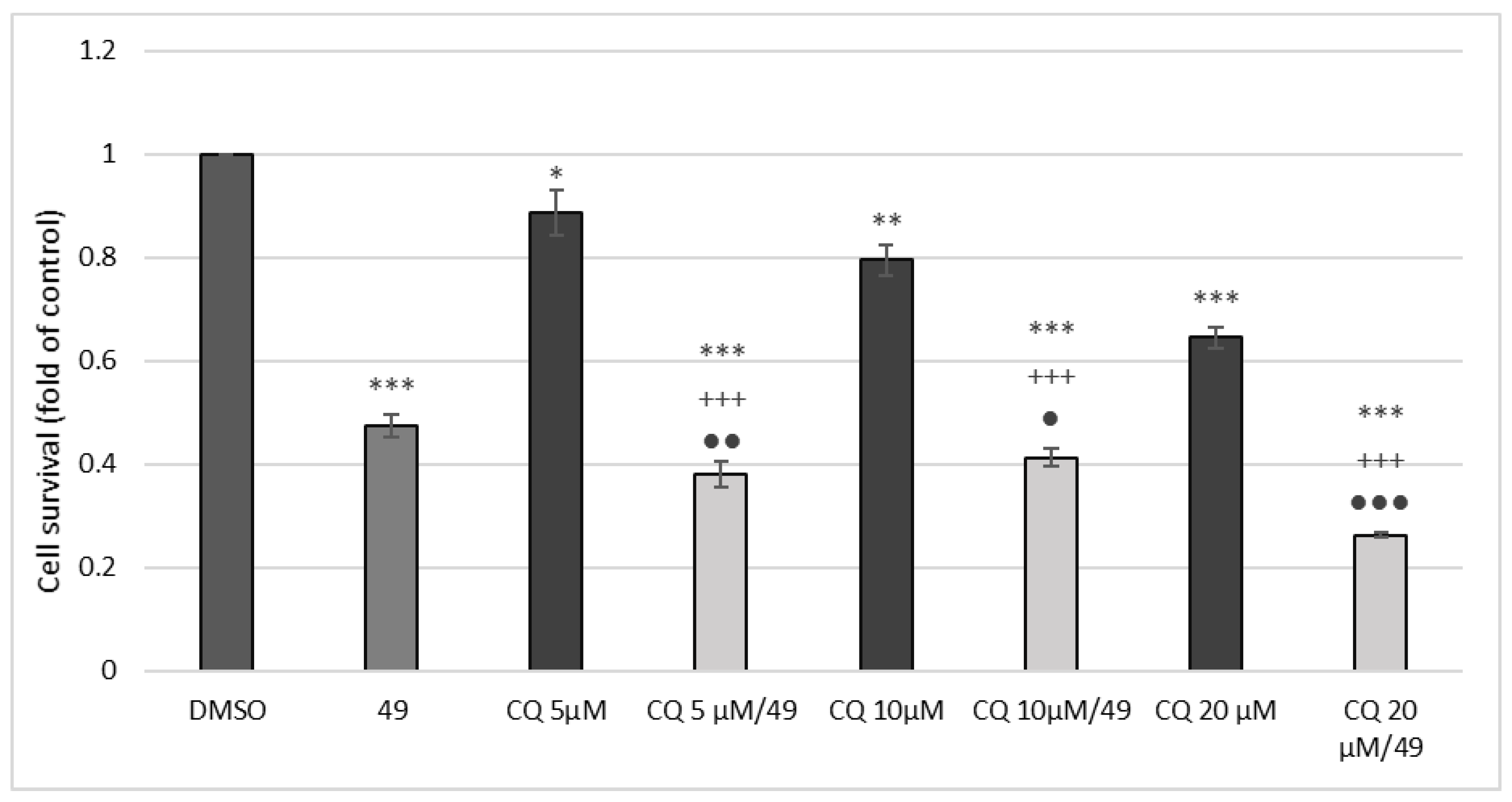
2.2.6. MTT Assay with Chloroquine Combination
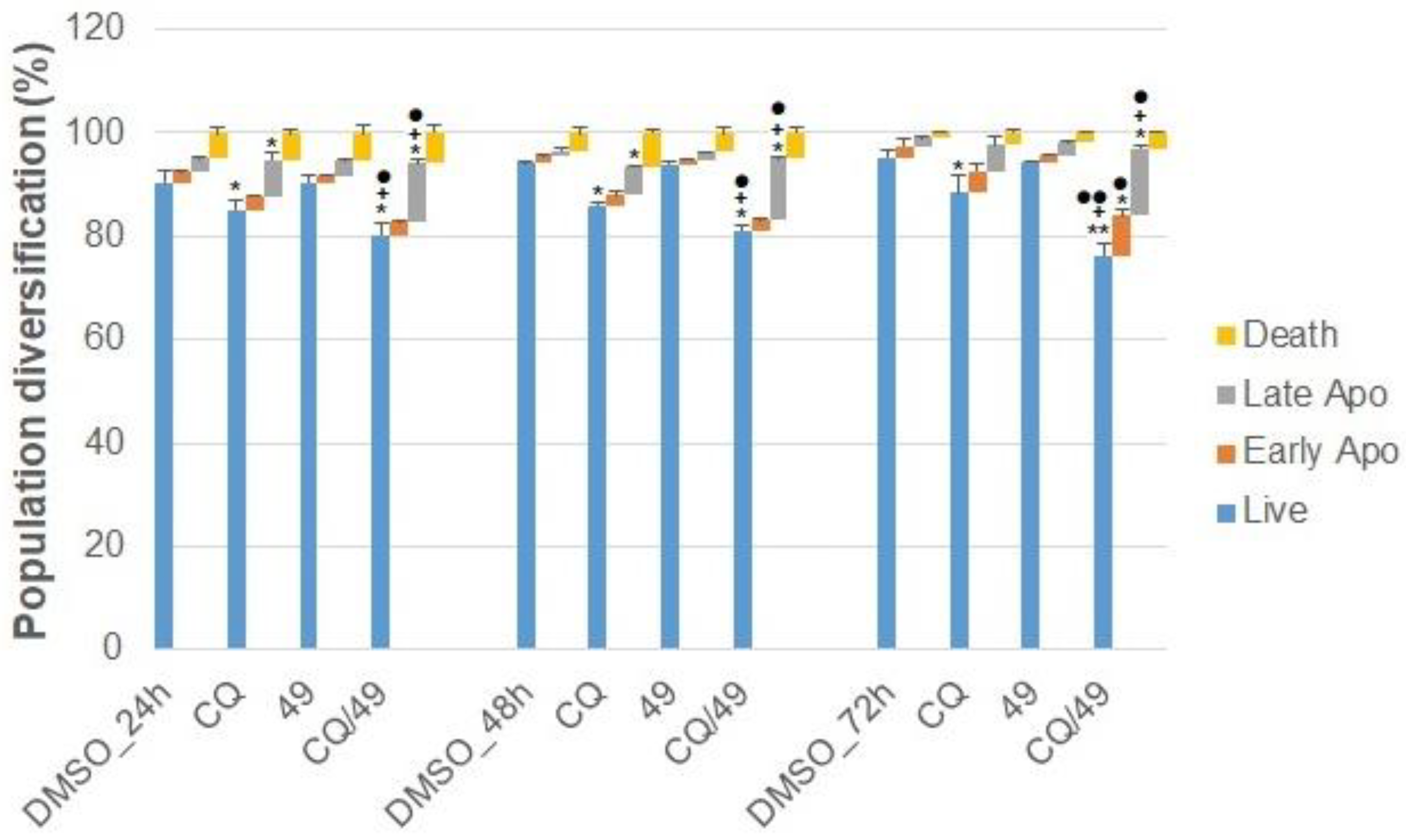
2.2.7. Effect of Chloroquine Co-Treatment on Cell Cycle
2.2.8. Effect of Chloroquine on Apoptosis
2.2.9. Effect of Compound 49 on Glutathione (GSH) Levels and Activity of GSH-Related Enzymes
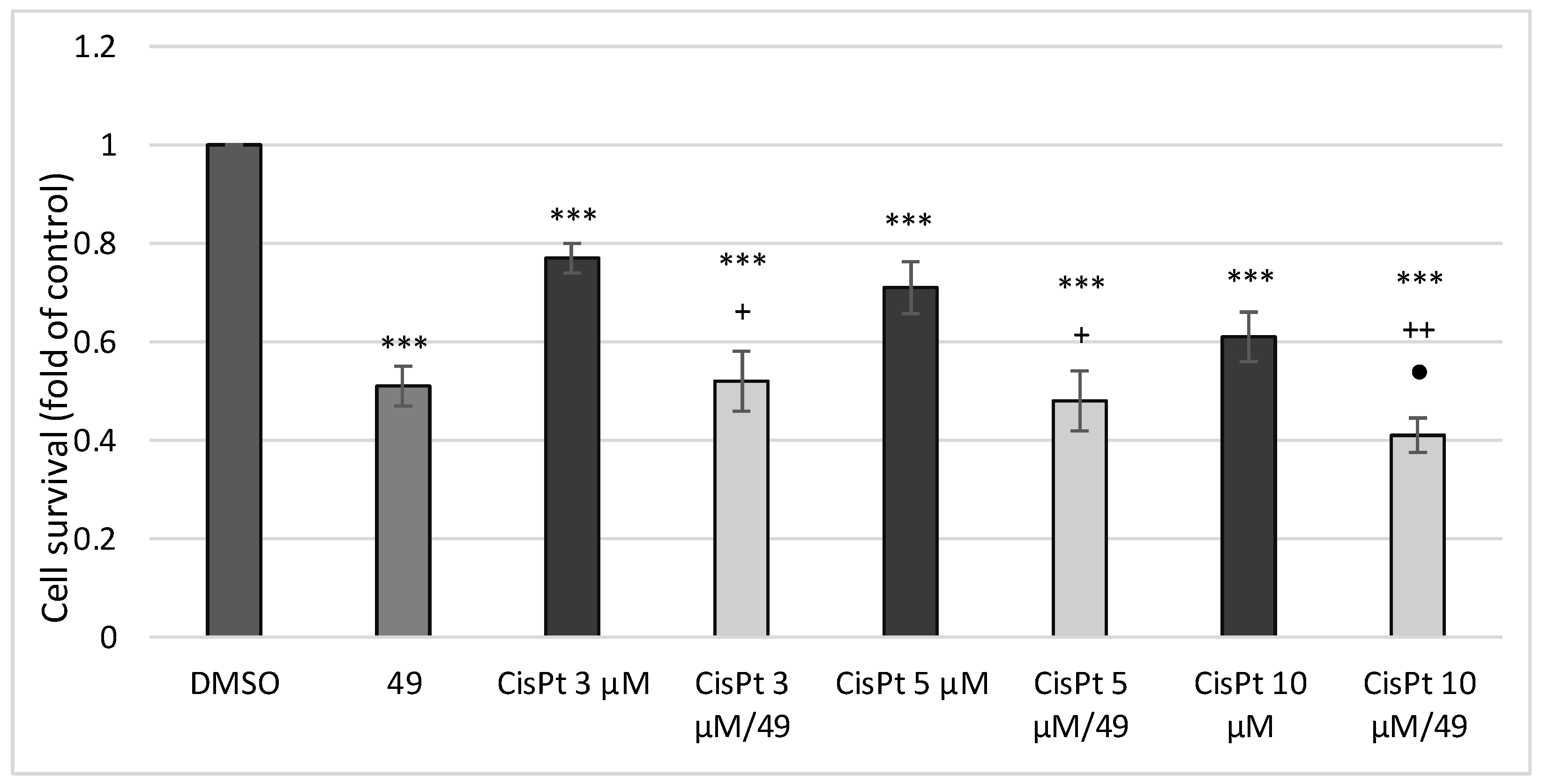
2.2.10. Effect of Simultaneous Treatment with Compound 49 and Cis-Platin (CisPt) on A549 Human Lung Cancer Cell Proliferation
3. Materials and Methods
3.1. General Remarks
3.2. Synthesis
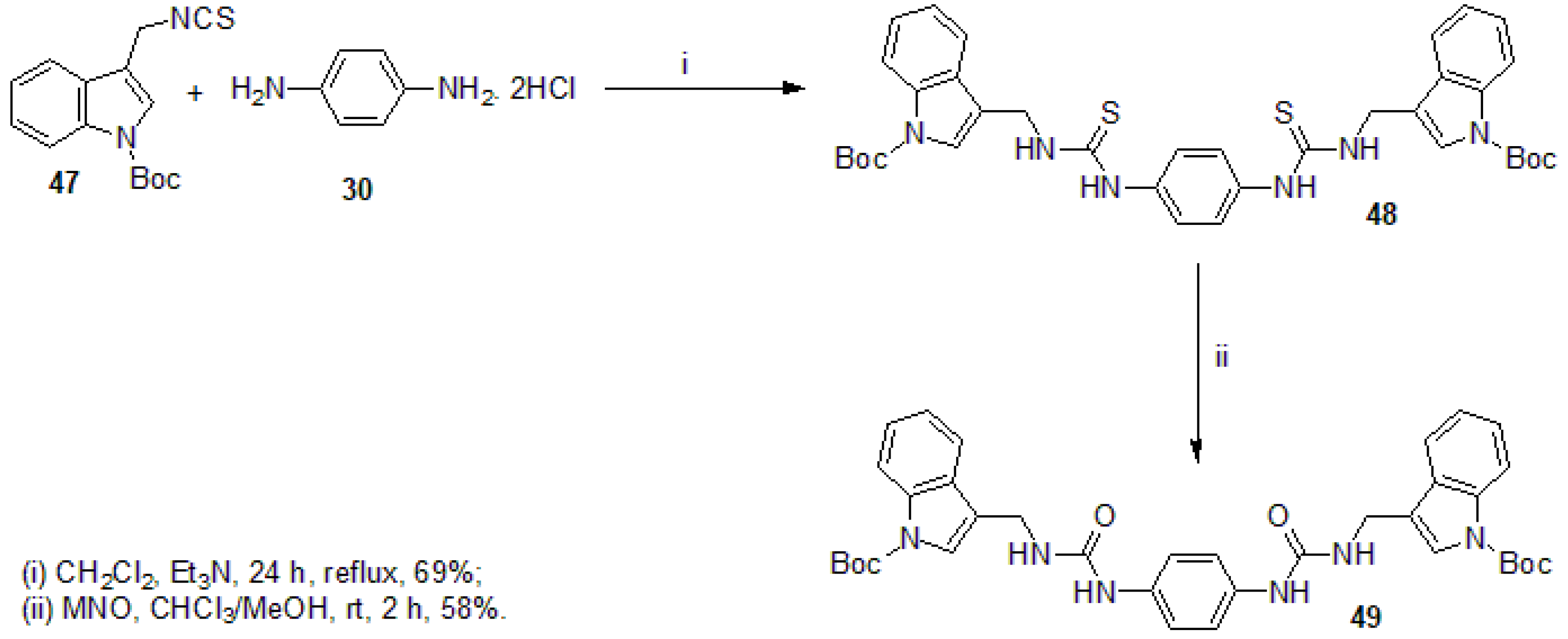
3.2.1. Synthesis of N,N′-(1,4-phenylene)bis{N′-[1-(tert-butoxycarbonyl)indol-3-yl]methyl (thiourea)} (48)
3.2.2. Synthesis of N,N′-(1,4-phenylene)bis{N′-[1-(tert-butoxycarbonyl)indol-3-yl]methyl (urea)} (49)
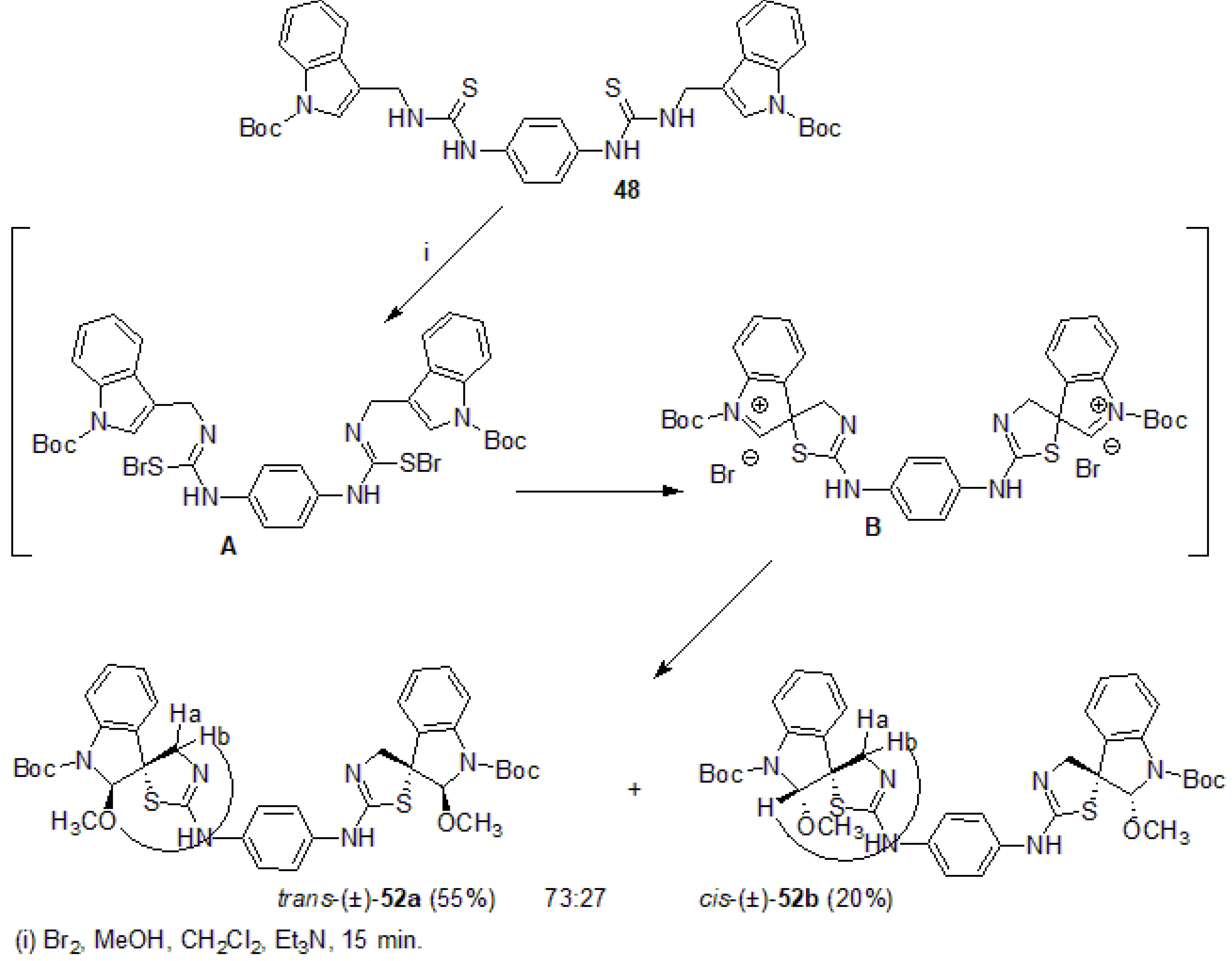
3.2.3. Synthesis of 2,2′-[1,4-phenylenebis(azanylylidene)]bis{3-[(1-(tert-butoxycarbonyl)-indol-3-yl)methyl]-1,3-thiazolidin-4-one} (51a)
3.2.4. Synthesis of trans-(±)- and cis-(±)-N,N′-bis[1-(tert-Butoxycarbonyl)-2-methoxy-spiro{indoline-3,5′-[4′,5′]dihydrothiazol-2′-yl}]benzene-1,4-diamine [(±)-52a, (±)-52b]
Trans-(±)-52a
Cis-(±)-52b
3.3. Biological Activity
3.3.1. Tested Compounds
3.3.2. Cell Culture
3.3.3. MTT Colorimetric Assay
3.3.4. BrdU Cell Proliferation Assay
3.3.5. Cell Cycle Analysis
3.3.6. Annexin V/PI Analysis
3.3.7. Western Blot Analysis
3.3.8. Measurement of Glutathione Content and Antioxidant Enzyme Activity
3.3.9. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bray, F.; Laversanne, M.; Weiderpass, E.; Soerjomataram, I. The ever-increasing importance of cancer as a leading cause of premature death worldwide. Cancer 2021, 127, 3029–3030. [Google Scholar] [CrossRef] [PubMed]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Dickens, E.; Ahmed, S. Principles of cancer treatment by chemotherapy. Surgery 2018, 36, 134–138. [Google Scholar] [CrossRef]
- Cragg, G.M.; Pezzuto, J.M. Natural Products as a Vital Source for the Discovery of Cancer Chemotherapeutic and Chemopreventive Agents. Med. Princ. Pract. Int. J. Kuwait Univ. Health Sci. Cent. 2016, 25 (Suppl. 2), 41–59. [Google Scholar] [CrossRef] [PubMed]
- Pedras, M.S.; Yaya, E.E. Phytoalexins from Brassicaceae: News from the front. Phytochemistry 2010, 71, 1191–1197. [Google Scholar] [CrossRef]
- Chripkova, M.; Zigo, F.; Mojzis, J. Antiproliferative Effect of Indole Phytoalexins. Molecules 2016, 21, 1626. [Google Scholar] [CrossRef]
- Budovska, M.; Krochtova, K.; Michalkova, R.; Mojzis, J. Aminoanalogues of isobrassinin, erucalexin and isocyclobrassinin: Synthesis and evaluation of the antiproliferative and cytotoxic properties. Tetrahedron 2022, 120, 132898. [Google Scholar] [CrossRef]
- Budovska, M.; Pilatova, M.; Varinska, L.; Mojzis, J.; Mezencev, R. The synthesis and anticancer activity of analogs of the indole phytoalexins brassinin, 1-methoxyspirobrassinol methyl ether and cyclobrassinin. Bioorg. Med. Chem. 2013, 21, 6623–6633. [Google Scholar] [CrossRef]
- Budovska, M.; Selesova, I.; Tischlerova, V.; Michalkova, R.; Mojzis, J. Design, synthesis, and biological evaluation of novel 5-bromo derivatives of indole phytoalexins. Mon. Chem.-Chem. Mon. 2020, 151, 1737–1758. [Google Scholar] [CrossRef]
- Chripkova, M.; Drutovic, D.; Pilatova, M.; Mikes, J.; Budovska, M.; Vaskova, J.; Broggini, M.; Mirossay, L.; Mojzis, J. Brassinin and its derivatives as potential anticancer agents. Toxicol. Vitr. Int. J. Publ. Assoc. BIBRA 2014, 28, 909–915. [Google Scholar] [CrossRef]
- Hong, T.; Ham, J.; Song, J.; Song, G.; Lim, W. Brassinin Inhibits Proliferation in Human Liver Cancer Cells via Mitochondrial Dysfunction. Cells 2021, 10, 332. [Google Scholar] [CrossRef] [PubMed]
- Kello, M.; Drutovic, D.; Chripkova, M.; Pilatova, M.; Budovska, M.; Kulikova, L.; Urdzik, P.; Mojzis, J. ROS-dependent antiproliferative effect of brassinin derivative homobrassinin in human colorectal cancer Caco2 cells. Molecules 2014, 19, 10877–10897. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.M.; Park, J.H.; Kim, K.D.; Nam, D.; Shim, B.S.; Kim, S.H.; Ahn, K.S.; Choi, S.H.; Ahn, K.S. Brassinin Induces Apoptosis in PC-3 Human Prostate Cancer Cells through the Suppression of PI3K/Akt/mTOR/S6K1 Signaling Cascades. Phytother. Res. 2014, 28, 423–431. [Google Scholar] [CrossRef] [PubMed]
- Tischlerova, V.; Kello, M.; Budovska, M.; Mojzis, J. Indole phytoalexin derivatives induce mitochondrial-mediated apoptosis in human colorectal carcinoma cells. World J. Gastroenterol. 2017, 23, 4341–4353. [Google Scholar] [CrossRef] [PubMed]
- Elsbaey, M.; Ahmed, K.F.M.; Elsebai, M.F.; Zaghloul, A.; Amer, M.M.A.; Lahloub, M.F.I. Cytotoxic constituents of Alocasia macrorrhiza. Z. Nat. C 2017, 72, 21–25. [Google Scholar] [CrossRef]
- Tian, X.L.; Liu, K.D.; Zu, X.Y.; Ma, F.Y.; Li, Z.; Lee, M.; Chen, H.Y.; Li, Y.; Zhao, Y.Z.; Liu, F.F.; et al. 3,3′-Diindolylmethane inhibits patient-derived xenograft colon tumor growth by targeting COX1/2 and ERK1/2. Cancer Lett. 2019, 448, 20–30. [Google Scholar] [CrossRef]
- Buduma, K.; Chinde, S.; Dommati, A.K.; Sharma, P.; Shukla, A.; Srinivas, K.; Arigari, N.K.; Khan, F.; Tiwari, A.K.; Grover, P.; et al. Synthesis and evaluation of anticancer and antiobesity activity of 1-ethoxy carbonyl-3,5-bis (3′-indolyl methylene)-4-pyperidone analogs. Bioorg. Med. Chem. Lett. 2016, 26, 1633–1638. [Google Scholar] [CrossRef]
- Das Mukherjee, D.; Kumar, N.M.; Tantak, M.P.; Das, A.; Ganguli, A.; Datta, S.; Kumar, D.; Chakrabarti, G. Development of Novel Bis(indolyl)-hydrazide-Hydrazone Derivatives as Potent Microtubule-Targeting Cytotoxic Agents against A549 Lung Cancer Cells. Biochemistry 2016, 55, 3020–3035. [Google Scholar] [CrossRef]
- Zhang, Y.; Hu, C. Anticancer activity of bisindole alkaloids derived from natural sources and synthetic bisindole hybrids. Arch. Pharm. 2020, 353, e2000092. [Google Scholar] [CrossRef]
- Kutschy, P.; Dzurilla, M.; Takasugi, M.; Torok, M.; Achbergerova, I.; Homzova, R.; Racova, M. New syntheses of indole phytoalexins and related compounds. Tetrahedron 1998, 54, 3549–3566. [Google Scholar] [CrossRef]
- Calianese, D.C.; Birge, R.B. Biology of phosphatidylserine (PS): Basic physiology and implications in immunology, infectious disease, and cancer. Cell Commun. Signal. 2020, 18, 41. [Google Scholar] [CrossRef] [PubMed]
- Sikder, S.; Mondal, A.; Das, C.; Kundu, T.K. Autophagy in Cancer: A Metabolic Perspective. Sub-Cell. Biochem. 2022, 100, 143–172. [Google Scholar] [CrossRef]
- Shimizu, S.; Takehara, T.; Hikita, H.; Kodama, T.; Tsunematsu, H.; Miyagi, T.; Hosui, A.; Ishida, H.; Tatsumi, T.; Kanto, T.; et al. Inhibition of autophagy potentiates the antitumor effect of the multikinase inhibitor sorafenib in hepatocellular carcinoma. Int. J. Cancer 2012, 131, 548–557. [Google Scholar] [CrossRef] [PubMed]
- Michalkova, R.; Kello, M.; Kudlickova, Z.; Gazdova, M.; Mirossay, L.; Mojzisova, G.; Mojzis, J. Programmed Cell Death Alterations Mediated by Synthetic Indole Chalcone Resulted in Cell Cycle Arrest, DNA Damage, Apoptosis and Signaling Pathway Modulations in Breast Cancer Model. Pharmaceutics 2022, 14, 503. [Google Scholar] [CrossRef]
- Katsuragi, Y.; Ichimura, Y.; Komatsu, M. p62/SQSTM1 functions as a signaling hub and an autophagy adaptor. FEBS J. 2015, 282, 4672–4678. [Google Scholar] [CrossRef] [PubMed]
- Ichimura, Y.; Komatsu, M. Selective degradation of p62 by autophagy. Semin. Immunopathol. 2010, 32, 431–436. [Google Scholar] [CrossRef]
- Ao, J.; Zeng, F.; Wang, L.; Qiu, L.; Cao, R.; Li, X. Design, synthesis and pharmacological evaluation of beta-carboline derivatives as potential antitumor agent via targeting autophagy. Eur. J. Med. Chem. 2022, 246, 114955. [Google Scholar] [CrossRef]
- Kaluzki, I.; Hailemariam-Jahn, T.; Doll, M.; Kaufmann, R.; Balermpas, P.; Zoller, N.; Kippenberger, S.; Meissner, M. Dimethylfumarate Inhibits Colorectal Carcinoma Cell Proliferation: Evidence for Cell Cycle Arrest, Apoptosis and Autophagy. Cells 2019, 8, 1329. [Google Scholar] [CrossRef]
- Zhang, Y.B.; Gong, J.L.; Xing, T.Y.; Zheng, S.P.; Ding, W. Autophagy protein p62/SQSTM1 is involved in HAMLET-induced cell death by modulating apotosis in U87MG cells. Cell Death Dis. 2013, 4, e550. [Google Scholar] [CrossRef]
- Kang, R.; Zeh, H.J.; Lotze, M.T.; Tang, D. The Beclin 1 network regulates autophagy and apoptosis. Cell Death Differ. 2011, 18, 571–580. [Google Scholar] [CrossRef]
- Chang, C.H.; Lee, C.Y.; Lu, C.C.; Tsai, F.J.; Hsu, Y.M.; Tsao, J.W.; Juan, Y.N.; Chiu, H.Y.; Yang, J.S.; Wang, C.C. Resveratrol-induced autophagy and apoptosis in cisplatin-resistant human oral cancer CAR cells: A key role of AMPK and Akt/mTOR signaling. Int. J. Oncol. 2017, 50, 873–882. [Google Scholar] [CrossRef]
- Fan, Y.; Li, J.; Yang, Y.; Zhao, X.; Liu, Y.; Jiang, Y.; Zhou, L.; Feng, Y.; Yu, Y.; Cheng, Y. Resveratrol modulates the apoptosis and autophagic death of human lung adenocarcinoma A549 cells via a p53-dependent pathway: Integrated bioinformatics analysis and experimental validation. Int. J. Oncol. 2020, 57, 925–938. [Google Scholar] [CrossRef] [PubMed]
- Zachari, M.; Ganley, I.G. The mammalian ULK1 complex and autophagy initiation. Essays Biochem. 2017, 61, 585–596. [Google Scholar] [CrossRef] [PubMed]
- Egan, D.F.; Shackelford, D.B.; Mihaylova, M.M.; Gelino, S.; Kohnz, R.A.; Mair, W.; Vasquez, D.S.; Joshi, A.; Gwinn, D.M.; Taylor, R.; et al. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science 2011, 331, 456–461. [Google Scholar] [CrossRef] [PubMed]
- Ko, C.P.; Lin, C.W.; Chen, M.K.; Yang, S.F.; Chiou, H.L.; Hsieh, M.J. Pterostilbene induce autophagy on human oral cancer cells through modulation of Akt and mitogen-activated protein kinase pathway. Oral Oncol. 2015, 51, 593–601. [Google Scholar] [CrossRef] [PubMed]
- Tamargo-Gomez, I.; Marino, G. AMPK: Regulation of Metabolic Dynamics in the Context of Autophagy. Int. J. Mol. Sci. 2018, 19, 3812. [Google Scholar] [CrossRef] [PubMed]
- Baeken, M.W.; Weckmann, K.; Diefenthaler, P.; Schulte, J.; Yusifli, K.; Moosmann, B.; Behl, C.; Hajieva, P. Novel Insights into the Cellular Localization and Regulation of the Autophagosomal Proteins LC3A, LC3B and LC3C. Cells 2020, 9, 2315. [Google Scholar] [CrossRef]
- Maranhao, S.S.; Moura, A.F.; Oliveira, A.C.A.; Lima, D.J.B.; Barros-Nepomuceno, F.W.A.; Paier, C.R.K.; Pinheiro, A.C.; Nogueira, T.C.M.; de Souza, M.V.N.; Pessoa, C. Synthesis of PJOV56, a new quinoxalinyl-hydrazone derivative able to induce autophagy and apoptosis in colorectal cancer cells, and related compounds. Bioorg. Med. Chem. Lett. 2020, 30, 126851. [Google Scholar] [CrossRef]
- Ramachandran, S.; Kaushik, I.S.; Srivastava, S.K. Pimavanserin: A Novel Autophagy Modulator for Pancreatic Cancer Treatment. Cancers 2021, 13, 5661. [Google Scholar] [CrossRef]
- Rok, J.; Rzepka, Z.; Kowalska, J.; Banach, K.; Beberok, A.; Wrzesniok, D. The Anticancer Potential of Doxycycline and Minocycline-A Comparative Study on Amelanotic Melanoma Cell Lines. Int. J. Mol. Sci. 2022, 23, 831. [Google Scholar] [CrossRef] [PubMed]
- Aman, Y.; Schmauck-Medina, T.; Hansen, M.; Morimoto, R.I.; Simon, A.K.; Bjedov, I.; Palikaras, K.; Simonsen, A.; Johansen, T.; Tavernarakis, N.; et al. Autophagy in healthy aging and disease. Nat. Aging 2021, 1, 634–650. [Google Scholar] [CrossRef] [PubMed]
- Hseu, Y.C.; Huang, Y.C.; Thiyagarajan, V.; Mathew, D.C.; Lin, K.Y.; Chen, S.C.; Liu, J.Y.; Hsu, L.S.; Li, M.L.; Yang, H.L. Anticancer activities of chalcone flavokawain B from Alpinia pricei Hayata in human lung adenocarcinoma (A549) cells via induction of reactive oxygen species-mediated apoptotic and autophagic cell death. J. Cell. Physiol. 2019, 234, 17514–17526. [Google Scholar] [CrossRef] [PubMed]
- Maiti, P.; Scott, J.; Sengupta, D.; Al-Gharaibeh, A.; Dunbar, G.L. Curcumin and Solid Lipid Curcumin Particles Induce Autophagy, but Inhibit Mitophagy and the PI3K-Akt/mTOR Pathway in Cultured Glioblastoma Cells. Int. J. Mol. Sci. 2019, 20, 399. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Chang, H.; Peng, X.; Bai, Q.; Yi, L.; Zhou, Y.; Zhu, J.; Mi, M. Resveratrol inhibits breast cancer stem-like cells and induces autophagy via suppressing Wnt/beta-catenin signaling pathway. PLoS ONE 2014, 9, e102535. [Google Scholar] [CrossRef]
- Balendiran, G.K.; Dabur, R.; Fraser, D. The role of glutathione in cancer. Cell Biochem. Funct. 2004, 22, 343–352. [Google Scholar] [CrossRef]
- Mezencev, R.; Kutschy, P.; Salayova, A.; Curillova, Z.; Mojzis, J.; Pilatova, M.; McDonald, J. Anticancer properties of 2-piperidyl analogues of the natural indole phytoalexin 1-methoxyspirobrassinol. Chemotherapy 2008, 54, 372–378. [Google Scholar] [CrossRef]
- Desideri, E.; Filomeni, G.; Ciriolo, M.R. Glutathione participates in the modulation of starvation-induced autophagy in carcinoma cells. Autophagy 2012, 8, 1769–1781. [Google Scholar] [CrossRef]
- Mancilla, H.; Maldonado, R.; Cereceda, K.; Villarroel-Espindola, F.; Montes de Oca, M.; Angulo, C.; Castro, M.A.; Slebe, J.C.; Vera, J.C.; Lavandero, S.; et al. Glutathione Depletion Induces Spermatogonial Cell Autophagy. J. Cell. Biochem. 2015, 116, 2283–2292. [Google Scholar] [CrossRef]
- Sarkhosh-Inanlou, R.; Molaparast, M.; Mohammadzadeh, A.; Shafiei-Irannejad, V. Sanguinarine enhances cisplatin sensitivity via glutathione depletion in cisplatin-resistant ovarian cancer (A2780) cells. Chem. Biol. Drug Des. 2020, 95, 215–223. [Google Scholar] [CrossRef]
- Troyano, A.; Fernandez, C.; Sancho, P.; de Blas, E.; Aller, P. Effect of glutathione depletion on antitumor drug toxicity (apoptosis and necrosis) in U-937 human promonocytic cells. The role of intracellular oxidation. J. Biol. Chem. 2001, 276, 47107–47115. [Google Scholar] [CrossRef]
- Xu, Y.; Han, X.X.; Li, Y.Y.; Min, H.; Zhao, X.; Zhang, Y.L.; Qi, Y.Q.; Shi, J.; Qi, S.; Bao, Y.P.; et al. Sulforaphane Mediates Glutathione Depletion via Polymeric Nanoparticles to Restore Cisplatin Chemosensitivity. ACS Nano 2019, 13, 13445–13455. [Google Scholar] [CrossRef] [PubMed]
- Kachadourian, R.; Leitner, H.M.; Day, B.J. Selected flavonoids potentiate the toxicity of cisplatin in human lung adenocarcinoma cells: A role for glutathione depletion. Int. J. Oncol. 2007, 31, 161–168. [Google Scholar] [CrossRef] [PubMed]
- Floreani, M.; Petrone, M.; Debetto, P.; Palatini, P. A comparison between different methods for the determination of reduced and oxidized glutathione in mammalian tissues. Free. Radic. Res. 1997, 26, 449–455. [Google Scholar] [CrossRef] [PubMed]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Comp. | Cell Line IC50 (μM) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Caco-2 | HCT116 | HeLa | MCF-7 | A549 | Jurkat | BLM | MDA-MB-231 | MCF10A | Cos-7 | |
| 48 | 51.7 ± 4.1 | 51.9 ± 4.7 | 65.3 ± 5.3 | 77.0 ± 2.1 | 73.1 ± 7.3 | 38.8 ± 2.9 | 77.1 ± 4.3 | 46 ± 2.5 | 67.3 ± 1.9 | 78.3 ± 2.6 |
| 49 | 8.6 ± 0.9 | 8.8 ± 0.6 | 8.9 ± 0.2 | >100 | 8.7 ± 0.1 | 98.8 ± 1.7 | >100 | >100 | 55.1 ± 4.1 | >100 |
| 51a | >100 | 97.7 ± 3.2 | >100 | >100 | 93.8 ± 1.0 | >100 | >100 | >100 | >100 | >100 |
| 52a | 6.1 ± 2.1 | 5.8 ± 0.3 | 6.4 ± 0.7 | 62.9 ± 3.9 | 5.95 ± 0.2 | 6.1 ± 0.7 | 7.4 ± 2.5 | 8.3 ± 3.2 | 10.1 ± 6.2 | 7.15 ± 0.3 |
| 49 | Selectivity index—A549 cells vs. non-cancer cells | 6.3 | 11.5 | |||||||
| SOD (μkat/mg Prot) | GPx (μkat/mg Prot) | GR (μkat/mg Prot) | GSH (nmol/mg Prot) | GST (μkat/mg Prot) | ||
|---|---|---|---|---|---|---|
| DMSO | 24 | 0.523 ± 0.29 | 0.299 ± 0.009 | 2.950 ± 0.47 | 1.707 ± 0.619 | 1.620 ± 0.09 |
| 48 | 0.604 ± 0.32 | 0.284 ± 0.031 | 2.440 ± 0.59 | 1.546 ± 0.123 | 1.200 ± 0.19 | |
| 72 | 0.466 ± 0.21 | 0.205 ± 0.108 | 3.950 ± 0.53 | 0.603 ± 0.043 | 1.330 ± 0.46 | |
| 49 | 24 | 0.408 ± 0.24 | 0.148 ± 0.021 *** | 8.200 ± 2.97 * | 0.395 ± 0.043 * | 3.540 ± 1.07 * |
| 48 | 0.526 ± 0.27 | 0.139 ± 0.028 ** | 11.270 ± 1.04 *** | 0.107 ± 0.015 *** | 2.580 ± 0.44 ** | |
| 72 | 0.523 ± 0.38 | 0.077 ± 0.038 | 10.100 ± 2.89 * | 0.228 ± 0.062 ** | 5.060 ± 0.52 *** |
| Primary Antibody | Mr (kDa) | Origin | Manufacturer |
|---|---|---|---|
| p62/SQSTM1 | 62 | Rabbit | Abcam, Cambridge, United Kingdom |
| Beclin-1 | 60 | Rabbit | Cell Signaling Technology, Danvers, MA, USA |
| Phospho-AMPKα | 62 | Rabbit | |
| Phospho-ULK Ser555 | 140–150 | Rabbit | |
| LC3A/B | 14/16 | Rabbit | |
| ATG7 | 78 | Rabbit | |
| β-actin | 45 | Rabbit | |
| Secondary antibody | Mr (kDa) | Origin | Manufacturer |
| Anti-rabbit IgG HRP | - | Goat | Cell Signaling Technology, Danvers, MA, USA |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Budovska, M.; Michalkova, R.; Kello, M.; Vaskova, J.; Mojzis, J. Design, Synthesis and Antiproliferative Evaluation of Bis-Indole Derivatives with a Phenyl Linker: Focus on Autophagy. Molecules 2023, 28, 251. https://doi.org/10.3390/molecules28010251
Budovska M, Michalkova R, Kello M, Vaskova J, Mojzis J. Design, Synthesis and Antiproliferative Evaluation of Bis-Indole Derivatives with a Phenyl Linker: Focus on Autophagy. Molecules. 2023; 28(1):251. https://doi.org/10.3390/molecules28010251
Chicago/Turabian StyleBudovska, Marianna, Radka Michalkova, Martin Kello, Janka Vaskova, and Jan Mojzis. 2023. "Design, Synthesis and Antiproliferative Evaluation of Bis-Indole Derivatives with a Phenyl Linker: Focus on Autophagy" Molecules 28, no. 1: 251. https://doi.org/10.3390/molecules28010251
APA StyleBudovska, M., Michalkova, R., Kello, M., Vaskova, J., & Mojzis, J. (2023). Design, Synthesis and Antiproliferative Evaluation of Bis-Indole Derivatives with a Phenyl Linker: Focus on Autophagy. Molecules, 28(1), 251. https://doi.org/10.3390/molecules28010251