2. Results and Discussion
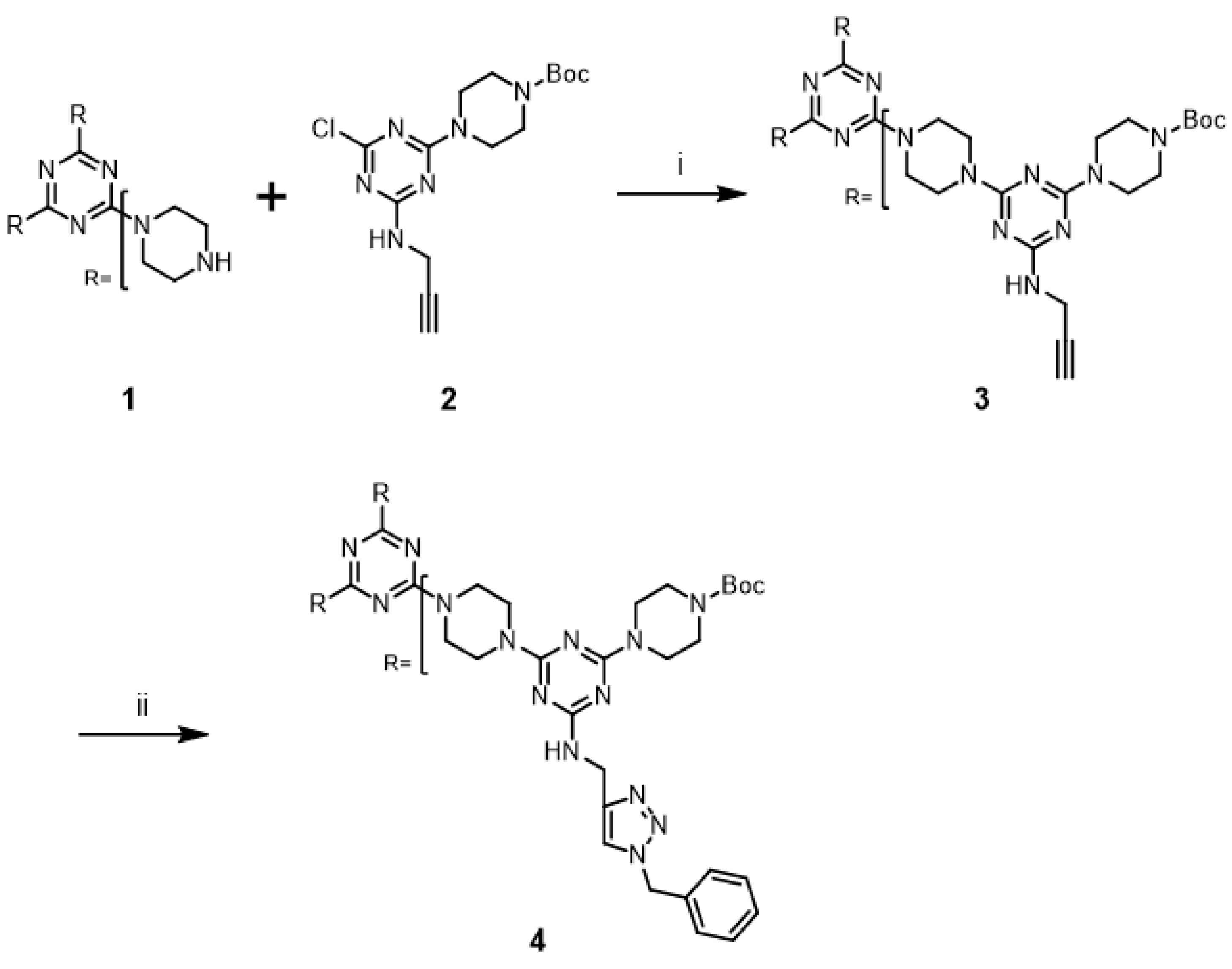
The “click-and-grow” strategy to prepare the first-generation (G1) dendrimer
3 containing three alkynes is outlined in
Scheme 1. A stoichiometric amount of monochlorotriazine
2 [
17] was treated with tris(piperazyl) triazine core
1 [
25] in the presence of excess base for three days by refluxing CHCl
3 to afford G1 dendrimer
3 in 91% yield. The desired dendrimer
3 was easily purified from unreacted
2 and incompletely substituted cores by silica gel column chromatography.
A well-known catalyst, CuSO
4/ascorbic acid [
26], was initially used for the Huisgen cycloaddition reaction of G1 dendrimer
3 with benzyl azide at room temperature. The progress of the reaction was monitored using thin layer chromatography (TLC) and mass spectrometry; the reaction remained incomplete after three days. This low reactivity was likely due to the solubility of dendrimer
3. While this catalytic reaction is typically performed in an aqueous solution, dendrimer
3 did not show sufficient solubility in mixed solvent systems (THF/water). The same cycloaddition reaction of G1 dendrimer
3 was performed with CuI as the copper catalyst in THF. However, the reaction remained incomplete after three days, but the solubility of dendrimer
3 was improved. In addition, a byproduct with 5-iodo-1,2,3-triazole ring [
27,
28] was generated. This cycloaddition of G1 dendrimer
3 was optimized using microwave irradiation based on the previously published reports; [
29] the results are summarized in
Table 1. The desired triazole dendrimer
4 was obtained in 85% yield when the reaction was performed with CuSO
4/ascorbic acid in THF/water under microwave irradiation for 15 min (Entry 1). The remaining copper salts were easily removed by washing with aqueous NaOH solution. The desired product was obtained by simple reprecipitation with MeOH from a clear solution of the crude product in CHCl
3. Dendrimer
4 was obtained in 94% yield when the reaction was performed with CuI in THF under identical microwave irradiation conditions (Entry 2). The byproduct with the 5-iodo-1,2,3-triazole ring was not generated in this case. These results suggested that microwave irradiation significantly improved the yield of the cycloaddition reaction of the triazine dendrimer. The same reaction was carried out using a pressure vessel in an oil bath at 110 °C without microwave irradiation. The desired reaction occurred, affording comparably high yields (Entries 3 and 4), albeit more slowly.
The synthesis and modification of the second-generation dendrimer
6 is shown in
Scheme 2. The deprotection of the Boc groups of dendrimer
4 was achieved with 50% trifluoroacetic acid (TFA) in CH
2Cl
2. Dendrimer
5 was extracted using CHCl
3 from a basic solution of NaOH, and then used without further purification. This material was treated with a stoichiometric amount of monochlorotriazine
2 in the presence of excess base for five days under reflux conditions to afford
6 in 97% yield. Dendrimer
6 was easily purified by silica gel column chromatography. Copper-catalyzed alkyne-azide cycloaddition reaction of
6 with benzyl azide was examined under microwave irradiation with CuSO
4/ascorbic acid and CuI, and the desired triazole dendrimer
7 was obtained in 91% yields, respectively (Entries 5 and 6). When the reactions were performed at 110 °C in a pressure vessel, the desired triazole dendrimer
7 was also obtained in high yield (Entries 7 and 8). However, a longer reaction time (2×) was required to complete the reaction compared to the G1 dendrimer
3. G2 dendrimer
7 was purified through simple precipitation by MeOH addition to the crude organic phase obtained from extraction as well as G1 dendrimer
4.
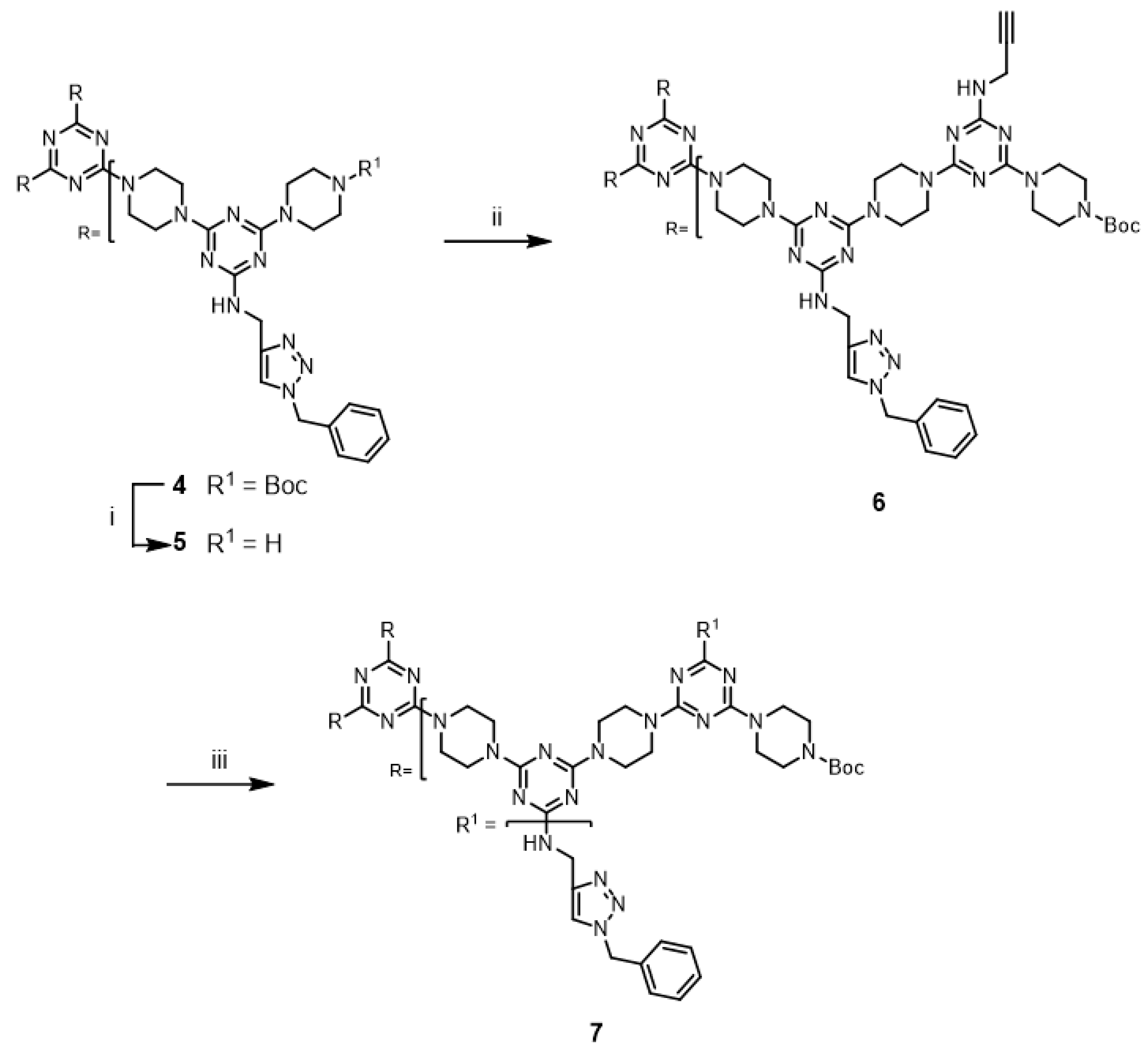
The successful cycloaddition-mediated derivatization of
6 led us to investigate the click chemistry of a third-generation dendrimer. Dendrimer
9 was prepared via an iterative extension of
7 (
Scheme 3) and purified using silica gel column chromatography. The copper-catalyzed alkyne-azide cycloaddition reaction of
9 with benzyl azide was investigated under microwave irradiation using CuSO
4/ascorbic acid and CuI; the desired triazole dendrimer
10 was obtained with low yields of 11% and 14%, respectively (Entries 9 and 10). When the reactions were performed at 110 °C in a pressure vessel, the desired triazole dendrimer
10 was also obtained (Entries 11 and 12). The click modification of G3 dendrimer
9 required a longer reaction time than that in the case of G2 dendrimer
6 for the complete disappearance of
9. TLC analysis showed evidence for the formation of polar, potentially polymeric species that could arise from alkyne–alkyne homocoupling reactions [
30]. In addition, triazole dendrimer
10 could not be purified by simple precipitation because of its solubility limitations in various solvents and the presence of multiple impurities. Dendrimer
10 was purified using silica gel column chromatography.
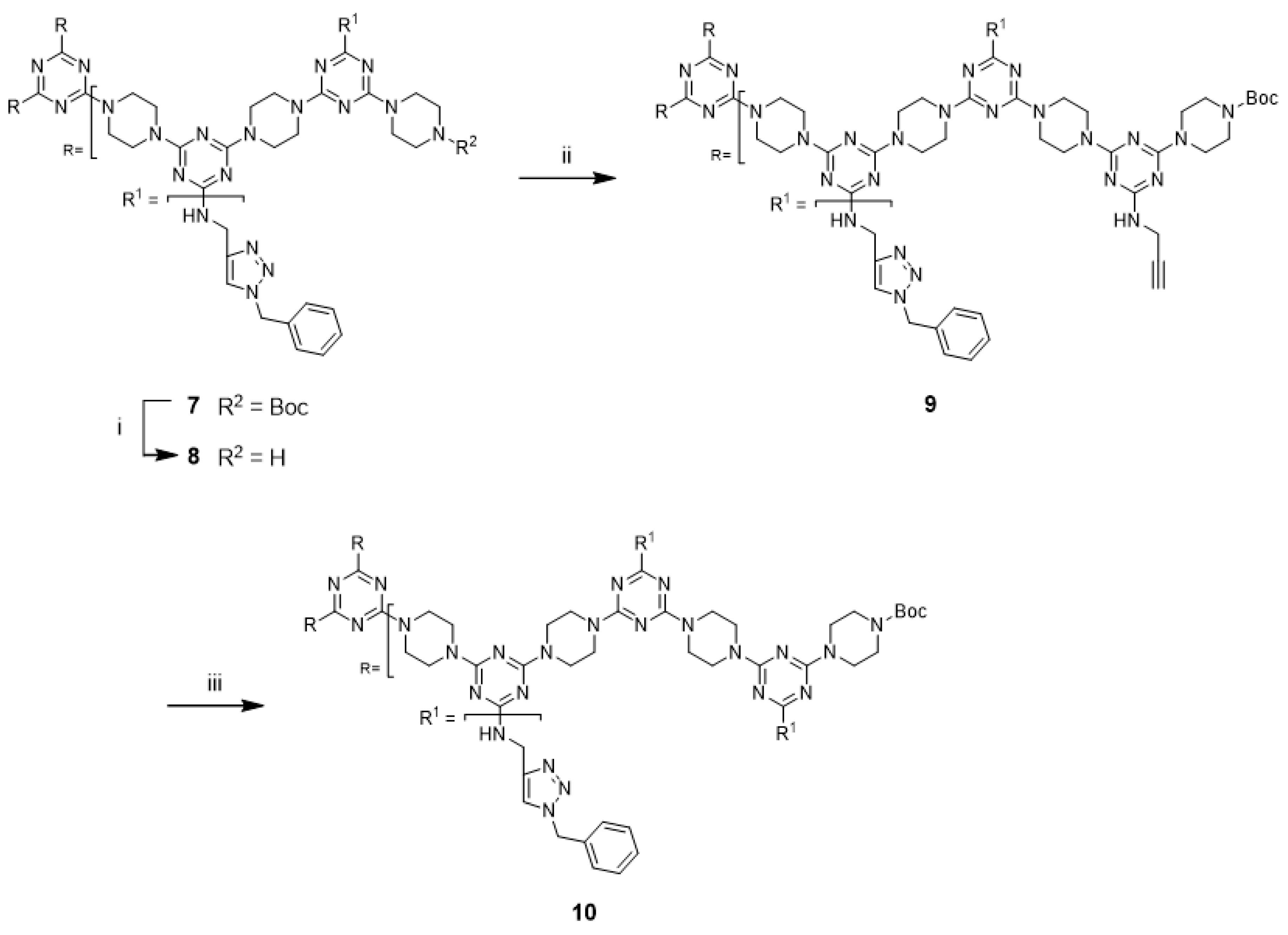
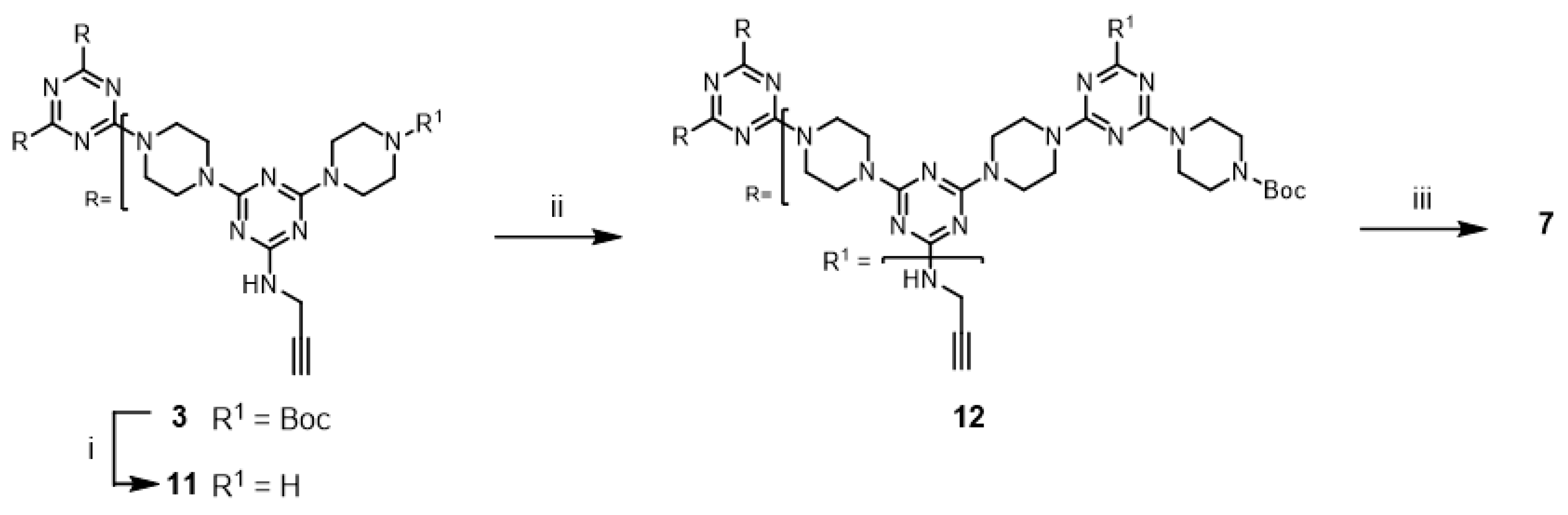
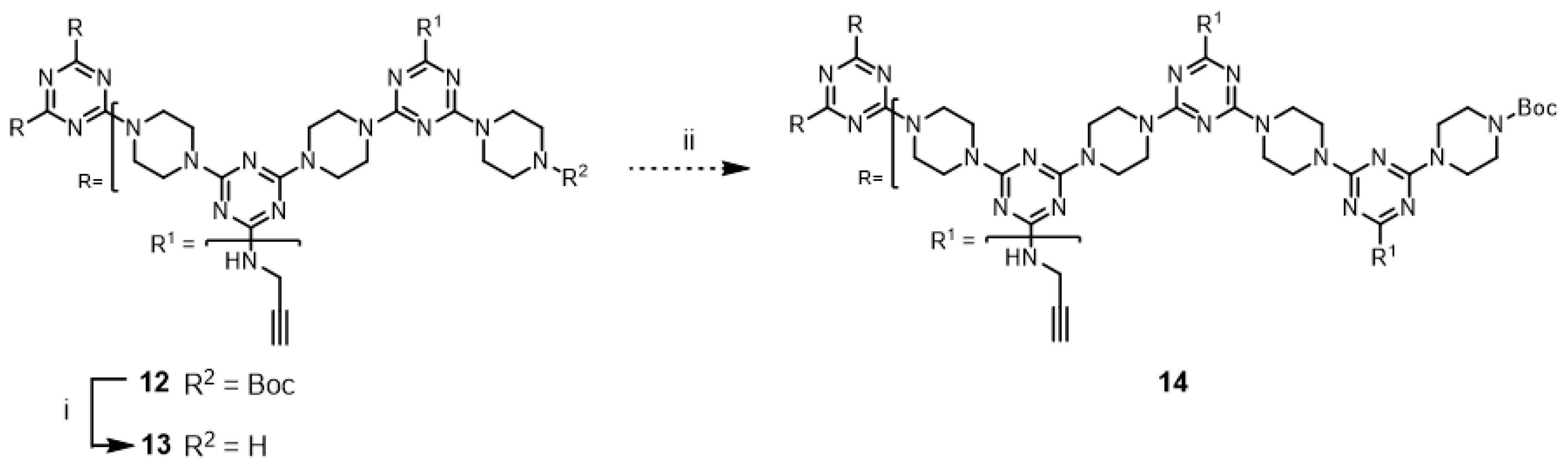
Although not explored in the syntheses described above, the “click-and-grow” strategy allows the incorporation of different azide-bearing groups at each generation of the dendrimer. Alternatively, a single azide-bearing group can be incorporated throughout the dendrimer if pendant alkynes are carried through the iterative growth of these targets and globally “clicked.” To explore the “grow-then-click” strategy, second- and third-generation dendrimers having six and nine alkynyl groups in the molecules, respectively, were targeted for synthesis. Dendrimer
12 was prepared from
3 in 76% overall yield using a similar method as that employed for preparing G2 dendrimer
6 (
Scheme 4). The reaction of
12 with benzyl azide under microwave irradiation with CuSO
4/ascorbic acid and CuI yielded the desired triazole dendrimer
7 in high yields (Entries 13 and 14). When the same reactions were carried out at 110 °C in a pressure vessel, the desired triazole dendrimer
7 was also obtained in high yields (Entries 15 and 16). The yields and reaction times of the cycloaddition reaction of dendrimer
12 were similar to those of dendrimer
6. However, the preparation of dendrimer
14 was more challenging than the synthesis of G2 dendrimer
12 (
Scheme 5). Although deprotection of
12 was achieved with 50% TFA in CH
2Cl
2, the low solubility of
13 in organic solvents prevented the synthesis of
14. TLC analysis indicated that a significant amount of
2 remained after one week. Notably, dendrimer
14 could not be synthesized even after changing the solvents to CHCl
3/MeOH (5:1), CHCl
3/THF, THF, dichloroethane, and dioxane. The poor solubility of dendrimer
13 led to the abandonment of this route.
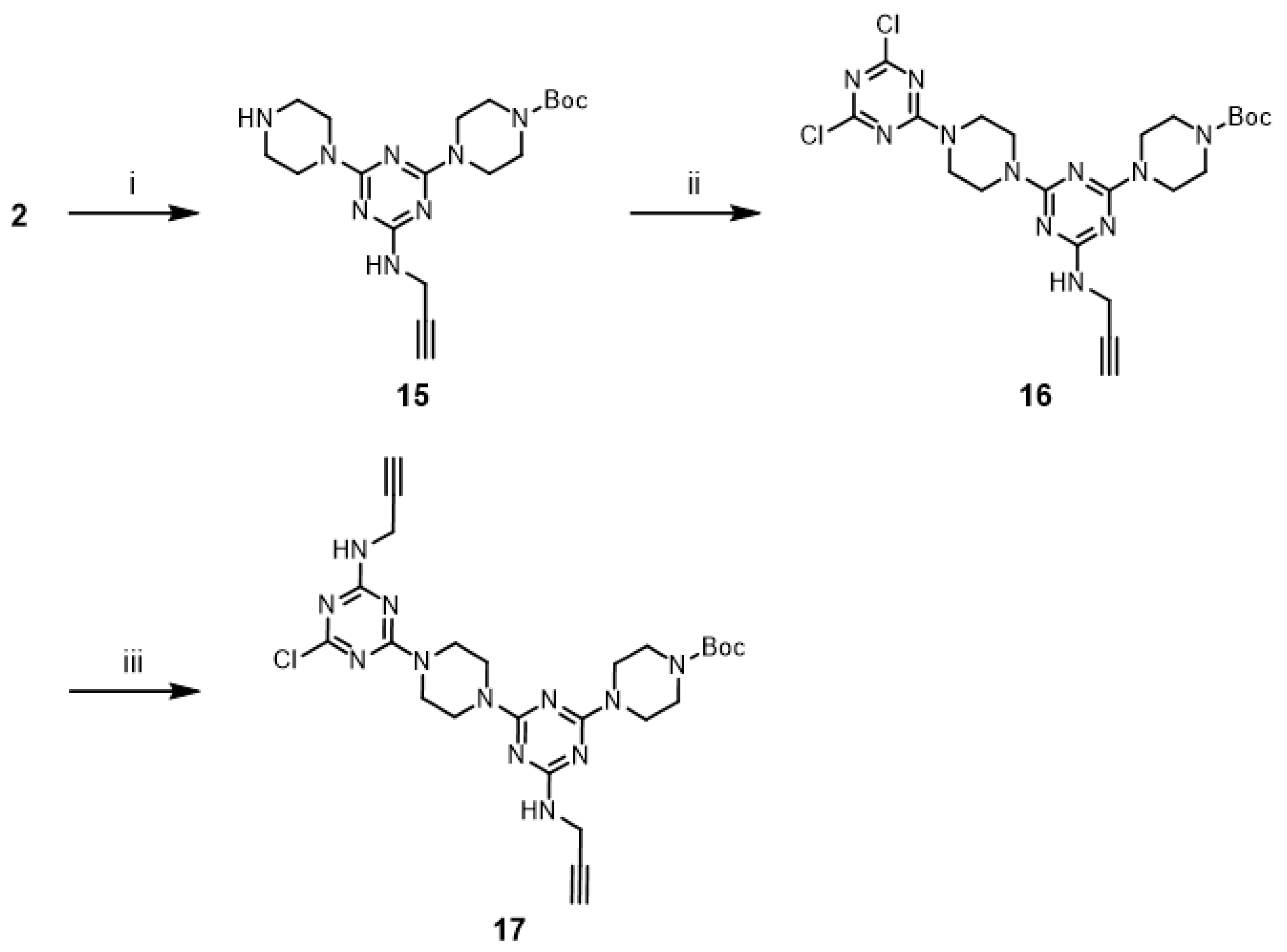
An alternative approach to prepare
14 was attempted. The reaction of G1 dendrimer
11 with second-generation dendron
17 was envisioned to afford G3 dendrimer
14. Dendron
17 was prepared from
2 in 65% overall yield (
Scheme 6). Monochlorotriazine
2 was treated with excess piperazine, which afforded mono-
N-substituted piperazine
15. Subsequently,
15 was converted into dichlorotriazine
16 with excess cyanuric chloride. The by-products of the reaction with excess reactants were observed in both cases. A stoichiometric amount of dendron
17 was treated with G1 dendrimer
11 for one week under reflux conditions using CHCl
3/THF (1:1) mixture. In this case, the reaction mixture was a clear solution at the beginning of the reaction, but precipitation gradually increased in the mixture. However, TLC analysis showed the presence of a large amount of remaining
17, while the desired G3 dendrimer
14 was not observed. These results suggested that the synthesis of dendrimer
14 was difficult because of solubility issues.
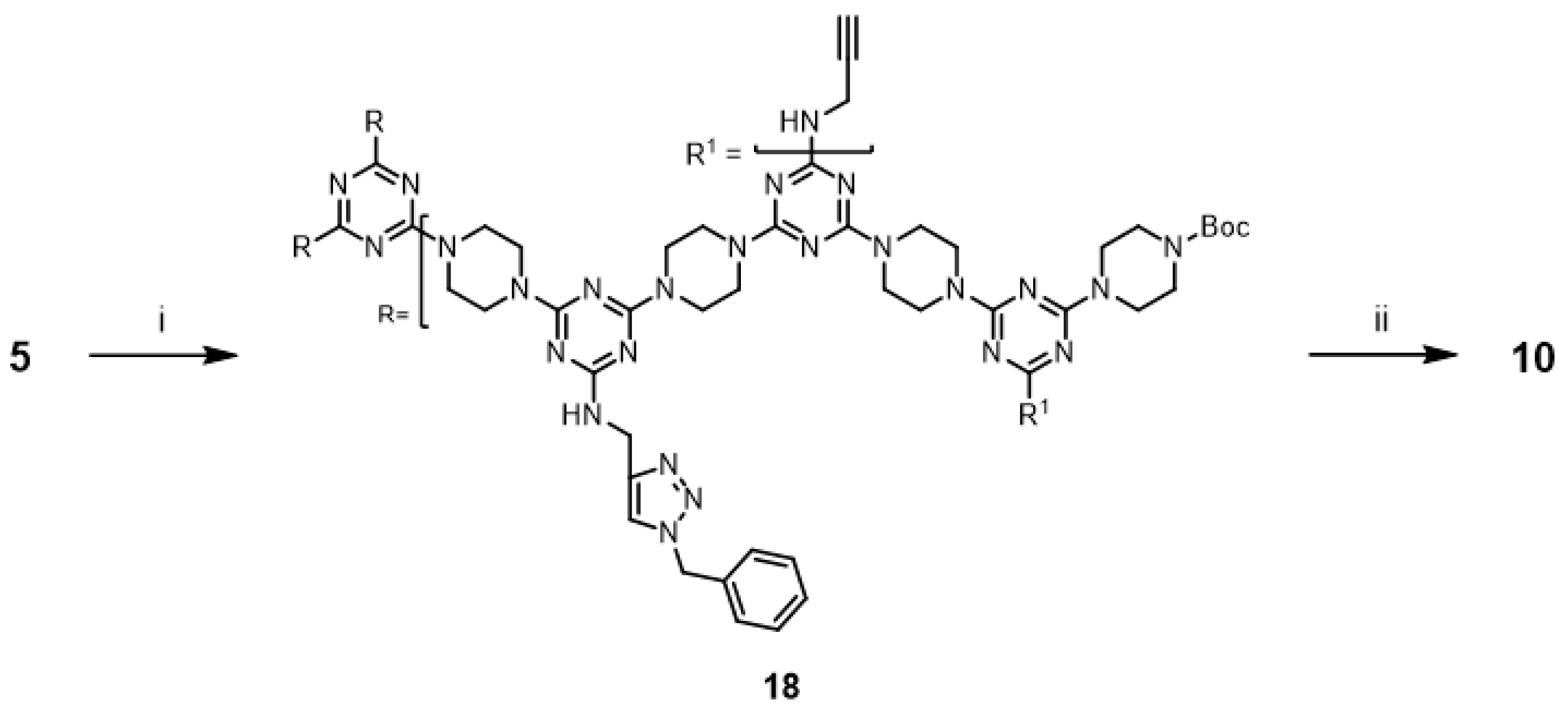
Additionally, the synthesis of G3 triazine dendrimers
9 and
10 with 1-benzyl-1,2,3-triazole rings was accomplished. This result led us to attempt a new strategy for synthesizing G3 dendrimer
18 with three 1-benzyl-1,2,3-triazole rings. Dendrimer
18 was prepared via an iterative extension of
17 (
Scheme 7) with dendrimer
5 and purified using silica gel column chromatography. The copper-catalyzed alkyne-azide cycloaddition reaction of
18 and benzyl azide was investigated under microwave irradiation with CuSO
4/ascorbic acid and CuI. The desired triazole dendrimer
10 was obtained with low yields of 11% (with CuSO
4/ascorbic acid) and 10% (with CuI) (Entries 17 and 18). When the reactions were performed at 110 °C in a pressure vessel, the desired triazole dendrimer
10 was also obtained (Entries 19 and 20).
3. Materials and Methods
3.1. General Experimental
All chemicals were obtained from TCI Fine Chemicals, Tokyo, Japan, Wako Pure Chemical Industries, Tokyo, Japan, Kanto Chemical, Tokyo, Japan and Sigma-Aldrich, St. Louis, MO, USA, and used without further purification. NMR spectra were recorded in CDCl
3 or CDCl
3/CD
3OD (5:1) on either a JEOL ECS-400 or a JEOL ECA-600 spectrometer, Tokyo, Japan.
1H NMR data are reported in ppm (δ) relative to TMS.
13C NMR data are reported in ppm (δ) relative to the central line of the triplet for CDCl
3 at 77.0 ppm. Mass spectra were recorded on a JEOL JMS-S3000 SpialTOF instrument, Tokyo, Japan. Microwave experiments were carried out using a CEM Discover Microwave Synthesizer (CEM Corporation, Tokyo, Japan), and the irradiation was performed at a maximum power of 150 W. Chromatographic separations were carried out on a silica gel column (Kanto Chemical 60N, 63–210 µm, Tokyo, Japan; or Chromatorex
® NH-DM1020, 100–200 mesh, Fuji Silysia Chemical Ltd., Tokyo, Japan). The NMR spectra are shown in
Supplementary Materials Pages S1–S15.
3.2. Synthesis of G1 Dendrimer 3
Compound 2 (4.75 g, 13.5 mmol) and DIPEA (7.1 mL, 41.8 mmol) were successively added to a solution of triazine core 1 (1.37 g, 4.11 mmol) in CHCl3 (30 mL), and the resulting mixture was refluxed for three days. After the reaction mixture was concentrated under reduced pressure, the residue was dissolved in CHCl3 (70 mL). The organic phase was washed with water (50 mL × 3), dried over Na2SO4, and concentrated under reduced pressure. The crude product was purified by silica gel column chromatography (gradient elution using CH2Cl2/EtOAc (2:1) until no detectable 2 was observed, as determined by UV spotting, to CHCl3/MeOH (10:1) to obtain the desired product) to afford 3 as a white solid (4.82 g, 91%). 1H NMR (600 MHz, CDCl3) δ 4.94 (t, J = 5.7 Hz, 3H), 4.19 (dd, J = 5.7, 2.5 Hz, 6H), 3.85–3.71 (m, 36H), 3.48–3.42 (m, 12H), 2.20 (t, J = 2.5 Hz, 3H), 1.49 (s, 27H). 13C NMR (150 MHz, CDCl3) δ 165.8, 165.4, 165.2, 154.8, 80.8, 79.9, 70.7, 43.1, 43.0, 42.9, 30.4, 28.4. HRMS (MALDI): calcd for C60H87N27NaO6, 1304.7225 (M + Na+); found, 1304.7240.
3.3. Synthesis of G1 Dendrimer 4
3.3.1. CuSO4/Ascorbic Acid
Benzyl azide (33.0 µL, 0.264 mmol), ascorbic acid (21 mg, 0.119 mmol), and copper (II) sulfate (3.7 mg, 0.0232 mmol) were successively added to a solution of compound 3 (101 mg, 0.0787 mmol) in THF/H2O (1:1 v/v, 5 mL). The resulting mixture was subjected to microwave irradiation at 110 °C for 15 min in a sealed vial or stirred at 110 °C for 1 h in a pressure vessel. After the reaction mixture was concentrated, the residue was dissolved in CHCl3 (70 mL), and the solution was washed with 5% aqueous NaOH (30 mL × 2) and water (30 mL × 2). The organic phase was dried over Na2SO4 and then evaporated. The crude product was purified by reprecipitation with MeOH from a clear solution of CHCl3 to afford 4 as an off-white solid. Yield; MW: (113 mg, 0.0672 mmol, 85%), pressure vessel: (110 mg, 0.0654 mmol, 83%).
3.3.2. CuI/DIPEA
Benzyl azide (33.0 µL, 0.264 mmol), DIPEA (40 µL, 0.235 mmol), and copper (I) iodide (4.9 mg, 0.0257 mmol) were successively added to a solution of compound 3 (101 mg, 0.0787 mmol) in THF (5 mL). The resulting mixture was subjected to microwave irradiation at 110 °C for 15 min in a sealed vial or stirred at 110 °C for 1 h in a pressure vessel. After the reaction mixture was concentrated, the residue was dissolved in CHCl3 (70 mL) and the solution was washed with 5% aqueous NaOH (30 mL × 2) and water (30 mL × 2). The organic phase was dried over Na2SO4 and then evaporated. The crude product was purified by reprecipitation with MeOH from a clear solution of CHCl3 to afford 4 as an off-white solid. Yield, MW: (125 mg, 0.0743 mmol, 94%); pressure vessel: (118 mg, 0.0702 mmol, 89%). 1H NMR (600 MHz, CDCl3) δ 7.44–7.29 (m, 12H), 7.27–7.20 (m, 6H), 5.48 (s, 6H), 5.23 (t, J = 6.1 Hz, 3H), 4.67 (d, J = 6.0 Hz, 6H), 3.83–3.65 (m, 36H), 3.49–3.32 (m, 12H), 1.49 (s, 27H). 13C NMR (150 MHz, CDCl3) δ 166.1, 165.4, 165.2, 154.8, 146.9, 134.6, 129.1, 128.8, 128.0, 121.5, 79.9, 54.1, 43.0, 43.0, 42.9, 36.6, 28.4. HRMS (MALDI): calcd for C81H108N36NaO6, 1703.9145 (M + Na+); found, 1703.9137.
3.4. Synthesis of G1 Dendrimer 5
TFA (10 mL) was added to a solution of compound 4 (309 mg, 0.184 mmol) in CH2Cl2 (10 mL), and the resulting mixture was stirred for 3 h at room temperature. After the reaction mixture was concentrated under reduced pressure, the residue was basified with 20% aqueous NaOH (50 mL). The aqueous solution was extracted with CHCl3 (50 mL × 6). The organic phase was washed with water (30 mL × 1), dried over Na2SO4, and concentrated under reduced pressure to afford 5 as a white solid (228 mg, 90%), which was used for the next reaction without further purification. 1H NMR (600 MHz, CDCl3) δ 7.39–7.31 (m, 12H), 7.26–7.20 (m, 6H), 5.47 (s, 6H), 5.24 (t, J = 6.1 Hz, 3H), 4.67 (d, J = 6.1 Hz, 6H), 3.96–3.55 (m, 39H), 2.88–2.79 (m, 12H).13C NMR (150 MHz, CDCl3) δ 166.1, 165.3, 165.2, 165.1, 147.0, 134.6, 129.1, 128.7, 128.0, 121.5, 54.1, 46.0, 44.2, 43.0, 43.0, 36.6. HRMS (MALDI); calcd for C66H84N36Na, 1403.7572 (M + Na+); found, 1403.7561.
3.5. Synthesis of G2 Dendrimer 6
Compound 2 (219 mg, 0.621 mmol) and DIPEA (301 µL, 1.77 mmol) were successively added to a solution of G1 dendrimer 5 (215 mg, 0.156 mmol) in CHCl3 (20 mL), and the resulting mixture was refluxed for 5 days. After the reaction mixture was concentrated under reduced pressure, the residue was dissolved in CHCl3 (50 mL). The organic phase was washed with water (50 mL × 3), dried over Na2SO4, and concentrated under reduced pressure. The crude product was purified by silica gel column chromatography (gradient elution using CH2Cl2/EtOAc (2:1) until no detectable 2 was observed, as determined by UV spotting, to CHCl3/MeOH (10:1) to obtain the desired product) to afford 6 as an off-white solid (352 mg, 97%). 1H NMR (400 MHz, CDCl3) δ 7.38–7.30 (m, 12H), 7.26–7.22 (m, 6H), 5.48 (s, 6H), 5.30–5.20 (m, 3H), 4.99–4.89 (m, 3H), 4.68 (d, J = 6.1 Hz, 6H), 4.20 (dd, J = 5.7, 2.5 Hz, 6H), 3.78–3.68 (m, 60H), 3.49–3.40 (m, 12H), 2.21 (t, J = 2.5 Hz, 3H), 1.48 (s, 27H). 13C NMR (100 MHz, CDCl3) δ 166.1, 165.8, 165.4, 165.2, 154.8, 146.9, 134.6, 129.1, 128.7, 128.0, 121.5, 80.8, 79.9, 70.7, 54.1, 43.0, 43.0, 36.6, 30.5, 28.4. HRMS (MALDI): calcd for C111H144N54NaO6, 2352.2515 (M + Na+); found, 2352.2524.
3.6. Synthesis of G2 Dendrimer 7 from Compound 6
3.6.1. CuSO4/Ascorbic Acid
Benzyl azide (16.5 µL, 0.132 mmol), ascorbic acid (10.4 mg, 0.0591 mmol), and copper (II) sulfate (1.9 mg, 0.0119 mmol) were successively added to a solution of compound 6 (93 mg, 0.0399 mmol) in THF/H2O (1:1 v/v, 5 mL). The resulting mixture was subjected to microwave irradiation at 110 °C for 30 min in a sealed vial or stirred at 110 °C for 2 h in a pressure vessel. After the reaction mixture was concentrated, the residue was dissolved in CHCl3 (70 mL), and the solution was washed with 5% aqueous NaOH (30 mL × 2) and water (30 mL × 2). The organic phase was dried over Na2SO4 and then evaporated. The crude product was purified by reprecipitation with MeOH from a clear solution of CHCl3 to afford 7 as an off-white solid. Yield; MW: (99.1 mg, 0.0363 mmol, 91%), pressure vessel: (92.2 mg, 0.0338 mmol, 85%).
3.6.2. CuI/DIPEA
Benzyl azide (17.7 µL, 0.142 mmol), DIPEA (22 µL, 0.129 mmol), and copper (I) iodide (2.6 mg, 0.0137 mmol) were successively added to a solution of compound 6 (100 mg, 0.0429 mmol) in THF (5 mL). The resulting mixture was subjected to microwave irradiation at 110 °C for 30 min in a sealed vial or stirred at 110 °C for 2 h in a pressure vessel. After the reaction mixture was concentrated, the residue was dissolved in CHCl3 (70 mL) and the solution was washed with 5% aqueous NaOH (30 mL × 2) and water (30 mL × 2). The organic phase was dried over Na2SO4 and then evaporated. The crude product was purified by reprecipitation with MeOH from a clear solution of CHCl3 to afford 7 as an off-white solid. Yield; MW: (107 mg, 0.0392 mmol, 91%), pressure vessel: (106 mg, 0.0388 mmol, 91%). 1H NMR (600 MHz, CDCl3) δ 7.45–7.29 (m, 24H), 7.26–7.19 (m, 12H), 5.48 (d, J = 3.8 Hz, 12H), 5.35–5.15 (m, 6H), 4.67 (dd, J = 11.0, 6.0 Hz, 12H), 3.86–3.63 (m, 60H), 3.49–3.30 (m, 12H), 1.48 (s, 27H). 13C NMR (150 MHz, CDCl3) δ 166.1, 165.4, 165.2, 154.8, 146.9, 134.6, 129.1, 128.7, 128.0, 121.5, 79.9, 54.1, 43.1, 43.0, 42.9, 42.9, 36.6, 28.4. HRMS (MALDI): calcd for C132H165N63NaO6, 2751.4435 (M + Na+); found, 2751.4466.
3.7. Synthesis of G2 Dendrimer 8
TFA (10 mL) was added to a solution of compound 7 (467 mg, 0.171 mmol) in CH2Cl2 (10 mL), and the resulting mixture was stirred for 3 h at room temperature. After the reaction mixture was concentrated under reduced pressure, the residue was basified with 20% aqueous NaOH (30 mL). The aqueous solution was extracted with CHCl3 (30 mL × 5). The organic phase was washed with water (30 mL × 1), dried over Na2SO4, and concentrated under reduced pressure to afford 8 as a white solid (385 mg, 93%), which was used for the next reaction without further purification. 1H NMR (400 MHz, CDCl3) δ 7.44–7.28 (m, 24H), 7.26–7.19 (m, 12H), 5.47 (d, J = 2.6 Hz, 12H), 5.26 (t, J = 6.1 Hz, 6H), 4.67 (t, J = 5.5 Hz, 12H), 3.99–3.49 (m, 63H), 2.91–2.73 (m, 12H). 13C NMR (100 MHz, CDCl3) δ 166.1, 165.4, 165.2, 165.1, 147.0, 134.7, 134.6, 129.1, 128.7, 128.7, 128.0, 121.6, 121.5, 54.1, 46.0, 44.2, 43.1, 43.0, 36.6. HRMS (MALDI): calcd for C117H141N63Na, 2451.2862 (M + Na+); found, 2451.2861.
3.8. Synthesis of G3 Dendrimer 9
Compound 2 (85 mg, 0.241 mmol) and DIPEA (122 µL, 0.718 mmol) were successively added to a solution of G2 dendrimer 8 (145 mg, 0.0597 mmol) in CHCl3 (20 mL), and the resulting mixture was refluxed for 7 days. After the reaction mixture was concentrated under reduced pressure, the residue was dissolved in CHCl3 (100 mL). The organic phase was washed with water (50 mL × 2), dried over Na2SO4, and concentrated under reduced pressure. The crude product was purified by silica gel column chromatography (gradient elution using CH2Cl2/EtOAc (2:1) until no detectable 2 was observed, as determined by UV spotting, to CHCl3/MeOH (15:1) to obtain the desired product) to afford 9 as a pale yellow solid (161 mg, 80%). 1H NMR (400 MHz, CDCl3) δ 7.38–7.31 (m, 24H), 7.26–7.20 (m, 12H), 5.48 (d, J = 4.3 Hz, 12H), 5.26–5.16 (m, 6H), 4.68 (t, J = 5.9 Hz, 12H), 4.19 (dd, J = 5.8, 2.6 Hz, 6H), 3.92–3.65 (m, 84H), 3.47–3.40 (m, 12H), 2.20 (t, J = 2.5 Hz, 3H), 1.48 (s, 27H). 13C NMR (100 MHz, CDCl3) δ 166.2, 165.9, 165.4, 165.2, 165.2, 154.8, 146.9, 134.7, 129.1, 128.8, 128.1, 121.5, 80.9, 79.9, 70.7, 54.1, 43.0, 36.6, 30.5, 28.4. HRMS (MALDI): calcd for C162H201N81NaO6, 3399.7805 (M + Na+); found, 3399.7780.
3.9. Synthesis of G3 Dendrimer 10 from Compound 9
3.9.1. CuSO4/Ascorbic Acid
Benzyl azide (11 µL, 0.0880 mmol), ascorbic acid (7.6 mg, 0.0432 mmol), and copper (II) sulfate (1.5 mg, 0.0094 mmol) were successively added to a solution of compound 9 (90 mg, 0.0266 mmol) in THF/H2O (1:1 v/v, 5 mL). The resulting mixture was subjected to microwave irradiation at 110 °C for 2 h in a sealed vial or stirred at 110 °C for 8 h in a pressure vessel. After the reaction mixture was concentrated, the residue was dissolved in CHCl3 (70 mL) and the solution was washed with 5% aqueous NaOH (30 mL × 2) and water (30 mL × 2). The organic phase was dried over Na2SO4 and then evaporated. The crude product was purified with column chromatography on silica gel (CHCl3/MeOH, 20:1) to afford 10 as a pale yellow solid. Yield; MW: (11.1 mg, 2.91 µmol, 11%), pressure vessel: (16.0 mg, 4.23 µmol, 16%).
3.9.2. CuI/DIPEA
Benzyl azide (11 µL, 0.0880 mmol), DIPEA (14 µL, 0.0823 mmol), and copper (I) iodide (2.0 mg, 0.0105 mmol) were successively added to a solution of compound 9 (90 mg, 0.0266 mmol) in THF (5 mL). The resulting mixture was subjected to microwave irradiation at 110 °C for 2 h in a sealed vial or stirred at 110 °C for 8 h in a pressure vessel. After the reaction mixture was concentrated, the residue was dissolved in CHCl3 (70 mL) and the solution was washed with 5% aqueous NaOH (30 mL × 2) and water (30 mL × 2). The organic phase was dried over Na2SO4 and then evaporated. The crude product was purified with column chromatography on silica gel (CHCl3/MeOH, 20:1) to afford 10 as a pale yellow solid. Yield; MW: (14.0 mg, 3.71 µmol, 14%), pressure vessel: (28.2 mg, 7.46 µmol, 28%). 1H NMR (400 MHz, CDCl3:CD3OD = 5:1 v/v) δ 7.79–7.15 (m, 54H), 5.49 (d, J = 3.8 Hz, 18H), 4.81–4.48 (m, 18H), 4.41–4.16 (m, 9H), 3.92–3.56 (m, 84H), 3.45–3.35 (m, 12H), 1.49 (s, 27H). 13C NMR (100 MHz, CDCl3:CD3OD = 5:1 v/v) δ 165.8, 165.2, 164.9, 154.9, 146.8, 134.3, 130.9, 128.9, 128.6, 127.9, 121.8, 80.2, 65.5, 54.0, 42.8, 36.0, 29.5, 28.1, 25.4. HRMS (MALDI): calcd for C183H222N90NaO6, 3798.9725 (M + Na+); found, 3798.9722.
3.10. Synthesis of G1 Dendrimer 11
TFA (20 mL) was added to a solution of compound 3 (843 mg, 0.657 mmol) in CH2Cl2 (20 mL), and the resulting mixture was stirred for 3 h at room temperature. After the reaction mixture was concentrated under reduced pressure, the residue was basified with 20% aqueous NaOH (50 mL). The aqueous solution was extracted with CHCl3 (70 mL × 6). The organic phase was washed with water (30 mL × 1), dried over Na2SO4, and concentrated under reduced pressure to afford 11 as a white solid (555 mg, 86%), which was used for the next reaction without further purification. 1H NMR (400 MHz, CDCl3) δ 4.97 (t, J = 5.7 Hz, 3H), 4.19 (dd, J = 5.5, 2.5 Hz, 6H), 3.88–3.66 (m, 39H), 2.92–2.82 (m, 12H), 2.19 (t, J = 2.5 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 165.8, 165.4, 165.2, 165.1, 81.0, 70.6, 46.0, 44.3, 43.1, 43.0, 30.4. HRMS (MALDI): calcd for C45H64N27, 982.5833 (M + H+); found, 982.5820.
3.11. Synthesis of G2 Dendrimer 12
Compound 2 (775 mg, 2.20 mmol) and DIPEA (1.0 mL 5.91 mmol) were successively added to a solution of G1 dendrimer 11 (542 mg, 0.552 mmol) in CHCl3 (20 mL), and the resulting mixture was refluxed for 5 days. After the reaction mixture was concentrated under reduced pressure, the residue was dissolved in CHCl3 (100 mL). The organic phase was washed with water (50 mL × 2), dried over Na2SO4, and concentrated under reduced pressure. The crude product was purified by silica gel column chromatography (gradient elution using CH2Cl2/EtOAc (2:1) until no detectable 2 was observed, as determined by UV spotting, to CHCl3/MeOH (10:1) to obtain the desired product) to afford 12 as an off-white solid (936 mg, 88%). 1H NMR (600 MHz, CDCl3) δ 4.95 (t, J = 6.1 Hz, 6H), 4.25–4.15 (m, 12H), 3.92–3.70 (m, 60H), 3.49–3.41 (m, 12H), 2.21 (t, J = 2.5 Hz, 6H), 1.48 (s, 27H). 13C NMR (150 MHz, CDCl3) δ 165.8, 165.4, 165.2, 154.8, 80.9, 80.9, 79.9, 70.7, 43.1, 43.0, 42.9, 30.5, 28.4. HRMS (MALDI): calcd for C90H123N45NaO6, 1953.0595 (M + Na+); found, 1953.0604.
3.12. Synthesis of G2 Dendrimer 7 from Compound 12
3.12.1. CuSO4/Ascorbic Acid
Benzyl azide (43.0 µL, 0.344 mmol), ascorbic acid (27.7 mg, 0.157 mmol), and copper (II) sulfate (5.3 mg, 0.0332 mmol) were successively added to a solution of compound 12 (101 mg, 0.0523 mmol) in THF/H2O (1:1 v/v, 5 mL). The resulting mixture was subjected to microwave irradiation at 110 °C for 30 min in a sealed vial or stirred at 110 °C for 2 h in a pressure vessel. After the reaction mixture was concentrated, the residue was dissolved in CHCl3 (70 mL) and the solution was washed with 5% aqueous NaOH (30 mL × 2) and water (30 mL × 2). The organic phase was dried over Na2SO4 and then evaporated. The crude product was purified by reprecipitation with MeOH from a clear solution of CHCl3 to afford 7 as an off-white solid. Yield; MW: (124 mg, 0.0454 mmol, 87%), pressure vessel: (126 mg, 0.0462 mmol, 88%).
3.12.2. CuI/DIPEA
Benzyl azide (43.0 µL, 0.344 mmol), DIPEA (53 µL, 0.312 mmol), and copper (I) iodide (6.8 mg, 0.0357 mmol) were successively added to a solution of compound 12 (101 mg, 0.0523 mmol) in THF (5 mL). The resulting mixture was subjected to microwave irradiation at 110 °C for 30 min in a sealed vial or stirred at 110 °C for 2 h in a pressure vessel. After the reaction mixture was concentrated, the residue was dissolved in CHCl3 (70 mL) and the solution was washed with 5% aqueous NaOH (30 mL × 2) and water (30 mL × 2). The organic phase was dried over Na2SO4 and then evaporated. The crude product was purified by reprecipitation with MeOH from a clear solution of CHCl3 to afford 7 as an off-white solid. Yield; MW: (123 mg, 0.0451 mmol, 86%), pressure vessel: (129 mg, 0.0472 mmol, 90%).
3.13. Synthesis of G2 Dendrimer 13
TFA (15 mL) was added to a solution of compound 12 (193 mg, 0.100 mmol) in CH2Cl2 (15 mL), and the resulting mixture was stirred for 3 h at room temperature. After the reaction mixture was concentrated under reduced pressure, the residue was basified with 20% aqueous NaOH (50 mL). The aqueous solution was extracted with CHCl3 (100 mL × 5). The organic phase was washed with water (30 mL × 1), dried over Na2SO4, and concentrated under reduced pressure to afford 13 as a white solid (127 mg, 78%), which was used for the next reaction without further purification. 1H NMR (600 MHz, CDCl3) δ 4.87 (t, J = 5.8 Hz, 6H), 4.21 (td, J = 5.5, 2.5 Hz, 12H), 3.85–3.72 (m, 63H), 2.90–2.85 (m, 12H), 2.22–2.19 (m, 6H). 13C NMR (150 MHz, CDCl3) δ 165.9, 165.4, 165.3, 165.1, 81.0, 80.9, 70.7, 70.7, 46.1, 44.3, 43.1, 43.1, 43.0, 30.5. HRMS (MALDI): calcd for C75H99N45Na, 1652.9022 (M + Na+); found, 1652.9012.
3.14. Synthesis of Compound 15
Piperazine anhydrous (5.45 g, 63.3 mmol) was added to a solution of compound 2 (2.23 g, 6.32 mmol) in CHCl3 (30 mL), and the resulting mixture was stirred for 1 h at room temperature. After the reaction mixture was concentrated under reduced pressure, the residue was dissolved in CHCl3 (70 mL). The organic phase was washed with water (35 mL × 2), dried over Na2SO4, and concentrated under reduced pressure. The crude product was purified by column chromatography on NH silica gel (CHCl3/EtOAc, 20:1) to afford 15 as a white solid (2.01 g, 79%). 1H NMR (400 MHz, CDCl3) δ 5.01 (t, J = 5.8 Hz, 1H), 4.17 (dd, J = 5.6, 2.5 Hz, 2H), 3.85–3.61 (m, 9H), 3.48–3.38 (m, 4H), 2.93–2.79 (m, 4H), 2.19 (t, J = 2.5 Hz, 1H), 1.48 (s, 9H). 13C NMR (100 MHz, CDCl3) δ 165.8, 165.2, 165.0, 154.8, 80.9, 79.8, 70.6, 46.0, 44.2, 42.9, 30.4, 28.4. HRMS (MALDI): calcd for C19H31N8O2, 403.2565 (M + H+); found, 403.2565.
3.15. Synthesis of Compound 16
Cyanuric chloride (1.10 g, 5.99 mmol) and DIPEA (510.0 µL, 3.00 mmol) were successively added to a solution of compound 15 (1.20 g, 2.99 mmol) in CHCl3 (15 mL), and the resulting mixture was stirred for 2 h at 0 °C. After the reaction mixture was concentrated under reduced pressure, the residue was dissolved in CHCl3 (70 mL). The organic phase was washed with water (25 mL × 2), dried over Na2SO4, and concentrated under reduced pressure. The crude product was purified by column chromatography on silica gel (CHCl3/EtOAc, 15:1) to afford 16 as a white solid (1.60 g, 97%). 1H NMR (400 MHz, CDCl3) δ 4.94 (t, J = 5.7 Hz, 1H), 4.19 (dd, J = 5.7, 2.5 Hz, 2H), 3.95–3.68 (m, 12H), 3.49–3.40 (m, 4H), 2.20 (t, J = 2.5 Hz, 1H), 1.49 (s, 9H). 13C NMR (100 MHz, CDCl3) δ 170.4, 165.8, 165.2, 165.1, 164.1, 154.8, 80.6, 80.0, 70.8, 44.0, 43.0, 42.6, 30.5, 28.4. HRMS (MALDI): calcd for C22H29Cl2N11NaO2, 572.1775 (M + Na+); found, 572.1784.
3.16. Synthesis of Compound 17
Propargylamine (140 µL, 2.19 mmol) and DIPEA (1.24 mL, 7.29 mmol) were added to a solution of compound 16 (801 mg, 1.46 mmol) in THF (20 mL), and the resulting mixture was stirred for 1.5 h at room temperature. After the reaction mixture was concentrated under reduced pressure, the residue was dissolved in CHCl3 (70 mL). The organic phase was washed with water (50 mL × 3), dried over Na2SO4, and concentrated under reduced pressure. The crude product was purified by column chromatography on silica gel (CHCl3/MeOH, 20:1) to afford 17 as a white solid (708 mg, 85%). 1H NMR (400 MHz, CDCl3) δ 6.33–6.20 (m, 1H), 4.99 (t, J = 5.3 Hz, 1H), 4.26–4.14 (m, 4H), 3.93–3.70 (m, 12H), 3.48–3.41 (m, 4H), 2.23 (t, J = 2.5 Hz, 1H), 2.20 (t, J = 2.5 Hz, 1H), 1.48 (s, 9H). 13C NMR (100 MHz, CDCl3) δ 169.1, 165.8, 165.2, 165.2, 164.4, 154.8, 80.7, 79.9, 79.5, 71.2, 70.7, 43.4, 42.9, 42.7, 30.6, 30.5, 28.4. HRMS (MALDI): calcd for C25H33ClN12NaO2, 591.2430 (M + Na+); found, 591.2436.
3.17. Synthesis of G3 Dendrimer 18
Compound 17 (542 mg, 0.952 mmol) and DIPEA (502 µL, 2.95 mmol) were successively added to a solution of G1 dendrimer 5 (326 mg, 0.236 mmol) in CHCl3 (20 mL), and the resulting mixture was refluxed for 8 days. After the reaction mixture was concentrated under reduced pressure, the residue was dissolved in CHCl3 (100 mL). The organic phase was washed with water (30 mL × 1), dried over Na2SO4, and concentrated under reduced pressure. The crude product was purified by silica gel column chromatography (gradient elution using (CH2Cl2/EtOAc 2:1) until no detectable 16 was observed, as determined by UV spotting, to (CHCl3/MeOH 15:1) to obtain the desired product) to afford 18 as a slightly yellow solid (638 mg, 91%). 1H NMR (600 MHz, CDCl3) δ 7.41–7.29 (m, 12H), 7.26–7.22 (m, 6H), 5.49 (s, 6H), 5.32–5.23 (m, 3H), 5.17–4.84 (m, 6H), 4.69 (d, J = 6.1 Hz, 6H), 4.23–4.16 (m, 12H), 3.88–3.70 (m, 84H), 3.47–3.41 (m, 12H), 2.23–2.18 (m, 6H), 1.48 (s, 27H). 13C NMR (150 MHz, CDCl3) δ 166.1, 165.8, 165.4, 165.2, 154.8, 147.0, 134.6, 129.1, 128.8, 128.0, 121.5, 80.9, 80.9, 79.9, 70.7, 54.1, 43.0, 43.0, 36.6, 30.5, 28.4. HRMS (MALDI): calcd for C141H180N72NaO6, 3000.5885 (M + Na+); found, 3000.5873.
3.18. Synthesis of G3 Dendrimer 10 from Compound 18
3.18.1. CuSO4/Ascorbic Acid
Benzyl azide (25.0 µL, 0.200 mmol), ascorbic acid (16.6 mg, 0.0943 mmol), and copper (II) sulfate (2.9 mg, 0.0182 mmol) were successively added to a solution of compound 18 (90 mg, 0.0302 mmol) in THF/H2O (1:1 v/v, 5 mL). The resulting mixture was subjected to microwave irradiation at 110 °C for 3 h in a sealed vial or stirred at 110 °C for 12 h in a pressure vessel. After the reaction mixture was concentrated, the residue was dissolved in CHCl3 (70 mL) and the solution was washed with 5% aqueous NaOH (30 mL × 2). The organic phase was dried over Na2SO4 and then evaporated. The crude product was purified with column chromatography on silica gel (CHCl3/MeOH, 20:1) to afford 10 as a yellow solid. Yield; MW: (12.3 mg, 3.26 µmol, 11%), pressure vessel: (12.6 mg, 3.33 µmol, 11%).
3.18.2. CuI/DIPEA
Benzyl azide (25.0 µL, 0.200 mmol), DIPEA (31.0 µL, 0.182 mmol), and copper (I) iodide (4.2 mg, 0.0221 mmol) were successively added to a solution of compound 18 (90 mg, 0.0302 mmol) in THF (5 mL). The resulting mixture was subjected to microwave irradiation at 110 °C for 3 h in a sealed vial or stirred at 110 °C for 12 h in a pressure vessel. After the reaction mixture was concentrated, the residue was dissolved in CHCl3 (70 mL) and the solution was washed with 5% aqueous NaOH (30 mL × 2). The organic phase was dried over Na2SO4 and then evaporated. The crude product was purified with column chromatography on silica gel (CHCl3/MeOH, 20:1) to afford 10 as a pale yellow solid. Yield; MW: (11.0 mg, 2.91 µmol, 10%), pressure vessel: (18.1 mg, 4.79 µmol, 16%).










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}