
Synthesis and Investigation of Novel CHCA-Derived Matrices for Matrix-Assisted Laser Desorption/Ionization Mass Spectrometric Analysis of Lipids

 , , ,
, , ,  , and
, and 
Abstract
:1. Introduction
2. Results and Discussion
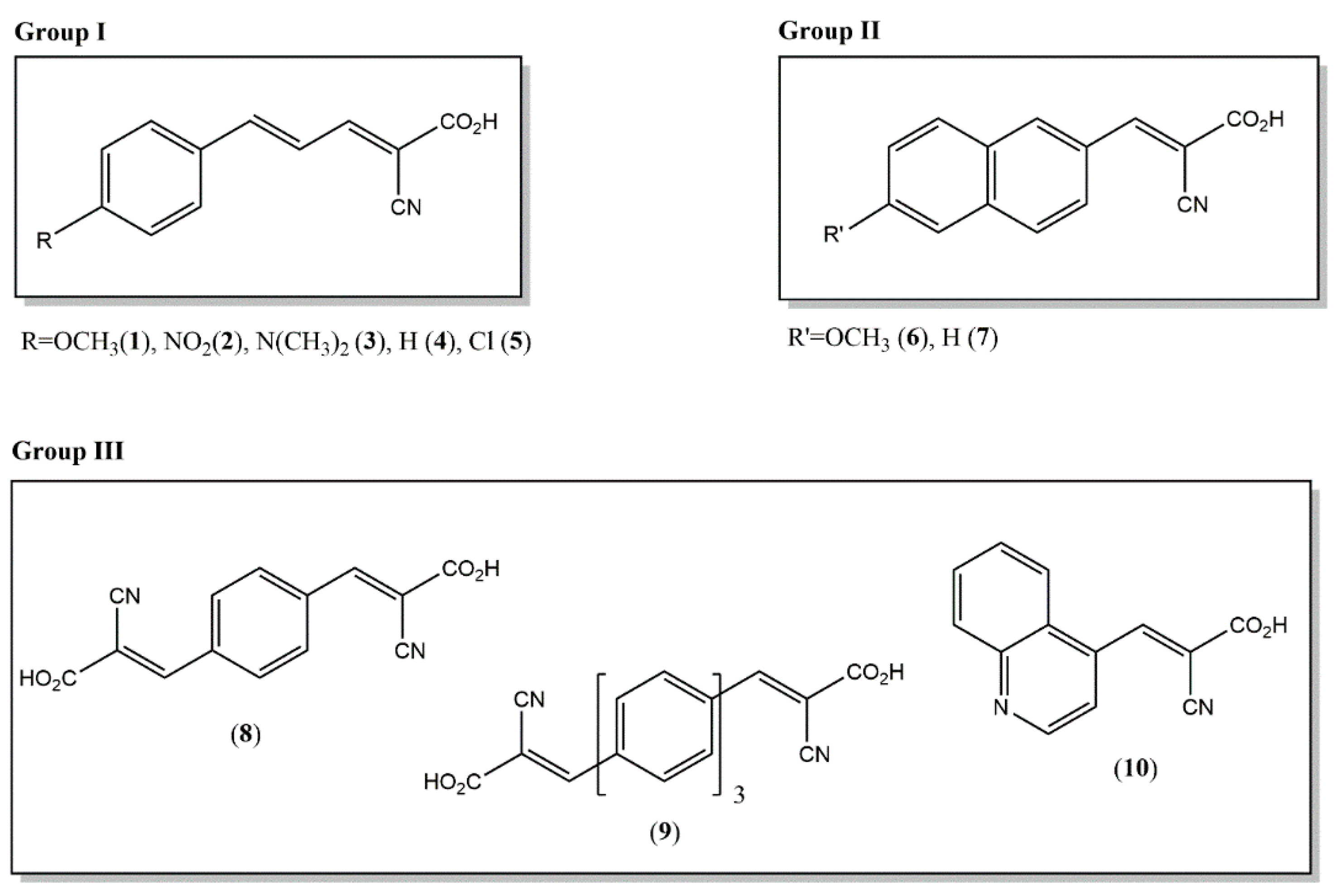
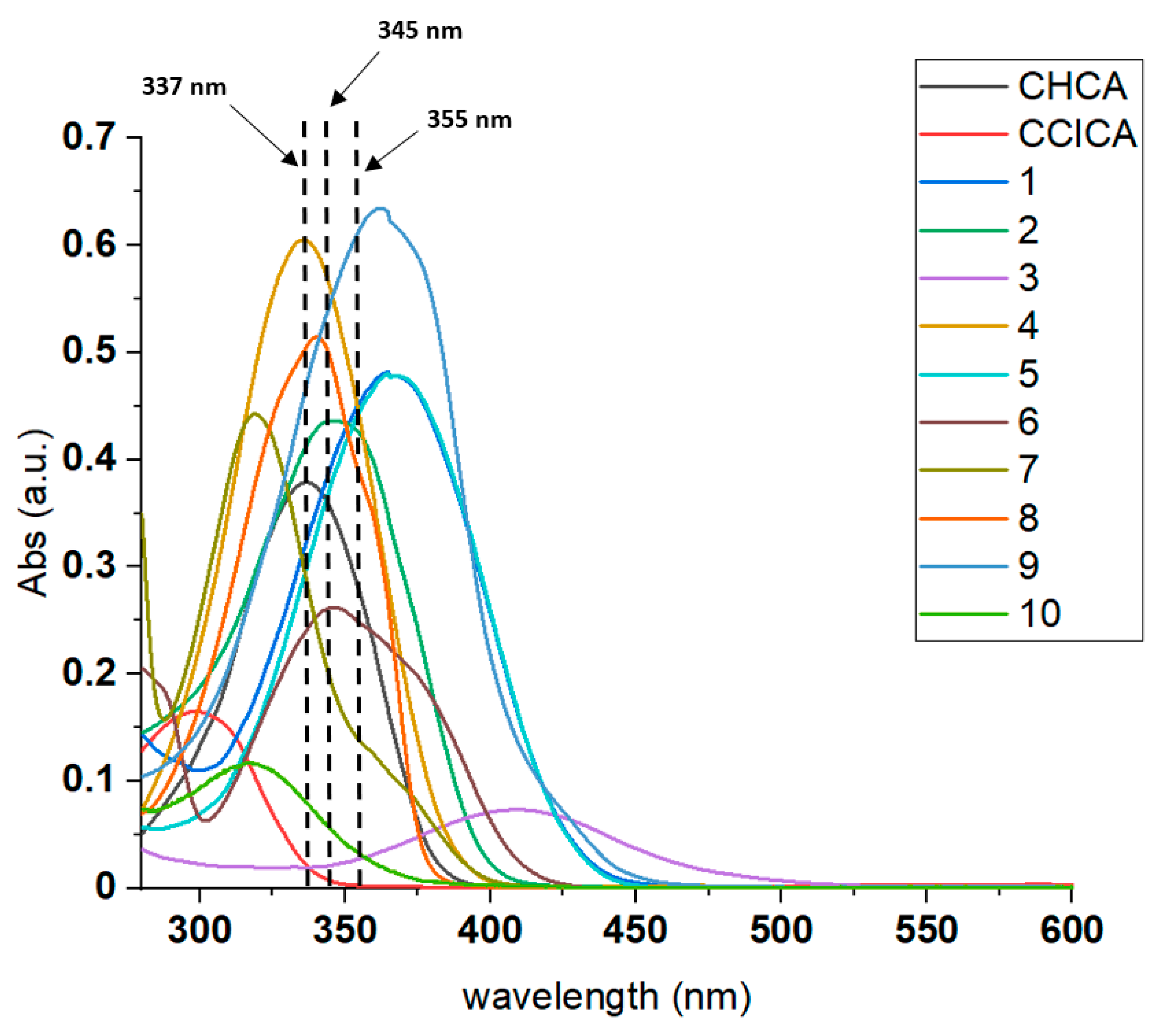
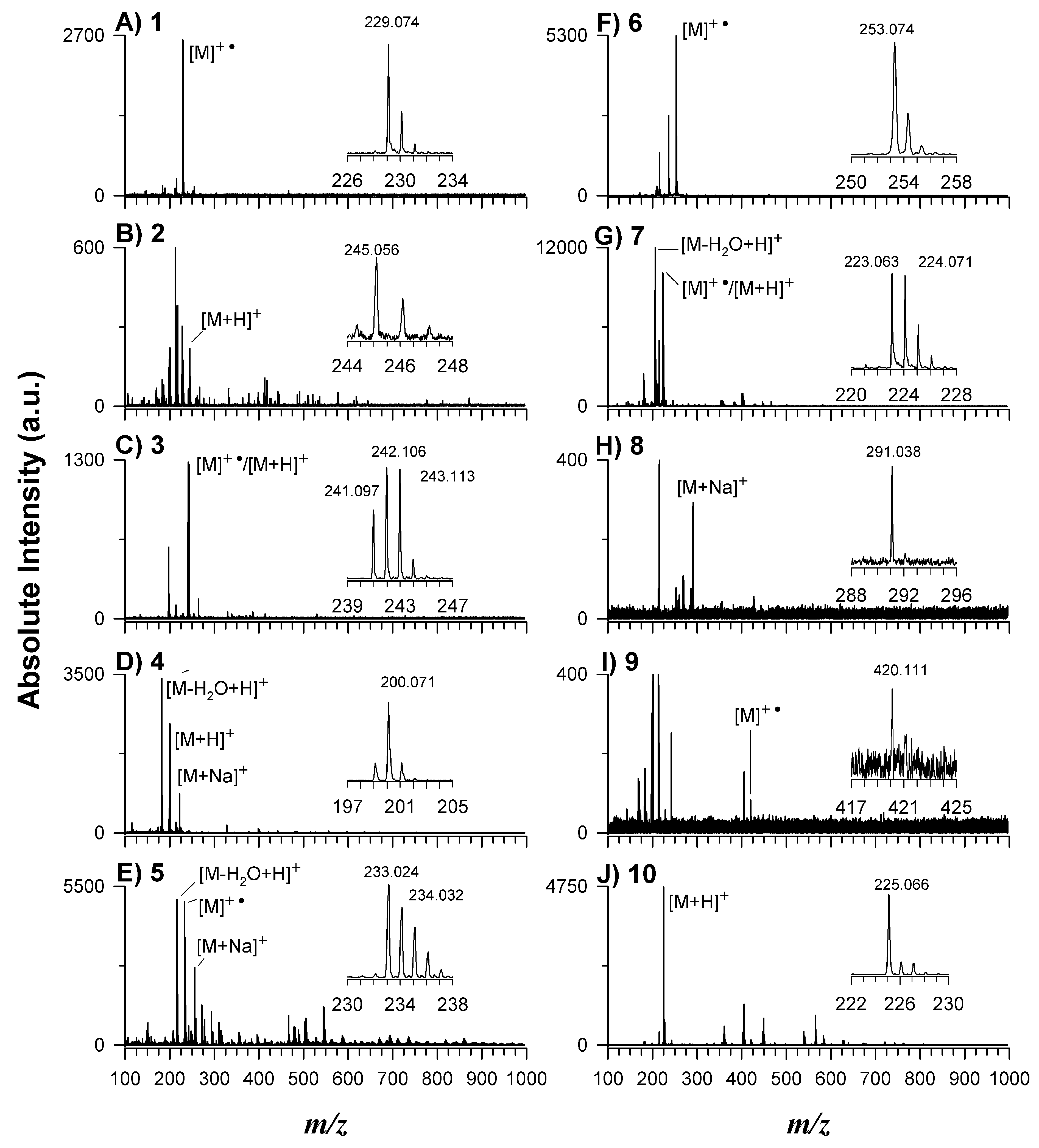
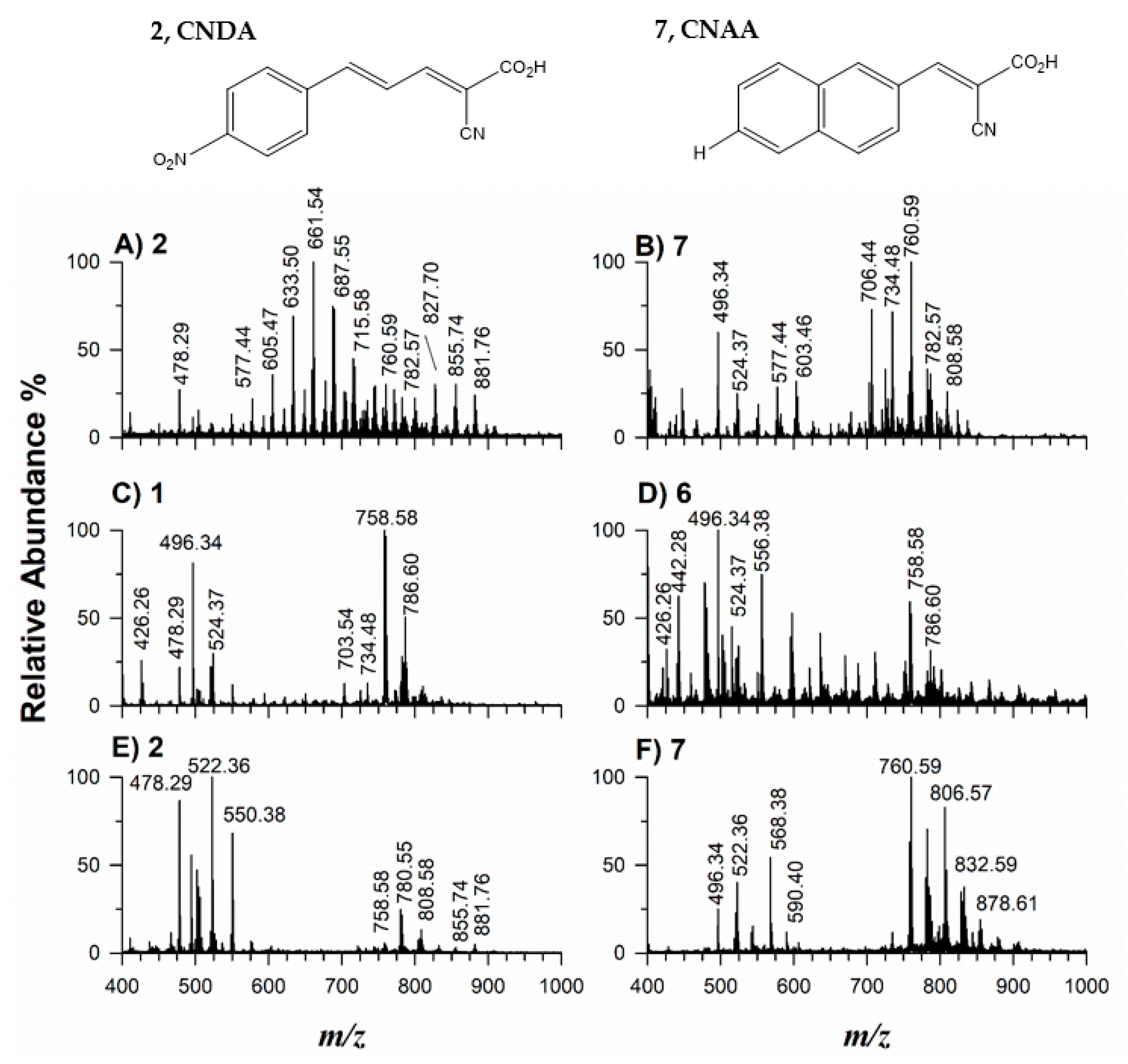
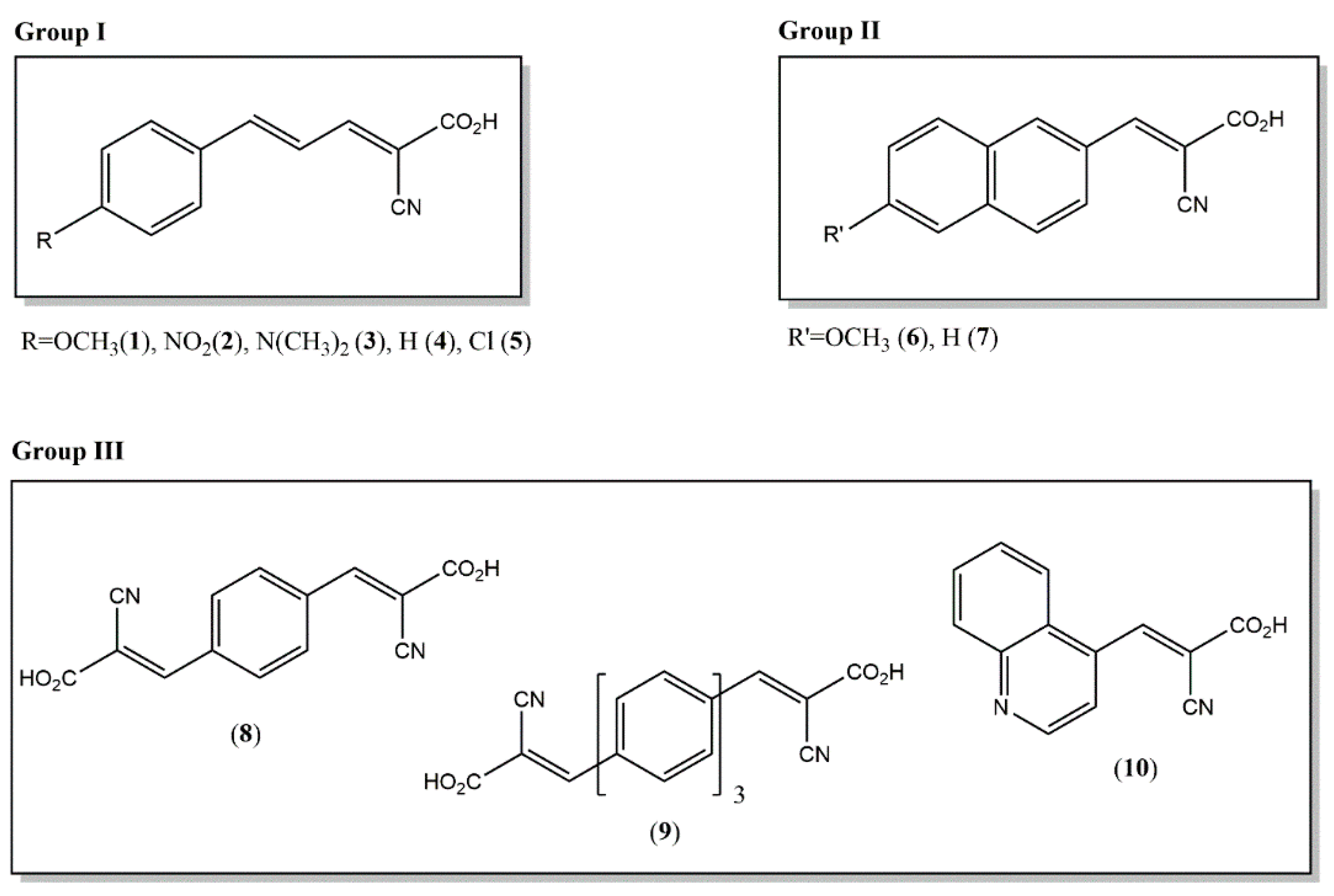
2.1. Synthesis and Characterization of Novel Matrices
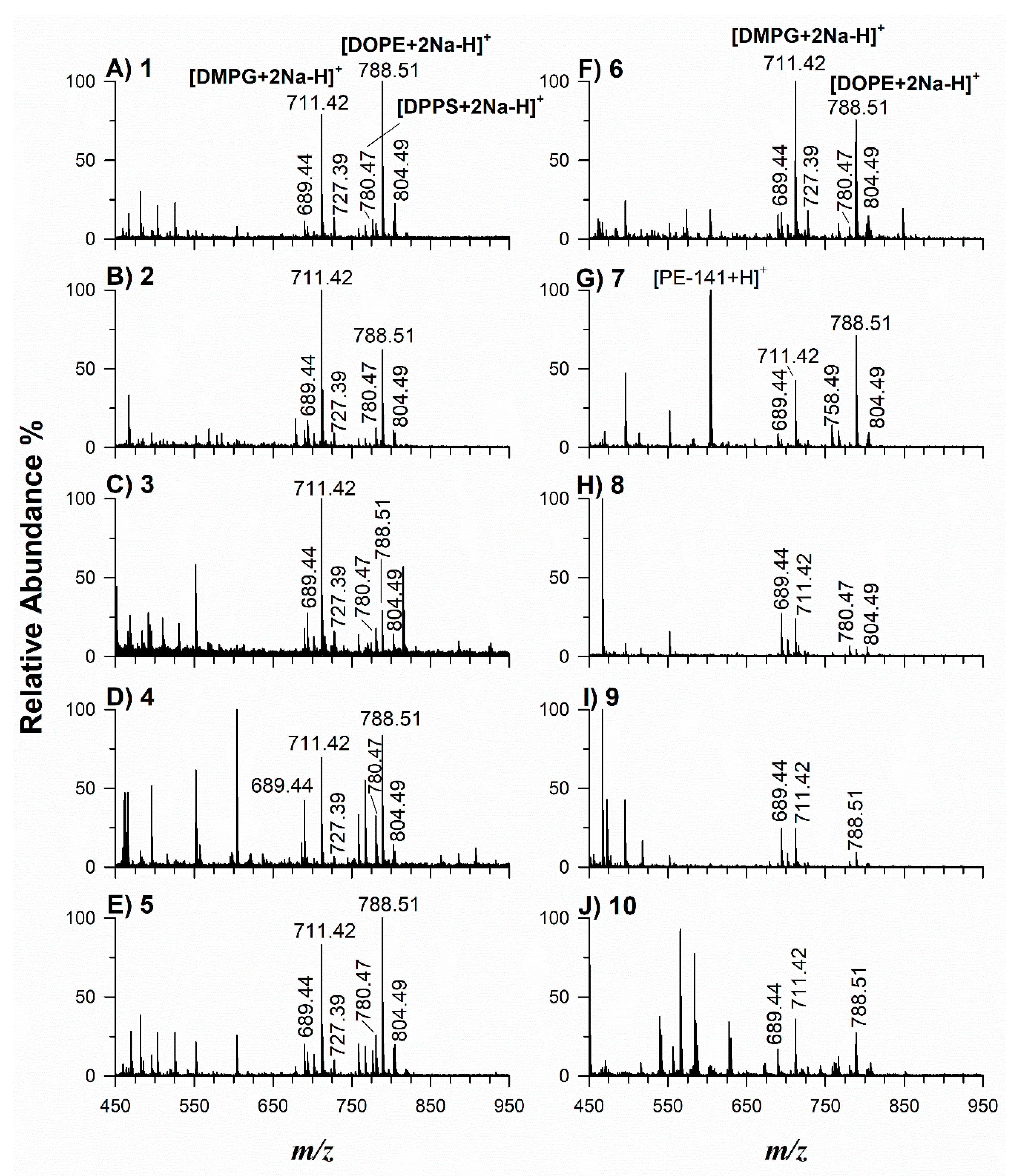
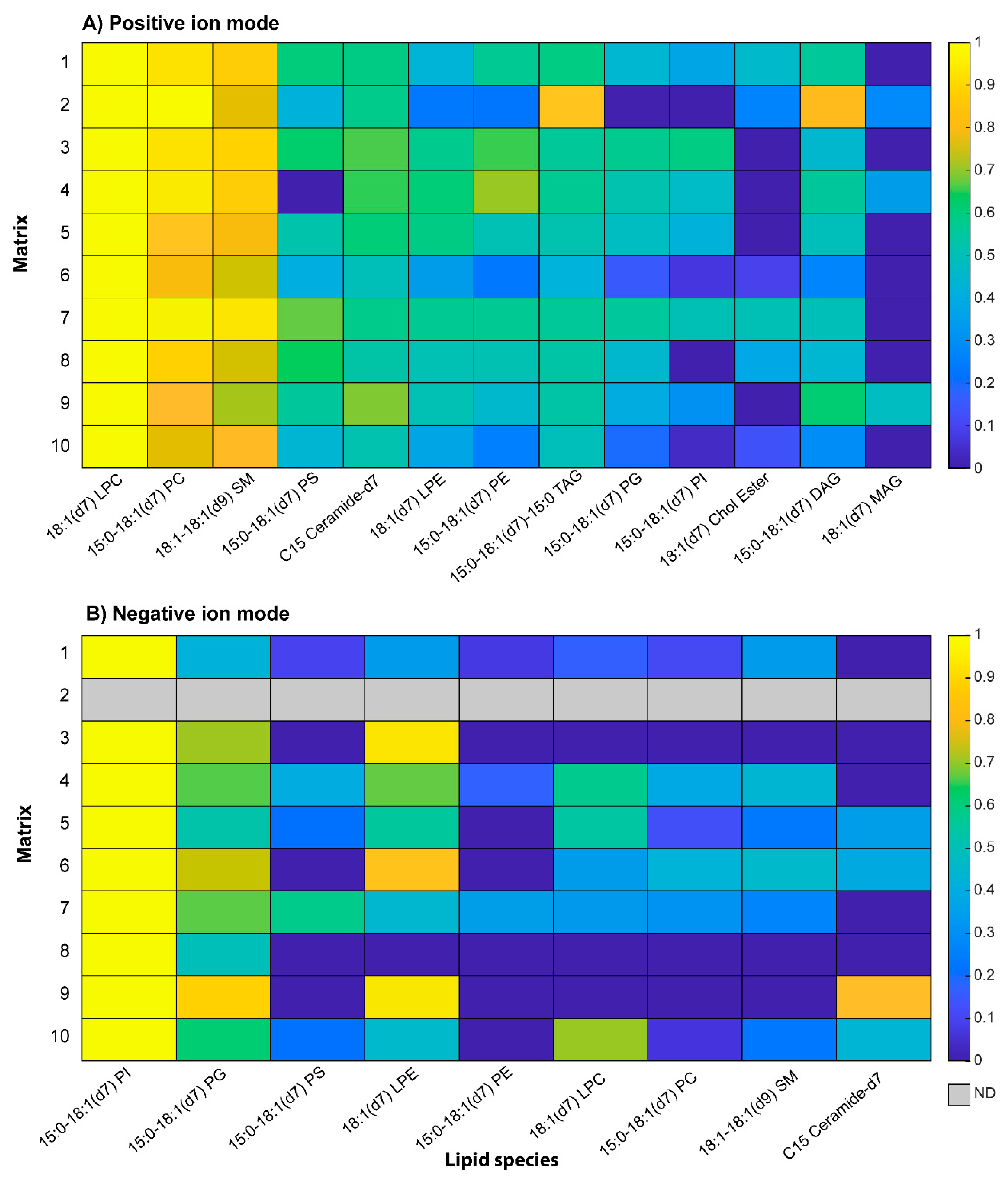
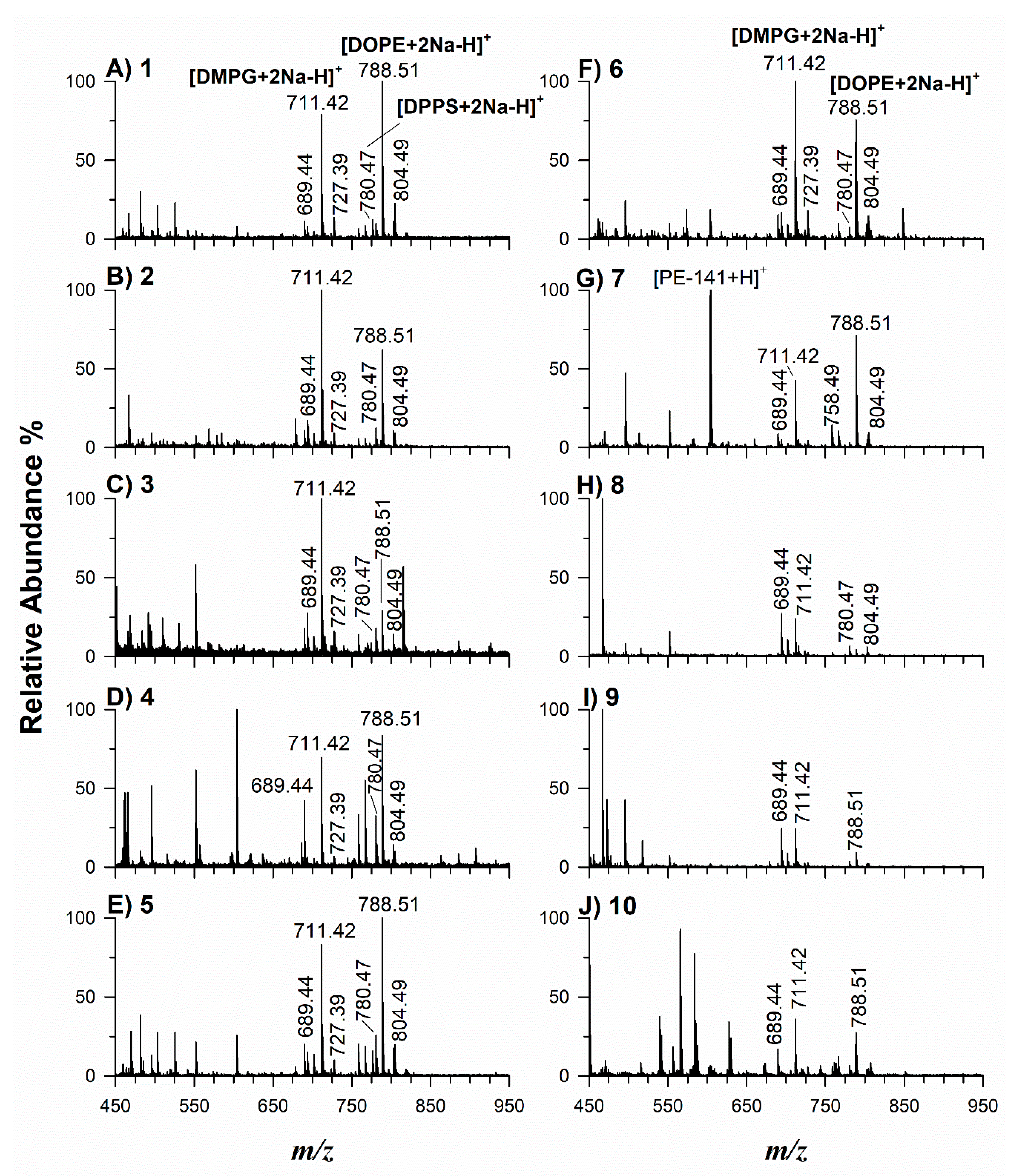
2.2. Application of Potential Matrices to Standard Lipid Mixtures
2.3. Application of Selected Matrices to Real Samples
3. Experimental Section
- 1.
- Protocol A
- Dissolution of the matrix (5 mg/mL) in ethanol.
- Heating the solution at about 40 °C for 2 h
- Filtration of the solution and subsequent dilution of the filtrate 50:50 (v/v) with ultra-pure H2O and shaking
- Incubation of the liquid in the refrigerator overnight to allow the precipitation of the solid
- Removal of the supernatant and subsequent drying of the purified solid
- Repetition of the procedure at least in triplicate
- 2.
- Protocol B
- Dissolution of the matrix (5 mg/mL) in a mixture of acetonitrile: water (70:30)
- Heating the solution to a temperature close to boiling
- Cooling at room temperature and subsequently in an ice bath to allow the precipitation of the crystals
- Removal of the precipitate by centrifugation or filtration and successive drying
- Repetition of the procedure at least in triplicate
4. Sample Preparation
5. Instrumentations
6. Methods for Quantum Computations
7. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bergman, N.; Shevchenko, D.; Bergquist, J. Approaches for the Analysis of Low Molecular Weight Compounds with Laser Desorption/Ionization Techniques and Mass Spectrometry. Anal. Bioanal. Chem. 2014, 406, 49–61. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.L.; Gross, M.L.; Hsu, F.-F. Ionic-Liquid Matrices for Improved Analysis of Phospholipids by MALDI-TOF Mass Spectrometry. J. Am. Soc. Mass Spectrom. 2005, 16, 679–682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Annese, C.; Fanizza, I.; Calvano, C.D.; D’Accolti, L.; Fusco, C.; Curci, R.; Williard, P.G. Selective Synthesis of Hydroxy Analogues of Valinomycin Using Dioxiranes. Org. Lett. 2011, 13, 5096–5099. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beavis, R.C.; Chaudhary, T.; Chait, B.T. α-Cyano-4-Hydroxycinnamic Acid as a Matrix for Matrixassisted Laser Desorption Mass Spectromtry. Org. Mass Spectrom. 1992, 27, 156–158. [Google Scholar] [CrossRef]
- Wu, K.J.; Shaler, T.A.; Becker, C.H. Time-of-Flight Mass Spectrometry Of Underivatized Single-Stranded DNA Oligomers by Matrix-Assisted Laser Desorption. Anal. Chem. 1994, 66, 1637–1645. [Google Scholar] [CrossRef]
- Nordhoff, E.; Kirpekar, F.; Karas, M.; Cramer, R.; Hahner, S.; Hillenkamp, F.; Kristiansen, K.; Roepstorff, P.; Lezius, A. Comparison of IR- and UV-Matrix-Assisted Laser Desorption/Ionization Mass Spectrometry of Oligodeoxynucleotides. Nucleic Acids Res. 1994, 22, 2460–2465. [Google Scholar] [CrossRef] [Green Version]
- Tholey, A.; Heinzle, E. Ionic (Liquid) Matrices for Matrix-Assisted Laser Desorption/Ionization Mass Spectrometry—Applications and Perspectives. Anal. Bioanal. Chem. 2006, 386, 24–37. [Google Scholar] [CrossRef]
- Monopoli, A.; Cotugno, P.; Cortese, M.; Calvano, C.D.; Ciminale, F.; Nacci, A. Selective N-Alkylation of Arylamines with Alkyl Chloride in Ionic Liquids: Scope and Applications. European J. Org. Chem. 2012, 3105–3111. [Google Scholar] [CrossRef]
- Towers, M.W.; Cramer, R. Ionic Liquids and Other Liquid Matrices for Sensitive MALDI MS Analysis. In Advances in MALDI and Laser-Induced Soft Ionization Mass Spectrometry; Springer International Publishing: Cham, Switzerland, 2016. [Google Scholar]
- Abdelhamid, H.N. Ionic Liquids for Mass Spectrometry: Matrices, Separation and Microextraction. TrAC Trends Anal. Chem. 2016, 77, 122–138. [Google Scholar] [CrossRef]
- Dong, W.; Shen, Q.; Baibado, J.T.; Liang, Y.; Wang, P.; Huang, Y.; Zhang, Z.; Wang, Y.; Cheung, H.-Y. Phospholipid Analyses by MALDI-TOF/TOF Mass Spectrometry Using 1,5-Diaminonaphthalene as Matrix. Int. J. Mass Spectrom. 2013, 343–344, 15–22. [Google Scholar] [CrossRef]
- Calvano, C.D.; Ventura, G.; Cataldi, T.R.I.; Palmisano, F. Improvement of Chlorophyll Identification in Foodstuffs by MALDI ToF/ToF Mass Spectrometry Using 1,5-Diaminonaphthalene Electron Transfer Secondary Reaction Matrix. Anal. Bioanal. Chem. 2015, 407, 6369–6379. [Google Scholar] [CrossRef] [PubMed]
- Calvano, C.D.; Capozzi, M.A.M.; Punzi, A.; Farinola, G.M.; Cataldi, T.R.I.; Palmisano, F. 1,5-Diaminonaphtalene Is a Highly Performing Electron-Transfer Secondary-Reaction Matrix for Laser Desorption Ionization Mass Spectrometry of Indolenine-Based Croconaines. ACS Omega 2018, 3, 17821–17827. [Google Scholar] [CrossRef] [PubMed]
- Shroff, R.; Svatoš, A. Proton Sponge: A Novel and Versatile MALDI Matrix for the Analysis of Metabolites Using Mass Spectrometry. Anal. Chem. 2009, 81, 7954–7959. [Google Scholar] [CrossRef] [PubMed]
- Calvano, C.D.; Cataldi, T.R.I.; Kögel, J.F.; Monopoli, A.; Palmisano, F.; Sundermeyer, J. Superbasic Alkyl-Substituted Bisphosphazene Proton Sponges: A New Class of Deprotonating Matrices for Negative Ion Matrix-Assisted Ionization/Laser Desorption Mass Spectrometry of Low Molecular Weight Hardly Ionizable Analytes. Rapid Commun. Mass Spectrom. 2016, 30, 1680–1686. [Google Scholar] [CrossRef] [PubMed]
- Calvano, C.D.; Cataldi, T.R.I.; Kögel, J.F.; Monopoli, A.; Palmisano, F.; Sundermeyer, J. Structural Characterization of Neutral Saccharides by Negative Ion MALDI Mass Spectrometry Using a Superbasic Proton Sponge as Deprotonating Matrix. J. Am. Soc. Mass Spectrom. 2017, 28, 1666–1675. [Google Scholar] [CrossRef] [PubMed]
- Shanta, S.R.; Zhou, L.-H.; Park, Y.S.; Kim, Y.H.; Kim, Y.; Kim, K.P. Binary Matrix for MALDI Imaging Mass Spectrometry of Phospholipids in Both Ion Modes. Anal. Chem. 2011, 83, 1252–1259. [Google Scholar] [CrossRef]
- Chen, Y.; Gao, D.; Bai, H.; Liu, H.; Lin, S.; Jiang, Y. Carbon Dots and 9AA as a Binary Matrix for the Detection of Small Molecules by Matrix-Assisted Laser Desorption/Ionization Mass Spectrometry. J. Am. Soc. Mass Spectrom. 2016, 27, 1227–1235. [Google Scholar] [CrossRef]
- Feenstra, A.D.; O’Neill, K.C.; Yagnik, G.B.; Lee, Y.J. Organic–Inorganic Binary Mixture Matrix for Comprehensive Laser-Desorption Ionization Mass Spectrometric Analysis and Imaging of Medium-Size Molecules Including Phospholipids, Glycerolipids, and Oligosaccharides. RSC Adv. 2016, 6, 99260–99268. [Google Scholar] [CrossRef]
- Han, C.; Li, S.; Yue, Q.; Li, N.; Yang, H.; Zhao, Z. Polydopamine-Capped AgNPs as a Novel Matrix Overcoming the Ion Suppression of Phosphatidylcholine for MALDI MS Comprehensive Imaging of Glycerophospholipids and Sphingolipids in Impact-Induced Injured Brain. Analyst 2019, 144, 6304–6312. [Google Scholar] [CrossRef]
- Giampà, M.; Lissel, M.B.; Patschkowski, T.; Fuchser, J.; Hans, V.H.; Gembruch, O.; Bednarz, H.; Niehaus, K. Maleic Anhydride Proton Sponge as a Novel MALDI Matrix for the Visualization of Small Molecules (<250 m/z) in Brain Tumors by Routine MALDI ToF Imaging Mass Spectrometry. Chem. Commun. 2016, 52, 9801–9804. [Google Scholar]
- Lorkiewicz, P.; Cecilia Yappert, M. 2-(2-Aminoethylamino)-5-Nitropyridine as a Basic Matrix for Negative-Mode Matrix-Assisted Laser Desorption/Ionization Analysis of Phospholipids. J. Mass Spectrom. 2009, 44, 137–143. [Google Scholar] [CrossRef] [PubMed]
- Jaskolla, T.W.; Onischke, K.; Schiller, J. 2,5-Dihydroxybenzoic Acid Salts for Matrix-Assisted Laser Desorption/Ionization Time-of-Flight Mass Spectrometric Lipid Analysis: Simplified Spectra Interpretation and Insights into Gas-Phase Fragmentation. Rapid Commun. Mass Spectrom. 2014, 28, 1353–1363. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, H.; Jurcic, K.; Wang, J.S.H.; Whitehead, S.N.; Yeung, K.K.C. 1,6-Diphenyl-1,3,5-Hexatriene (DPH) as a Novel Matrix for MALDI MS Imaging of Fatty Acids, Phospholipids, and Sulfatides in Brain Tissues. Anal. Chem. 2017, 89, 12828–12836. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, H.; Zhou, Y.; Wang, J.; Xiong, C.; Xue, J.; Zhan, L.; Nie, Z. N -Phenyl-2-Naphthylamine as a Novel MALDI Matrix for Analysis and in Situ Imaging of Small Molecules. Anal. Chem. 2018, 90, 729–736. [Google Scholar] [CrossRef] [PubMed]
- Le, C.H.; Han, J.; Borchers, C.H. Dithranol as a MALDI Matrix for Tissue Imaging of Lipids by Fourier Transform Ion Cyclotron Resonance Mass Spectrometry. Anal. Chem. 2012, 84, 8391–8398. [Google Scholar] [CrossRef]
- Cheng, X.; Ye, X.; Liu, D.; Zhao, N.; Gao, H.; Wang, P.; Ge, G.; Zhang, X. N -Butyl-4-Hydroxy-1,8-Naphthalimide: A New Matrix for Small Molecule Analysis by Matrix-Assisted Laser Desorption/Ionization Mass Spectrometry. Rapid Commun. Mass Spectrom. 2017, 31, 1779–1784. [Google Scholar] [CrossRef]
- Teuber, K.; Fedorova, M.; Hoffmann, R.; Schiller, J. 2,4-Dinitrophenylhydrazine as a New Reactive Matrix to Analyze Oxidized Phospholipids by MALDI-TOF Mass Spectrometry. Anal. Lett. 2012, 45, 968–976. [Google Scholar] [CrossRef]
- Bahr, U.; Jaskolla, T.W. Employing ‘Second Generation’ Matrices. In Advances in MALDI and Laser-Induced Soft Ionization Mass Spectrometry; Springer International Publishing: Cham, Switzerland, 2016; pp. 3–35. ISBN 9783319048192. [Google Scholar]
- Qiao, Z.; Lissel, F. MALDI Matrices for the Analysis of Low Molecular Weight Compounds: Rational Design, Challenges and Perspectives. Chem. Asian J. 2021, 16, 868–878. [Google Scholar] [CrossRef]
- Shroff, R.; Rulíšek, L.; Doubský, J.; Svatoš, A. Acid–Base-Driven Matrix-Assisted Mass Spectrometry for Targeted Metabolomics. Proc. Natl. Acad. Sci. USA 2009, 106, 10092–10096. [Google Scholar] [CrossRef] [Green Version]
- Calvano, C.D.; Ventura, G.; Palmisano, F.; Cataldi, T.R.I. 4-Chloro-α-Cyanocinnamic Acid Is an Efficient Soft Matrix for Cyanocobalamin Detection in Foodstuffs by Matrix-Assisted Laser Desorption/Ionization Mass Spectrometry (MALDI MS). J. Mass Spectrom. 2016, 51, 841–848. [Google Scholar] [CrossRef]
- Ventura, G.; Arnesano, F.; Calvano, C.D.; Palmisano, F.; Cataldi, T.R.I. Cyanocobalamin Conjugates of Cisplatin and Diaminocyclohexane-Platinum(ii): Matrix-Assisted Laser Desorption Ionization Mass Spectrometry Characterization Using 4-Chloro-α-Cyanocinnamic Acid as the Matrix. RSC Adv. 2017, 7, 53658–53666. [Google Scholar] [CrossRef] [Green Version]
- Fülöp, A.; Porada, M.B.; Marsching, C.; Blott, H.; Meyer, B.; Tambe, S.; Sandhoff, R.; Junker, H.-D.; Hopf, C. 4-Phenyl-α-Cyanocinnamic Acid Amide: Screening for a Negative Ion Matrix for MALDI-MS Imaging of Multiple Lipid Classes. Anal. Chem. 2013, 85, 9156–9163. [Google Scholar] [CrossRef] [PubMed]
- Monopoli, A.; Calvano, C.D.; Nacci, A.; Palmisano, F. Boronic Acid Chemistry in MALDI MS: A Step Forward in Designing a Reactive Matrix with Molecular Recognition Capabilities. Chem. Commun. 2014, 50, 4322–4324. [Google Scholar] [CrossRef]
- He, H.; Qin, L.; Zhang, Y.; Han, M.; Li, J.; Liu, Y.; Qiu, K.; Dai, X.; Li, Y.; Zeng, M.; et al. 3,4-Dimethoxycinnamic Acid as a Novel Matrix for Enhanced In Situ Detection and Imaging of Low-Molecular-Weight Compounds in Biological Tissues by MALDI-MSI. Anal. Chem. 2019, 91, 2634–2643. [Google Scholar] [CrossRef] [PubMed]
- Tambe, S.; Blott, H.; Fülöp, A.; Spang, N.; Flottmann, D.; Bräse, S.; Hopf, C.; Junker, H.-D. Structure-Performance Relationships of Phenyl Cinnamic Acid Derivatives as MALDI-MS Matrices for Sulfatide Detection. Anal. Bioanal. Chem. 2017, 409, 1569–1580. [Google Scholar] [CrossRef]
- Soltwisch, J.; Jaskolla, T.W.; Hillenkamp, F.; Karas, M.; Dreisewerd, K. Ion Yields in UV-MALDI Mass Spectrometry As a Function of Excitation Laser Wavelength and Optical and Physico-Chemical Properties of Classical and Halogen-Substituted MALDI Matrixes. Anal. Chem. 2012, 84, 6567–6576. [Google Scholar] [CrossRef]
- Zhou, Q.; Fülöp, A.; Hopf, C. Recent Developments of Novel Matrices and On-Tissue Chemical Derivatization Reagents for MALDI-MSI. Anal. Bioanal. Chem. 2021, 413, 2599–2617. [Google Scholar] [CrossRef]
- Leopold, J.; Popkova, Y.; Engel, K.; Schiller, J. Recent Developments of Useful MALDI Matrices for the Mass Spectrometric Characterization of Lipids. Biomolecules 2018, 8, 173. [Google Scholar] [CrossRef] [Green Version]
- Jones, G. The Knoevenagel Condensation. In Organic Reactions; John Wiley & Sons: Hoboken, NJ, USA, 2011; pp. 204–599. [Google Scholar]
- Zenobi, R.; Knochenmuss, R. Ion Formation in MALDI Mass Spectrometry. Mass Spectrom. Rev. 1998, 17, 337–366. [Google Scholar] [CrossRef]
- Beavis, R.C. Matrix-Assisted Ultraviolet Laser Desorption: Evolution and Principles. Org. Mass Spectrom. 1992, 27, 653–659. [Google Scholar] [CrossRef]
- Knochenmuss, R.; Zenobi, R. MALDI Ionization: The Role of In-Plume Processes. Chem. Rev. 2003, 103, 441–452. [Google Scholar] [CrossRef] [PubMed]
- Karas, M.; Bachmann, D.; Hillenkamp, F. Influence of the Wavelength in High-Irradiance Ultraviolet Laser Desorption Mass Spectrometry of Organic Molecules. Anal. Chem. 1985, 57, 2935–2939. [Google Scholar] [CrossRef]
- Lu, I.-C.; Lee, C.; Lee, Y.-T.; Ni, C.-K. Ionization Mechanism of Matrix-Assisted Laser Desorption/Ionization. Annu. Rev. Anal. Chem. 2015, 8, 21–39. [Google Scholar] [CrossRef] [PubMed]
- Asbury, G.R.; Al-Saad, K.; Siems, W.F.; Hannan, R.M.; Hill, H.H. Analysis of Triacylglycerols and Whole Oils by Matrix-Assisted Laser Desorption/Ionization Time of Flight Mass Spectrometry. J. Am. Soc. Mass Spectrom. 1999, 10, 983–991. [Google Scholar] [CrossRef] [Green Version]
- Leopold, J.; Popkova, Y.; Engel, K.M.; Schiller, J. Visualizing Phosphatidylcholine via Mass Spectrometry Imaging: Relevance to Human Health. Expert Rev. Proteomics 2018, 15, 791–800. [Google Scholar] [CrossRef]
- Jaskolla, T.W.; Lehmann, W.D.; Karas, M. 4-Chloro-α-Cyanocinnamic Acid Is an Advanced, Rationally Designed MALDI Matrix. Proc. Natl. Acad. Sci. USA 2008, 105, 12200–12205. [Google Scholar] [CrossRef] [Green Version]
- Jaskolla, T.; Fuchs, B.; Karas, M.; Schiller, J. The New Matrix 4-Chloro-α-Cyanocinnamic Acid Allows the Detection of Phosphatidylethanolamine Chloramines by MALDI-TOF Mass Spectrometry. J. Am. Soc. Mass Spectrom. 2009, 20, 867–874. [Google Scholar] [CrossRef] [Green Version]
- Picariello, G.; Sacchi, R.; Addeo, F. One-step Characterization of Triacylglycerols from Animal Fat by MALDI-TOF MS. Eur. J. Lipid Sci. Technol. 2007, 109, 511–524. [Google Scholar] [CrossRef]
- Schiller, J.; Zschörnig, O.; Petkovic´, M.; Müller, M.; Arnhold, J.; Arnold, K. Lipid Analysis of Human HDL and LDL by MALDI-TOF Mass Spectrometry and 31P-NMR. J. Lipid Res. 2001, 42, 1501–1508. [Google Scholar] [CrossRef]
- Yu, Z.; Kastenmüller, G.; He, Y.; Belcredi, P.; Möller, G.; Prehn, C.; Mendes, J.; Wahl, S.; Roemisch-Margl, W.; Ceglarek, U.; et al. Differences between Human Plasma and Serum Metabolite Profiles. PLoS ONE 2011, 6, e21230. [Google Scholar] [CrossRef]
- Signor, L.; Boeri Erba, E. Matrix-Assisted Laser Desorption/Ionization Time of Flight (MALDI-TOF) Mass Spectrometric Analysis of Intact Proteins Larger than 100 KDa. J. Vis. Exp. 2013, 79, e50635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bligh, E.G.; Dyer, W.J. A Rapid Method of Total Lipid Extraction and Purification. Can. J. Biochem. Physiol. 1959, 37, 911–917. [Google Scholar] [CrossRef] [PubMed]
- Weininger, D. SMILES, a Chemical Language and Information System. 1. Introduction to Methodology and Encoding Rules. J. Chem. Inf. Model. 1988, 28, 31–36. [Google Scholar] [CrossRef]
- RDKit. Available online: https://www.rdkit.org/ (accessed on 7 January 2022).
- Aprà, E.; Bylaska, E.J.; de Jong, W.A.; Govind, N.; Kowalski, K.; Straatsma, T.P.; Valiev, M.; van Dam, H.J.J.; Alexeev, Y.; Anchell, J.; et al. NWChem: Past, Present, and Future. J. Chem. Phys. 2020, 152, 184102. [Google Scholar] [CrossRef] [PubMed]
- Stephens, P.J.; Devlin, F.J.; Chabalowski, C.F.; Frisch, M.J. Ab Initio Calculation of Vibrational Absorption and Circular Dichroism Spectra Using Density Functional Force Fields. J. Phys. Chem. 1994, 98, 11623–11627. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced Basis Sets of Split Valence, Triple Zeta Valence and Quadruple Zeta Valence Quality for H to Rn: Design and Assessment of Accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297. [Google Scholar] [CrossRef]
- Mirabelli, M.F.; Zenobi, R. Observing Proton Transfer Reactions Inside the MALDI Plume: Experimental and Theoretical Insight into MALDI Gas-Phase Reactions. J. Am. Soc. Mass Spectrom. 2017, 28, 1676–1686. [Google Scholar] [CrossRef]
- Hara, K.; Kurashige, M.; Ito, S.; Shinpo, A.; Suga, S.; Sayama, K.; Arakawa, H. Novel polyene dyes for highly efficient dye-sensitized solar cells. Chem. Commun. 2003, 2, 252–253. [Google Scholar] [CrossRef]
- Porta, T.; Grivet, C.; Knochenmuss, R.; Varesio, E.; Hopfgartner, G. Alternative chca-based matrices for the analysis of low molecular weight compounds by uv-maldi-tandem mass spectrometry. J. Mass Spectrom. 2011, 46, 144–152. [Google Scholar] [CrossRef]
- Monopoli, A.; Nacci, T.A.; Cataldi, C.D. Calvano. Molecules 2020, 25, 6054. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound n. | LogP | Molecular Formula (M) | Theoretical Mass M (Da) | ε (345 nm) | Proton Affinity (PA, kJ/mol) |
|---|---|---|---|---|---|
| 1 | 2.22 | C13H11NO3 | 229.0739 | 26227 | 872.9 |
| 2 | 2.00 | C12H8N2O4 | 244.0484 | 29067 | 802.3 |
| 3 | 2.63 | C14H14N2O2 | 242.1055 | 1467 | 892.6 |
| 4 | 2.34 | C12H9NO2 | 199.0633 | 29263 | 838.5 |
| 5 | 2.90 | C12H8ClNO2 | 233.0244 | 27530 | 874.1 |
| 6 | 2.70 | C15H11NO3 | 253.0739 | 20077 | 857.3 |
| 7 | 2.83 | C14H9NO2 | 223.0633 | 13067 | 857.7 |
| 8 | 1.62 | C14H8N2O4 | 268.0484 | 65467 | 784.5 |
| 9 | 4.97 | C26H16N2O4 | 420.1110 | 76826 | 835.2 |
| 10 | 1.91 | C13H8N2O2 | 224.0586 | 3667 | 830.0 |
| ClCCA | 2.39 | C10H6ClNO2 | 207.0087 | 23570 | 804.6 |
| CHCA | 1.44 | C10H7NO3 | 189.0426 | 4333 | 863.2 |
| Lipid Species | [M+H]+ m/z | [M+Na]+ m/z | [M-H]− m/z |
|---|---|---|---|
| 15:0–18:1(d7)PC | 753.61 | 775.59 | 736.60 [M-CH3-H]− |
| 18:1(d7)LPC | 529.40 | 551.38 | 512.38 [M-CH3-H]− |
| 15:0–18:1(d7)PE | 711.57 | 733.55 | 709.55 |
| 18:1(d7)LPE | 487.53 | 509.52 | 485.33 |
| 15:0–18:1(d7)PG | 742.54 | 764.52 | 740.55 |
| 15:0–18:1(d7)PI | 830.56 | 852.55 | 828.56 |
| 15:0–18:1(d7)PS | 755.55 | 777.54 | 753.54 |
| 15:0–18:1(d7)–15:0 TAG | 812.75 | 834.74 | 810.76 |
| 15:0–18:1(d7) DAG | 588.55 | 610.54 | 586.54 |
| 18:1(d7) MAG | 364.34 | 386.33 | 362.33 |
| 18:1(d7) Chol Ester | 658.65 | 680.63 | 656.64 |
| 18:1–18:1(d9)SM | 738.65 | 760.63 | 722.63 [M-CH3-H]− |
| C15 ceramide-d7 | 531.55 | 553.54 | 529.533 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Monopoli, A.; Ventura, G.; Aloia, A.; Ciriaco, F.; Nacci, A.; Cataldi, T.R.I.; Calvano, C.D. Synthesis and Investigation of Novel CHCA-Derived Matrices for Matrix-Assisted Laser Desorption/Ionization Mass Spectrometric Analysis of Lipids. Molecules 2022, 27, 2565. https://doi.org/10.3390/molecules27082565
Monopoli A, Ventura G, Aloia A, Ciriaco F, Nacci A, Cataldi TRI, Calvano CD. Synthesis and Investigation of Novel CHCA-Derived Matrices for Matrix-Assisted Laser Desorption/Ionization Mass Spectrometric Analysis of Lipids. Molecules. 2022; 27(8):2565. https://doi.org/10.3390/molecules27082565
Chicago/Turabian StyleMonopoli, Antonio, Giovanni Ventura, Andrea Aloia, Fulvio Ciriaco, Angelo Nacci, Tommaso R. I. Cataldi, and Cosima D. Calvano. 2022. "Synthesis and Investigation of Novel CHCA-Derived Matrices for Matrix-Assisted Laser Desorption/Ionization Mass Spectrometric Analysis of Lipids" Molecules 27, no. 8: 2565. https://doi.org/10.3390/molecules27082565
APA StyleMonopoli, A., Ventura, G., Aloia, A., Ciriaco, F., Nacci, A., Cataldi, T. R. I., & Calvano, C. D. (2022). Synthesis and Investigation of Novel CHCA-Derived Matrices for Matrix-Assisted Laser Desorption/Ionization Mass Spectrometric Analysis of Lipids. Molecules, 27(8), 2565. https://doi.org/10.3390/molecules27082565