
α-Glucosidase Inhibitory and Antimicrobial Benzoylphloroglucinols from Garcinia schomburgakiana Fruits: In Vitro and In Silico Studies

 , ,
, ,
Abstract
:1. Introduction
2. Results and Discussion
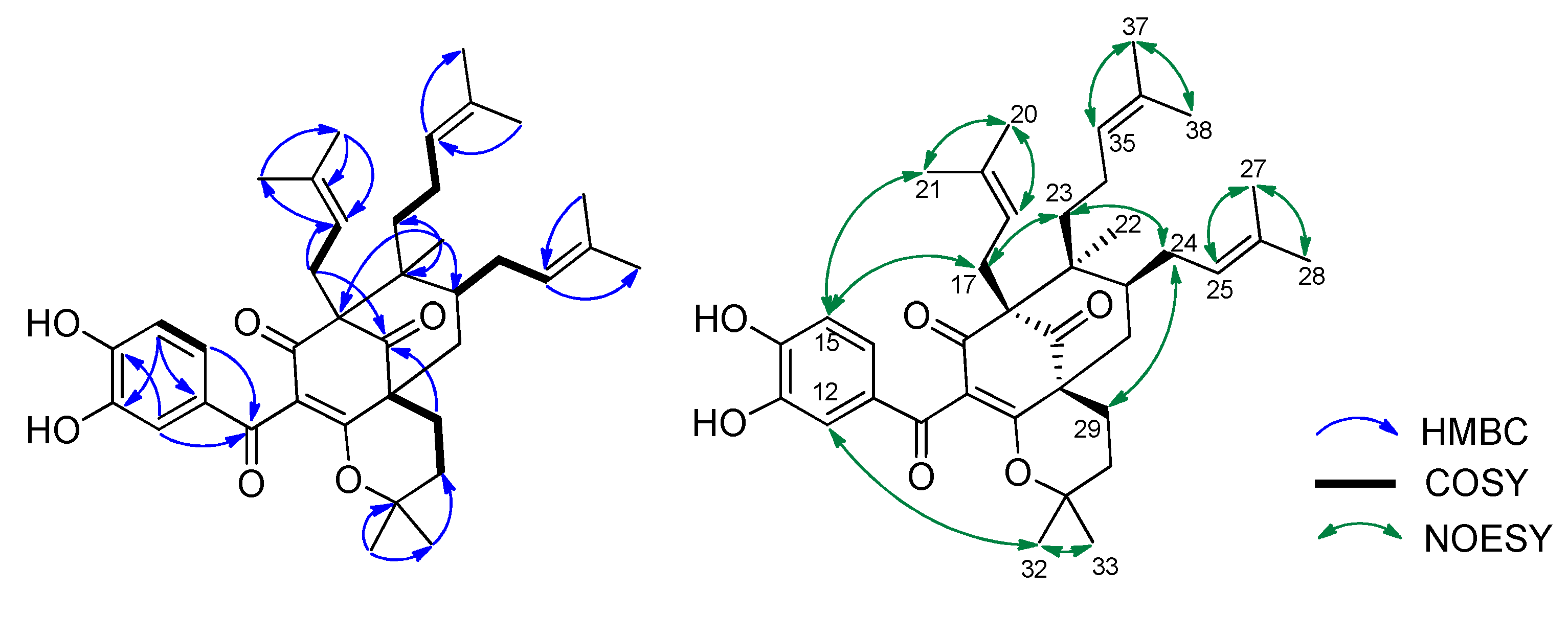
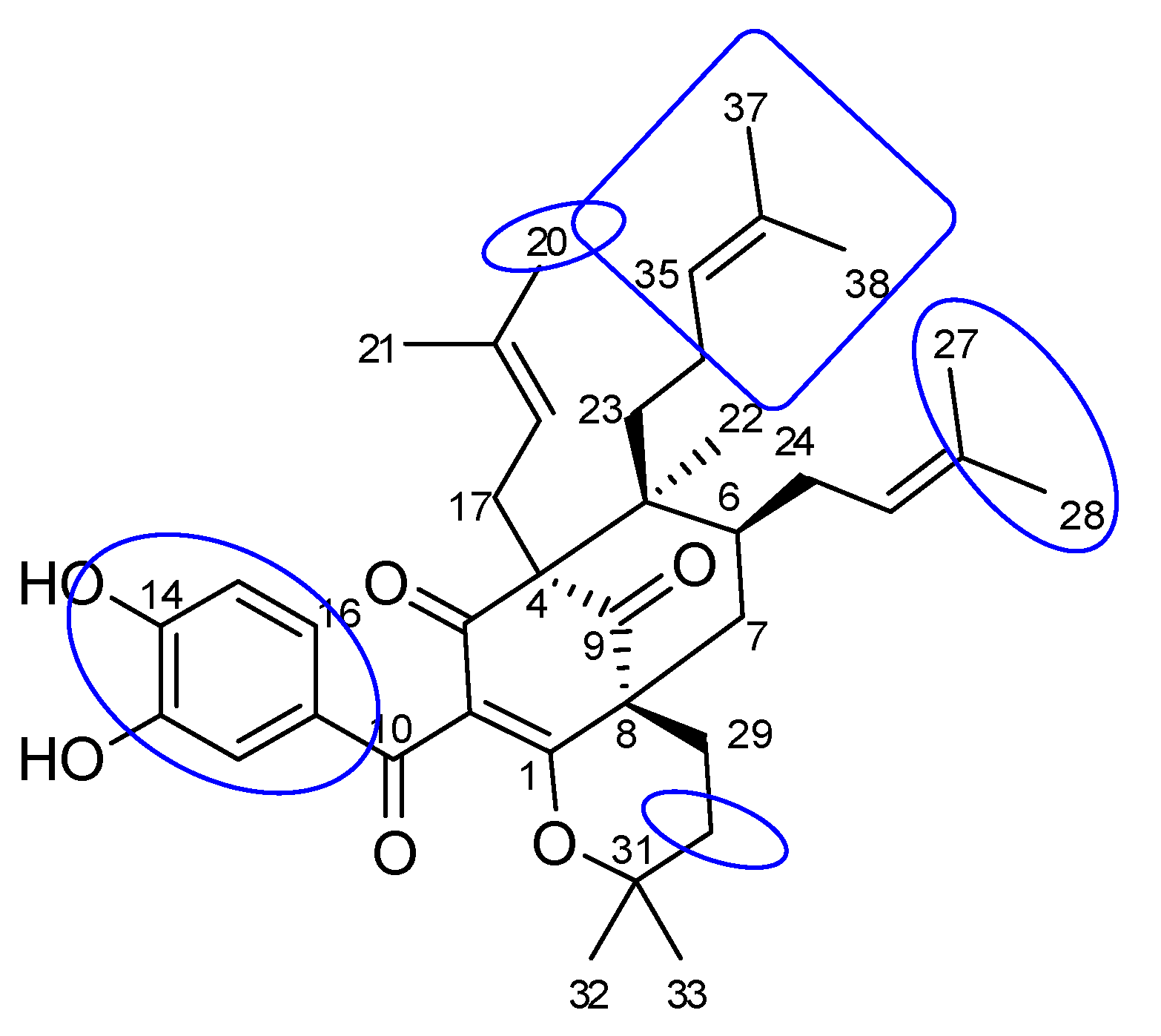
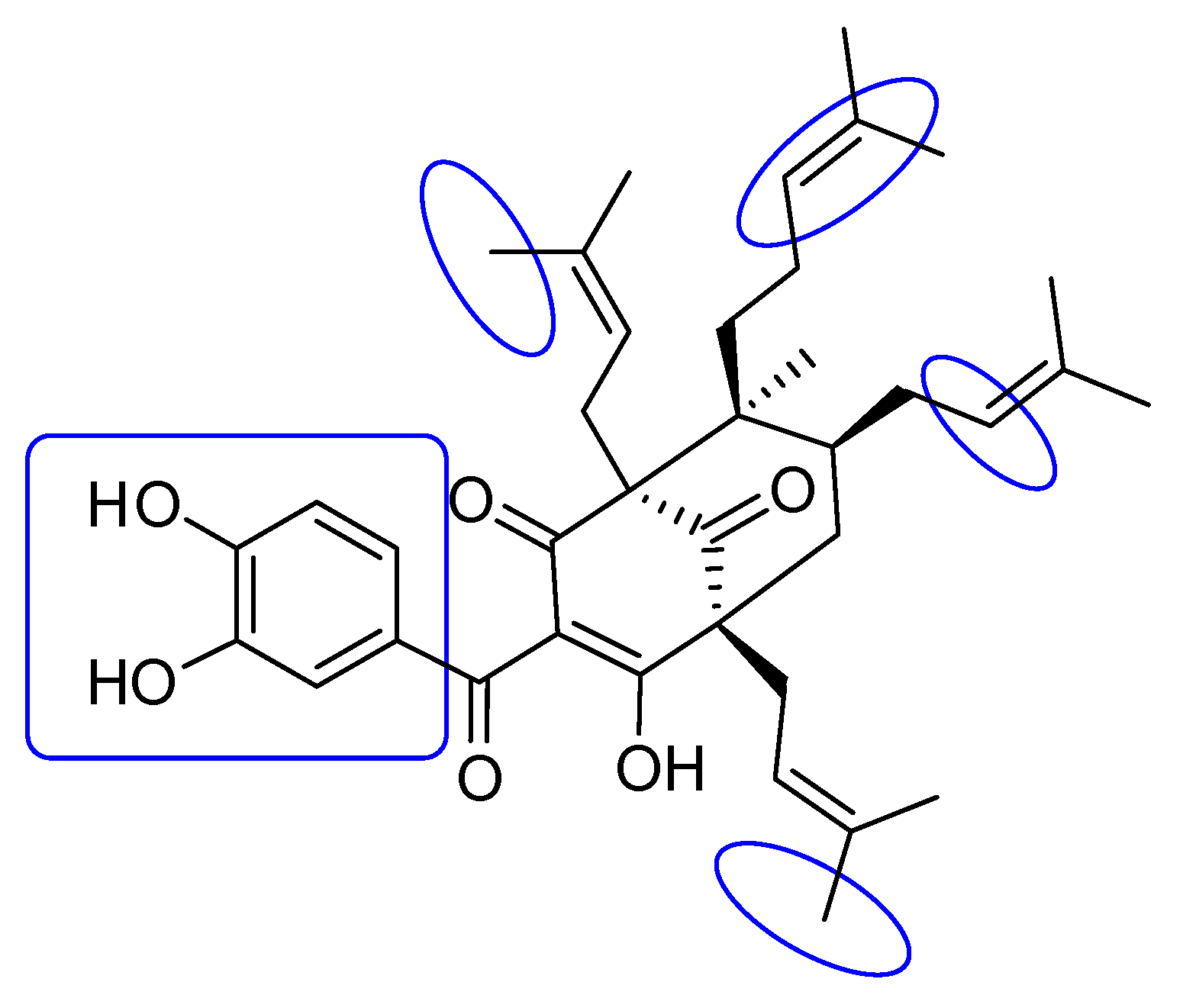
2.1. Phytochemical Identification
2.2. Biological Activities of Isolated Compounds
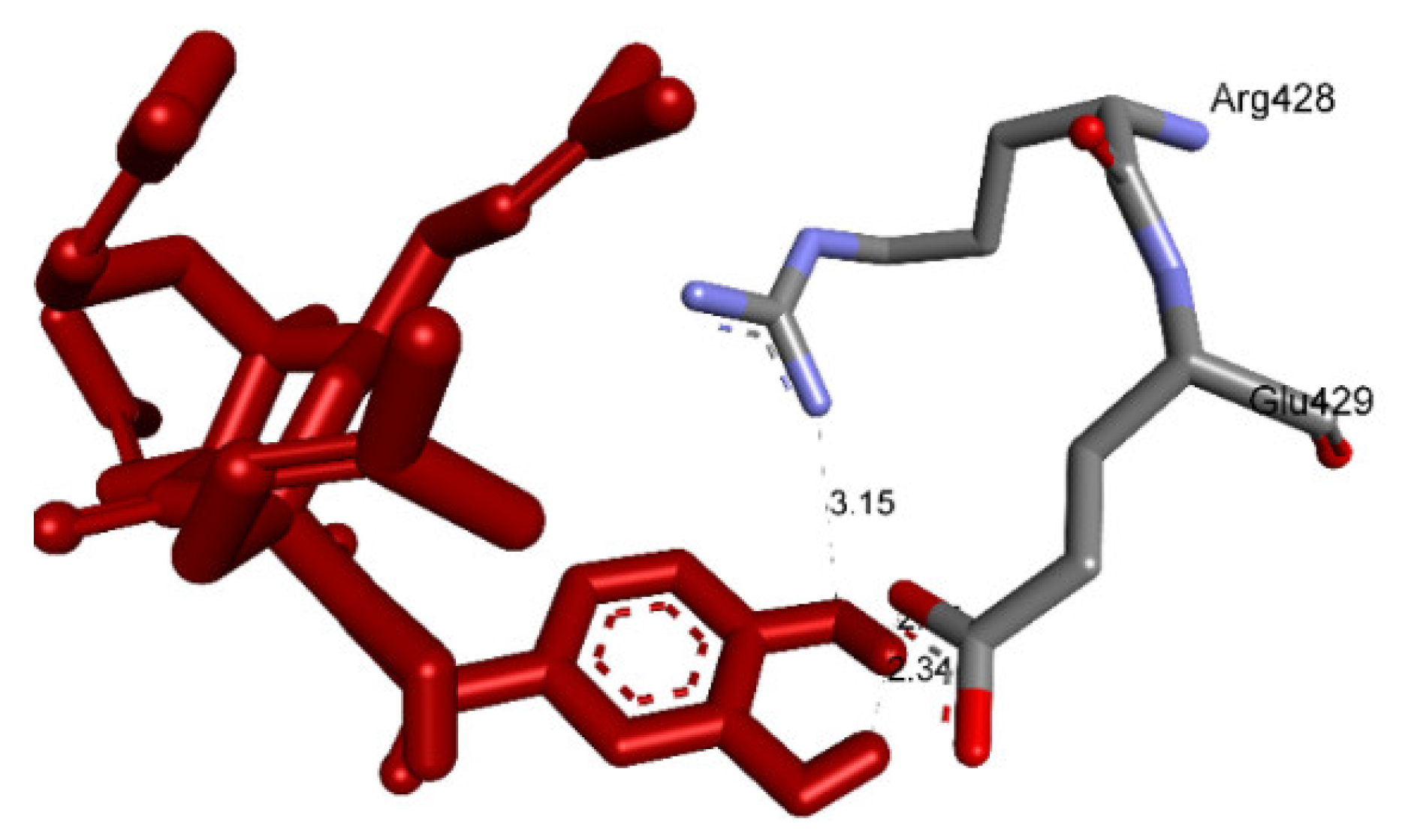
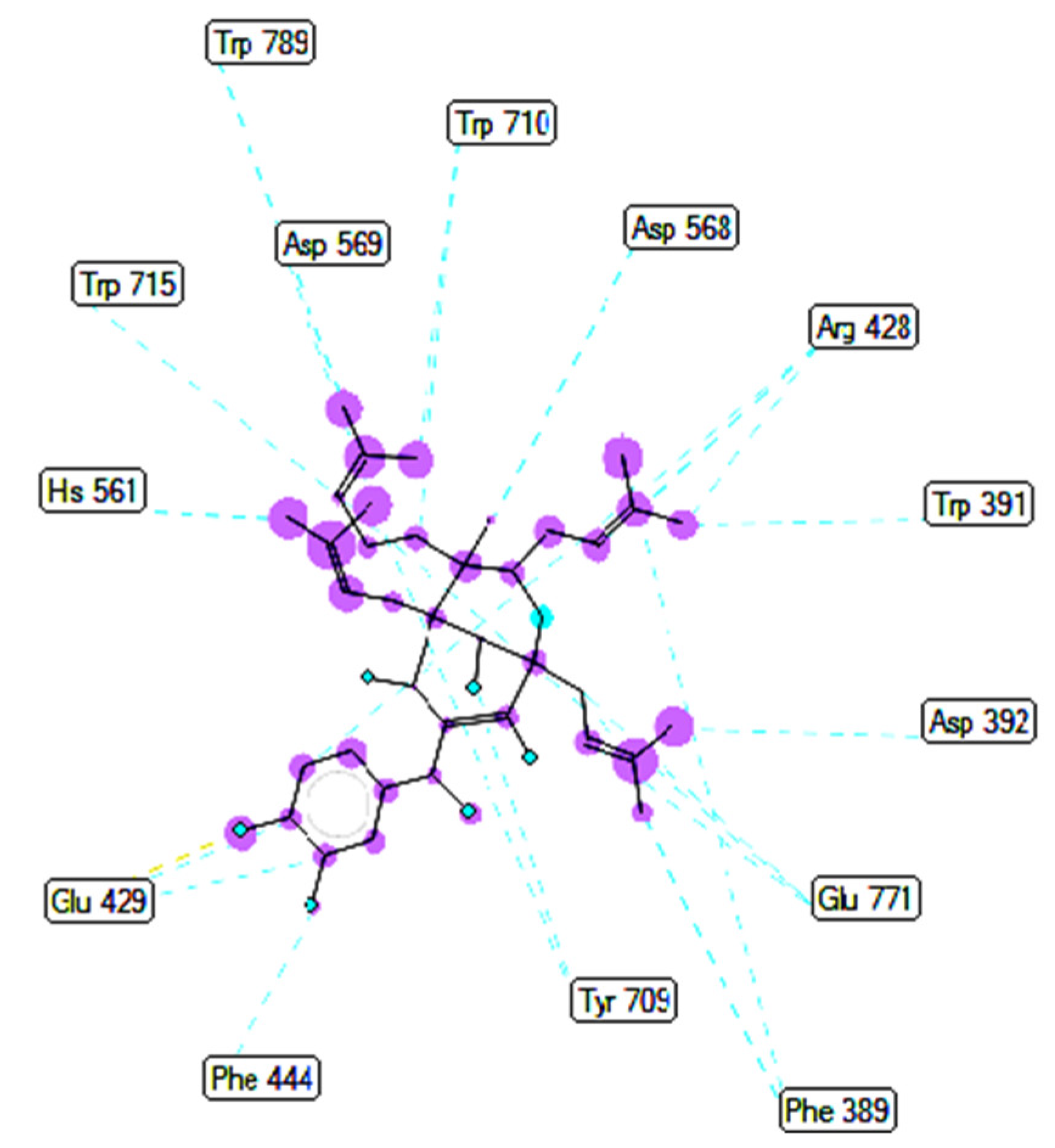
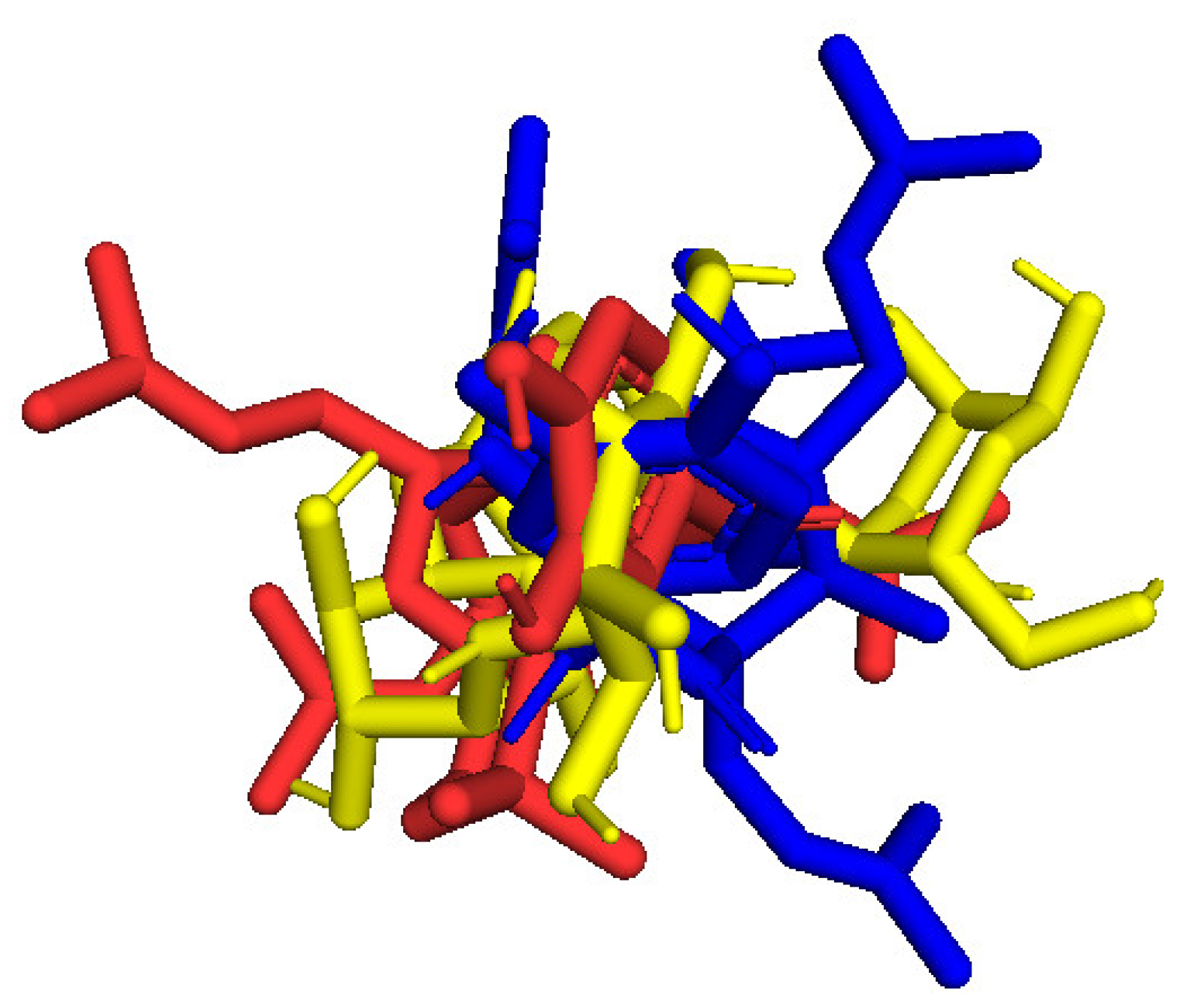
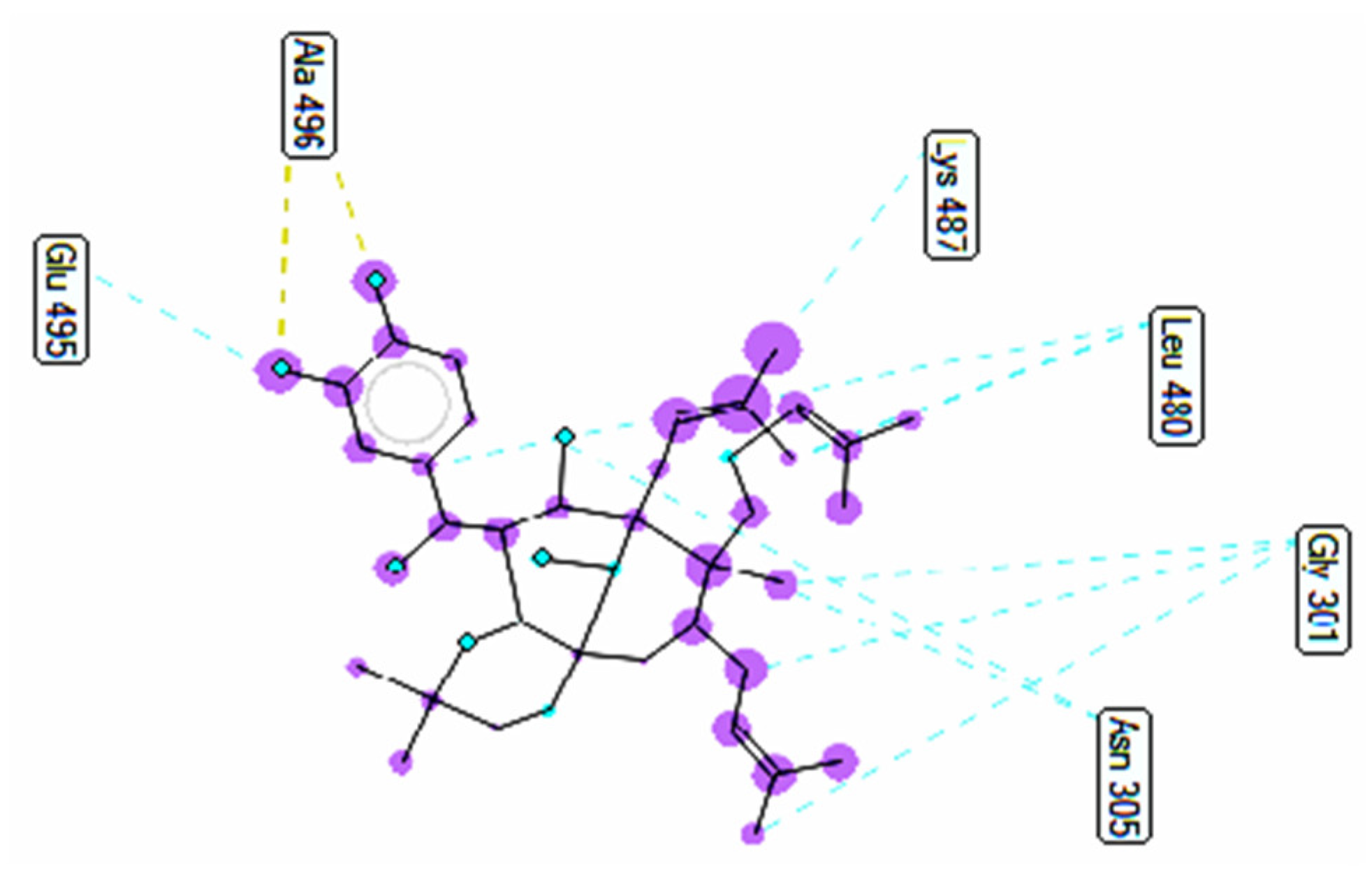
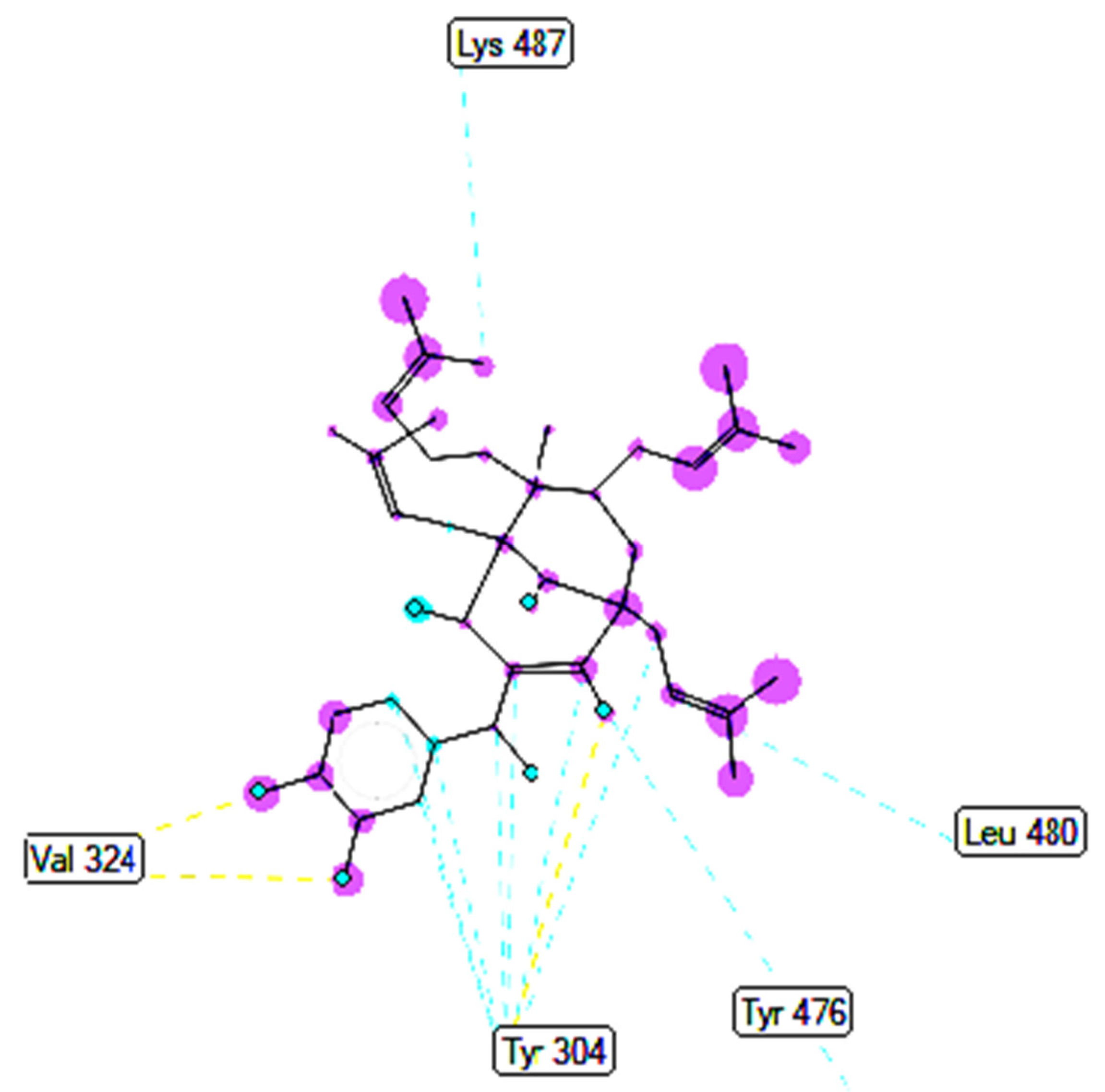
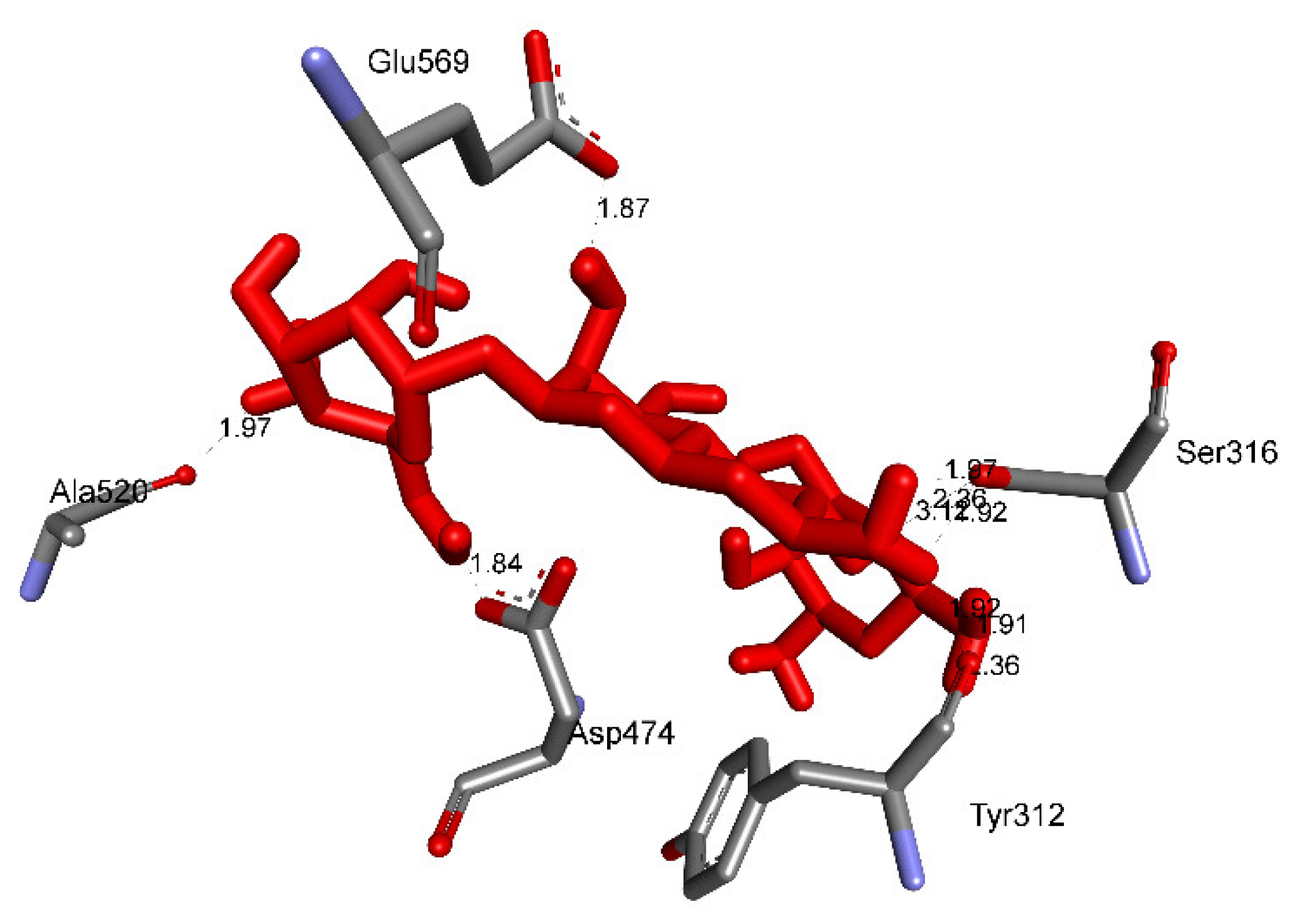
2.3. Molecular Docking Studies of Compounds 1 and 2
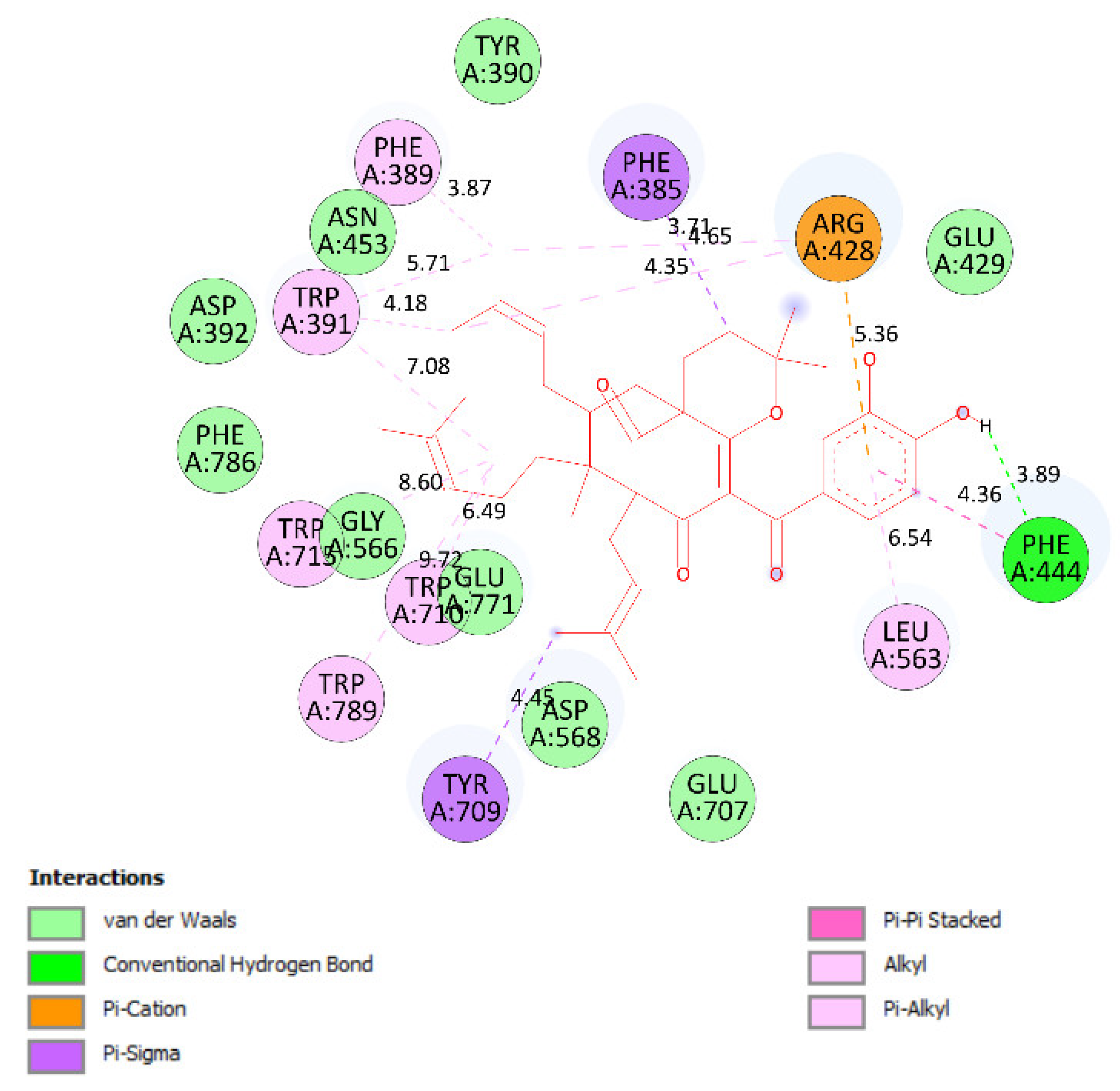
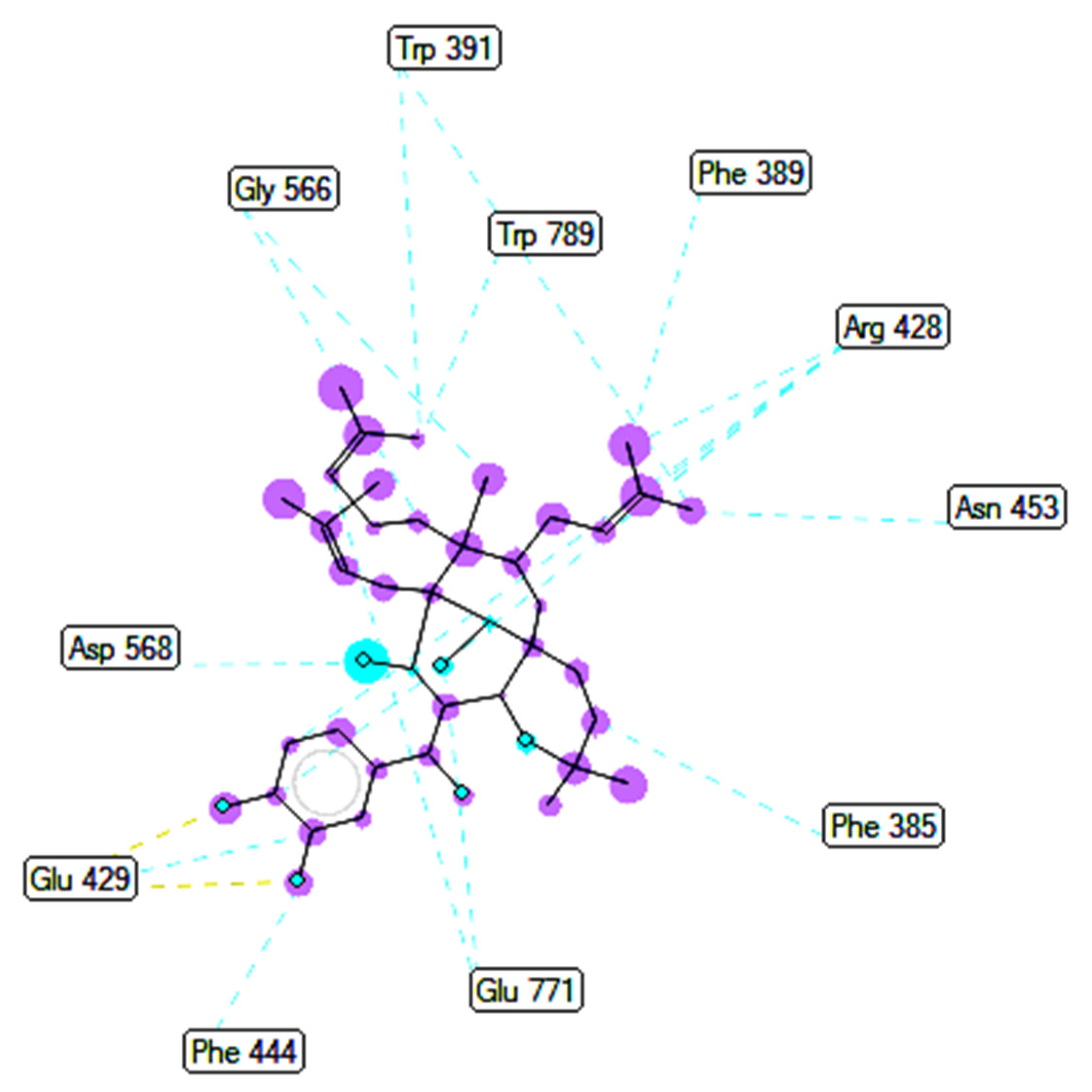
2.3.1. In Silico α-Glucosidase Enzyme Inhibition
2.3.2. In Silico Antimicrobial Activity
2.3.3. In Silico Physicochemical Properties, Drug-Likeness, and Pharmacokinetic Predictions
3. Materials and Methods
3.1. Source of the Plant Material
3.2. Isolation
3.3. α-Glucosidase Inhibition Assay
3.4. Antibacterial Activity Assay
3.5. Cytotoxicity Assay
3.6. Molecular Docking Studies and ADMET
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Zimmet, P.; Alberti, K.G.M.M.; Shaw, J. Global and Societal Implications of the Diabetes Epidemic. Nature 2001, 414, 782–787. [Google Scholar] [CrossRef] [PubMed]
- Tzoulaki, I.; Molokhia, M.; Curcin, V.; Little, M.P.; Millett, C.J.; Ng, A.; Hughes, R.I.; Khunti, K.; Wilkins, M.R.; Majeed, A.; et al. Risk of Cardiovascular Disease and All Cause Mortality among Patients with Type 2 Diabetes Prescribed Oral Antidiabetes Drugs: Retrospective Cohort Study Using UK General Practice Research Database. BMJ 2009, 339, b4731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rocha, S.; Ribeiro, D.; Fernandes, E.; Freitas, M. A Systematic Review on Anti-Diabetic Properties of Chalcones. Curr. Med. Chem. 2020, 27, 2257–2321. [Google Scholar] [CrossRef] [PubMed]
- Chandran, R.; Parimelazhagan, T.; Shanmugam, S.; Thankarajan, S. Antidiabetic Activity of Syzygium calophyllifolium in Streptozotocin-Nicotinamide Induced Type-2 Diabetic Rats. Biomed. Pharmacother. 2016, 82, 547–554. [Google Scholar] [CrossRef] [PubMed]
- Hollander, P. Safety Profile of Acarbose, an α-Glucosidase Inhibitor. Drugs 1992, 44, 47–53. [Google Scholar] [CrossRef] [PubMed]
- Chiasson, J.-L.; Josse, R.G.; Gomis, R.; Hanefeld, M.; Karasik, A.; Laakso, M. Acarbose for Prevention of Type 2 Diabetes Mellitus: The STOP-NIDDM Randomised Trial. Lancet 2002, 359, 2072–2077. [Google Scholar] [CrossRef]
- Kumar, S.; Narwal, S.; Kumar, V.; Prakashukne, O.; Viswanathan, V.; Phadatare, A. Alpha-Glucosidase Inhibitors from Plants: A Natural Approach to Treat Diabetes. Pharmacogn. Rev. 2011, 5, 19–29. [Google Scholar] [CrossRef] [Green Version]
- Antimicrobial Benzyltetrahydroisoquinoline-Derived Alkaloids from the Leaves of Doryphora aromatica. J. Nat. Prod. 2021, 84, 676–682. [CrossRef]
- Ingersoll, D.W.; Bronstein, P.M.; Bonventre, J. Chemical Modulation of Agonistic Display in Betta Splendens. J. Comp. Physiol. Psychol. 1976, 90, 198–202. [Google Scholar] [CrossRef]
- Bell, E.W.; Zhang, Y. DockRMSD: An Open-Source Tool for Atom Mapping and RMSD Calculation of Symmetric Molecules through Graph Isomorphism. J. Cheminform. 2019, 11, 40. [Google Scholar] [CrossRef] [Green Version]
- Tran, C.; Dao, T.; Tran, T.; Mai, D.; Tran, T. Alpha-Glucosidase Inhibitory Diterpenes from Euphorbia antiquorum Growing in Vietnam. Molecules 2021, 26, 2257. [Google Scholar] [CrossRef] [PubMed]
- Sichaem, J.; Aree, T.; Lugsanangarm, K.; Tip-Pyang, S. Identification of Highly Potent α-Glucosidase Inhibitory and Antioxidant Constituents from Zizyphus rugosa Bark: Enzyme Kinetic and Molecular Docking Studies with Active Metabolites. Pharm. Biol. 2017, 55, 1436–1441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, H.T.; Pham, D.D.; Nguyen, H.T.; Do, T.H.; Nguyen, N.H.; Duong, T.H. Design, Modification, and Bio-Evaluation of Salazinic Acid Derivatives. Arab. J. Chem. 2021, 15, 103535. [Google Scholar]
- Marti, G.; Eparvier, V.; Litaudon, M.; Grellier, P.; Guéritte, F. A New Xanthone from the Bark Extract of Rheedia Acuminata and Antiplasmodial Activity of Its Major Compounds. Molecules 2010, 15, 7106–7114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aljohny, B.O.; Rauf, A.; Anwar, Y.; Naz, S.; Wadood, A. Antibacterial, Antifungal, Antioxidant, and Docking Studies of Potential Dinaphthodiospyrols from Diospyros lotus Linn Roots. ACS Omega 2021, 6, 5878–5885. [Google Scholar] [CrossRef] [PubMed]
- Aamir, M.; Singh, V.K.; Dubey, M.K.; Meena, M.; Kashyap, S.P.; Katari, S.K.; Upadhyay, R.S.; Umamaheswari, A.; Singh, S. In silico Prediction, Characterization, Molecular Docking, and Dynamic Studies on Fungal SDRs as Novel Targets for Searching Potential Fungicides against Fusarium Wilt in Tomato. Front. Pharmacol. 2018, 9, 1038. [Google Scholar] [CrossRef] [PubMed]
- Eze, F.U.; Okoro, U.C.; Ugwu, D.I.; Okafor, S.N. Biological Activity Evaluation of Some New Benzenesulphonamide Derivatives. Front. Chem. 2019, 7, 634. [Google Scholar] [CrossRef]
- Staker, B.L.; Feese, M.D.; Cushman, M.; Pommier, Y.; Zembower, D.; Stewart, L.; Burgin, A.B. Structures of Three Classes of Anticancer Agents Bound to the Human Topoisomerase I-DNA Covalent Complex. J. Med. Chem. 2005, 48, 2336–2345. [Google Scholar] [CrossRef]
- Aliye, M.; Dekebo, A.; Tesso, H.; Abdo, T.; Eswaramoorthy, R.; Melaku, Y. Molecular Docking Analysis and Evaluation of the Antibacterial and Antioxidant Activities of the Constituents of Ocimum cufodontii. Sci. Rep. 2021, 11, 10101. [Google Scholar] [CrossRef]
- Zhang, H.J.; Wang, X.Z.; Cao, Q.; Gong, G.H.; Quan, Z.S. Design, Synthesis, Anti-Inflammatory Activity, and Molecular Docking Studies of Perimidine Derivatives Containing Triazole. Bioorg. Med. Chem. Lett. 2017, 27, 4409–4414. [Google Scholar] [CrossRef]
- 21 Lien Do, T.M.; Duong, T.-H.; Nguyen, V.-K.; Phuwapraisirisan, P.; Doungwichitrkul, T.; Niamnont, N.; Jarupinthusophon, S.; Sichaem, J. Schomburgkixanthone, a Novel Bixanthone from the Twigs of Garcinia schomburgkiana. Nat. Prod. Res. 2021, 35, 3613–3618. [Google Scholar] [CrossRef] [PubMed]
- Jarapula, R.; Gangarapu, K.; Manda, S.; Rekulapally, S. Synthesis, in Vivo Anti-Inflammatory Activity, and Molecular Docking Studies of New Isatin Derivatives. Int. J. Med. Chem. 2016, 2016, 2181027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaennakam, S.; Sukandar, E.R.; Juntagoot, T.; Siripong, P.; Tip-pyang, S. Four New Xanthones and Their Cytotoxicity from the Stems of Garcinia schomburgkiana. J. Nat. Med. 2021, 75, 871–876. [Google Scholar] [CrossRef] [PubMed]
- Le, D.H.; Nishimura, K.; Takenaka, Y.; Mizushina, Y.; Tanahashi, T. Polyprenylated Benzoylphloroglucinols with DNA Polymerase Inhibitory Activity from the Fruits of Garcinia schomburgkiana. J. Nat. Prod. 2016, 79, 1798–1807. [Google Scholar] [CrossRef]
- Marti, G.; Eparvier, V.; Moretti, C.; Prado, S.; Grellier, P.; Hue, N.; Thoison, O.; Delpech, B.; Guéritte, F.; Litaudon, M. Antiplasmodial Benzophenone Derivatives from the Root Barks of Symphonia globulifera (Clusiaceae). Phytochemistry 2010, 71, 964–974. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Yu, T.; Gao, X.-M.; Zhou, Y.; Qiao, C.-F.; Peng, Y.; Chen, S.-L.; Luo, K.Q.; Xu, H.-X. Apoptotic Effects of Polyprenylated Benzoylphloroglucinol Derivatives from the Twigs of Garcinia multiflora. J. Nat. Prod. 2010, 73, 1355–1359. [Google Scholar] [CrossRef] [PubMed]
- Casero, C.; Machín, F.; Méndez-Álvarez, S.; Demo, M.; Ravelo, Á.G.; Pérez-Hernández, N.; Joseph-Nathan, P.; Estévez-Braun, A. Structure and Antimicrobial Activity of Phloroglucinol Derivatives from Achyrocline satureioides. J. Nat. Prod. 2015, 78, 93–102. [Google Scholar] [CrossRef]
- Nganou, B.K.; Simo Konga, I.; Fankam, A.G.; Bitchagno, G.T.M.; Sonfack, G.; Nayim, P.; Celik, I.; Koyutürk, S.; Kuete, V.; Tane, P. Guttiferone BL with Antibacterial Activity from the Fruits of Allanblackia gabonensis. Nat. Prod. Res. 2019, 33, 2638–2646. [Google Scholar] [CrossRef]
- Crockett, S.L.; Kunert, O.; Pferschy-Wenzig, E.-M.; Jacob, M.; Schuehly, W.; Bauer, R. Phloroglucinol and Terpenoid Derivatives from Hypericum cistifolium and H. galioides (Hypericaceae). Front. Plant Sci. 2016, 7, 961. [Google Scholar] [CrossRef] [Green Version]
- Hidalgo-Figueroa, S.; Rodríguez-Luévano, A.; Almanza-Pérez, J.C.; Giacoman-Martínez, A.; Ortiz-Andrade, R.; León-Rivera, I.; Navarrete-Vázquez, G. Synthesis, Molecular Docking, Dynamic Simulation and Pharmacological Characterization of Potent Multifunctional Agent (Dual GPR40-PPARγ Agonist) for the Treatment of Experimental Type 2 Diabetes. Eur. J. Pharmacol. 2021, 907, 174244. [Google Scholar] [CrossRef]
- Duong, T.-H.; Nguyen, H.T.; Nguyen, C.H.; Tran, N.-M.-A.; Danova, A.; Tran, T.-M.-D.; Vu-Huynh, K.L.; Musa, V.; Jutakanoke, R.; Nguyen, N.-H.; et al. Identification of Highly Potent α-Glucosidase Inhibitors from Artocarpus Integer and Molecular Docking Studies. Chem. Biodivers. 2021, 18, e2100499. [Google Scholar] [CrossRef] [PubMed]
- Sichaem, J.; Nguyen, H.-H.; Nguyen, V.-H.; Mac, D.-H.; Mai, D.-T.; Nguyen, H.-C.; Tran, T.-N.-M.; Pham, N.-K.-T.; Nguyen, H.-H.; Niamnont, N.; et al. A New Labdane-Type Diterpenoid from the Leaves of Vitex negundo L. Nat. Prod. Res. 2021, 35, 2329–2334. [Google Scholar] [CrossRef] [PubMed]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No | 1 | 2 | ||
|---|---|---|---|---|
| δH | δC | δH | δC | |
| 1 | 179.0 | 199.3 | ||
| 2 | 116.6 | 116.9 | ||
| 3 | 189.8 | 198.6 | ||
| 4 | 72.7 | 69.6 | ||
| 5 | 49.1 | 49.4 | ||
| 6 | 2.14 (m) | 37.8 | 1.61 (m) | 37.8 |
| 7 | 2.54 (m); 1.44 (m) | 39.7 | 2.05 (m); 1.46 (m) | 40.0 |
| 8 | 49.0 | 64.7 | ||
| 9 | 208.4 | 207.0 | ||
| 10 | 191.7 | 195.6 | ||
| 11 | 131.3 | 129.0 | ||
| 12 | 7.38 (d, 2.0) | 116.1 | 7.21 (d, 2.0) | 116.6 |
| 13 | 150.6 | 150.4 | ||
| 14 | 146.8 | 144.5 | ||
| 15 | 6.85 (d, 8.5) | 115.5 | 6.82 (d, 8.5) | 114.0 |
| 16 | 7.19 (dd, 8.5, 2.0) | 125.6 | 7.21 (dd, 8.5, 2.0) | 124.5 |
| 17 | 2.66 (dd, 13.5, 7.5); 2.50 (m) | 24.7 | 2.81 (dd, 14.5, 9.0); 2.69 (m) | 25.6 |
| 18 | 4.84 (brt) | 122.2 | 4.94 (brt) | 120.0 |
| 19 | 133.1 | 132.9 | ||
| 20 | 1.64 (s) | 25.8 | 1.67 (s) | 25.0 |
| 21 | 1.59 (s) | 17.8 | 1.59 (s) | 17.4 |
| 22 | 0.82 (s) | 15.5 | 0.88 (s) | 16.9 |
| 23 | 1.81 (m); 1.78 (m) | 36.0 | 1.87 (m); 1.78 (m) | 36.0 |
| 24 | 2.14 (m); 1.98 (m) | 24.0 | 2.14 (m); 2.02 (m) | 24.0 |
| 25 | 5.04 (brt) | 123.7 | 5.03 (brt) | 124.5 |
| 26 | 133.8 | 131.0 | ||
| 27 | 1.75 (s) | 25.0 | 1.67 (s) | 25.0 |
| 28 | 1.66 (s) | 17.2 | 1.61 (s) | 17.2 |
| 29 | 2.53 (m) | 25.8 | 2.57 (m); 2.53 (m) | 30.6 |
| 30 | 1.43 (m) | 29.1 | 5.10 (brt) | 120.4 |
| 31 | 82.0 | 133.8 | ||
| 32 | 1.25 (s) | 31.4 | 1.72 (s) | 25.3 |
| 33 | 1.01 (s) | 26.7 | 1.69 (s) | 17.3 |
| 34 | 2.26 (m); 1.91 (m) | 31.0 | 2.14 (m); 2.02 (m) | 31.8 |
| 35 | 5.21 (brt) | 125.6 | 5.20 (brt) | 122.6 |
| 36 | 131.7 | 132.9 | ||
| 37 | 1.63 (s) | 25.2 | 1.72 (s) | 25.2 |
| 38 | 1.59 (s) | 17.3 | 1.67 (s) | 17.3 |
| Entry | Active Pose | Affinity Energy (a) | Ki (b) | The Number of Hydrogen Bonds (c) | The Property and Bond Length (d) |
|---|---|---|---|---|---|
| Compound 1 | 148/200 | −10.51 | 0.02 | 2 | A:Arg428:N−Compound 1:O (3.01 Å) Compound 1:H−A:Glu429:O (2.39Å) |
| Compound 2 | 41/200 | −10.12 | 0.04 | 3 | A:Arg428:N−Compound 2:O (3.16 Å) Compound 2:H−A:Glu 429:O (2.15 Å) Compound 2:H−A:Glu 429:O (2.34 Å) |
| Acarbose | 170/200 | −5.22 | 149.6 | 10 | A: Tyr709:O−Acarbose:O (3.08 Å) A:Trp710:N−Acarbose:O (2.69 Å) Acarbose:H−A:Tyr709:OH (2.26 Å) Acarbose:H−A:Glu771:O (2.14 Å) Acarbose:H−A:Gly566:O (2.12 Å) Acarbose:H−A:Asp 392:O (1.97 Å) Acarbose:H−A:Glu771:O (2.23 Å) Acarbose:H−A:Glu771:O (2.43 Å) Acarbose:H−A:Asp392:O (2.48 Å) Acarbose 1:H−A:Asp392:O(2.08 Å) |
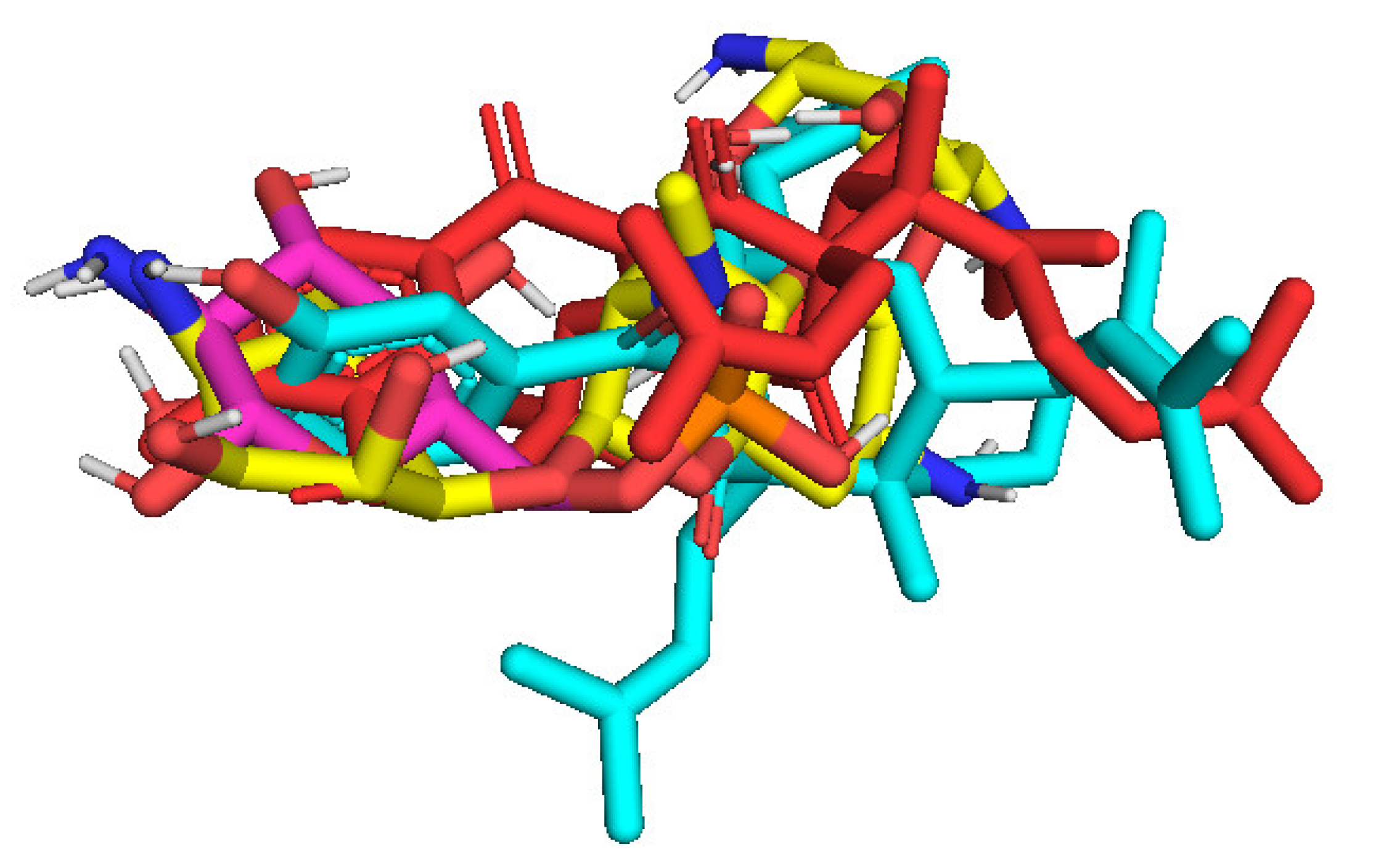
| RMSD (Å) | Pose 148 | Pose 41 | Pose 170 |
|---|---|---|---|
| Pose 170, a reference pose | 2.28 | 4.094 | 0 |
| Entry | Active Pose | Affinity Energy (a) | Ki (b) | The Number of Hydrogen Bonds (c) | The Property and Bond Length (d) |
|---|---|---|---|---|---|
| Compound 1 | 158 | −8.56 | 0.53 | 3 | X:Ala 496:N−Compound 1:O (3.2 Å) Compound 1:H−X:Ala496:O (1.92 Å) Compound 1:H−X:Ala 496:O (2.03 Å) |
| Compound 2 | 35 | −6.24 | 26.84 | X:Tyr304:OH−Compound 2:O (3.04 Å) Compound 2:H−X:Val 324:O (1.93 Å) Compound 2:H−X:Val 324:O (2.14 Å) | |
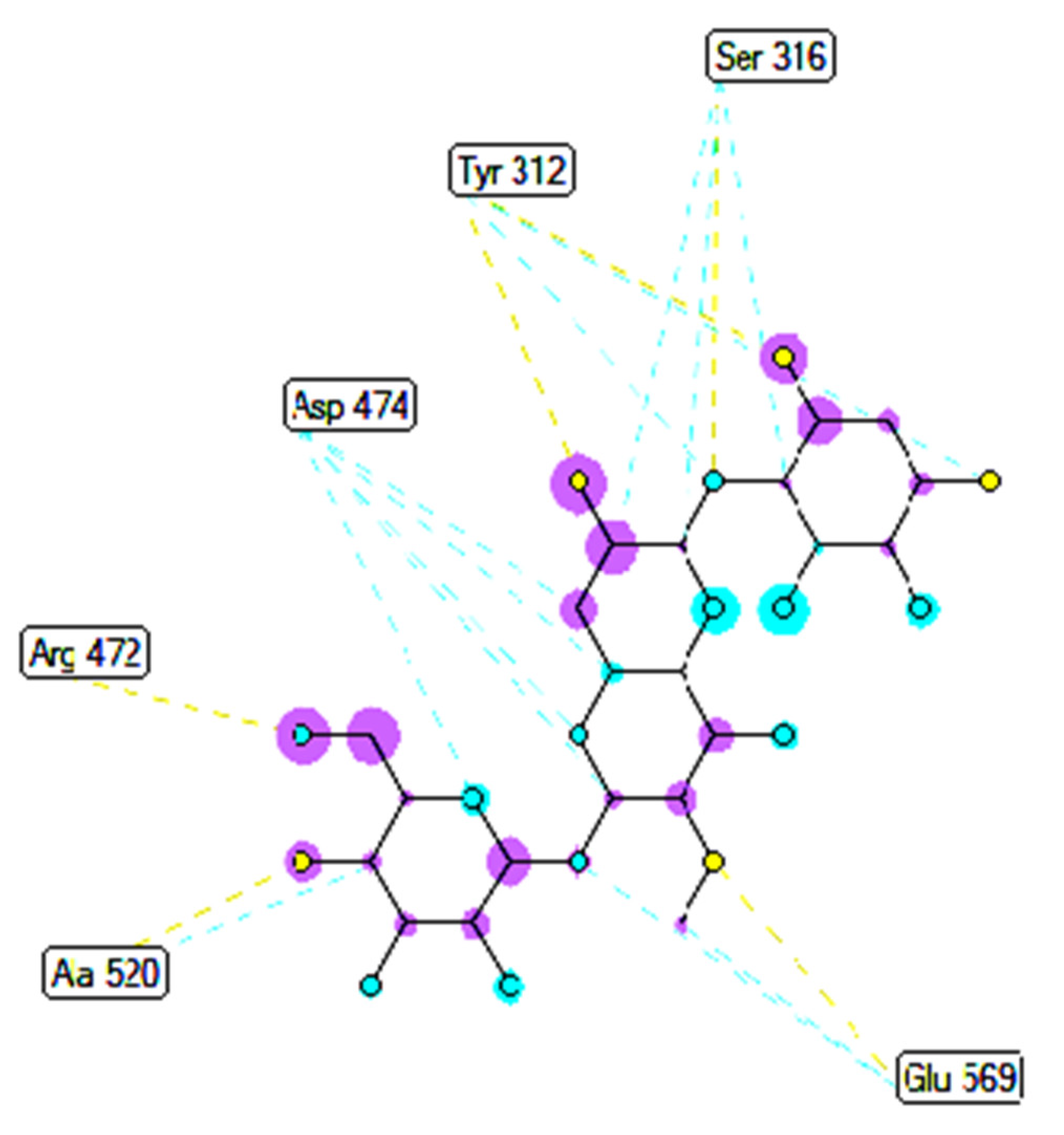
| Apramycin | 172 | −6.94 | 8.17 | 10 | X:Ser316:O−Apramycin:N (2.36 Å) X:Ser316:O−Apramycin:O (3.12 Å) Apramycin:H−X:Ala 520:O (1.96 Å) Apramycin:H−X:Asp474:OD1 (1.84 Å) Apramycin:H−X:Glu569:OE2 (1.87 Å) Apramycin:H−X:Tyr312:O (1.91 Å) Apramycin:H−X:Ser316:OG (1.92 Å) Apramycin:H −X:Ser316:OG (1.97 Å) Apramycin:H−X:Tyr312:O (1.91 Å) Apramycin:H−X:Tyr312:O (2.36 Å) |
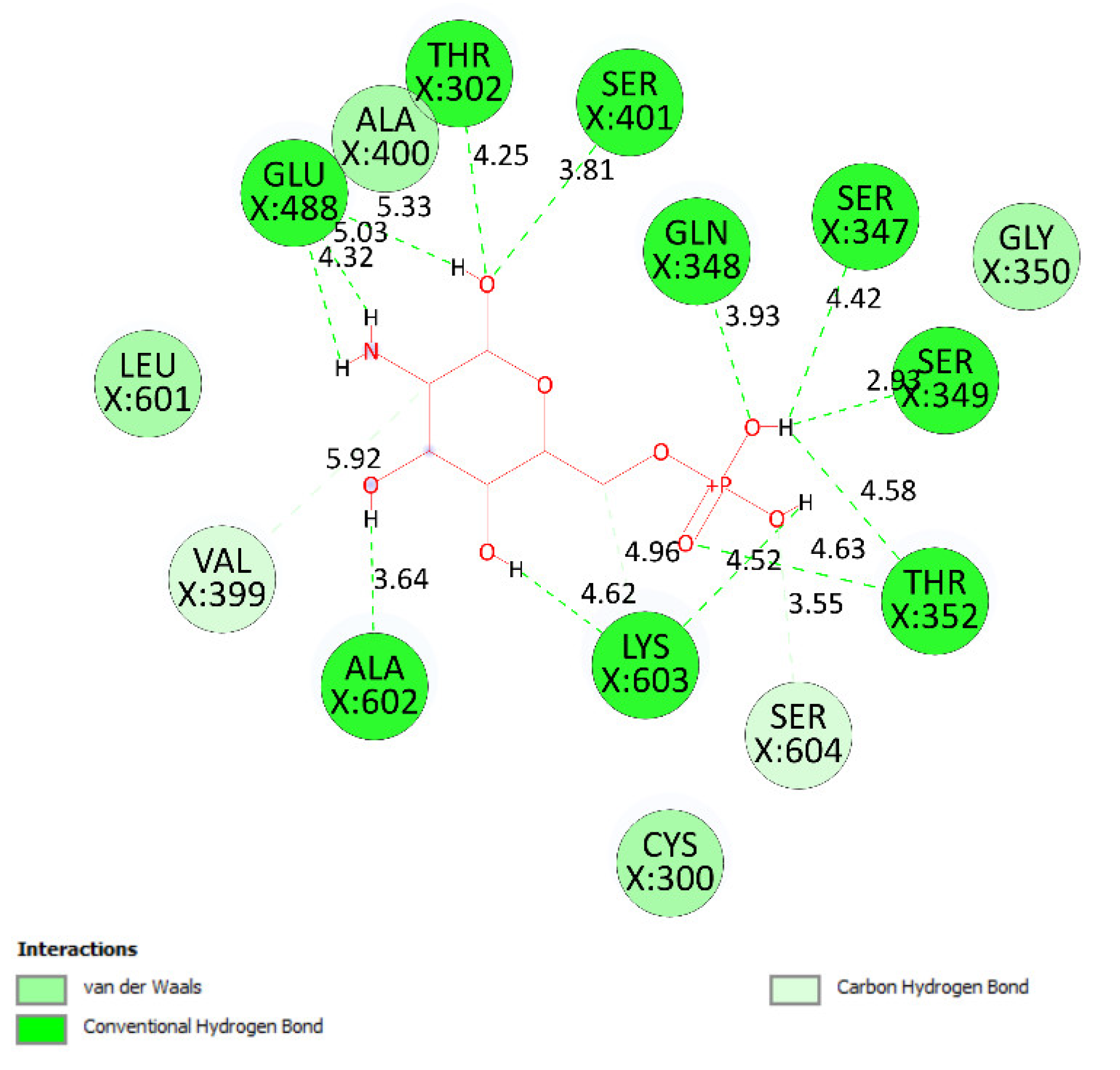
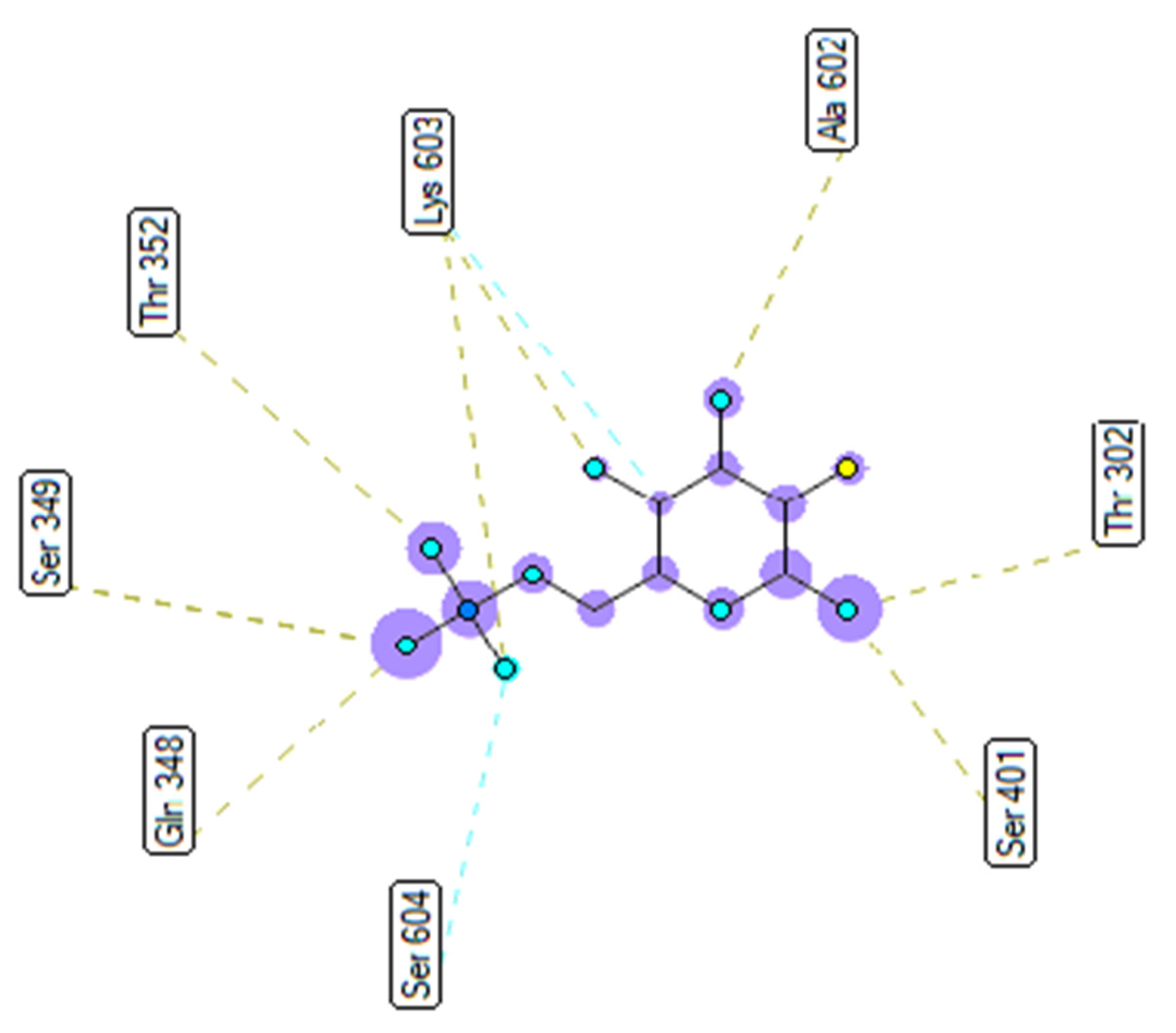
| Small ligand | 83 | −5.38 | 114 | 13 | X:Thr 302:O−Small ligand:O (2.86 Å) X:Gln 348:N−Small ligand:O (3.11 Å) X:Ser 349:N−Small ligand:O (2.95 Å) X:Ser 349:O−Small ligand:O (2.66 Å) X:Thr 352:O−Small ligand:O (3.00 Å) X:Ser 401:N−Small ligand:O (3.06 Å) Small ligand:H−X:Glu 488:O (2.03 Å) Small ligand:H−X:Glu 488:O (1.88 Å) Small ligand:H−X:Glu 488:O (2.05 Å) Small ligand:H−X:Ala 602:O (1.88 Å) Small ligand:H−X:Ser 349:O (1.90 Å) Small ligand:H−X:Lys 603:O (2.15 Å) Small ligand:H−X:Lys 603:O (1.92 Å) |
| MSD (Å) | Pose 158 | Pose 35 | Pose 83 | Pose 172 |
|---|---|---|---|---|
| Pose 172, a standard drug, a reference pose | 3.32 | 2.166 | 2.839 | 0 |
| Property | Value | Comment |
|---|---|---|
| Molecular weight | 602.36 | Contain hydrogen atoms. Optimal: 100–600 |
| Volume | 657.955 | Van der Waals volume |
| nHA | 6 | Number of hydrogen bond acceptors. Optimal: 0–12 |
| nHD | 2 | Number of hydrogen bond donors. Optimal: 0–7 |
| nRot | 9 | Number of rotatable bonds. Optimal: 0–11 |
| MaxRing | 12 | Number of atoms in the biggest ring. Optimal: 0–18 |
| nHet | 6 | Number of heteroatoms. Optimal: 1–15 |
| fChar | 0 | Formal charge. Optimal: −4–4 |
| nRig | 27 | Number of rigid bonds. Optimal: 0–30 |
| Flexibility | 0.333 | Flexibility = nRot/nRig |
| Stereocenters | 4 | Optimal: ≤2 |
| Optimal: ≤2 | 100.9 | Topological polar surface area. Optimal: 0–140 |
| logS | −4.218 | Log of the aqueous solubility. Optimal: −4–0.5 log mol/L |
| logP | 8.424 | Log of the octanol/water partition coefficient. Optimal: 0–3 |
| logD | 5.451 | logP at physiological pH 7.4. Optimal: 1–3 |
| Property | Value | Comment |
|---|---|---|
| QED | 0.096 | A measure of drug-likeness based on the concept of desirability; attractive: >0.67; unattractive: 0.49~0.67; too complex: <0.34. |
| SAscore | 6.097 | Synthetic accessibility score is designed to estimate ease of synthesis of drug-like molecules. SAscore ≥ 6, difficult to synthesize; SAscore < 6, easy to synthesize. |
| Fsp3 | 0.553 | The number of sp3 hybridized carbons/total carbon count, correlating with melting point and solubility. Fsp3 ≥ 0.42 is considered a suitable value. |
| MCE-18 | 153.763 | MCE-18 stands for medicinal chemistry evolution. MCE-18 ≥ 45 is considered a suitable value. |
| NPscore | 2.295 | Natural-product-likeness score. This score is typically in the range of −5 to 5. The higher the score is, the higher the probability is that the molecule is an NP. |
| Lipinski Rule | Rejected | MW ≤ 500; logP ≤ 5; Hacc ≤ 10; Hdon ≤ 5. If two properties are out of range, a poor absorption or permeability is possible; one property being out of range is acceptable. |
| Pfizer Rule | Accepted | logP > 3; TPSA < 75; compounds with a high log P (>3) and low TPSA (<75) are likely to be toxic. |
| GSK Rule | Rejected | MW ≤ 400; logP ≤ 4; compounds satisfying the GSK rule may have a more favorable ADMET profile. |
| Golden Triangle | Rejected | 200 ≤ MW ≤ 500; −2 ≤logD ≤ 5; compounds satisfying the Golden Triangle rule may have a more favorable ADMET profile. |
| PAINS | 1 alert | Pan-assay interference compounds, frequent hitters, α-screen artifacts and reactive compound. |
| ALARM NMR | 4 alerts | Thiol reactive compounds. |
| BMS | 0 alerts | Undesirable, reactive compounds. |
| Chelator Rule | 2 alerts | Chelating compounds. |
| Property | Value | Comment |
|---|---|---|
| Caco-2 Permeability | −4.852 | Optimal: higher than −5.15 log unit |
| MDCK Permeability | 1.6e−05 | Low permeability: <2 × 10−6 cm/s Medium permeability: 2–20 × 10−6 cm/s High passive permeability: >20 × 10−6 cm/s |
| Pgp-inhibitor | 0.175 | Category 1: inhibitor; Category 0: non-inhibitor; the output value is the probability of being Pgp-inhibitor |
| Pgp-substrate | 0.021 | Category 1: substrate; Category 0: non-substrate; the output value is the probability of being Pgp-substrate |
| HIA | 0.038 | Human intestinal absorption; Category 1: HIA+(HIA < 30%); Category 0: HIA-(HIA < 30%); the output value is the probability of being HIA+ |
| Property | Value | Comment |
|---|---|---|
| PPB | 95.95% | Plasma protein binding; optimal: <90%. Drugs with high protein binding may have a low therapeutic index. |
| VD | 0.994 | Volume distribution; optimal: 0.04–20 L/kg. |
| BBB Penetration | 0.012 | Blood–brain barrier penetration; Category 1: BBB+; Category 0: BBB−; the output value is the probability of being BBB+. |
| Fu | 7.588% | The fraction unbound in plasma; low: <5%; middle: 5~20%; high: >20%. |
| Property | Value | Comment |
|---|---|---|
| CYP1A2 inhibitor | 0.1 | Category 1: inhibitor; Category 0: non-inhibitor; the output value is the probability of being an inhibitor. |
| CYP1A2 substrate | 0.26 | Category 1: substrate; Category 0: non-substrate; the output value is the probability of being a substrate |
| CYP2C19 inhibitor | 0.802 | Category 1: inhibitor; Category 0: non-inhibitor; the output value is the probability of being an inhibitor |
| CYP2C19 substrate | 0.352 | Category 1: substrate; Category 0: non-substrate; the output value is the probability of being a substrate |
| CYP2C9 inhibitor | 0.762 | Category 1: inhibitor; Category 0: non-inhibitor; the output value is the probability of being an inhibitor. |
| CYP2C9 substrate | 0.882 | Category 1: substrate; Category 0: non-substrate; the output value is the probability of being a substrate. |
| CYP2D6 inhibitor | 0.905 | Category 1: inhibitor; Category 0: non-inhibitor; the output value is the probability of being an inhibitor. |
| CYP2D6 substrate | 0.022 | Category 1: substrate; Category 0: non-substrate; the output value is the probability of being a substrate. |
| CYP3A4 inhibitor | 0.866 | Category 1: inhibitor; Category 0: non-inhibitor; the output value is the probability of being an inhibitor. |
| CYP3A4 substrate | 0.854 | Category 1: substrate; Category 0: non-substrate; the output value is the probability of being a substrate. |
| Property | Value | Comment |
|---|---|---|
| CL | 20.163 | Clearance; high: >15 mL/min/kg; moderate: 5–15 mL/min/kg; low: <5 mL/min/kg. |
| T1/2 | 0.027 | Category 1: long half-life; Category 0: short half-life; long half-life: >3 h; short half-life: <3 h; the output value is the probability of having a long half-life. |
| Property | Value | Comment |
|---|---|---|
| hERG Blockers | 0.004 | Category 1: active; Category 0: inactive; the output value is the probability of being active. |
| H-HT | 0.956 | Human hepatotoxicity; Category 1: H-HT positive (+); Category 0: H-HT negative (−); the output value is the probability of being toxic. |
| DILI | 0.853 | Drug-induced liver injury. Category 1: drugs with a high risk of DILI; Category 0: drugs with no risk of DILI. The output value is the probability of being toxic. |
| AMES Toxicity | 0.014 | Category 1: AMES positive (+); Category 0: AMES negative (−); the output value is the probability of being toxic. |
| Rat Oral Acute Toxicity | 0.432 | Category 0: low toxicity; Category 1: high toxicity; the output value is the probability of being highly toxic. |
| FDAMDD | 0.037 | Maximum recommended daily dose; Category 1: FDAMDD (+); Category 0: FDAMDD (−); the output value is the probability of being positive. |
| Skin Sensitization | 0.013 | Category 1: sensitizer; Category 0: non-sensitizer; the output value is the probability of being a sensitizer. |
| Carcinogencity | 0.539 | Category 1: carcinogens; Category 0: non-carcinogens; the output value is the probability of being toxic. |
| Eye corrosion | 0.003 | Category 1: corrosive; Category 0: noncorrosive; the output value is the probability of being corrosive. |
| Eye irritation | 0.025 | Category 1: irritants; Category 0: non-irritants; the output value is the probability of being an irritant. |
| Respiratory Toxicity | 0.942 | Category 1: respiratory toxicants; Category 0: respiratory non-toxicants; the output value is the probability of being toxic. |
| Property | Value | Comment |
|---|---|---|
| Bioconcentration Factors | 0.444 | Bioconcentration factors are used for considering secondary poisoning potential and assessing risks to human health via the food chain. The unit is −log10((mg/L)/(1000 × MW)). |
| IGC50 | 4.596 | Tetrahymena pyriformis 50 percent growth inhibition concentration; the unit is −log10((mg/L)/(1000 × MW)). |
| LC50FM | 6.123 | 96-h fathead minnow 50 percent lethal concentration; the unit is −log10((mg/L)/(1000 × MW)). |
| LC50DM | 6.105 | 48-h daphnia magna 50 percent lethal concentration; the unit is −log10((mg/L)/(1000 × MW)). |
| Property | Value | Comment |
|---|---|---|
| Acute Toxicity Rule | 0 alerts | 20 substructures; acute toxicity during oral administration |
| Genotoxic Carcinogenicity Rule | 1 alert | 117 substructures; carcinogenicity or mutagenicity |
| Nongenotoxic Carcinogenicity Rule | 0 alerts | 23 substructures; carcinogenicity through nongenotoxic mechanisms |
| Skin Sensitization Rule | 12 alerts | 155 substructures; skin irritation |
| Aquatic Toxicity Rule | 3 alerts | 99 substructures; toxicity to liquid (water) |
| Nonbiodegradable Rule | 3 alerts | 19 substructures; nonbiodegradable |
| SureChEMBL Rule | 0 alerts | 164 substructures; MedChem unfriendly status |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nguyen, H.T.; Nguyen, T.-T.; Duong, T.-H.; Tran, N.-M.-A.; Nguyen, C.H.; Nguyen, T.-H.-A.; Sichaem, J. α-Glucosidase Inhibitory and Antimicrobial Benzoylphloroglucinols from Garcinia schomburgakiana Fruits: In Vitro and In Silico Studies. Molecules 2022, 27, 2574. https://doi.org/10.3390/molecules27082574
Nguyen HT, Nguyen T-T, Duong T-H, Tran N-M-A, Nguyen CH, Nguyen T-H-A, Sichaem J. α-Glucosidase Inhibitory and Antimicrobial Benzoylphloroglucinols from Garcinia schomburgakiana Fruits: In Vitro and In Silico Studies. Molecules. 2022; 27(8):2574. https://doi.org/10.3390/molecules27082574
Chicago/Turabian StyleNguyen, Huy Truong, Thanh-Trung Nguyen, Thuc-Huy Duong, Nguyen-Minh-An Tran, Chuong Hoang Nguyen, Thi-Hong-Anh Nguyen, and Jirapast Sichaem. 2022. "α-Glucosidase Inhibitory and Antimicrobial Benzoylphloroglucinols from Garcinia schomburgakiana Fruits: In Vitro and In Silico Studies" Molecules 27, no. 8: 2574. https://doi.org/10.3390/molecules27082574
APA StyleNguyen, H. T., Nguyen, T.-T., Duong, T.-H., Tran, N.-M.-A., Nguyen, C. H., Nguyen, T.-H.-A., & Sichaem, J. (2022). α-Glucosidase Inhibitory and Antimicrobial Benzoylphloroglucinols from Garcinia schomburgakiana Fruits: In Vitro and In Silico Studies. Molecules, 27(8), 2574. https://doi.org/10.3390/molecules27082574