
Defining the Role of Isoeugenol from Ocimum tenuiflorum against Diabetes Mellitus-Linked Alzheimer’s Disease through Network Pharmacology and Computational Methods

 , , , ,
, , , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Construction of Phytocompound Library
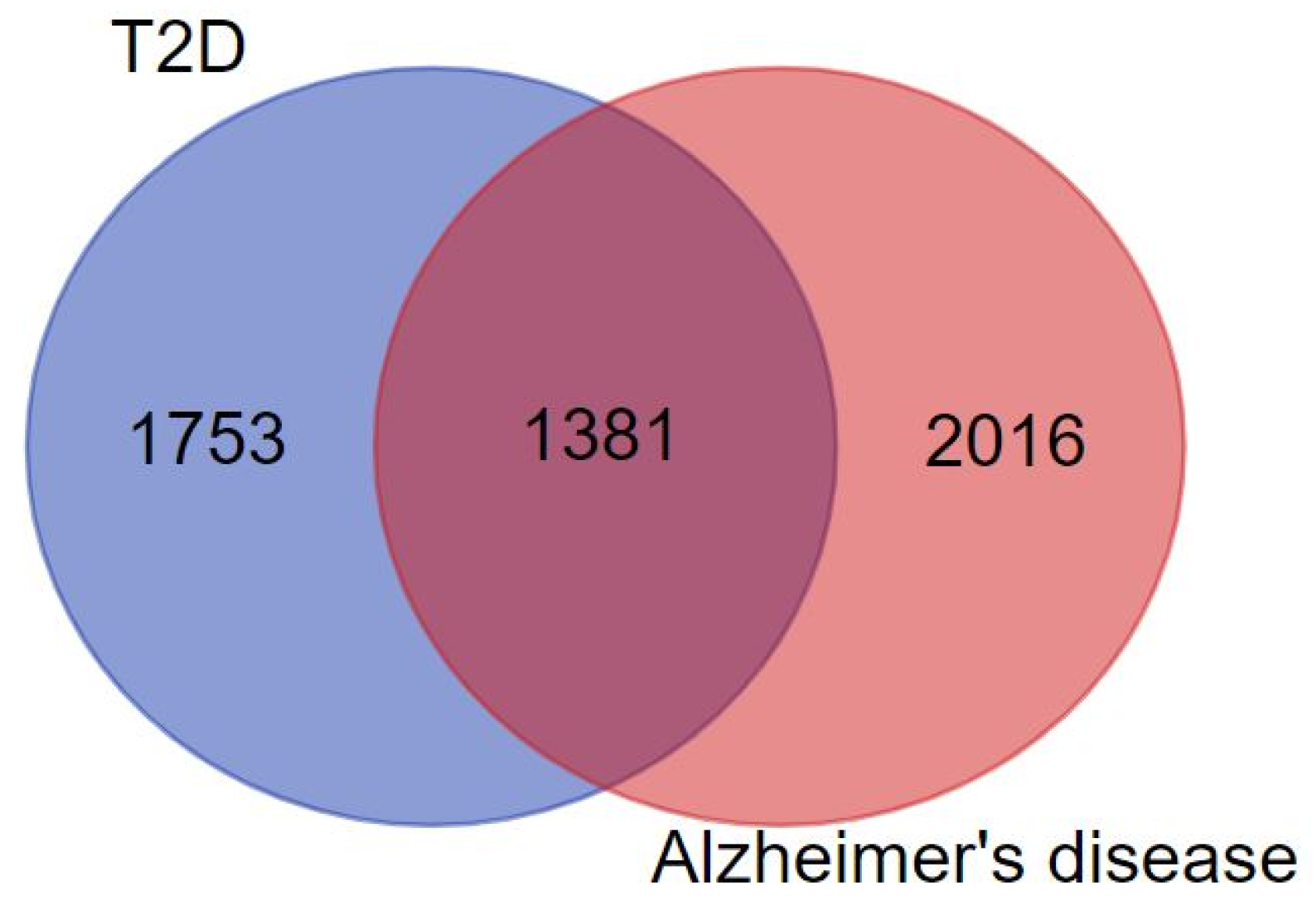
2.2. Prediction of Target Genes
2.3. Construction of PPI Network
2.4. C-T Network and Enrichment Analysis
2.5. Molecular Docking Simulation
2.6. Molecular Dynamics Simulation
2.7. Binding Free Energy Calculations
3. Results
3.1. Target Prediction and Active Compound Library
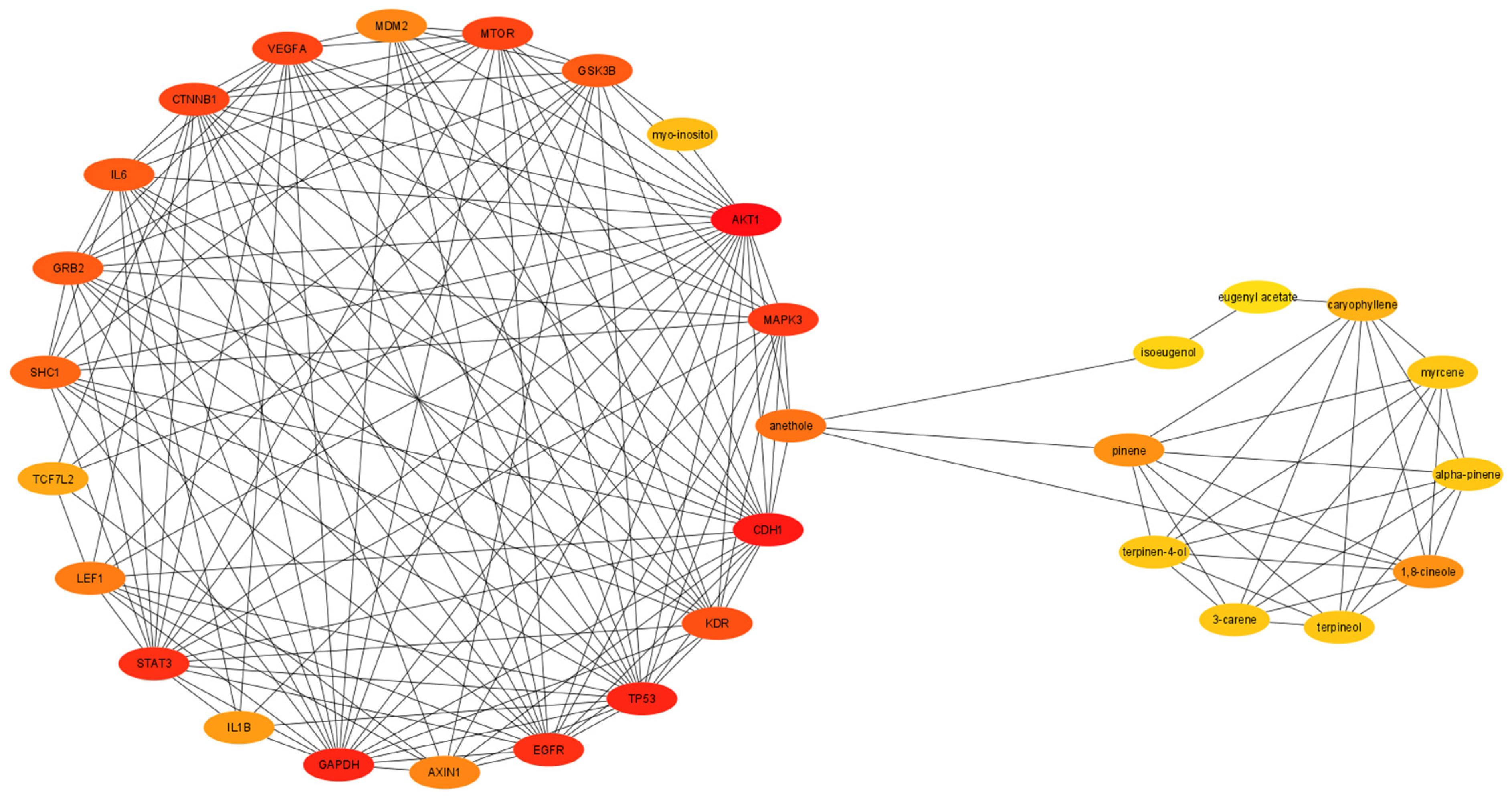
3.2. Construction of PPI Network
3.3. Network Analysis
3.4. Gene Ontology and Pathway Enrichment Analysis
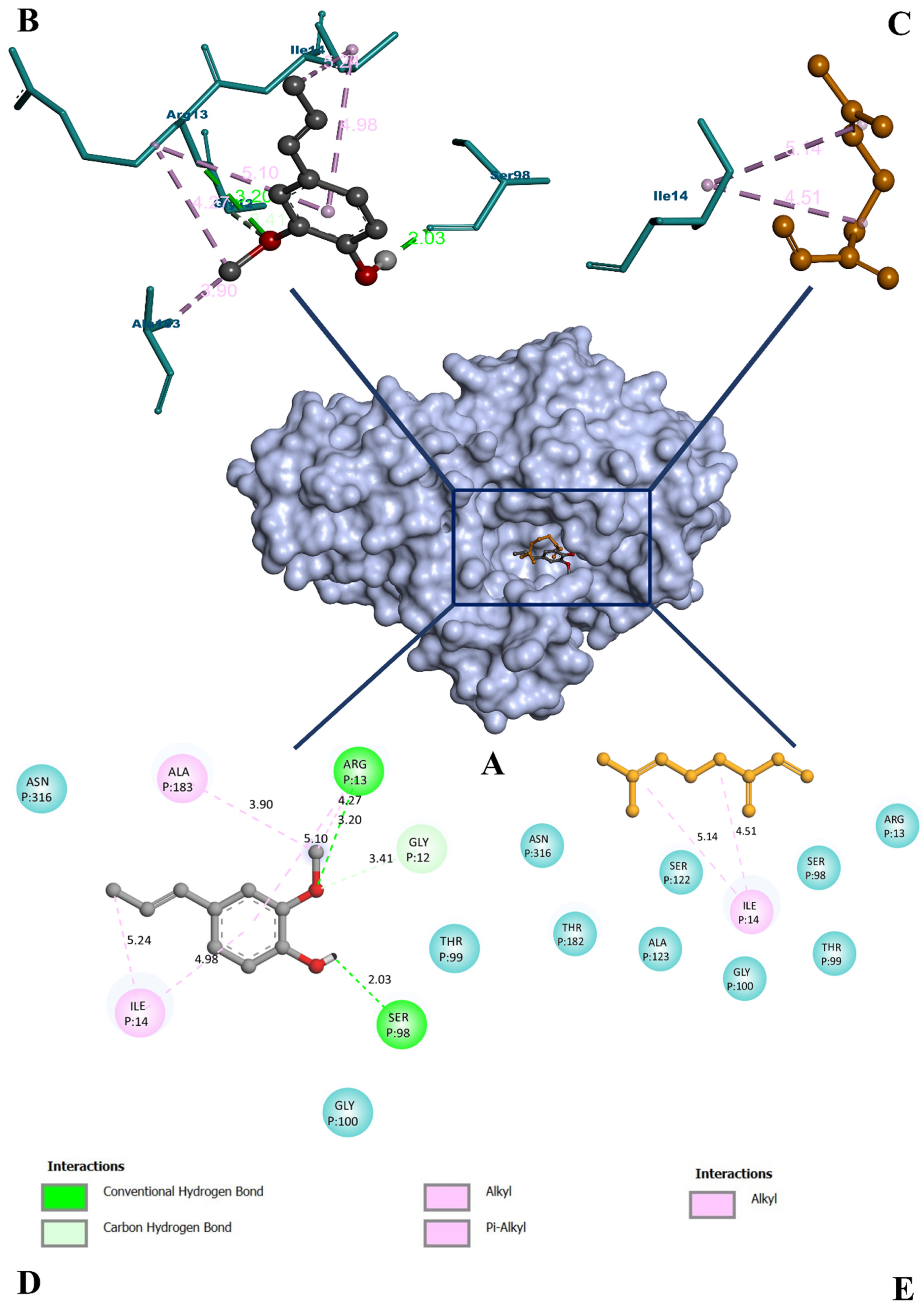
3.5. Molecular Docking Study
3.6. Molecular Dynamics Simulation
3.7. Binding Free Energy Calculations
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Sun, Y.; Ma, C.; Sun, H.; Wang, H.; Peng, W.; Zhou, Z.; Wang, H.; Pi, C.; Shi, Y.; He, X. Metabolism: A novel shared link between T2D and AD. J. Diabetes Res. 2020, 1, 2020. [Google Scholar]
- Chornenkyy, Y.; Wang, W.X.; Wei, A.; Nelson, P.T. Alzheimer’s disease and type 2 T2D are distinct diseases with potential overlapping metabolic dysfunction upstream of observed cognitive decline. Brain Pathol. 2019, 29, 3–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tumminia, A.; Vinciguerra, F.; Parisi, M.; Frittitta, L. Type 2 T2D and AD: Role of insulin signalling and therapeutic implications. Int. J. Mol. Sci. 2018, 19, 3306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, T.T.; Ta, Q.T.; Nguyen, T.K.; Nguyen, T.T.; Van Giau, V. Type 3 diabetes and its role implications in AD. Int. J. Mol. Sci. 2020, 21, 3165. [Google Scholar] [CrossRef] [PubMed]
- Arnold, S.E.; Arvanitakis, Z.; Macauley-Rambach, S.L.; Koenig, A.M.; Wang, H.Y.; Ahima, R.S.; Craft, S.; Gandy, S.; Buettner, C.; Stoeckel, L.E.; et al. Brain insulin resistance in type 2 diabetes and Alzheimer disease: Concepts and conundrums. Nat. Rev. Neurol. 2018, 14, 168–181. [Google Scholar] [CrossRef] [PubMed]
- Grover, J.K.; Yadav, S.; Vats, V. Medicinal plants of India with anti-diabetic potential. J. Ethnopharmacol. 2002, 81, 81–100. [Google Scholar] [CrossRef]
- Patil, S.M.; Kumari, V.C.; Sumana, K.; Sujay, S.; Tejaswini, M.; Shirahatti, P.S.; Ramu, R. Sustainable development of plant tissue culture industry: The Indian scenario. J. Appl. Biol. Biotechnol. 2021, 9, 18–27. [Google Scholar]
- Cohen, M.M. Tulsi—Ocimum sanctum: A herb for all reasons. J. Ayurveda. Integr. Med. 2014, 5, 251–259. [Google Scholar] [CrossRef] [Green Version]
- Mahajan, N.; Rawal, S.; Verma, M.; Poddar, M.; Alok, S. A phytopharmacological overview on Ocimum species with special emphasis on Ocimum sanctum. Biomed. Prev. Nutr. 2013, 3, 185–192. [Google Scholar] [CrossRef]
- Mohan, L.; Amberkar, M.V.; Kumari, M. Ocimum sanctum Linn. (TULSI)-an overview. Int. J. Pharm. Sci. Rev. Res. 2011, 7, 51–53. [Google Scholar]
- Pattanayak, P.; Behera, P.; Das, D.; Panda, S.K. Ocimum sanctum Linn. A reservoir plant for therapeutic applications: An overview. Pharmacogn Rev. 2010, 4, 95–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mondal, S.; Mirdha, B.R.; Mahapatra, S.C. The science behind sacredness of Tulsi (Ocimum sanctum Linn.). Indian J. Physiol. Pharmacol. 2009, 53, 291–306. [Google Scholar] [PubMed]
- Wang, Z.; Liu, J.; Yu, Y.; Chen, Y.; Wang, Y. Modular pharmacology: The next paradigm in drug discovery. Expert. Opin. Drug. Discov. 2012, 7, 667–677. [Google Scholar] [CrossRef] [PubMed]
- Keith, C.T.; Borisy, A.A.; Stockwell, B.R. Multicomponent therapeutics for networked systems. Nat. Rev. Drug Discov. 2005, 4, 71–78. [Google Scholar] [CrossRef]
- Schadt, E.E.; Friend, S.H.; Shaywitz, D.A. A network view of disease and compound screening. Nat. Rev. Drug Discov. 2009, 8, 286–295. [Google Scholar] [CrossRef]
- Ocaña, A.; Pandiella, A. Personalized therapies in the cancer “omics” era. Mol. Cancer 2010, 9, 202. [Google Scholar] [CrossRef] [Green Version]
- Mohanraj, K.; Karthikeyan, B.S.; Vivek-Ananth, R.P.; Chand, R.B.; Aparna, S.R.; Mangalapandi, P.; Samal, A. IMPPAT: A curated database of Indian Medicinal Plants, Phytochemistry and Therapeutics. Sci. Rep. 2018, 8, 4329. [Google Scholar] [CrossRef] [Green Version]
- Paul, S.; Andrew, M.; Owen, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar]
- Gu, S.; Xue, Y.; Gao, Y.; Shen, S.; Zhang, Y.; Chen, K.; Xue, S.; Pan, J.; Tang, Y.; Zhu, H.; et al. Mechanisms of indigo naturalis on treating ulcerative colitis explored by GEO gene chips combined with network pharmacology and molecular docking. Sci. Rep. 2020, 10, 15204. [Google Scholar] [CrossRef]
- Bindea, G.; Galon, J.; Mlecnik, B. CluePedia Cytoscape plugin: Pathway insights using integrated experimental and in silico data. Bioinformatics 2013, 29, 661–663. [Google Scholar] [CrossRef]
- Patil, S.M.; Martiz, R.M.; Ramu, R.; Shirahatti, P.S.; Prakash, A.; Kumar, B.P.; Kumar, N. Evaluation of flavonoids from banana pseudostem and flower (quercetin and catechin) as potent inhibitors of α-glucosidase: An in-silico perspective. J. Biomol. Struct. Dyn. 2021, 7, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maradesha, T.; Patil, S.M.; Al-Mutairi, K.A.; Ramu, R.; Madhunapantula, S.V.; Alqadi, T. Inhibitory effect of polyphenols from the whole green jackfruit flour against α-glucosidase, α-amylase, aldose reductase and glycation at multiple stages and their interaction: Inhibition kinetics and molecular simulations. Molecules 2022, 27, 1888. [Google Scholar] [CrossRef] [PubMed]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An open chemical toolbox. J. Cheminform. 2011, 3, 33–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patil, S.M.; Maruthi, K.R.; Bajpe, N.S.; Vyshali, V.M.; Sushmitha, S.; Chagalamari, A.; Ramith, R. Comparative molecular docking and simulation analysis of molnupiravir and remdesivir with SARS-CoV-2 RNA dependent RNA polymerase (RdRp). Bioinformation 2021, 7, 932–939. [Google Scholar]
- Jenkins, J.L.; Tanner, J.J. High-resolution structure of human D-glyceraldehyde-3-phosphate dehydrogenase. Acta. Crystallogr. D Biol. Crystallogr. 2006, 62, 290–301. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Cheminform. 2010, 31, 455–461. [Google Scholar] [CrossRef] [Green Version]
- Ganavi, D.; Ramu, R.; Kumar, V.; Patil, S.M.; Martiz, R.M.; Shirahatti, P.S.; Sathyanarayana, R.; Poojary, B.; Holla, B.S.; Poojary, V.; et al. In vitro and in silico studies of fluorinated 2,3-disubstituted thiazolidinone-pyrazoles as potential α-amylase inhibitors and antioxidant agents. Arch. Pharm. 2021, 12, e2100342. [Google Scholar] [CrossRef]
- Gurupadaswamy, H.D.; Ranganatha, V.L.; Ramu, R.; Patil, S.M.; Khanum, S.A. Competent synthesis of biaryl analogs via asymmetric Suzuki–Miyaura cross-coupling for the development of anti-inflammatory and analgesic agents. J. Iran. Chem. Soc. 2022, 1, 1–16. [Google Scholar] [CrossRef]
- Patil, S.M.; Martiz, R.M.; Ramu, R.; Shirahatti, P.S.; Prakash, A.; Chandra, J.S.; Ranganatha, L.V. In silico identification of novel benzophenone-coumarin derivatives as SARS-CoV-2 RNA dependent RNA polymerase (RdRp) inhibitors. J. Biomol. Struct. Dyn. 2021, 10, 1–17. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Vanommeslaeghe, K.; Hatcher, E.; Acharya, C.; Kundu, S.; Zhong, S.; Shim, J.; Darian, E.; Guvench, O.; Lopes, P.; Vorobyov, I.; et al. CHARMM general force field: A force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J. Comput. Chem. 2010, 31, 671–690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poojary, B.; Kumar, V.; Arunodaya, H.S.; Chandra, S.; Ramu, R.; Patil, S.M.; Baliga, A.; Rai, V.M.; Vishwanatha, U.; Vishwanatha, P.; et al. Potential fluorinated anti-MRSA thiazolidinone derivatives with antibacterial, antitubercular activity and molecular docking studies. Chem. Biodivers. 2021, 19, e202100532. [Google Scholar]
- Kumar, V.; Ramu, R.; Shirahatti, P.S.; Kumari, V.C.; Sushma, P.; Mandal, S.P.; Patil, S.M. α-glucosidase, α-amylase inhibition, kinetics and docking studies of novel (2-chloro-6-(trifluoromethyl) benzyloxy) arylidene) based rhodanine and rhodanine acetic acid derivatives. ChemistrySelect 2021, 6, 9637–9644. [Google Scholar] [CrossRef]
- Kumari, R.; Kumar, R.; Lynn, A. g_mmpbsa-a GROMACS tool for high-throughput MM-PBSA calculations. J. Chem. Inf. Model. 2014, 54, 1951–1962. [Google Scholar] [CrossRef]
- Genhedenk, S.; Ryde, U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin. Drug. Discov. 2015, 10, 449–461. [Google Scholar] [CrossRef]
- Baglietto-Vargas, D.; Shi, J.; Yaeger, D.M.; Ager, R.; LaFerla, F.M. Diabetes and AD crosstalk. Neurosci. Biobehav. Rev. 2016, 64, 272–287. [Google Scholar] [CrossRef]
- Jayaraman, A.; Pike, C.J. AD and type 2 diabetes: Multiple mechanisms contribute to interactions. Curr. Diabetes Rep. 2014, 14, 476. [Google Scholar] [CrossRef] [Green Version]
- Barbagallo, M.; Dominguez, L.J. Type 2 T2D and AD. World J. Diabetes 2014, 5, 889. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Song, D.; Leng, S.X. Link between type 2 diabetes and AD: From epidemiology to mechanism and treatment. Clin. Interv. Aging 2015, 10, 549. [Google Scholar] [CrossRef] [Green Version]
- Parasuraman, S.; Balamurugan, S.; Christapher, P.V.; Petchi, R.R.; Yeng, W.Y.; Sujithra, J.; Vijaya, C. Evaluation of antidiabetic and antihyperlipidemic effects of hydroalcoholic extract of leaves of Ocimum tenuiflorum (Lamiaceae) and prediction of biological activity of its phytoconstituents. Pharmacogn. Res. 2015, 7, 156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanumanthachar, J.; Milind, P. Evaluation of nootropic potential of Ocimum sanctum Linn. in mice. Ind. J. Exp. Biol. 2006, 44, 133–136. [Google Scholar]
- Raghavendra, M.; Maiti, R.; Kumar, S.; Acharya, S.B. Role of Ocimum sanctum in the experimental model of AD in rats. Int. J. Green Pharm. 2009, 3, 14–19. [Google Scholar] [CrossRef]
- Du, X.; Matsumura, T.; Edelstein, D.; Rossetti, L.; Zsengellér, Z.; Szabó, C.; Brownlee, M. Inhibition of GAPDH activity by poly (ADP-ribose) polymerase activates three major pathways of hyperglycemic damage in endothelial cells. J. Clin. Investig. 2003, 112, 1049–1057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanwar, M.; Kowluru, R.A. Role of glyceraldehyde 3-phosphate dehydrogenase in the development and progression of diabetic retinopathy. Diabetes 2009, 58, 227–234. [Google Scholar] [CrossRef] [Green Version]
- Suarez, S.; Penn, J.S. GAPDH/Siah1 signaling mediates apoptosis in high glucose-treated human retinal pericytes. Investig. Ophthalmol. Vis. Sci. 2015, 56, 924. [Google Scholar]
- Lazarev, V.F.; Guzhova, I.V.; Margulis, B.A. Glyceraldehyde-3-phosphate dehydrogenase is a multifaceted therapeutic target. Pharmaceutics 2020, 12, 416. [Google Scholar] [CrossRef]
- Kay, A.M.; Simpson, C.L.; Stewart, J.A. The role of AGE/RAGE signaling in diabetes-mediated vascular calcification. J. Diab. Res. 2016, 10, 2016. [Google Scholar] [CrossRef] [Green Version]
- Atanasov, A.G.; Zotchev, S.B.; Dirsch, V.M.; Supuran, C.T. Natural products in drug discovery: Advances and opportunities. Nat. Rev. Drug. Discov. 2021, 20, 200–216. [Google Scholar] [CrossRef]
- Ganesan, A. The impact of natural products upon modern drug discovery. Curr. Opin. Chem. Biol. 2008, 12, 306–317. [Google Scholar] [CrossRef]
- Butterfield, D.A.; Hardas, S.S.; Lange, M.L. Oxidatively modified glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and Alzheimer’s disease: Many pathways to neurodegeneration. J. Alzheimers Dis. 2010, 20, 369–393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yadav, S.; Pandey, S.K.; Singh, V.K.; Goel, Y.; Kumar, A.; Singh, S.M. Molecular docking studies of 3-bromopyruvate and its derivatives to metabolic regulatory enzymes: Implication in designing of novel anticancer therapeutic strategies. PLoS ONE 2017, 12, e0176403. [Google Scholar] [CrossRef] [PubMed]
- Galbiati, A.; Zana, A.; Conti, P. Covalent inhibitors of GAPDH: From unspecific warheads to selective compounds. Eur. J. Med. Chem. 2020, 207, 12740. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sl. No | Compound Names | Oral Bioavailability (OB ≥ 30%) | Blood–Brain Barrier (BBB) | Drug Half-Life (HL < 3 h) | Lipinski’s Rule (LR) of Five | Intestinal Epithelial Permeability (Caco-2 Cells) | Drug-Induced Liver Injury (DILI) | Clearness (CL > 15 mL/min/kg) | Molecular Weight (MW 100~600) | Hydrogen Bond Acceptor (0~12) | Hydrogen Bond Donor (0~7) | TPSA (0~140) | PAINS |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | Cyclo (L-val-L-Leu) | Pass | Pass | 0.671 | Accepted | −4.711 | Negative | 5.356 | 212.150 | 4 | 2 | 58.200 | 0 |
| 2 | α-Cubebene | Fail | Pass | 0.052 | Accepted | −4.408 | Negative | 18.693 | 204.190 | 0 | 0 | 0.000 | 0 |
| 3 | β-Caryophyllone | Moderate | Pass | 0.227 | Accepted | −4.750 | Negative | 14.774 | 220.180 | 1 | 0 | 17.070 | 0 |
| 4 | Phytosterols | Pass | Pass | 0.013 | Accepted | −4.756 | Negative | 16.686 | 414.390 | 1 | 1 | 20.230 | 0 |
| 5 | UNII-0V56HXQ8N5 | Moderate | Moderate | 0.059 | Accepted | −4.357 | Negative | 19.832 | 204.190 | 0 | 0 | 0.000 | 0 |
| 6 | (−)-Alloaromadendrene | Pass | Pass | 0.040 | Accepted | −4.577 | Negative | 13.563 | 204.190 | 0 | 0 | 0.000 | 0 |
| 7 | (−)-Camphene | Pass | Pass | 0.077 | Accepted | −4.463 | Negative | 9.346 | 136.130 | 0 | 0 | 0.000 | 0 |
| 8 | (−)-cis-Carveol | Fail | Pass | 0.378 | Accepted | −4.328 | Negative | 12.624 | 152.120 | 1 | 1 | 20.230 | 0 |
| 9 | (−)-Linalool | Pass | Pass | 0.493 | Accepted | −4.375 | Negative | 9.738 | 154.140 | 1 | 1 | 20.230 | 0 |
| 10 | (+)-α-Phellandrene | Pass | Pass | 0.617 | Accepted | −4.383 | Negative | 12.660 | 136.130 | 0 | 0 | 0.000 | 0 |
| 11 | (+)-δ-Cadinene | Moderate | Moderate | 0.051 | Accepted | −4.469 | Positive | 7.421 | 204.190 | 0 | 0 | 0.000 | 0 |
| 12 | (+)-endo-β-Bergamotene | Pass | Pass | 0.063 | Accepted | −4.466 | Negative | 16.946 | 204.190 | 0 | 0 | 0.000 | 0 |
| 13 | (1S,2R,4S)-(−)-Bornyl acetate | Pass | Pass | 0.243 | Accepted | −4.552 | Moderate | 6.063 | 196.150 | 2 | 0 | 26.300 | 0 |
| 14 | (1S)-1,7,7-Trimethylbicyclo[2.2.1]heptan-2-one | Pass | Pass | 0.701 | Accepted | −4.582 | Negative | 13.808 | 152.120 | 1 | 0 | 17.070 | 0 |
| 15 | (E)-β-Farnesene | Fail | Fail | 0.156 | Accepted | −4.537 | Moderate | 13.186 | 204.190 | 0 | 0 | 0.000 | 0 |
| 16 | (E)-α-Bisabolene | Fail | Fail | 0.092 | Accepted | −4.502 | Negative | 17.581 | 204.190 | 0 | 0 | 0.000 | 0 |
| 17 | (E)-β-Ocimene | Pass | Pass | 0.678 | Accepted | −4.434 | Negative | 14.171 | 136.130 | 0 | 0 | 0.000 | 0 |
| 18 | 1-Octen-3-ol | Fail | Fail | 0.672 | Accepted | −4.256 | Negative | 7.650 | 128.120 | 1 | 1 | 20.230 | 0 |
| 19 | 1S-α-Pinene | Pass | Pass | 0.114 | Accepted | −4.303 | Negative | 15.022 | 136.130 | 0 | 0 | 0.000 | 0 |
| 20 | 2,3-Dimethylaniline | Pass | Pass | 0.583 | Accepted | −4.255 | Negative | 10.496 | 121.090 | 1 | 2 | 26.020 | 0 |
| 21 | 2,5-Dimethoxybenzoic acid | Pass | Moderate | 0.885 | Accepted | −4.853 | Positive | 7.488 | 182.060 | 4 | 1 | 55.760 | 0 |
| 22 | 3-Carene | Pass | Pass | 0.132 | Accepted | −4.307 | Negative | 16.061 | 136.130 | 0 | 0 | 0.000 | 0 |
| 23 | 4-Terpineol | Pass | Pass | 0.447 | Accepted | −4.217 | Negative | 14.345 | 154.140 | 1 | 1 | 20.230 | 0 |
| 24 | Acetic acid | Pass | Pass | 0.791 | Accepted | −5.218 | Negative | 1.609 | 60.020 | 2 | 1 | 37.300 | 0 |
| 25 | Acetyleugenol | Pass | Pass | 0.843 | Accepted | −4.453 | Moderate | 8.457 | 206.090 | 3 | 0 | 35.530 | 0 |
| 26 | α-Fenchene | Pass | Pass | 0.099 | Accepted | −4.460 | Negative | 10.559 | 136.130 | 0 | 0 | 0.000 | 0 |
| 27 | α-Humulene | Fail | No | 0.095 | Accepted | −4.425 | Negative | 8.432 | 204.190 | 0 | 0 | 0.000 | 0 |
| 28 | α-Terpineol | Pass | Pass | 0.527 | Accepted | −4.193 | Negative | 8.942 | 154.140 | 1 | 1 | 20.230 | 0 |
| 29 | Apigenin | Fail | No | 0.856 | Accepted | −4.847 | Positive | 7.022 | 270.050 | 5 | 3 | 90.900 | 0 |
| 30 | Apigenin 7-glucuronide | Fail | No | 0.715 | Rejected | −6.376 | Positive | 1.194 | 446.080 | 11 | 6 | 187.120 | 0 |
| 31 | β-Cadinene | Fail | Moderate | 0.060 | Accepted | −4.392 | Negative | 17.975 | 204.190 | 0 | 0 | 0.000 | 0 |
| 32 | β-Carotene | Moderate | No | 0.076 | Rejected | −6.003 | Negative | 0.229 | 536.440 | 0 | 0 | 0.000 | 0 |
| 33 | β-Caryophyllene | Pass | Pass | 0.048 | Accepted | −4.517 | Negative | 9.943 | 204.190 | 0 | 0 | 0.000 | 0 |
| 34 | β-Pinene | Pass | Pass | 0.107 | Accepted | −4.460 | Negative | 10.097 | 136.130 | 0 | 0 | 0.000 | 0 |
| 35 | Bis-acetic acid | Fail | No | 0.998 | Rejected | −7.722 | Positive | 12.869 | 1700.170 | 46 | 25 | 777.980 | 1 alert |
| 36 | Carotene | Pass | No | 0.036 | Rejected | −5.634 | Negative | 0.671 | 536.440 | 0 | 0 | 0.000 | 0 |
| 37 | Carvacrol | Fail | Pass | 0.671 | Accepted | −4.436 | Negative | 11.335 | 150.100 | 1 | 1 | 20.230 | 1 alert |
| 38 | cis-Anethole | Pass | Moderate | 0.638 | Accepted | −4.440 | Negative | 11.146 | 148.090 | 1 | 0 | 9.230 | 0 |
| 39 | Decanal | Fail | Pass | 0.456 | Accepted | −4.551 | Negative | 5.049 | 156.150 | 1 | 0 | 17.070 | 0 |
| 40 | Dehydro-p-cymene | Pass | Pass | 0.568 | Accepted | −4.344 | Moderate | 10.755 | 132.090 | 0 | 0 | 0.000 | 0 |
| 41 | Dipentene | Fail | Pass | 0.233 | Accepted | −4.320 | Negative | 11.517 | 136.130 | 0 | 0 | 0.000 | 0 |
| 42 | Estragole | Moderate | Moderate | 0.577 | Accepted | −4.308 | Negative | 12.054 | 148.090 | 1 | 0 | 9.230 | 0 |
| 43 | Eucalyptol | Pass | Pass | 0.352 | Accepted | −4.414 | Negative | 8.066 | 154.140 | 1 | 0 | 9.230 | 0 |
| 44 | Eugenol | Fail | Pass | 0.887 | Accepted | −4.373 | Negative | 14.042 | 164.080 | 2 | 1 | 29.460 | 0 |
| 45 | γ-Selinene | Pass | Pass | 0.088 | Accepted | −4.577 | Negative | 13.350 | 204.190 | 0 | 0 | 0.000 | 0 |
| 46 | Geranyl acetate | Pass | Pass | 0.506 | Accepted | −4.420 | Moderate | 9.707 | 196.150 | 2 | 0 | 26.300 | 0 |
| 47 | Isoeugenol | Pass | Moderate | 0.880 | Accepted | −4.579 | Negative | 13.435 | 164.080 | 2 | 1 | 29.460 | 0 |
| 48 | L-Ascorbic acid | Fail | Fail | 0.928 | Accepted | −5.917 | Positive | 9.964 | 176.030 | 6 | 5 | 114.290 | 0 |
| 49 | Linolenic acid | Fail | Fail | 0.710 | Accepted | −4.631 | Negative | 4.877 | 278.220 | 2 | 1 | 37.300 | 0 |
| 50 | Luteolin-7-O-glucuronide | Fail | Fail | 0.855 | Rejected | −6.471 | Positive | 1.614 | 462.080 | 12 | 7 | 207.350 | 1 alert |
| 51 | Methyleugenol | Moderate | Pass | 0.848 | Accepted | −4.338 | Negative | 11.466 | 178.100 | 2 | 0 | 18.460 | 0 |
| 52 | Molludistin | Fail | Fail | 0.290 | Accepted | −5.776 | Positive | 3.398 | 416.110 | 9 | 5 | 149.820 | 0 |
| 53 | Myrcene | Pass | Pass | 0.453 | Accepted | −4.402 | Moderate | 13.108 | 136.130 | 0 | 0 | 0.000 | 0 |
| 54 | Nerol | Fail | Pass | 0.737 | Accepted | −4.299 | Positive | 12.604 | 154.140 | 1 | 1 | 20.230 | 0 |
| 55 | Octadeca-9,12-dienoic acid | Moderate | Fail | 0.628 | Accepted | −4.733 | Negative | 3.327 | 280.240 | 2 | 1 | 37.300 | 0 |
| 56 | Octadecanoate | Fail | Fail | 0.476 | Accepted | −5.068 | Negative | 2.425 | 284.270 | 2 | 1 | 37.300 | 0 |
| 57 | Oleic acid | Moderate | Fail | 0.546 | Accepted | −4.922 | Negative | 2.573 | 282.260 | 2 | 1 | 37.300 | 0 |
| 58 | Orientin | Fail | Fail | 0.724 | Rejected | −6.208 | Positive | 5.042 | 448.100 | 11 | 8 | 201.280 | 1 alert |
| 59 | Palmitic acid | Fail | Fail | 0.610 | Accepted | −5.027 | Negative | 2.377 | 256.240 | 2 | 1 | 37.300 | 0 |
| 60 | Thymol | Fail | Pass | 0.682 | Accepted | −4.387 | Negative | 9.444 | 150.100 | 1 | 1 | 20.230 | 0 |
| 61 | Ursolic acid | Moderate | Pass | 0.017 | Accepted | −5.221 | Negative | 3.671 | 456.360 | 3 | 2 | 57.530 | 0 |
| Name | Betweenness Centrality | Closeness Centrality | Degree Centrality | Number of Undirected Edges |
|---|---|---|---|---|
| GAPDH | 0.046608989 | 0.625061425 | 535 | 535 |
| AKT1 | 0.038310118 | 0.625368732 | 535 | 535 |
| ACTB | 0.035911388 | 0.625061425 | 534 | 534 |
| ALB | 0.036238133 | 0.620487805 | 524 | 524 |
| INS | 0.047353182 | 0.619581101 | 515 | 515 |
| TNF | 0.026923424 | 0.610951009 | 497 | 497 |
| IL6 | 0.025073263 | 0.610657705 | 491 | 491 |
| TP53 | 0.028814967 | 0.591627907 | 428 | 428 |
| IL1B | 0.012693258 | 0.581618656 | 405 | 405 |
| VEGFA | 0.00953204 | 0.574525745 | 380 | 380 |
| EGFR | 0.018377788 | 0.576086957 | 374 | 374 |
| STAT3 | 0.010150212 | 0.569127517 | 364 | 364 |
| CTNNB1 | 0.01889698 | 0.56735058 | 350 | 350 |
| MAPK3 | 0.010595927 | 0.563081009 | 333 | 333 |
| Sl. No. | Name of the Compound | Binding Affinity (kcal/mol) | Total No. of Non-Bonding Interactions | Total No. of Hydrogen Bonds |
|---|---|---|---|---|
| 1 | 1S-α-Pinene | −4.3 | 5 | 0 |
| 2 | 3-Carene | −4.3 | 5 | 0 |
| 3 | 4-Terpineol | −4.4 | 5 | 1 |
| 4 | Acetyleugenol | −5.0 | 6 | 1 |
| 5 | α-Terpineol | −4.4 | 5 | 1 |
| 6 | β-Caryophyllene | −5.6 | 3 | 0 |
| 7 | cis-Anethole | −4.1 | 4 | 1 |
| 8 | Eucalyptol | −4.3 | 4 | 1 |
| 9 | Geranyl acetate | −5.9 | 6 | 1 |
| 10 | Isoeugenol | −6.0 | 7 | 2 |
| 11 | Myrcene | −3.6 | 2 | 0 |
| Types of Binding Free Energy | GAPDH–Isoeugenol Complex | GAPDH–Myrcene Complex | ||
|---|---|---|---|---|
| Values (kj/mol) | Standard Deviation (kj/mol) | Values (kj/mol) | Standard Deviation (kj/mol) | |
| Van der Waal energy | −159.277 | ±7.426 | −124.482 | ±11.201 |
| Electrostatic energy | 0.803 | ±1.153 | 1.251 | ±3.091 |
| Polar solvation energy | 26.262 | ±4.667 | 20.381 | ±6.330 |
| SASA energy | −11.247 | ±0.590 | −9.201 | ±2.998 |
| Binding energy | −143.458 | ±8.685 | −110.921 | ±10.120 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martiz, R.M.; Patil, S.M.; Abdulaziz, M.; Babalghith, A.; Al-Areefi, M.; Al-Ghorbani, M.; Mallappa Kumar, J.; Prasad, A.; Mysore Nagalingaswamy, N.P.; Ramu, R. Defining the Role of Isoeugenol from Ocimum tenuiflorum against Diabetes Mellitus-Linked Alzheimer’s Disease through Network Pharmacology and Computational Methods. Molecules 2022, 27, 2398. https://doi.org/10.3390/molecules27082398
Martiz RM, Patil SM, Abdulaziz M, Babalghith A, Al-Areefi M, Al-Ghorbani M, Mallappa Kumar J, Prasad A, Mysore Nagalingaswamy NP, Ramu R. Defining the Role of Isoeugenol from Ocimum tenuiflorum against Diabetes Mellitus-Linked Alzheimer’s Disease through Network Pharmacology and Computational Methods. Molecules. 2022; 27(8):2398. https://doi.org/10.3390/molecules27082398
Chicago/Turabian StyleMartiz, Reshma Mary, Shashank M. Patil, Mohammed Abdulaziz, Ahmed Babalghith, Mahmoud Al-Areefi, Mohammed Al-Ghorbani, Jayanthi Mallappa Kumar, Ashwini Prasad, Nagendra Prasad Mysore Nagalingaswamy, and Ramith Ramu. 2022. "Defining the Role of Isoeugenol from Ocimum tenuiflorum against Diabetes Mellitus-Linked Alzheimer’s Disease through Network Pharmacology and Computational Methods" Molecules 27, no. 8: 2398. https://doi.org/10.3390/molecules27082398
APA StyleMartiz, R. M., Patil, S. M., Abdulaziz, M., Babalghith, A., Al-Areefi, M., Al-Ghorbani, M., Mallappa Kumar, J., Prasad, A., Mysore Nagalingaswamy, N. P., & Ramu, R. (2022). Defining the Role of Isoeugenol from Ocimum tenuiflorum against Diabetes Mellitus-Linked Alzheimer’s Disease through Network Pharmacology and Computational Methods. Molecules, 27(8), 2398. https://doi.org/10.3390/molecules27082398