
The Phosphorus Bond, or the Phosphorus-Centered Pnictogen Bond: The Covalently Bound Phosphorus Atom in Molecular Entities and Crystals as a Pnictogen Bond Donor





Abstract
:1. Introduction
2. Computational Details
3. Illustrative Crystalline Systems
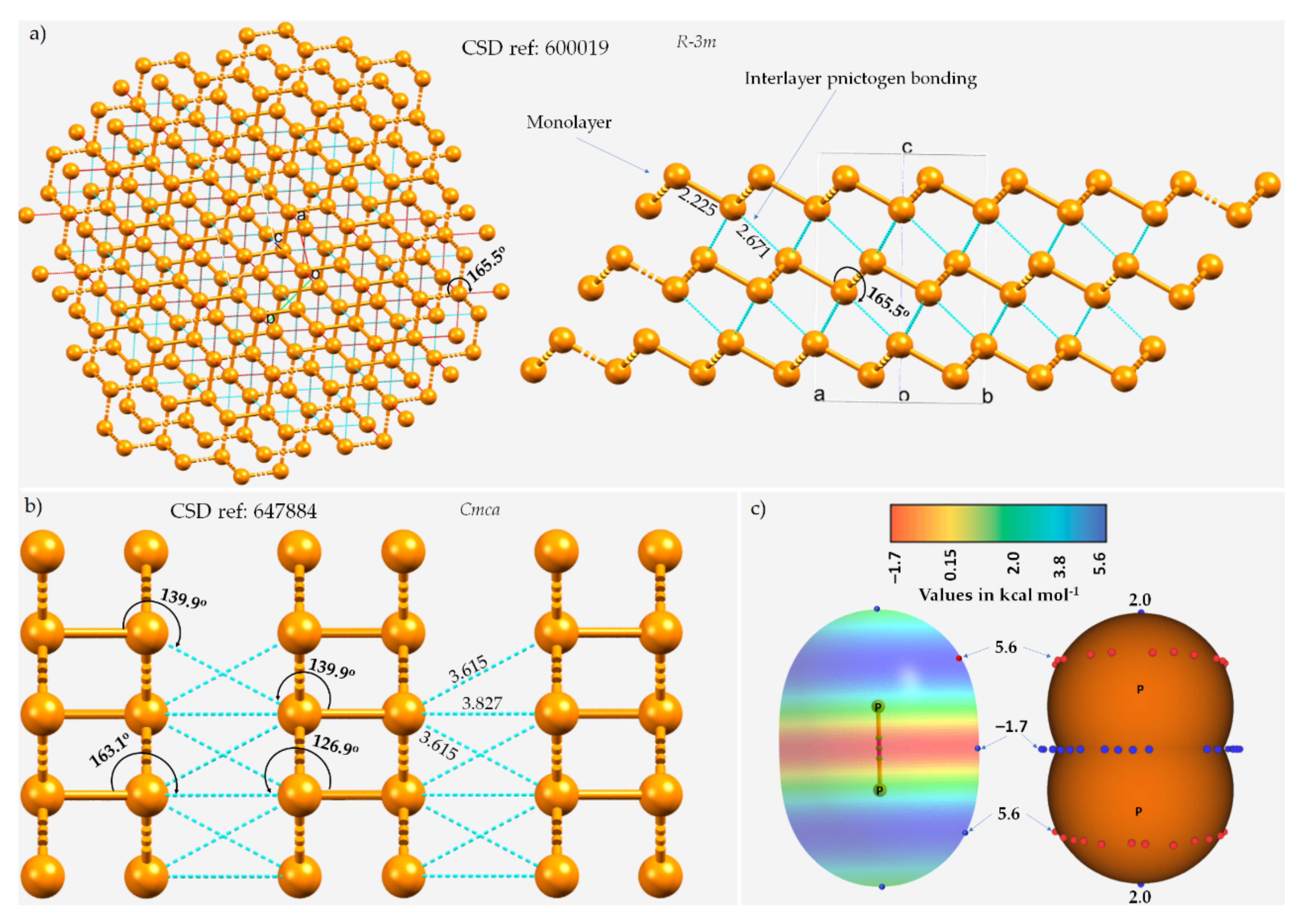
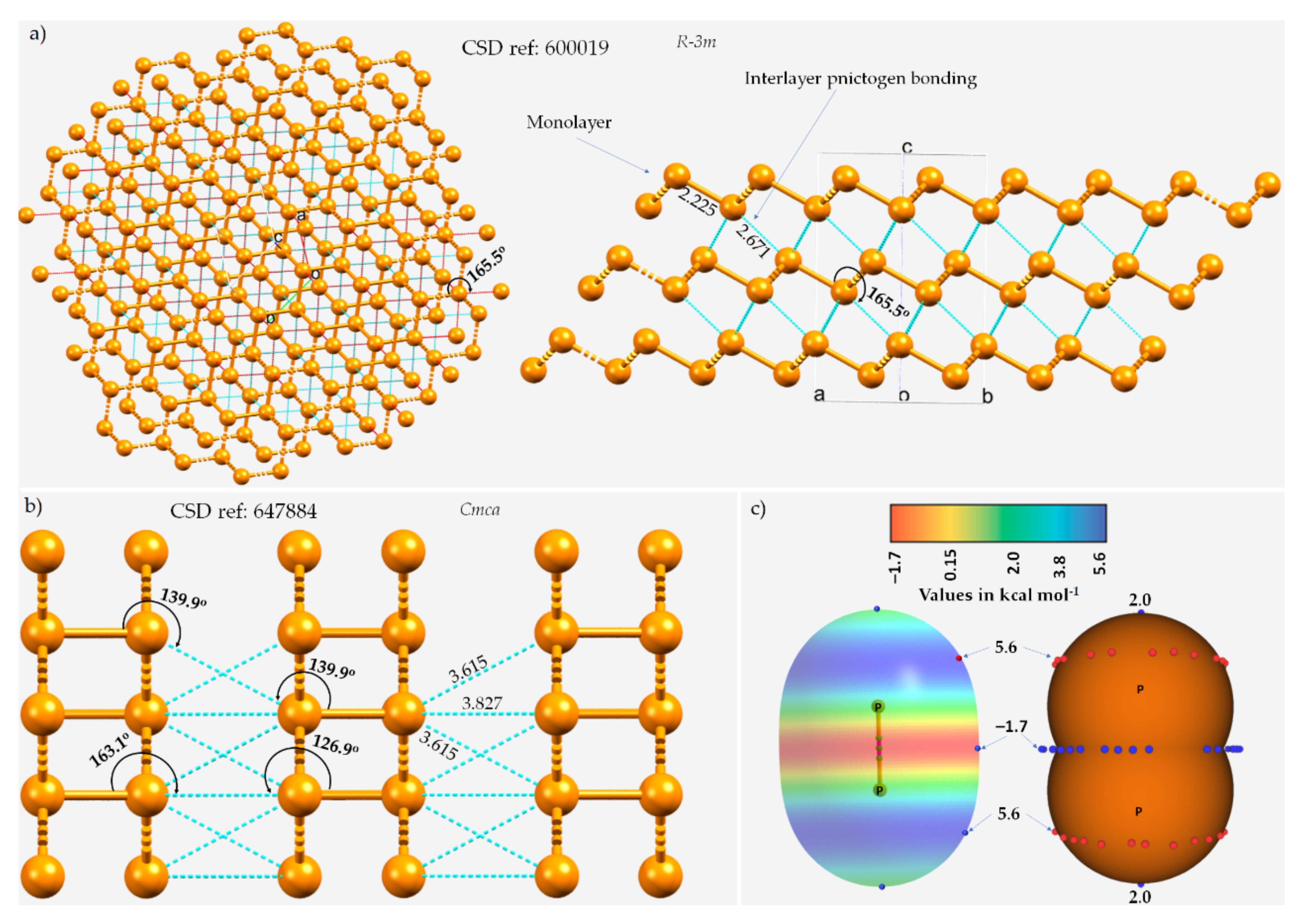
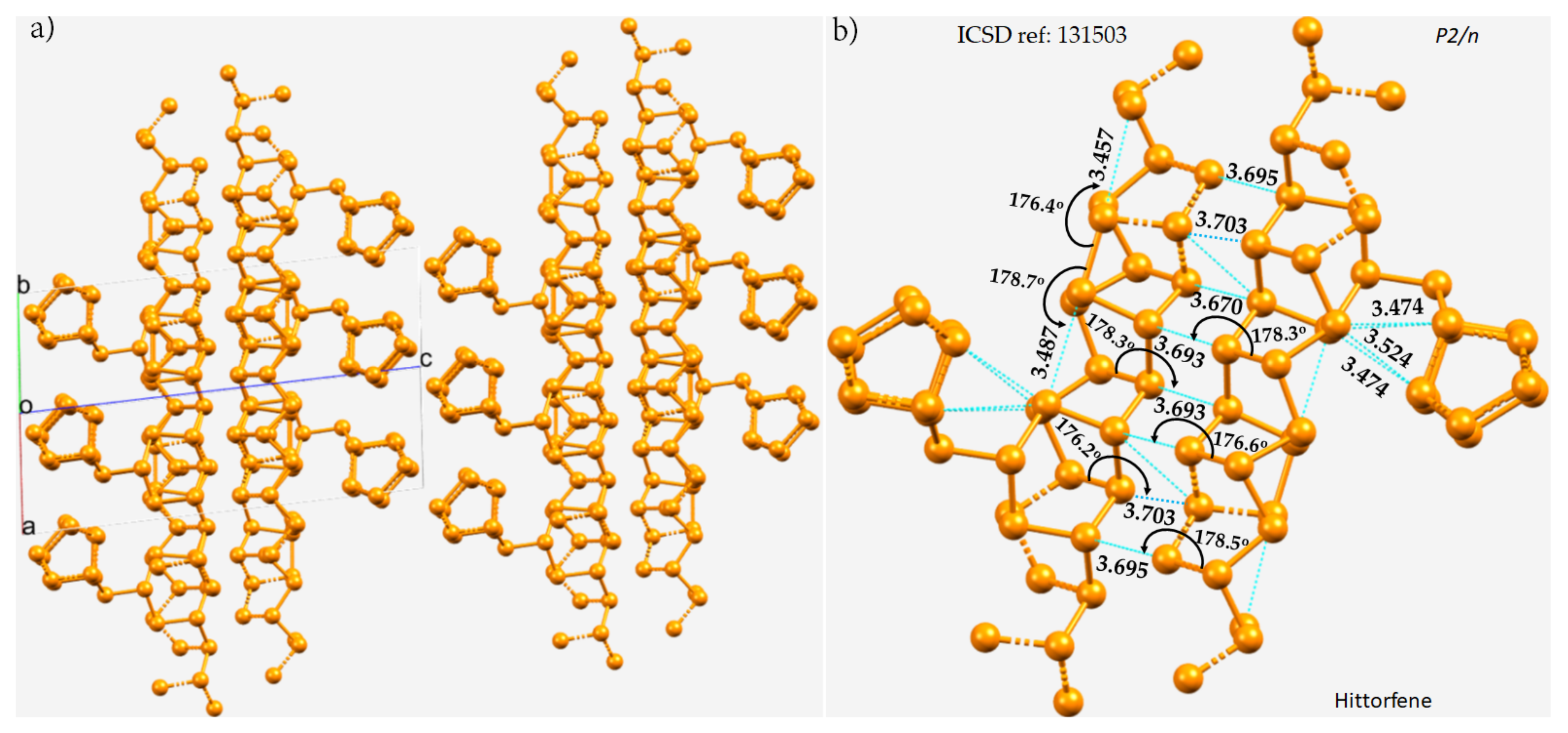
3.1. Polymorphs of Phosphorus
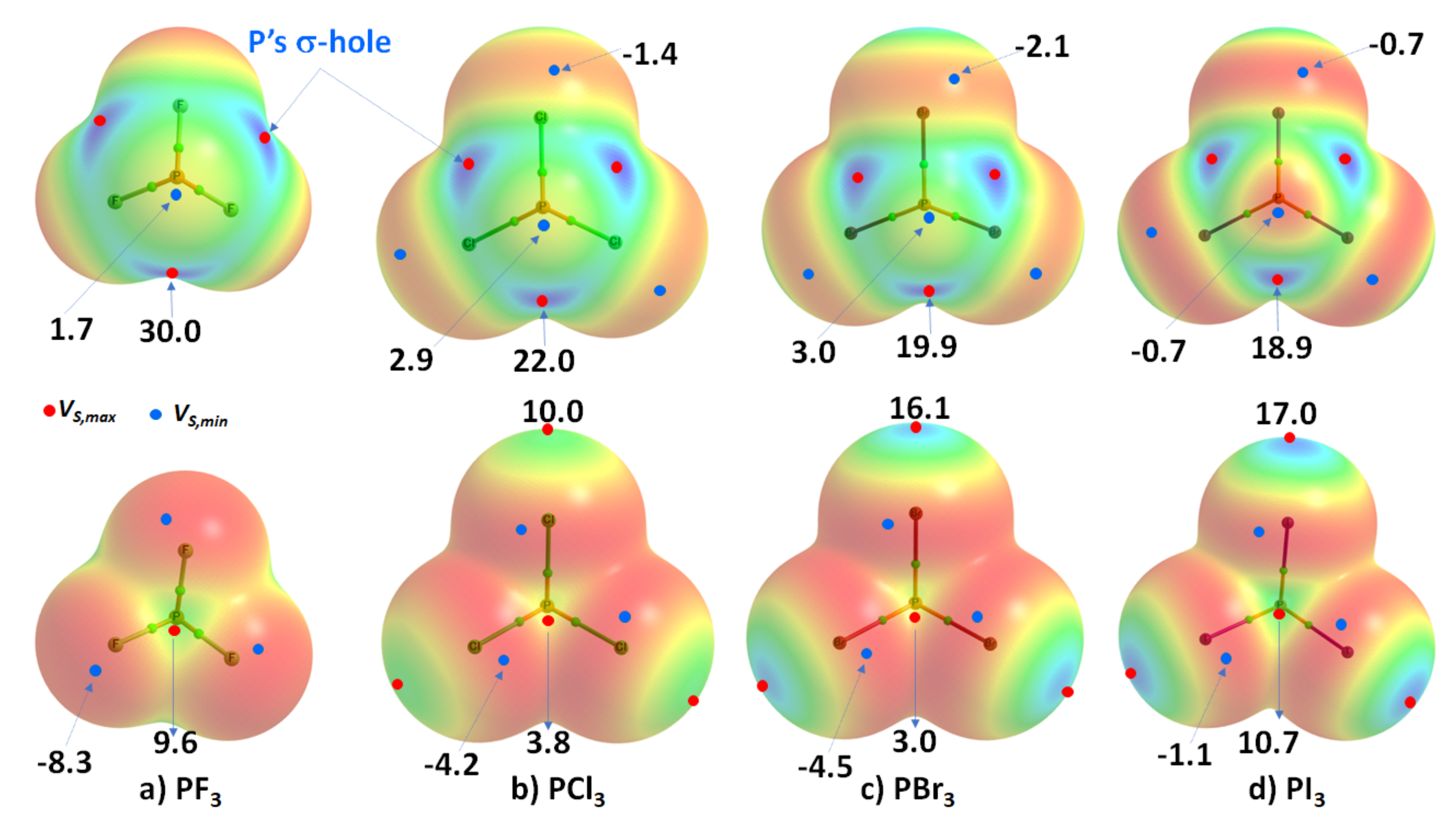
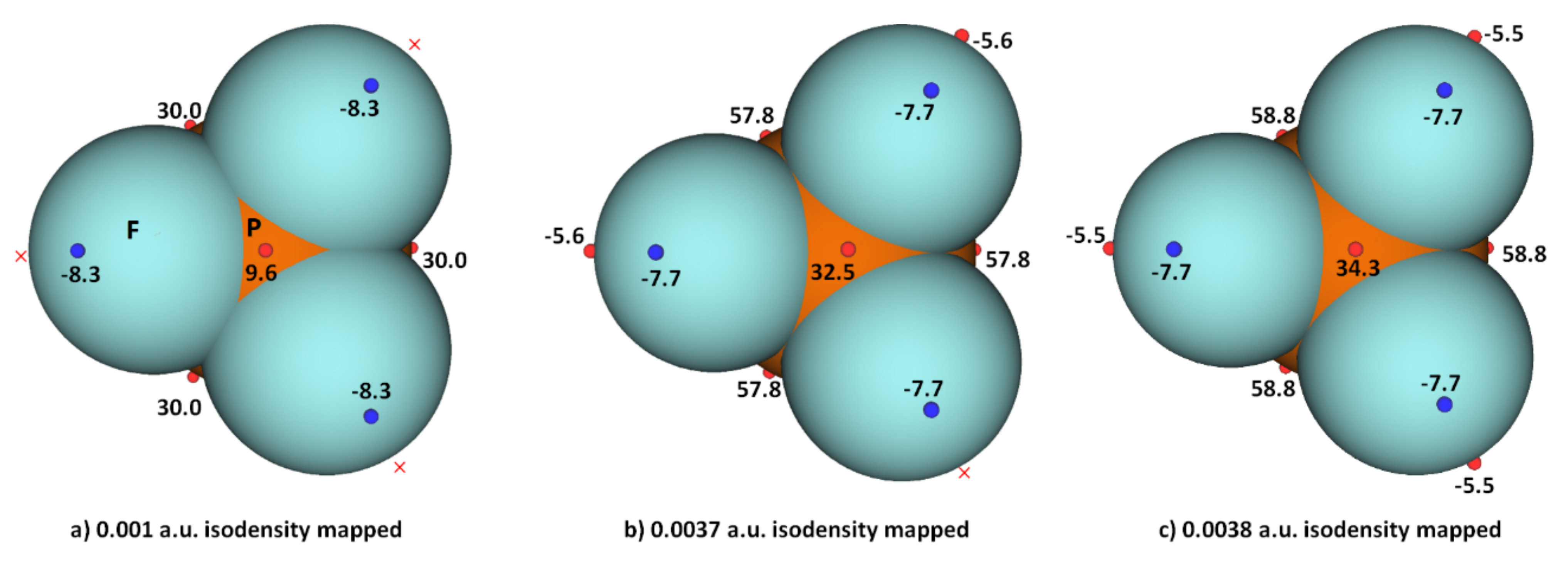
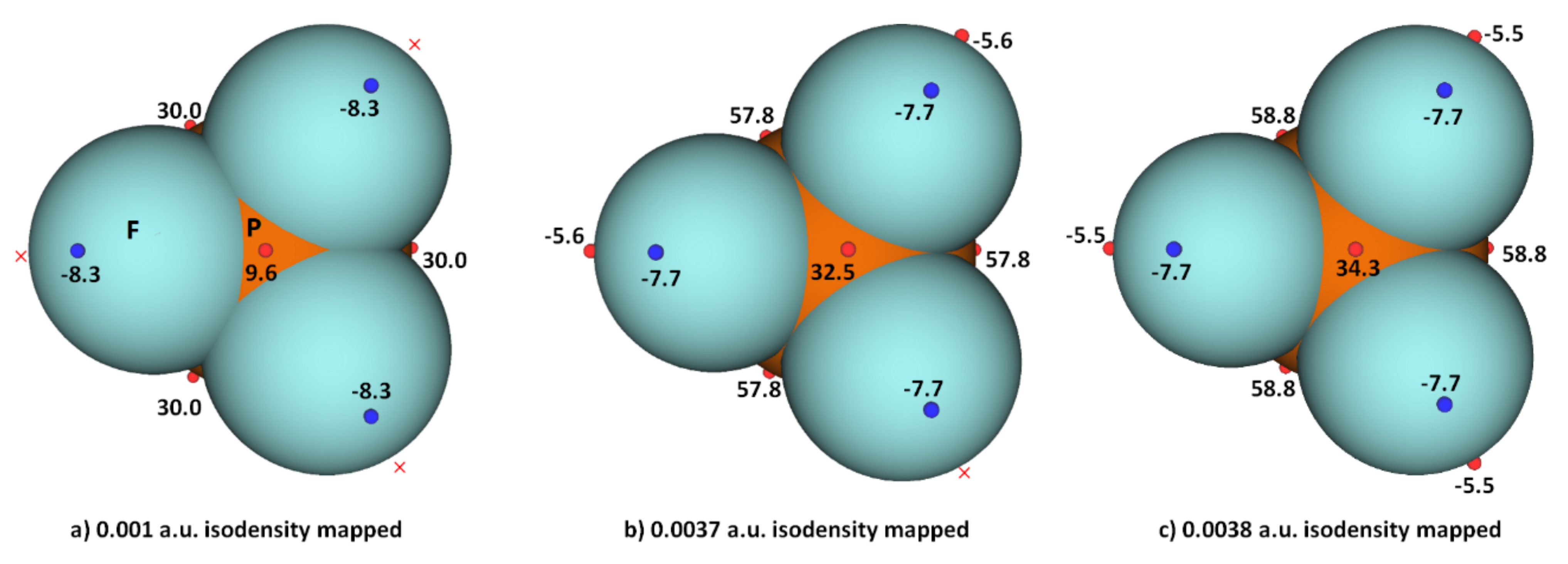
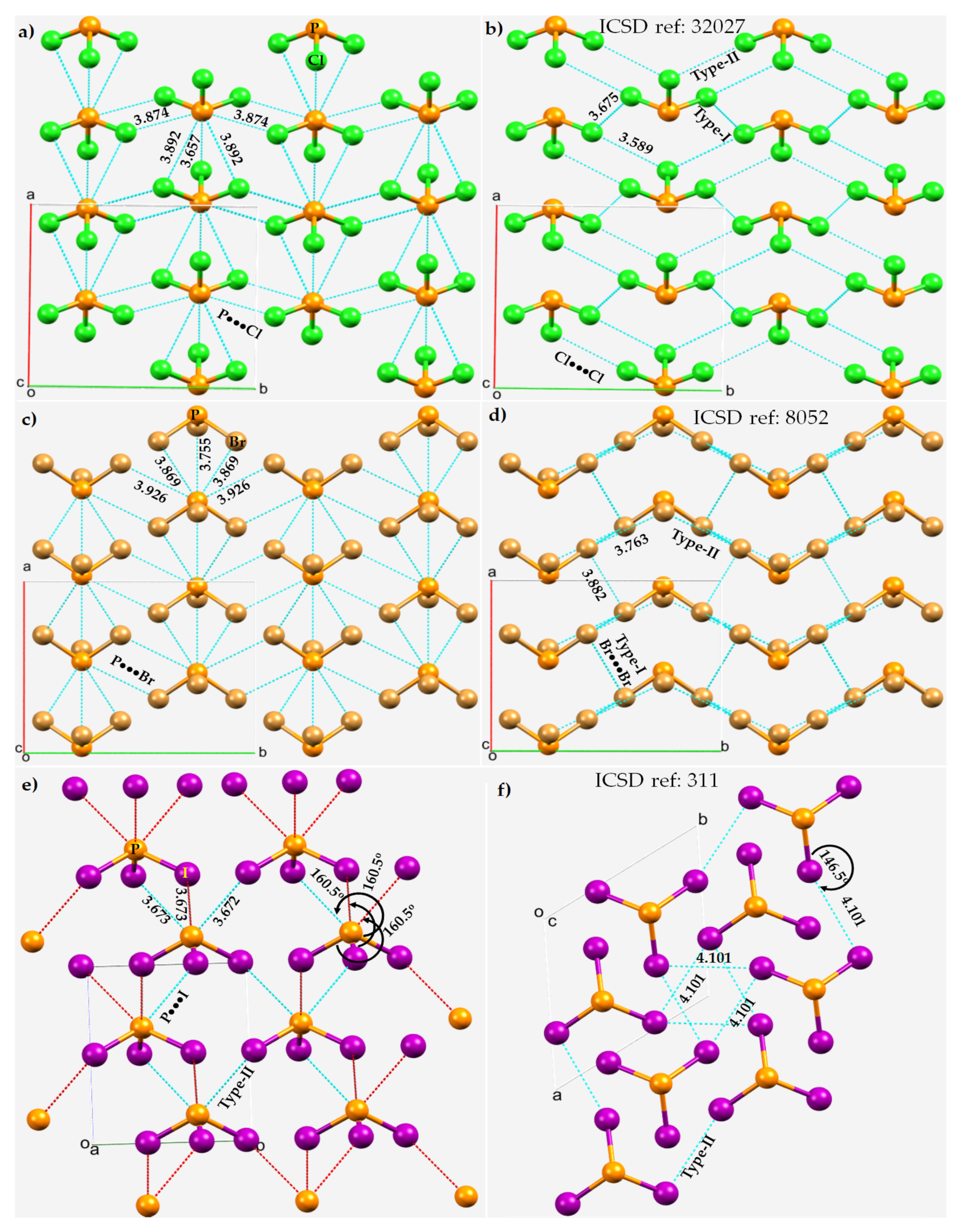
3.2. Phosphorus Trihalides
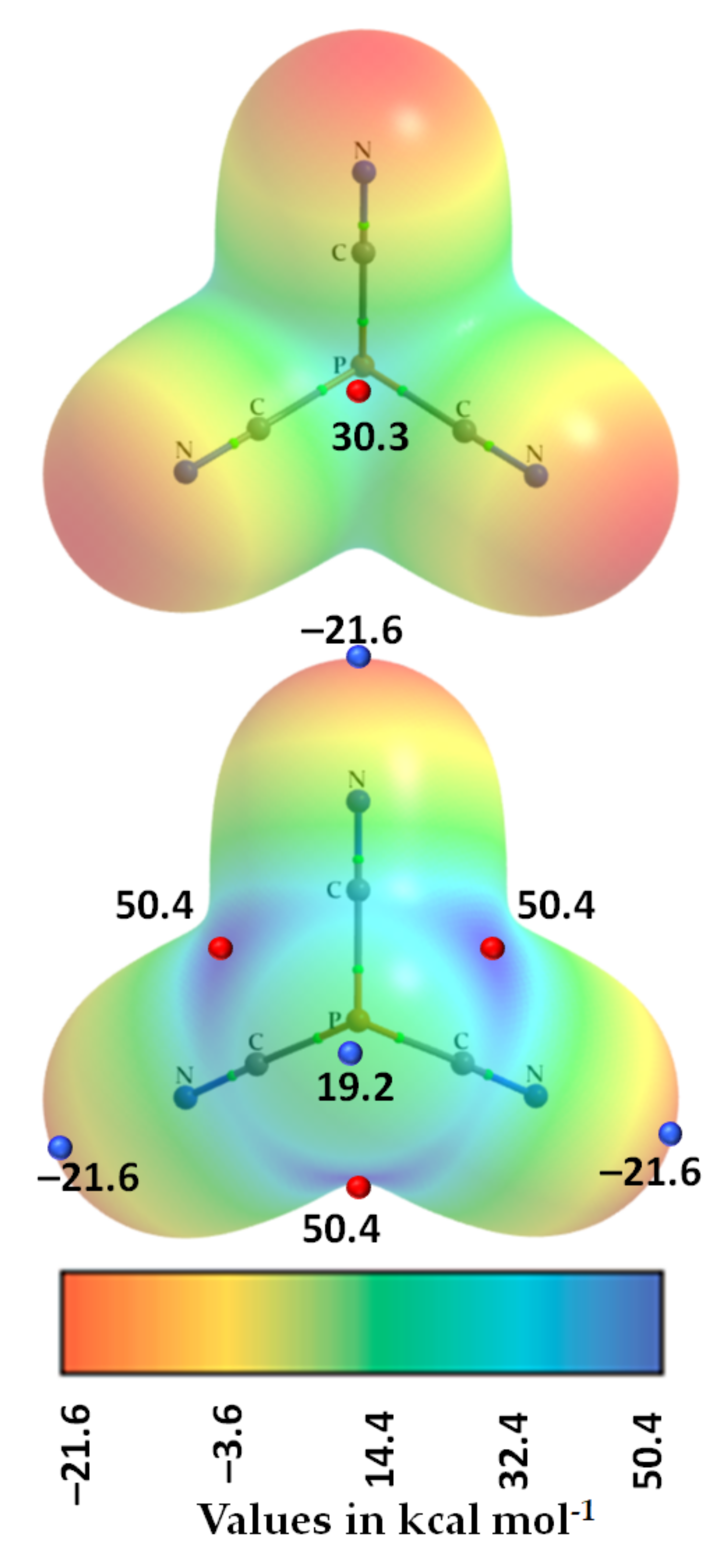
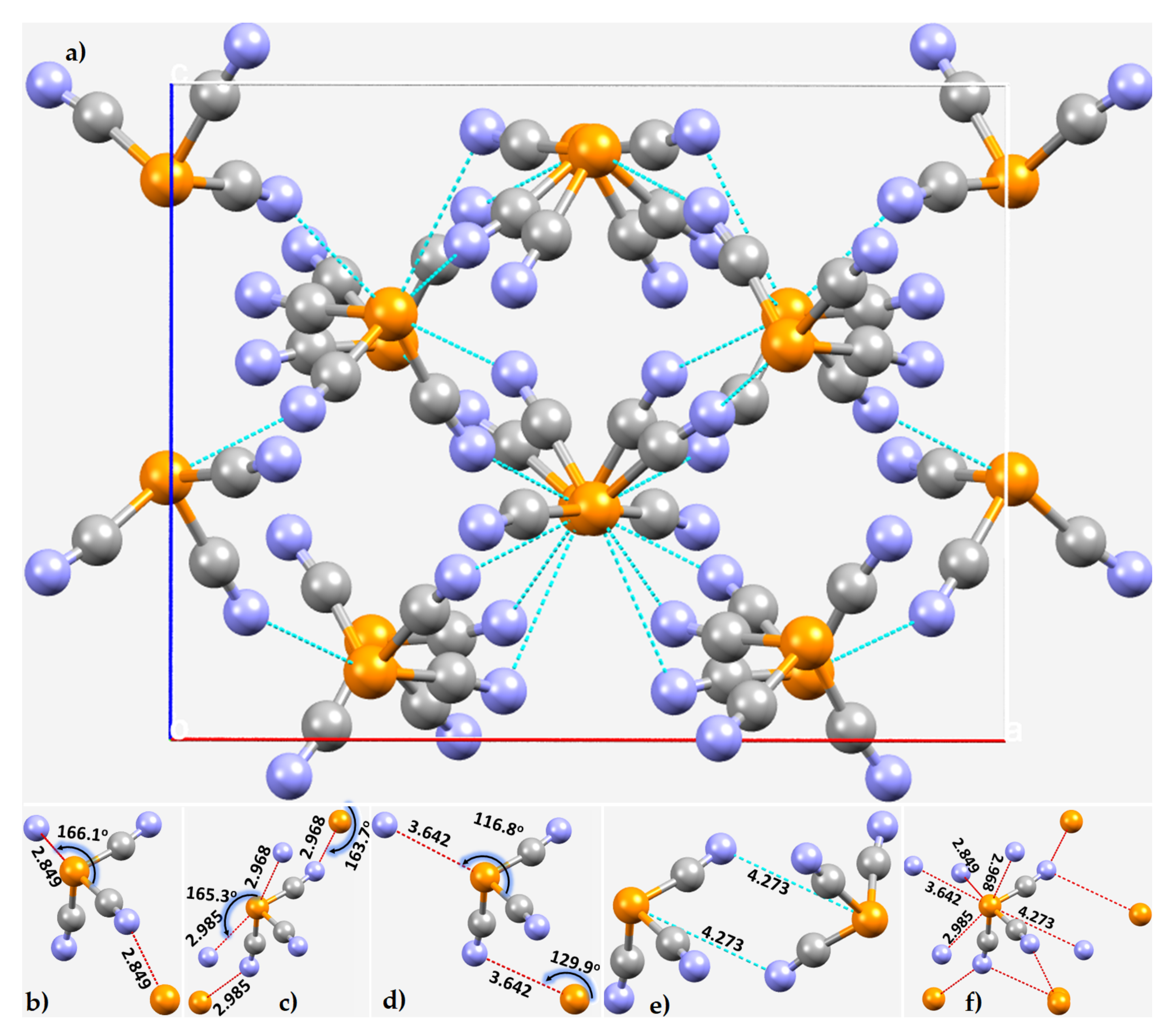
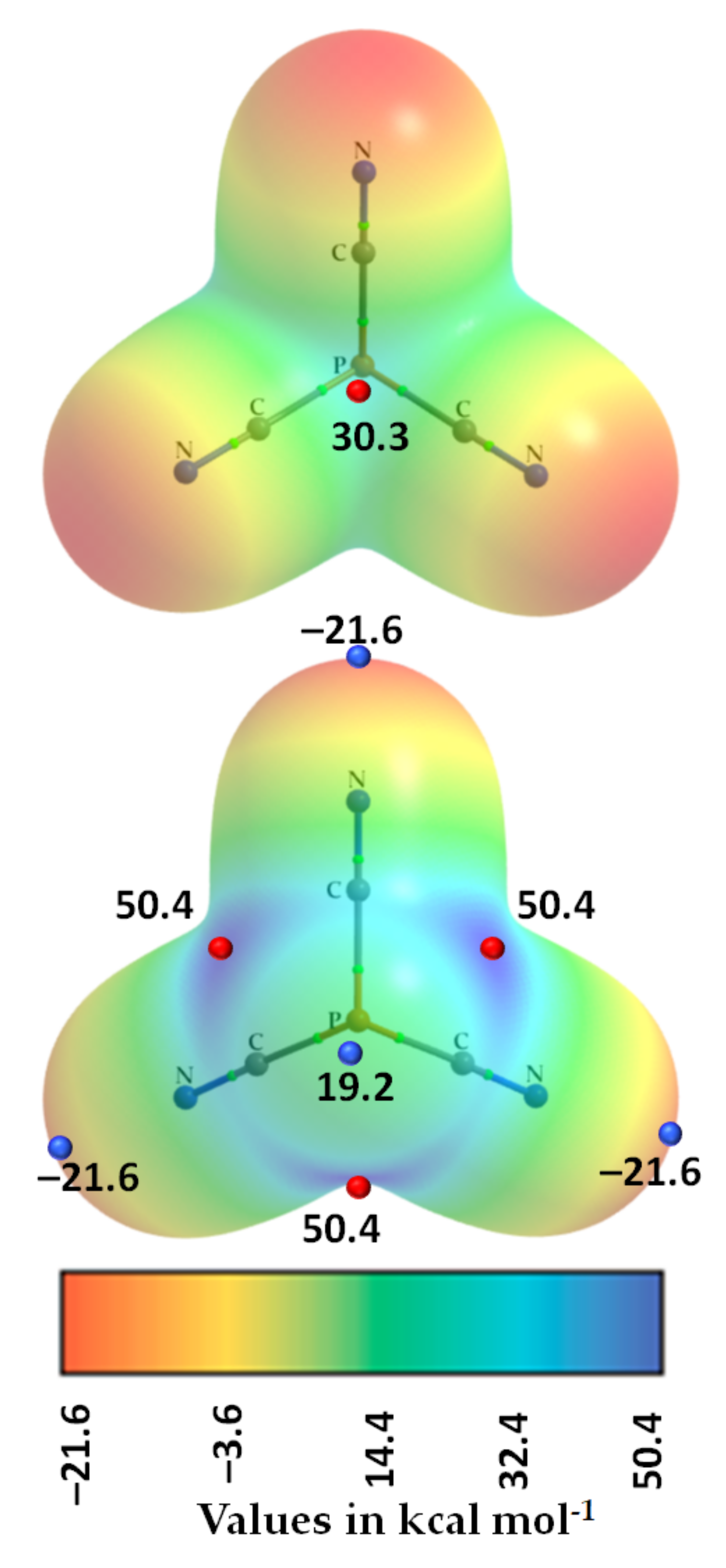
3.3. Phosphorus Tricyanide
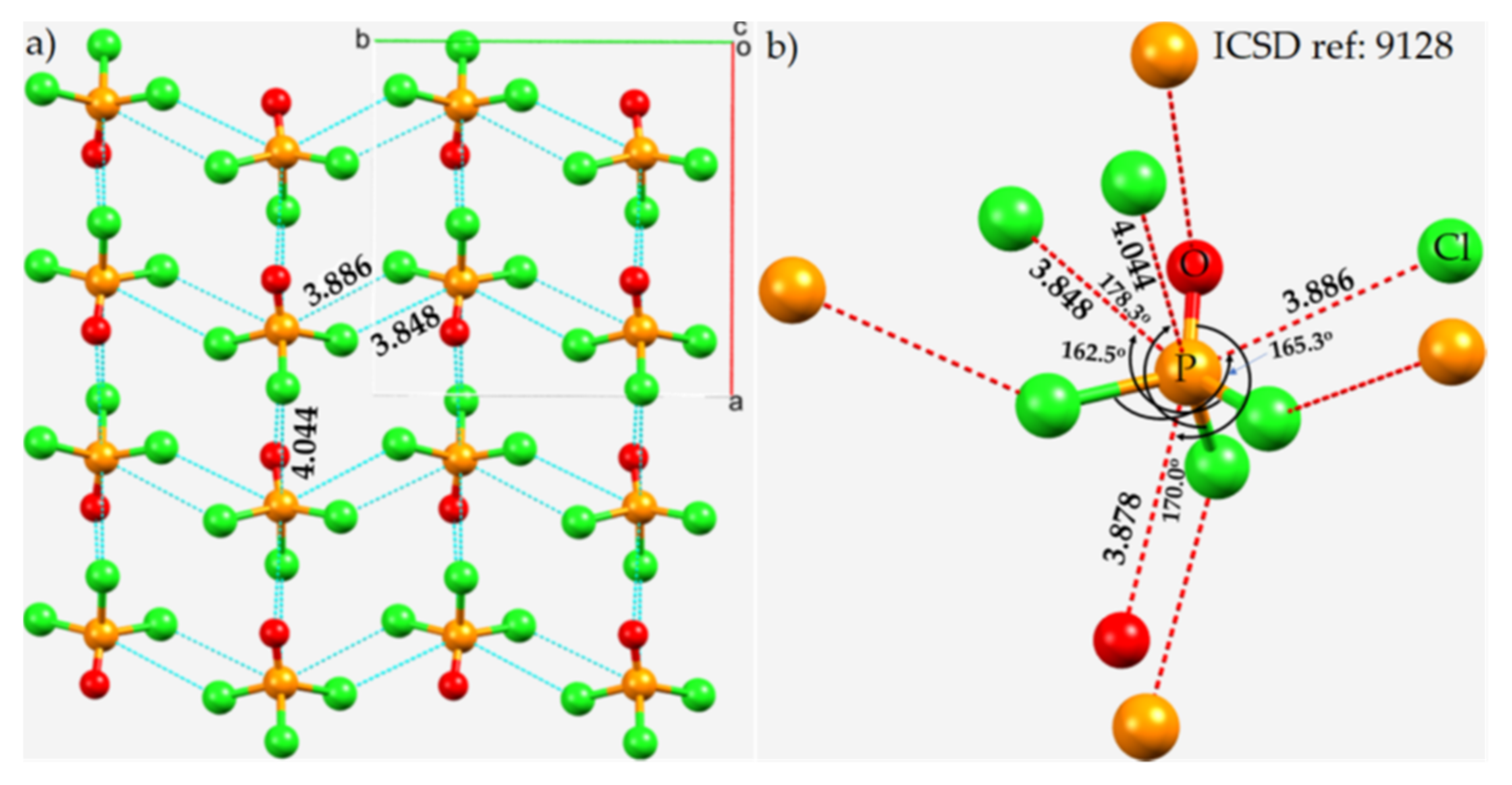
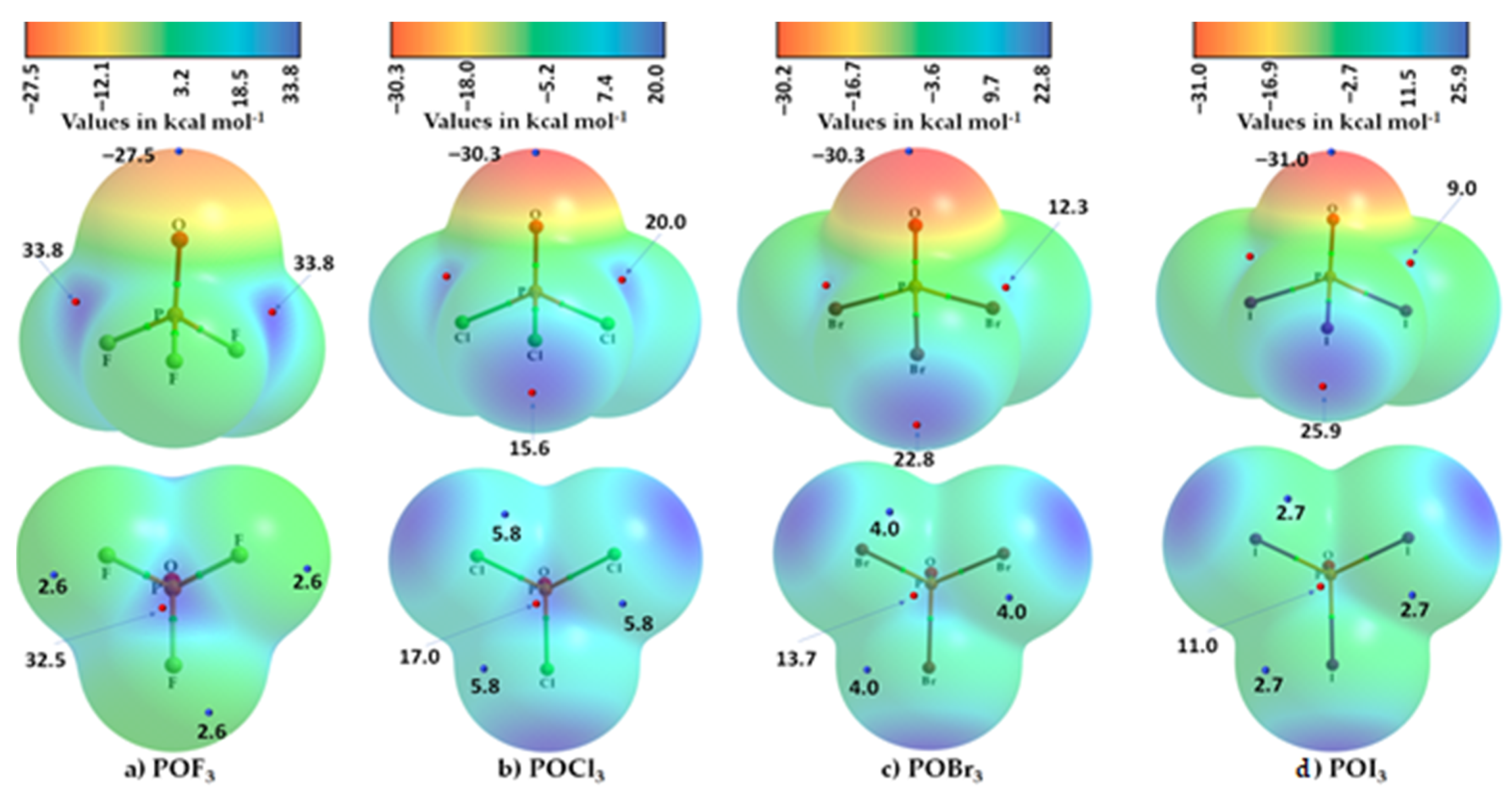
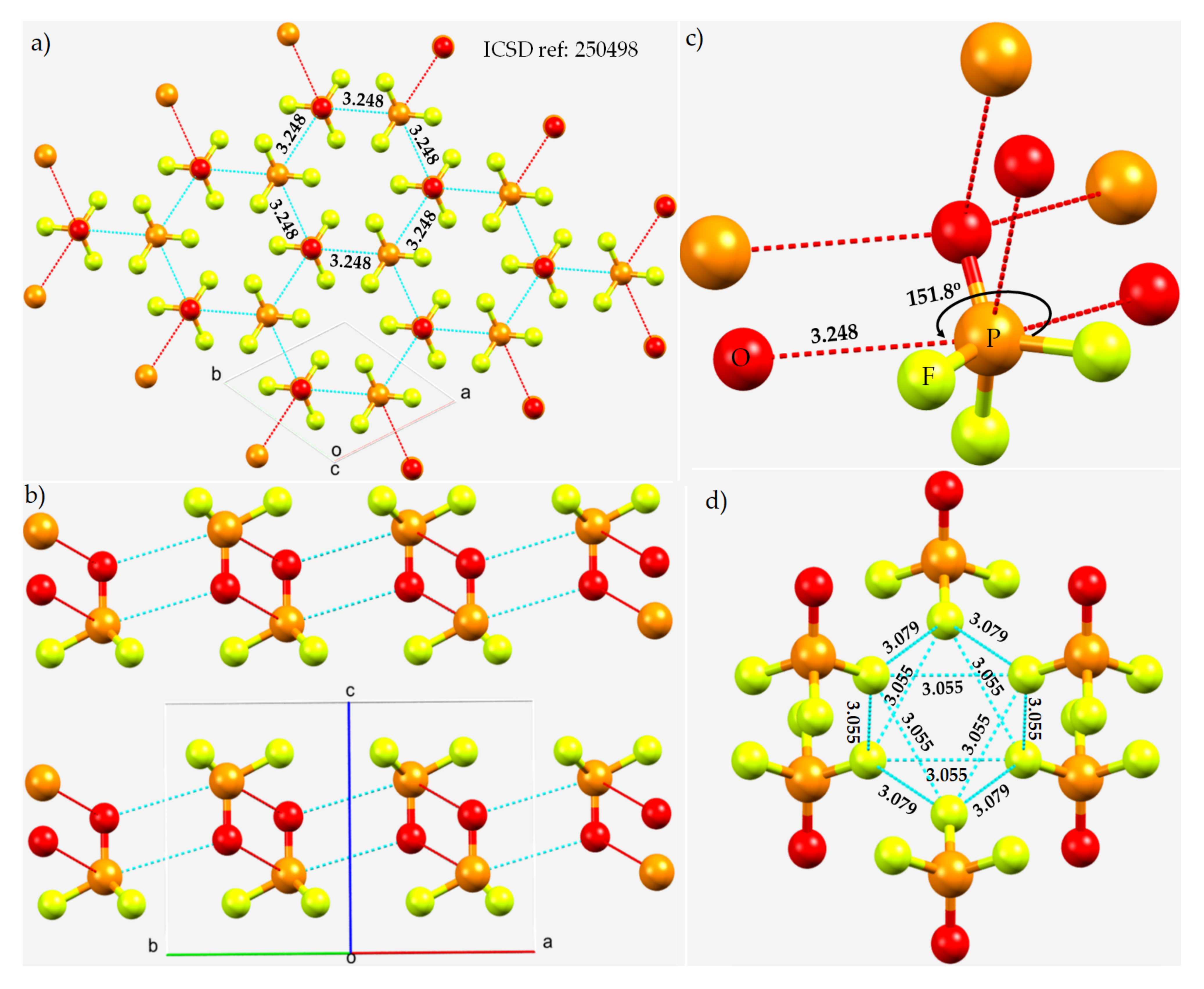
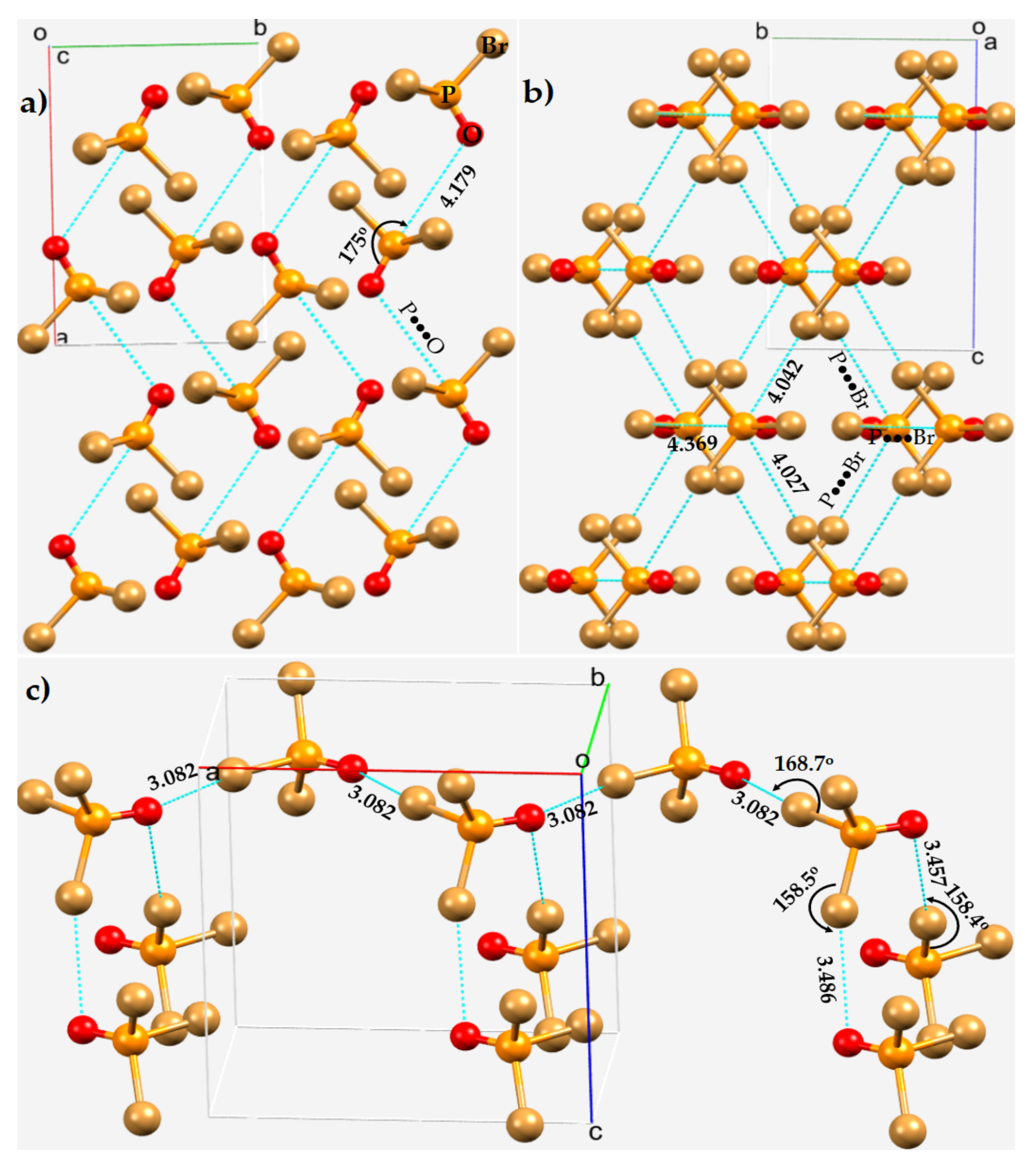
3.4. Phosphoryl Halides
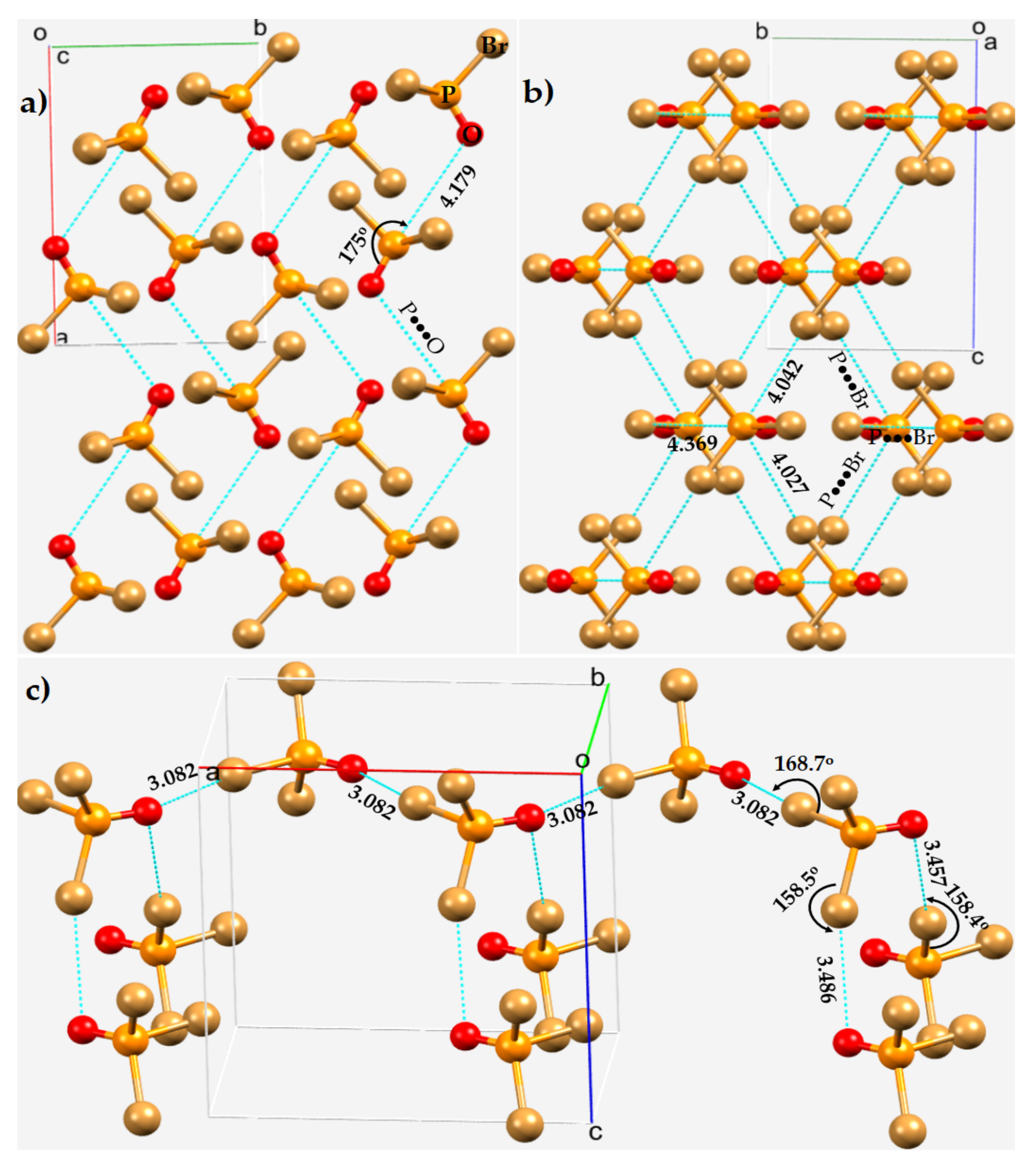
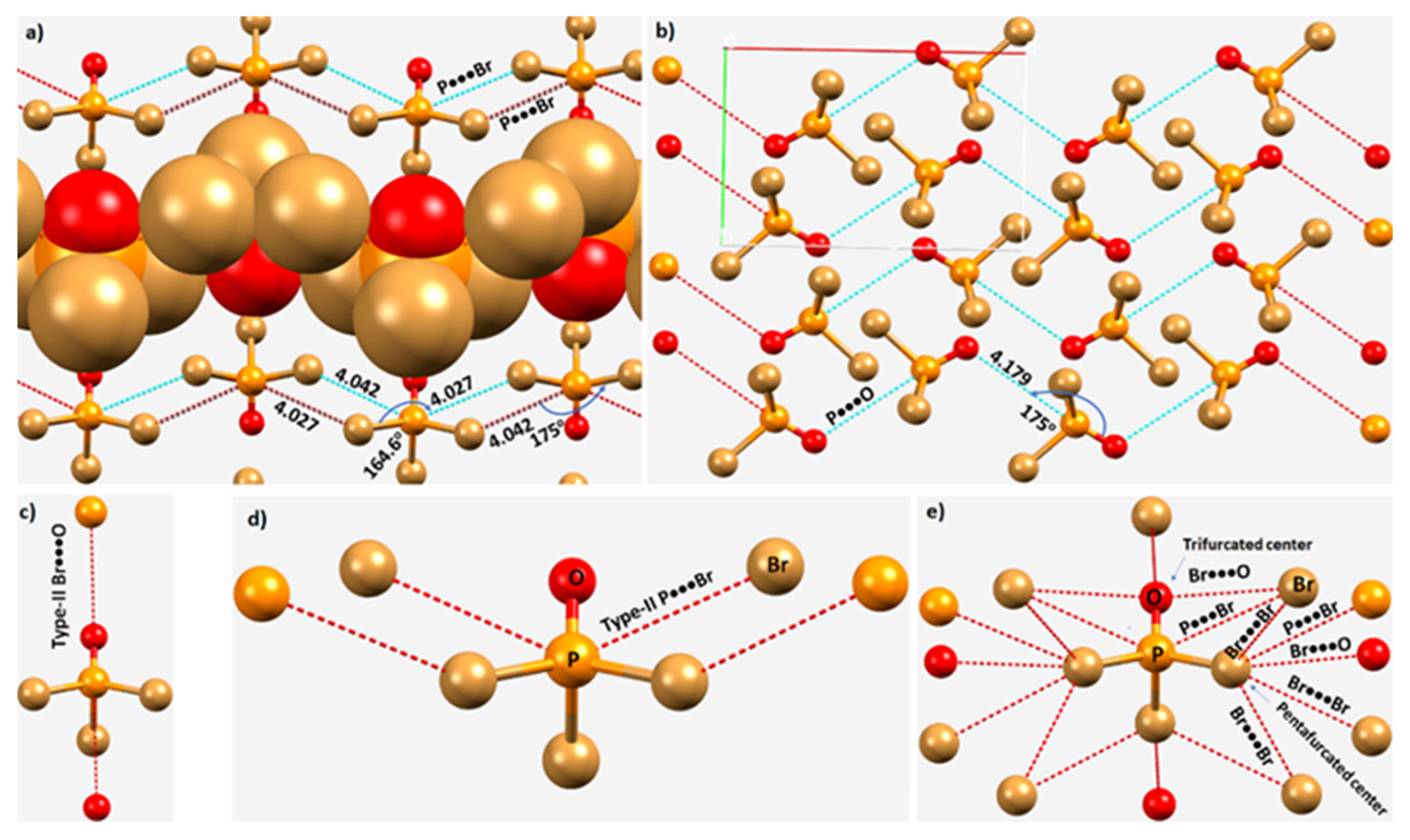
3.5. Diphosphorus Tetraiodide, P2I4
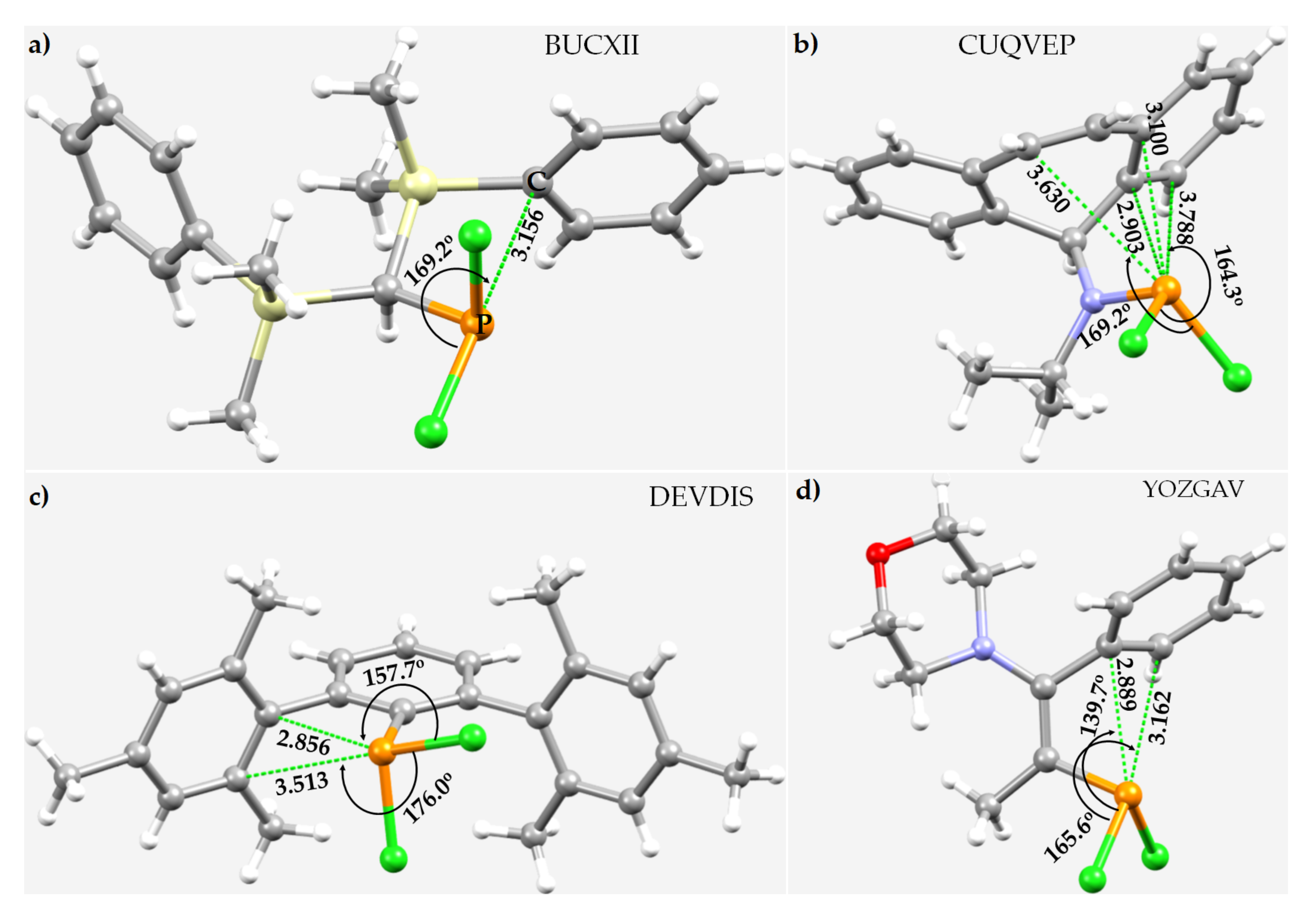
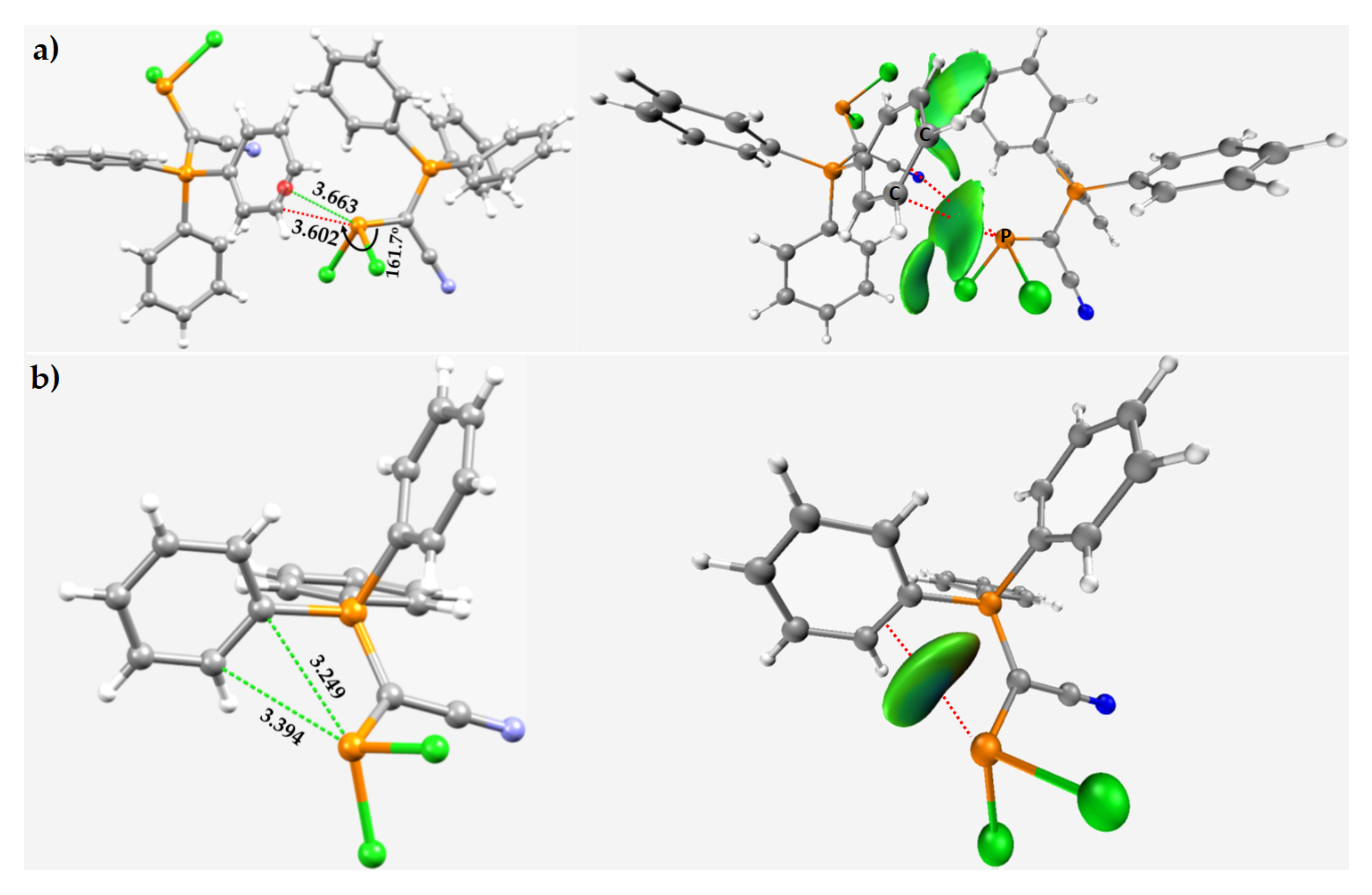
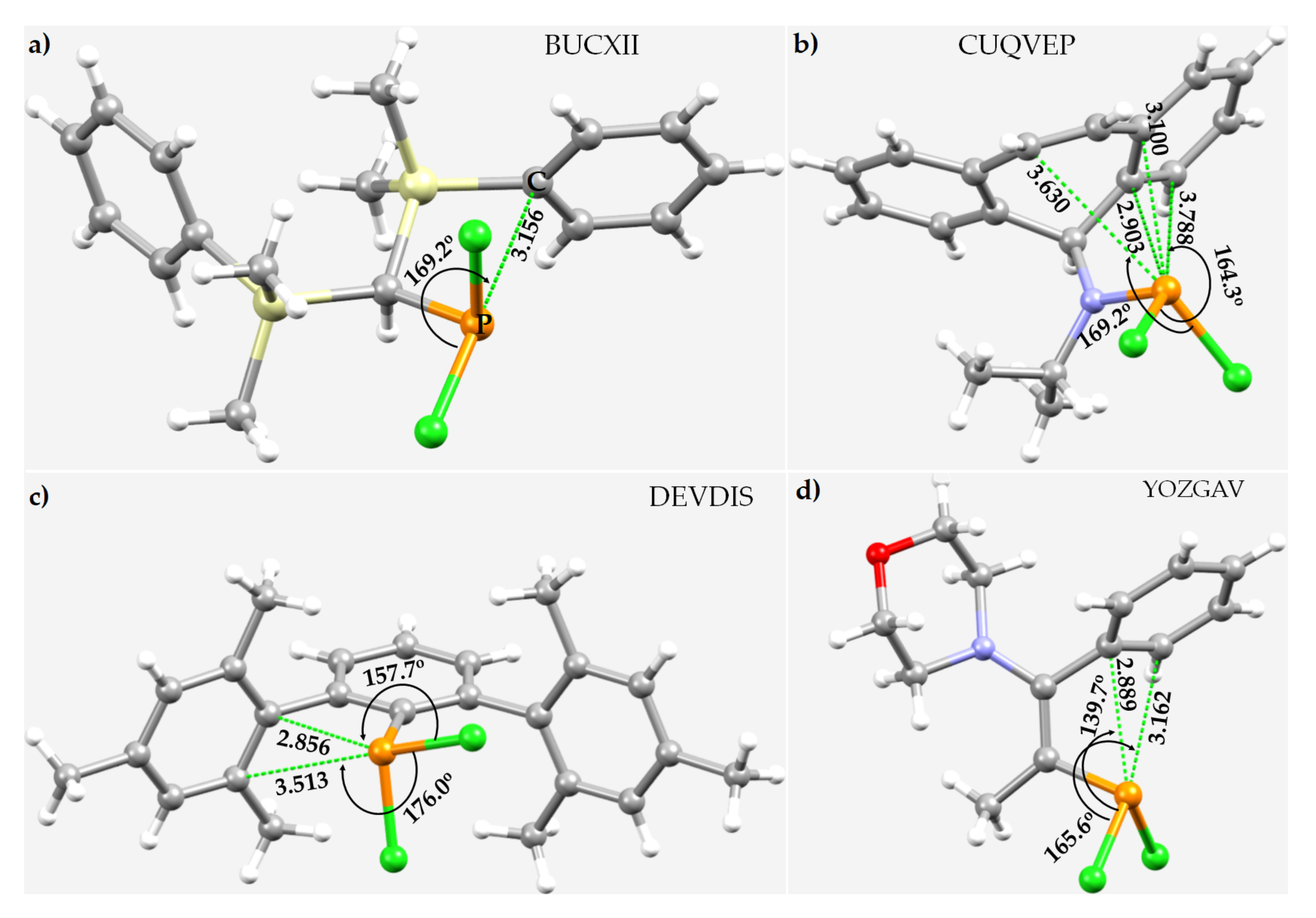
3.6. Miscellaneous Phosphorus Compounds
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Etter, M.C. Encoding and decoding hydrogen-bond patterns of organic compounds. Acc. Chem. Res. 1990, 23, 120–126. [Google Scholar] [CrossRef]
- Etter, M.C.; Urbanczyk-Lipkowska, Z.; Zia-Ebrahimi, M.; Panunto, T.W. Hydrogen bond-directed cocrystallization and molecular recognition properties of diarylureas. J. Am. Chem. Soc. 1990, 112, 8415–8426. [Google Scholar] [CrossRef]
- Johnson, E.R.; Keinan, S.; Mori-Sánchez, P.; Contreras-García, J.; Cohen, A.J.; Yang, W. Revealing Noncovalent Interactions. J. Am. Chem. Soc. 2010, 132, 6498–6506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Desiraju, G.R. A Bond by Any Other Name. Angew. Chem. Int. Ed. 2011, 50, 52–59. [Google Scholar] [CrossRef]
- Desiraju, G.R.; Steiner, T. The Weak Hydrogen Bond in Structural Chemistry and Biology (International Union of Crystallography Monographs on Crystallography, 9); Oxford University Press: Oxford, UK; New York, NY, USA, 1999. [Google Scholar]
- Ishikita, H.; Saito, K. Proton transfer reactions and hydrogen-bond networks in protein environments. J. Roy. Soc. Interface 2014, 11, 20130518. [Google Scholar] [CrossRef]
- Dongfang, J.; Tingting, S.; Huan, C.; Xufeng, S.; Yu Yanmin, Y. Non-covalent interactions in molecular recognition of porphyrins. Mater. Rev. 2018, 32, 3068–3075. [Google Scholar]
- Motherwell, W.B.; Moreno, R.B.; Pavlakos, I.; Arendorf, J.R.T.; Arif, T.; Tizzard, G.J.; Coles, S.J.; Aliev, A.E. Noncovalent Interactions of π Systems with Sulfur: The Atomic Chameleon of Molecular Recognition. Angew. Chem. Int. Ed. 2018, 57, 1193–1198. [Google Scholar] [CrossRef] [Green Version]
- Mahmudov, K.T.; Gurbanov, A.V.; Guedes da Silva, M.F.C.; Pombeiro, A.J.L. Noncovalent Interactions in C–H Bond Functionalization. In Noncovalent Interactions in Catalysis; Chapter 1; Mahmudov, K.T., Kopylovich, M.N., Guedes da Silva, M.F.C., Pombeiro, A.J.L., Eds.; Royal Society of Chemistry: London, UK, 2019; pp. 1–25. [Google Scholar] [CrossRef]
- Berger, G.; Frangvillea, P.; Meyer, F. Halogen bonding for molecular recognition: New developments in materials and biological science. Chem. Commun. 2020, 56, 4970–4981. [Google Scholar] [CrossRef]
- Yan, W.; Zheng, M.; Xu, C.; Chen, F.-E. Harnessing noncovalent interaction of chalcogen bond in organocatalysis: From the catalyst point of view. Green Synth. Cat. 2021, 2, 329–336. [Google Scholar] [CrossRef]
- Paraja, M.; Gini, A.; Sakai, N.; Matile, S. Pnictogen-Bonding Catalysis: An Interactive Tool to Uncover Unorthodox Mechanisms in Polyether Cascade Cyclizations. Chem. Eur. J. 2020, 26, 15471–15476. [Google Scholar] [CrossRef]
- Liu, Y.F.; Zhang, L.; Wei, W. Effect of noncovalent interaction on the self-assembly of a designed peptide and its potential use as a carrier for controlled bFGF release. Int. J. Nanomed. 2017, 12, 659–670. [Google Scholar] [CrossRef] [Green Version]
- Subramani, K.; Ahmed, W. Self-Assembly of Proteins and Peptides and Their Applications in Bionanotechnology and Dentistry. In Emerging Nanotechnologies in Dentistry; Chapter 13; Subramani, K., Ahmed, W., Eds.; William Andrew Publishing: Boston, MA, USA, 2012; pp. 209–224. [Google Scholar]
- Fung, S.Y.; Hong, Y.; Keyes-Baig, C.; Chen, P. 12—Self-assembly of peptides and its potential applications. In Molecular Interfacial Phenomena of Polymers and Biopolymers; Chen, P., Ed.; Woodhead Publishing, CRC Press: Boca Raton, FL, USA, 2005; pp. 421–474. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D.G. Density Functionals for Noncovalent Interaction Energies of Biological Importance. J. Chem. Theory Comp. 2007, 3, 289–300. [Google Scholar] [CrossRef]
- Corminboeuf, C. Minimizing Density Functional Failures for Non-Covalent Interactions beyond van der Waals Complexes. Acc. Chem. Res. 2014, 47, 3217–3224. [Google Scholar] [CrossRef]
- Burns, L.A.; Vázquez-Mayagoitia, Á.; Sumpter, B.G.; Sherrill, C.D. Density-functional approaches to noncovalent interactions: A comparison of dispersion corrections (DFT-D), exchange-hole dipole moment (XDM) theory, and specialized functionals. J. Chem. Phys. 2011, 134, 84107. [Google Scholar] [CrossRef]
- Gao, T.; Li, H.; Li, W.; Li, L.; Fang, C.; Li, H.; Hu, L.; Lu, Y.; Su, Z.-M. A machine learning correction for DFT non-covalent interactions based on the S22, S66 and X40 benchmark databases. J. Cheminform. 2016, 8, 24. [Google Scholar] [CrossRef] [Green Version]
- Mahmudov, K.T.; Gurbanov, A.V.; Aliyeva, V.A.; Resnati, G.; Pombeiro, A.J.L. Pnictogen bonding in coordination chemistry. Coord. Chem. Rev. 2020, 418, 213381. [Google Scholar] [CrossRef]
- Desiraju, G.R. Hydrogen bridges in crystal engineering: Interactions without borders. Acc. Chem. Res. 2002, 35, 565–573. [Google Scholar] [CrossRef]
- Ganguly, P.; Desiraju, G.R. Van der Waals and Polar Intermolecular Contact Distances: Quantifying Supramolecular Synthons. Chem. Asian J. 2008, 3, 868–880. [Google Scholar] [CrossRef]
- Arunan, E.; Desiraju, G.R.; Klein, R.A.; Sadlej, J.; Scheiner, S.; Alkorta, I.; Clary, D.C.; Crabtree, R.H.; Dannenberg, J.J.; Hobza, P.; et al. Definition of the hydrogen bond (IUPAC Recommendations 2011). Pure Appl. Chem. 2011, 83, 1637–1641. [Google Scholar] [CrossRef]
- Desiraju, G.R.; Shing Ho, P.; Kloo, L.; Legon, A.C.; Marquardt, R.; Metrangolo, P.; Politzer, P.; Resnati, G.; Rissanen, K. Definition of the halogen bond (IUPAC Recommendations 2013). Pure Appl. Chem. 2013, 85, 1711–1713. [Google Scholar] [CrossRef]
- Bauzá, A.; Mooibroek, T.J.; Frontera, A. Tetrel-Bonding Interaction: Rediscovered Supramolecular Force? Angew. Chem. Int. Ed. 2013, 52, 12317–12321. [Google Scholar] [CrossRef]
- Taylor, P.G.; Bassindale, A.R.; El Aziz, Y.; Pourny, M.; Stevenson, R.; Hursthouse, M.B.; Coles, S.J. Further studies of fluoride ion entrapment in octasilsesquioxane cages; X-ray crystal structure studies and factors that affect their formation. Dalton Trans. 2012, 41, 2048–2059. [Google Scholar] [CrossRef]
- Varadwaj, P.R.; Varadwaj, A.; Jin, B.Y.Y. Significant evidence of C···O and C···C long-range contacts in several heterodimeric complexes of CO with CH3-X, should one refer to them as carbon and dicarbon bonds! Phys. Chem. Chem. Phys. 2014, 16, 17238–17252. [Google Scholar] [CrossRef]
- Aakeroy, C.B.; Bryce, D.L.; Desiraju, R.G.; Frontera, A.; Legon, A.C.; Nicotra, F.; Rissanen, K.; Scheiner, S.; Terraneo, G.; Metrangolo, P.; et al. Definition of the chalcogen bond (IUPAC Recommendations 2019). Pure Appl. Chem. 2019, 91, 1889–1892. [Google Scholar] [CrossRef]
- Frontera, A.; Bauza, A. On the Importance of Pnictogen and Chalcogen Bonding Interactions in Supramolecular Catalysis. Int. J. Mol. Sci. 2021, 22, 12550. [Google Scholar] [CrossRef]
- Trubenstein, H.J.; Moaven, S.; Vega, M.; Unruh, D.K.; Cozzolino, A.F. Pnictogen bonding with alkoxide cages: Which pnictogen is best? New J. Chem. 2019, 43, 14305–14312. [Google Scholar] [CrossRef]
- Lindquist-Kleissler, B.; Wenger, J.S.; Johnstone, T.C. Analysis of Oxygen–Pnictogen Bonding with Full Bond Path Topological Analysis of the Electron Density. Inorg. Chem. 2021, 60, 1846–1856. [Google Scholar] [CrossRef]
- De Azevedo Santos, L.; van der Lubbe, S.C.C.; Hamlin, T.A.; Ramalho, T.C.; Matthias Bickelhaupt, F. A Quantitative Molecular Orbital Perspective of the Chalcogen Bond. ChemistryOpen 2021, 10, 391–401. [Google Scholar] [CrossRef]
- Bauzá, A.; Frontera, A. Aerogen Bonding Interaction: A New Supramolecular Force? Angew. Chem. Int. Ed. 2015, 54, 7340–7343. [Google Scholar] [CrossRef]
- Steiner, T.; Desiraju, G.R. Distinction between the weak hydrogen bond and the van der Waals interaction. Chem. Commun. 1998, 891–892. [Google Scholar] [CrossRef]
- Frontera, A.; Bauzá, A. On the Importance of σ–Hole Interactions in Crystal Structures. Crystals 2021, 11, 1205. [Google Scholar] [CrossRef]
- Holthoff, J.M.; Engelage, E.; Weiss, R.; Huber, S.M. “Anti-Electrostatic” Halogen Bonding. Angew. Chem. Int. Ed. 2020, 59, 11150–11157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loy, C.; Holthoff, J.M.; Weiss, R.; Huber, S.M.; Rosokha, S.V. “Anti-electrostatic” halogen bonding in solution. Chem. Sci. 2021, 12, 8246–8251. [Google Scholar] [CrossRef] [PubMed]
- Carré, F.; Chuit, C.; Corriu, R.J.P.; Monforte, P.; Nayyar, N.K.; Reyé, C. Intramolecular coordination at phosphorus: Donor-acceptor interaction in three- and four-coordinated phosphorus compounds. J. Organomet. Chem. 1995, 499, 147–154. [Google Scholar] [CrossRef]
- Ashe, A.J. Thermochromic Distibines and Dibismuthines. In Advances in Organometallic Chemistry; Stone, F.G.A., West, R., Eds.; Academic Press: San Diego, CA, USA, 1990; Volume 30, pp. 77–97. [Google Scholar]
- Del Bene, J.; Alkorta, I.; Elguero, J. The Pnicogen Bond in Review: Structures, Binding Energies, Bonding Properties, and Spin-Spin Coupling Constants of Complexes Stabilized by Pnicogen Bonds. In Noncovalent Forces. Challenges and Advances in Computational Chemistry and Physics; Scheiner, S., Ed.; Springer: Cham, Switzerland, 2015; Volume 19. [Google Scholar]
- Joshi, P.R.; Sankaran, K. P⋯N type pnicogen bonding in phosphorus trichloride–pyridine adduct: A matrix isolation infrared, DFT and ab initio study. J. Mol. Str. 2020, 1217, 128408. [Google Scholar] [CrossRef]
- Mokrai, R.; Barrett, J.; Apperley, D.C.; Batsanov, A.S.; Benkő, Z.; Heift, D. Weak Pnictogen Bond with Bismuth: Experimental Evidence Based on Bi−P Through-Space Coupling. Chem. Eur. J. 2019, 25, 4017–4024. [Google Scholar] [CrossRef] [Green Version]
- De Azevedo Santos, L.; Hamlin, T.A.; Ramalho, T.C.; Bickelhaupt, F.M. The pnictogen bond: A quantitative molecular orbital picture. Phys. Chem. Chem. Phys. 2021, 23, 13842–13852. [Google Scholar] [CrossRef]
- Greenwood, N.N.; Earnshaw, A. Chemistry of the Elements, 2nd ed.; Elsevier: Oxford, UK, 1997. [Google Scholar]
- International Crystal Structure Database (ICSD). Available online: https://icsd.products.fiz-karlsruhe.de/en (accessed on 25 January 2022).
- Belsky, A.; Hellenbrandt, M.; Karen, V.L.; Luksch, P. New developments in the Inorganic Crystal Structure Database (ICSD): Accessibility in support of materials research and design. Acta Crystallogr. B 2002, 58, 364–369. [Google Scholar] [CrossRef] [Green Version]
- Groom, C.R.; Bruno, I.J.; Lightfoot, M.P.; Ward, S.C. The Cambridge Structural Database. Acta Cryst. 2016, B72, 171–179. [Google Scholar] [CrossRef]
- Politzer, P.; Murray, J.S. The use and misuse of van der Waals radii. Struct. Chem. 2021, 32, 623–629. [Google Scholar] [CrossRef]
- Huber, S.M.; Scanlon, J.D.; Jimenez-Izal, E.; Ugalde, J.M.; Infante, I. On the directionality of halogen bonding. Phys. Chem. Chem. Phys. 2013, 15, 10350–10357. [Google Scholar] [CrossRef]
- Cavallo, G.; Metrangolo, P.; Milani, R.; Pilati, T.; Priimagi, A.; Resnati, G.; Terraneo, G. The halogen bond. Chem. Rev. 2016, 116, 2478–2601. [Google Scholar] [CrossRef] [Green Version]
- Varadwaj, P.R.; Varadwaj, A.; Marques, H.M. Halogen Bonding: A Halogen-Centered Noncovalent Interaction Yet to Be Understood. Inorganics 2019, 7, 40. [Google Scholar] [CrossRef] [Green Version]
- Murray, J.S.; Politzer, P. Molecular Surfaces, van der Waals Radii and Electrostatic Potentials in Relation to Noncovalent Interactions. Croat. Chem. Acta 2009, 82, 267–275. [Google Scholar]
- Lefebvre, C.; Rubez, G.; Khartabil, H.; Boisson, J.-C.; Contreras-García, J.; Hénon, E. Accurately extracting the signature of intermolecular interactions present in the NCI plot of the reduced density gradient versus electron density. Phys. Chem. Chem. Phys. 2017, 19, 17928–17936. [Google Scholar] [CrossRef]
- Lefebvre, C.; Khartabil, H.; Boisson, J.-C.; Contreras-García, J.; Piquemal, J.-P.; Hénon, E. The Independent Gradient Model: A New Approach for Probing Strong and Weak Interactions in Molecules from Wave Function Calculations. ChemPhysChem 2018, 19, 724–735. [Google Scholar] [CrossRef]
- Pritchard, B.P.; Altarawy, D.; Didier, B.; Gibson, T.D.; Windus, T.L. New Basis Set Exchange: An Open, Up-to-Date Resource for the Molecular Sciences Community. J. Chem. Inf. Model. 2019, 59, 4814–4820. [Google Scholar] [CrossRef]
- Schuchardt, K.L.; Didier, B.T.; Elsethagen, T.; Sun, L.; Gurumoorthi, V.; Chase, J.; Li, J.; Windus, T.L. Basis Set Exchange: A Community Database for Computational Sciences. J. Chem. Inf. Model. 2007, 47, 1045–1052. [Google Scholar] [CrossRef] [Green Version]
- Politzer, P.; Murray, J.S.; Clark, T.; Resnati, G. The σ-hole revisited. Phys. Chem. Chem. Phys. 2017, 19, 32166–32178. [Google Scholar] [CrossRef]
- Murray, J.S.; Lane, P.; Politzer, P. Expansion of the σ-hole concept. J. Mol. Model. 2009, 15, 723–729. [Google Scholar] [CrossRef]
- Clark, T.; Hennemann, M.; Murray, J.S.; Politzer, P. Halogen bonding: The σ-hole. J. Mol. Model. 2007, 13, 291–296. [Google Scholar] [CrossRef]
- Varadwaj, A.; Marques, H.M.; Varadwaj, P.R. Is the Fluorine in Molecules Dispersive? Is Molecular Electrostatic Potential a Valid Property to Explore Fluorine-Centered Non-Covalent Interactions? Molecules 2019, 24, 379. [Google Scholar] [CrossRef] [Green Version]
- Varadwaj, A.; Marques, H.M.; Varadwaj, P.R. Nature of halogen-centered intermolecular interactions in crystal growth and design: Fluorine-centered interactions in dimers in crystalline hexafluoropropylene as a prototype. J. Comp. Chem. 2019, 40, 1836–1860. [Google Scholar] [CrossRef]
- Varadwaj, P.R.; Varadwaj, A.; Marques, H.M. Does Chlorine in CH3Cl Behave as a Genuine Halogen Bond Donor? Crystals 2020, 10, 146. [Google Scholar] [CrossRef] [Green Version]
- Bader, R.F.W.; Carroll, M.T.; Cheeseman, J.R.; Chang, C. Properties of atoms in molecules: Atomic volumes. J. Am. Chem. Soc. 1987, 109, 7968–7979. [Google Scholar] [CrossRef]
- Politzer, P.; Murray, J.S. σ-Hole Interactions: Perspectives and Misconceptions. Crystals 2017, 7, 212. [Google Scholar] [CrossRef] [Green Version]
- Murray, J.S.; Resnati, G.; Politzer, P. Close contacts and noncovalent interactions in crystals. Faraday Discuss. 2017, 203, 113–130. [Google Scholar] [CrossRef]
- Wang, H.; Wang, W.; Jin, W.J. σ-Hole Bond vs π-Hole Bond: A Comparison Based on Halogen Bond. Chem. Rev. 2016, 116, 5072–5104. [Google Scholar] [CrossRef]
- Grabowski, S.J. Hydrogen bonds, and σ-hole and π-hole bonds—mechanisms protecting doublet and octet electron structures. Phys. Chem. Chem. Phys. 2017, 19, 29742–29759. [Google Scholar] [CrossRef]
- Pal, R.; Nagendra, G.; Samarasimhareddy, M.; Sureshbabu, V.V.; Guru Row, T.N. Observation of a reversible isomorphous phase transition and an interplay of “σ-holes” and “π-holes” in Fmoc-Leu-ψ[CH2-NCS]. Chem. Commun. 2015, 51, 933–936. [Google Scholar] [CrossRef]
- Peter, P.; Murray, J.S. σ-holes and π-holes: Similarities and differences. J. Comp. Chem. 2018, 39, 464–471. [Google Scholar] [CrossRef]
- Báuza, A.; Frontera, A.; Mooibroek, T.J. π-Hole Interactions Involving Nitro Compounds: Directionality of Nitrate Esters. Cryst. Growth Des. 2016, 16, 5520–5524. [Google Scholar] [CrossRef]
- Mooibroek, T.J. Coordinated nitrate anions can be directional π-hole donors in the solid state: A CSD study. CrystEngComm 2017, 19, 4485–4488. [Google Scholar] [CrossRef]
- Grabowski, S.J. π-Hole Bonds: Boron and Aluminum Lewis Acid Centers. ChemPhysChem 2015, 16, 1470–1479. [Google Scholar] [CrossRef]
- Jabłoński, M. Ten years of charge-inverted hydrogen bonds. Struct. Chem. 2020, 31, 61–80. [Google Scholar] [CrossRef]
- Jabłoński, M. Hydride-Triel Bonds. J. Comp. Chem. 2018, 39, 1177–1191. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09; Rev. C.01; Gaussian, Inc.: Wallinford, CT, USA, 2016. [Google Scholar]
- Macrae, C.F.; Bruno, I.J.; Chisholm, J.A.; Edgington, P.R.; McCabe, P.; Pidcock, E.; Rodriguez-Monge, L.; Taylor, R.; van de Streek, J.; Wood, P.A. Mercury 4.0: From visualization to analysis, design and prediction. J. Appl. Cryst. 2008, 41, 466–470. [Google Scholar] [CrossRef]
- Dennington, R.; Keith, T.; Millam, J. GaussView, V. 5; 5.0.9; Semichem, Inc.: Shawnee Mission, KS, USA, 2009. [Google Scholar]
- Keith, T.A. AIMAll (V. 19.10.12); TK Gristmill Software: Overland Park, KS, USA, 2019; Available online: http://aim.tkgristmill.com (accessed on 29 December 2021).
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comp. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD—Visual Molecular Dynamics. J. Molec. Graphics 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Xiao, H.; Hao, F.; Liao, X.; Shi, X.; Zhang, Y.; Chen, X. Prediction of a Two-Dimensional Phosphorus Nitride Monolayer (v2). Available online: https://arxiv.org/abs/1603.01957v2 (accessed on 29 December 2021).
- Schusteritsch, G.; Uhrin, M.; Pickard, C.J. Single-Layered Hittorf’s Phosphorus: A Wide-Bandgap High Mobility 2D Material. Nano Lett. 2016, 16, 2975–2980. [Google Scholar] [CrossRef] [Green Version]
- Tan, X.; Ji, Y.; Dong, H.; Liu, M.; Hou, T.; Li, Y. A novel metal-free two-dimensional material for photocatalytic water splitting—Phosphorus nitride (γ-PN). RSC Adv. 2017, 7, 50239–50245. [Google Scholar] [CrossRef] [Green Version]
- Xia, F.; Wang, H.; Hwang, J.C.M.; Neto, A.H.C.; Yang, L. Black phosphorus and its isoelectronic materials. Nat. Rev. Phys. 2019, 1, 306–317. [Google Scholar] [CrossRef]
- Eskandari, K.; Zariny, H. Halogen bonding: A lump-hole interaction. Chem. Phys. Lett. 2010, 492, 9–13. [Google Scholar] [CrossRef]
- Zhang, L.; Huang, H.; Zhang, B.; Gu, M.; Zhao, D.; Zhao, X.; Li, L.; Zhou, J.; Wu, K.; Cheng, Y.; et al. Structure and Properties of Violet Phosphorus and Its Phosphorene Exfoliation. Angew Chem. Int. Edn. 2020, 59, 1074–1080. [Google Scholar] [CrossRef]
- Krebs, H.; Holz, W.; Worms, K.H. Über die Struktur und die Eigenschaften der Halbmetalle, X. Eine Neue Rhombische Arsenmodifikation und Ihre Mischkristallbildung mit Schwarzem Phosphor. Chem. Ber. 1957, 90, 1031–1037. [Google Scholar] [CrossRef]
- Zelezny, W.F.; Baenziger, N.C. The Crystal Structure of Tetrachlorophosphonium Dichloroiodide. J. Am. Chem. Soc. 1952, 74, 6151–6152. [Google Scholar] [CrossRef]
- Clark, D.; Powell, H.M.; Wells, A.F. 134. The crystal structure of phosphorus pentachloride. J. Chem. Soc. 1942, 642–645. [Google Scholar] [CrossRef]
- Powell, H.M.; Clark, D.; Wells, A.F. Crystal Structure of Phosphorus Pentachloride. Nature 1940, 145, 149. [Google Scholar] [CrossRef]
- Van Driel, M.; Mac Gillavry, C.H. The crystal structure of phosphorus pentabromide. Rec. Trav. Chim. Pays-Bas 1943, 62, 167–171. [Google Scholar] [CrossRef]
- Leung, Y.C.; Waser, J.; Houten, S.v.; Vos, A.; Wiegers, G.A.; Wiebenga, E.H. The crystal structure of P4S3. Acta Crystallogr. 1957, 10, 574–582. [Google Scholar] [CrossRef]
- Van Houten, S.; Wiebenga, E.H. The crystal structure of P4S5. Acta Crystallogr. 1957, 10, 156–160. [Google Scholar] [CrossRef] [Green Version]
- Vos, A.; Wiebenga, E.H. The crystal structures of P4S10 and P4S7. Acta Crystallogr. 1955, 8, 217–223. [Google Scholar] [CrossRef] [Green Version]
- Wright, D.A.; Penfold, B.R. The crystal and molecular structure of phosphorus thioiodide. Acta Crystallogr. 1959, 12, 455–460. [Google Scholar] [CrossRef]
- Emerson, K. The crystal and molecular structure of sulfur dicyanide. Acta Crystallogr. 1966, 21, 970–974. [Google Scholar] [CrossRef]
- Chandra, S.; Suryaprasad, B.; Ramanathan, N.; Sundararajan, K. Dominance of unique P⋯π phosphorus bonding with π donors: Evidence using matrix isolation infrared spectroscopy and computational methodology. Phys. Chem. Chem. Phys. 2020, 22, 20771–20791. [Google Scholar] [CrossRef]
- Legon, A.C. Tetrel, pnictogen and chalcogen bonds identified in the gas phase before they had names: A systematic look at non-covalent interactions. Phys. Chem. Chem. Phys. 2017, 19, 14884–14896. [Google Scholar] [CrossRef]
- Varadwaj, A.; Varadwaj, P.R.; Jin, B.-Y. Fluorines in tetrafluoromethane as halogen bond donors: Revisiting address the nature of the fluorine’s σ-hole. Int. J. Quantum Chem. 2015, 115, 453–470. [Google Scholar] [CrossRef]
- Politzer, P.; Murray, J.S.; Yepes, D.; Jaque, P. Driving and retarding forces in a chemical reaction. J. Mol. Model. 2014, 20, 2351. [Google Scholar] [CrossRef]
- Esrafili, M.D.; Ghanbari, M.; Mohammadian-Sabet, F. Substituent effects on cooperativity of pnicogen bonds. J. Mol. Model. 2014, 20, 2436. [Google Scholar] [CrossRef]
- Morino, Y.; Kuchitsu, K.; Moritani, T. Molecular structure of phosphorus trifluoride studied by gas electron diffraction. Inorg. Chem. 1969, 8, 867–871. [Google Scholar] [CrossRef]
- Hartl, H.; Rama, M. Die Kristallstruktur von Phosphortrichlorid bei −110 °C/The Crystal Structure of Phosphorus Trichloride at −110 °C. Zeitschrift für Naturforschung B 1979, 34, 1035–1036. [Google Scholar] [CrossRef]
- Enjalbert, R.; Savariault, J.M.; Legros, J.P. Etude Structurale a 123 K du Trichlorure de Phosphore PCl3. Comptes Rendus de l’Académie des Sciences 1980, 290, 239–241. [Google Scholar]
- Enjalbert, R.; Galy, J. Structure cristalline du tribromure de phosphore a 193 K. Acta Crystallogr. B 1979, 35, 546–550. [Google Scholar] [CrossRef]
- Lance, E.T.; Haschke, J.M.; Peacor, D.R. Crystal and molecular structure of phosphorus triiodide. Inorg. Chem. 1976, 15, 780–781. [Google Scholar] [CrossRef]
- Emerson, K.; Britton, D. The crystal and molecular structure of phosphorus tricyanide. Acta Crystallogr. 1964, 17, 1134–1139. [Google Scholar] [CrossRef]
- Williams, Q.; Sheridan, J.; Gordy, W. Microwave Spectra and Molecular Structures of POF3, PSF3, POCl3, and PSCl3. J. Chem. Phys. 1952, 20, 164–167. [Google Scholar] [CrossRef]
- NIST Chemistry WebBook, SRD 69. Available online: https://webbook.nist.gov/chemistry/ (accessed on 3 January 2022).
- Cavell, R.G. Chemistry of phosphorus fluorides. Part II. Secondary alkylamino derivatives of phosphoryl fluoride. Can. J. Chem. 1967, 45, 1309–1319. [Google Scholar] [CrossRef]
- Feller, M.; Lux, K.; Kornath, A. Crystal Structure and Spectroscopic Investigations of POF3. Zeitschrift für Anorganische und Allgemeine Chemie 2014, 640, 53–56. [Google Scholar] [CrossRef]
- Olie, K. The crystal structure of POCl3. Acta Crystallogr. B 1971, 27, 1459–1460. [Google Scholar] [CrossRef]
- Templeton, L.K.; Templeton, D.H. The crystal structure of POBr3: Space group and refinement by least squares. Acta Crystallogr. B 1971, 27, 1678–1679. [Google Scholar] [CrossRef]
- Olie, K.; Mijlhoff, F.C. The crystal structure of POBr3 and intermolecular bonding. Acta Crystallogr. B 1969, 25, 974–977. [Google Scholar] [CrossRef]
- Varadwaj, P.R.; Varadwaj, A.; Jin, B.-Y. Halogen bonding interaction of chloromethane withseveral nitrogen donating molecules: Addressing thenature of the chlorine surface σ-hole. Phys. Chem. Chem. Phys. 2014, 16, 19573–19589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varadwaj, A.; Varadwaj, P.R.; Marques, H.M.; Yamashita, K. Revealing Factors Influencing the Fluorine-Centered Non-Covalent Interactions in Some Fluorine-substituted Molecular Complexes: Insights from First-Principles Studies. ChemPhysChem 2018, 19, 1486–1499. [Google Scholar] [CrossRef] [PubMed]
- Varadwaj, A.; Varadwaj, P.R.; Jin, B.-Y. Can an entirely negative fluorine in a molecule, viz. perfluorobenzene, interact attractively with the entirely negative site (s) on another molecule (s)? Like liking like! RSC Adv. 2016, 6, 19098–19110. [Google Scholar] [CrossRef]
- Varadwaj, P.R.; Varadwaj, A.; Marques, H.M.; Yamashita, K. Can Combined Electrostatic and Polarization Effects Alone Explain the F···F Negative-Negative Bonding in Simple Fluoro-Substituted Benzene Derivatives? A First-Principles Perspective. Computation 2018, 6, 51. [Google Scholar] [CrossRef] [Green Version]
- Leung, Y.C.; Waser, J. The Crystal Structure of Phosphorus Diiodide, P2I4. J. Phys. Chem. 1956, 60, 539–543. [Google Scholar] [CrossRef]
- Groh, M.F.; Müller, U.; Ahmed, E.; Rothenberger, A.; Ruck, M. Substitution of Conventional High-temperature Syntheses of Inorganic Compounds by Near-room-temperature Syntheses in Ionic Liquids. Z. Naturforsch. B 2013, 68, 1108–1122. [Google Scholar] [CrossRef]
- Zak, Z.; Cernik, M. Diphosphorus Tetraiodide at 120K. Acta Crystallogr. C 1996, 52, 290–291. [Google Scholar] [CrossRef]
- Schwarz, C.; Scherpf, T.; Rodstein, I.; Weismann, J.; Feichtner, K.-S.; Gessner, V.H. Ylide-Functionalization via Metalated Ylides: Synthesis and Structural Properties. ChemistryOpen 2019, 8, 621–626. [Google Scholar] [CrossRef] [Green Version]
- Jones, P.G.; Daniliuc, C.G.; du Mont, W.-W.; Riecke, A. CSD Private Communication; CCDC 1053040; CCDC: Cambridge, UK, 2015. [Google Scholar]
- Liedtke, J.; Loss, S.; Widauer, C.; Grützmacher, H. Phosphiranes as Ligands for Platinum Catalysed Hydrosilylations. Tetrahedron 2000, 56, 143–156. [Google Scholar] [CrossRef]
- Overländer, C.; Tirrée, J.J.; Nieger, M.; Niecke, E.; Moser, C.; Spirk, S.; Pietschnig, R. Preparation and molecular structure of 2,6-dimesitylphenyldichlorophosphane. Appl. Organomet. Chem. 2007, 21, 46–48. [Google Scholar] [CrossRef]
- Schmidpeter, A.; Nöth, H.; Jochem, G.; Schrödel, H.-P.; Karaghiosoff, K. Ylidyl-dihalogenphosphane—Strukturbilder einer sich anbahnenden Dissoziation. Chem. Ber. 1995, 128, 379–393. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Local Extrema on the Surface of Specific Atom/Bond | PF3 | PF3 | PCl3 | PBr3 | PI3 |
|---|---|---|---|---|---|
| 0.001 a.u. | 0.0028 a.u. | 0.001 a.u. | 0.001 a.u. | 0.001 a.u. | |
| Vs,min On X (lateral portions) | −7.7 | −7.5 | −4.2 | −4.4 | −3.7 |
| Vs,min On X (lateral portions) | −7.7 | −7.5 | −4.2 | −4.4 | −3.7 |
| Vs,min On X (lateral portions) | −7.7 | −7.5 | −4.2 | −4.4 | −3.7 |
| Vs,min on P (opposite to the triangular face formed by three X atoms) | 0.03 | 0.1 | 1.4 | 2.3 | 1.6 |
| VS,max (on P−X bond extensions) | - | −5.3 | 10.5 | 14.4 | 19.1 |
| VS,max (on P−X bond extensions) | - | −5.3 | 10.5 | 14.3 | 19.1 |
| VS,max (on P−X bond extensions) | 29.1 | −5.3 | 10.5 | 14.4 | 19.1 |
| VS,max (on X−P bond extensions) | 29.1 | 48.9 | 20.7 | 18.5 | 14.9 |
| VS,max (on X−P bond extensions) | 29.1 | 48.9 | 20.8 | 18.5 | 14.8 |
| VS,max (on X−P bond extensions) | 9.5 | 48.9 | 20.8 | 18.4 | 14.8 |
| VS,max (on the centroid of the triangular face formed by three X atoms) | - | 26.0 | 3.0 | 2.3 | 2.4 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Varadwaj, P.R.; Varadwaj, A.; Marques, H.M.; Yamashita, K. The Phosphorus Bond, or the Phosphorus-Centered Pnictogen Bond: The Covalently Bound Phosphorus Atom in Molecular Entities and Crystals as a Pnictogen Bond Donor. Molecules 2022, 27, 1487. https://doi.org/10.3390/molecules27051487
Varadwaj PR, Varadwaj A, Marques HM, Yamashita K. The Phosphorus Bond, or the Phosphorus-Centered Pnictogen Bond: The Covalently Bound Phosphorus Atom in Molecular Entities and Crystals as a Pnictogen Bond Donor. Molecules. 2022; 27(5):1487. https://doi.org/10.3390/molecules27051487
Chicago/Turabian StyleVaradwaj, Pradeep R., Arpita Varadwaj, Helder M. Marques, and Koichi Yamashita. 2022. "The Phosphorus Bond, or the Phosphorus-Centered Pnictogen Bond: The Covalently Bound Phosphorus Atom in Molecular Entities and Crystals as a Pnictogen Bond Donor" Molecules 27, no. 5: 1487. https://doi.org/10.3390/molecules27051487
APA StyleVaradwaj, P. R., Varadwaj, A., Marques, H. M., & Yamashita, K. (2022). The Phosphorus Bond, or the Phosphorus-Centered Pnictogen Bond: The Covalently Bound Phosphorus Atom in Molecular Entities and Crystals as a Pnictogen Bond Donor. Molecules, 27(5), 1487. https://doi.org/10.3390/molecules27051487