
New Blood Coagulation Factor XIIa Inhibitors: Molecular Modeling, Synthesis, and Experimental Confirmation

 , ,
, ,
Abstract
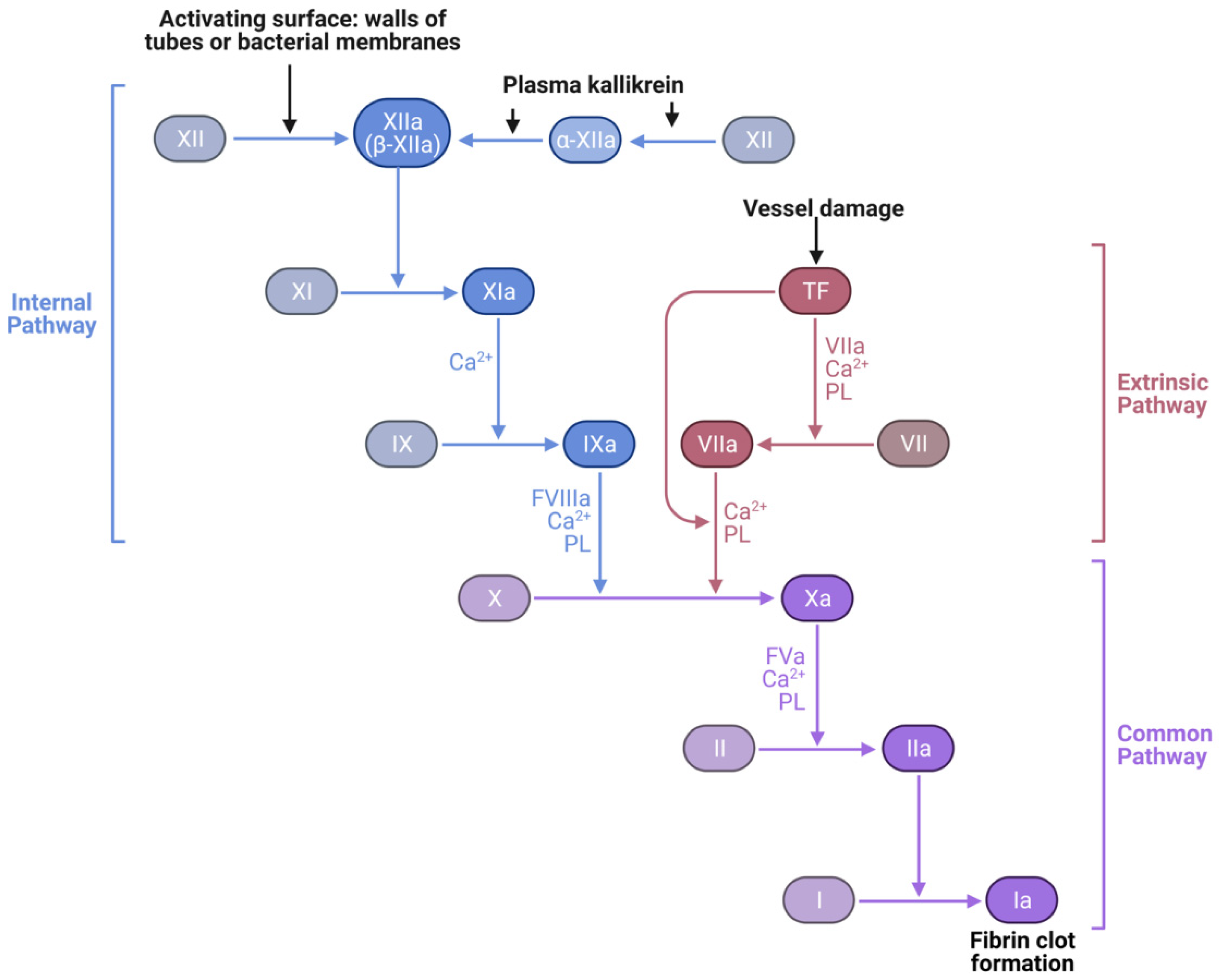
:1. Introduction
2. Results and Discussion
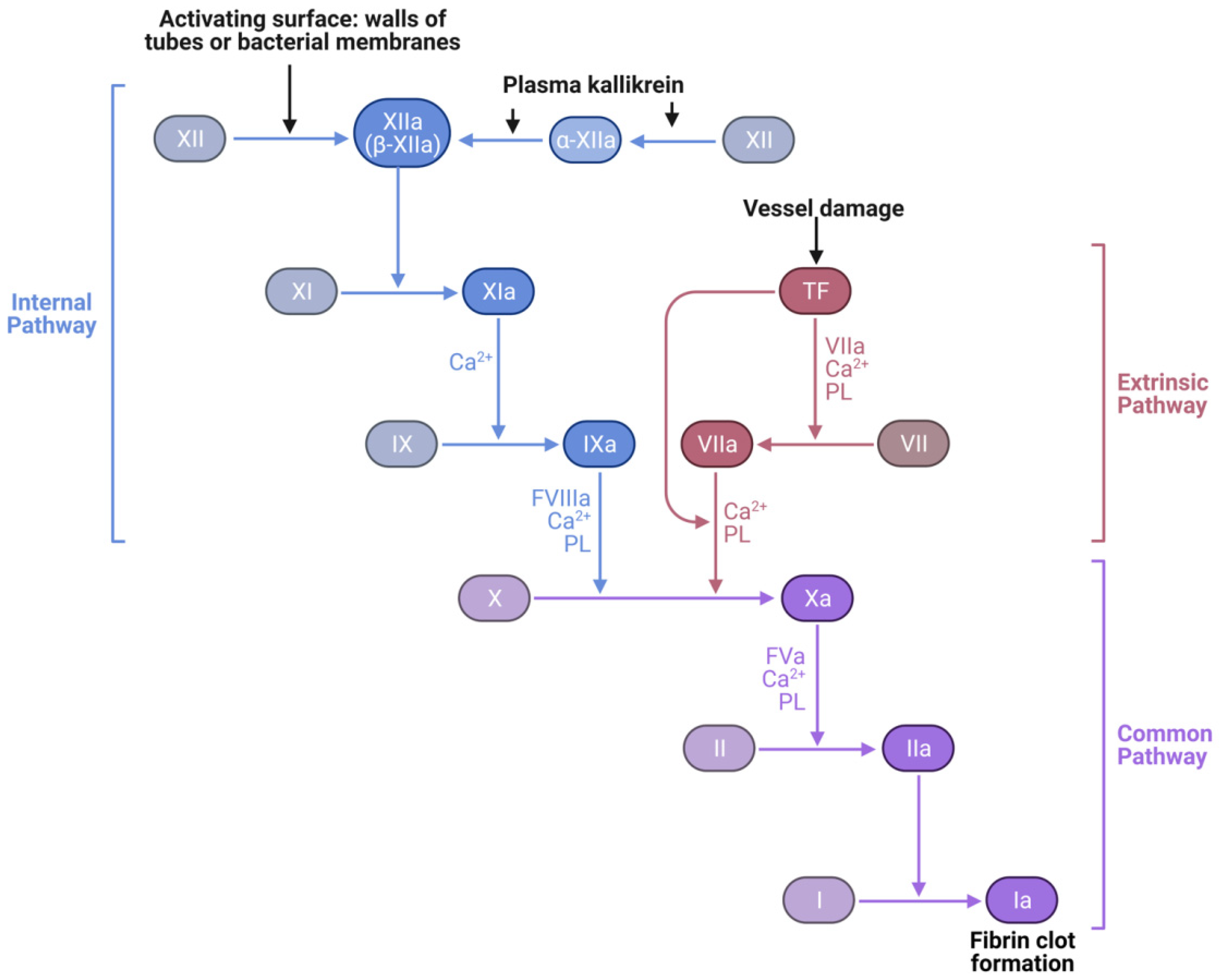
2.1. Factor XIIa Structure Preparation
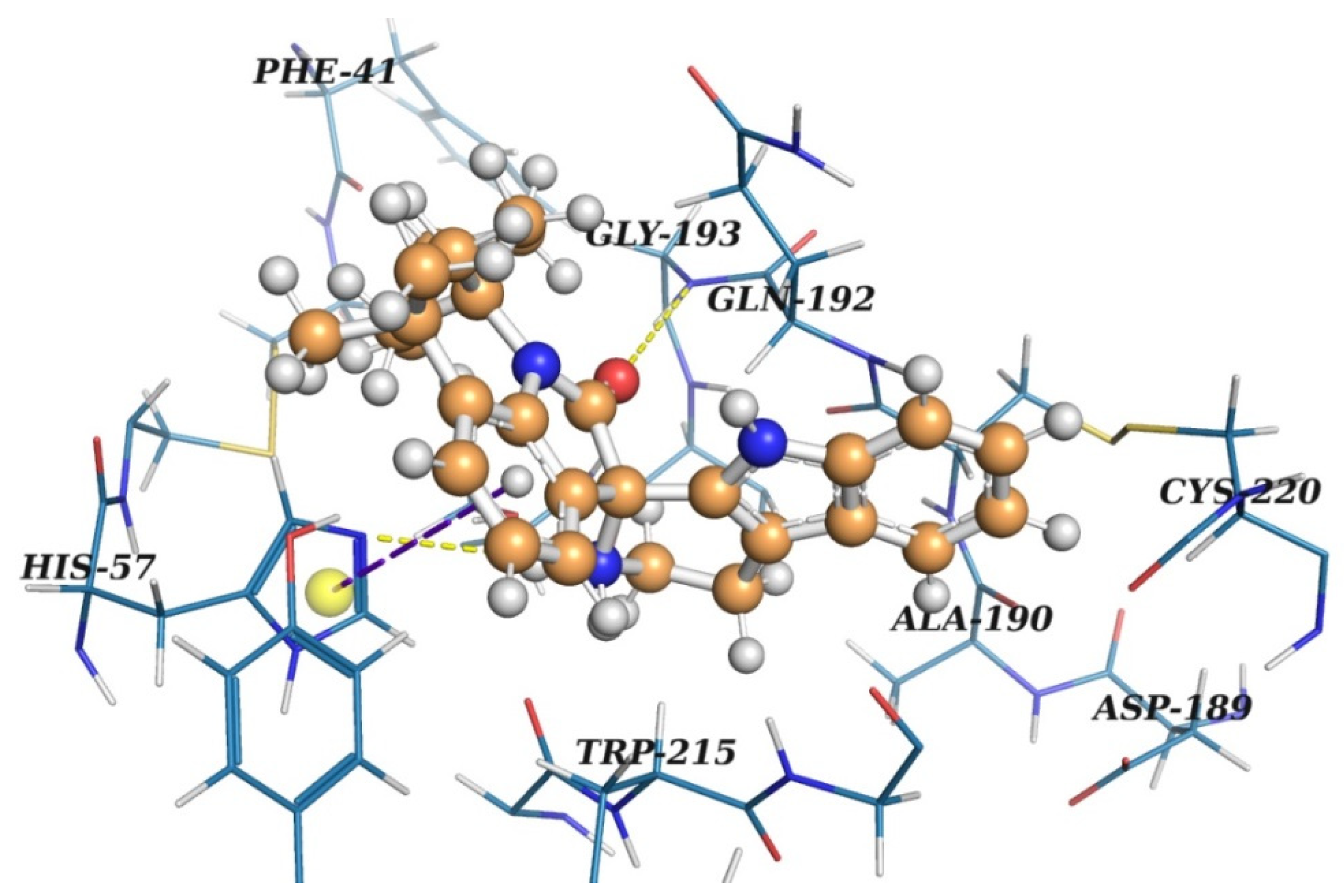
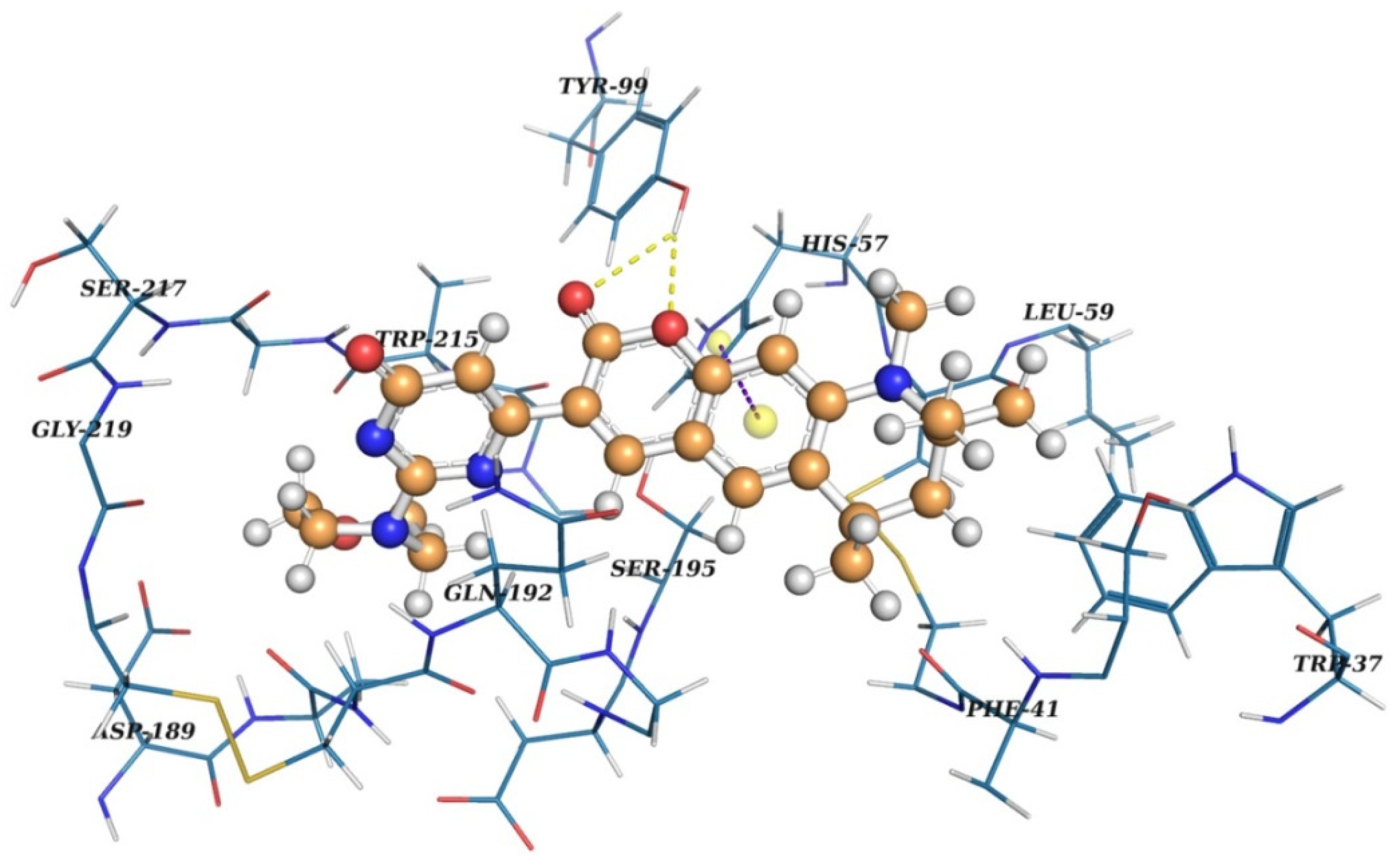
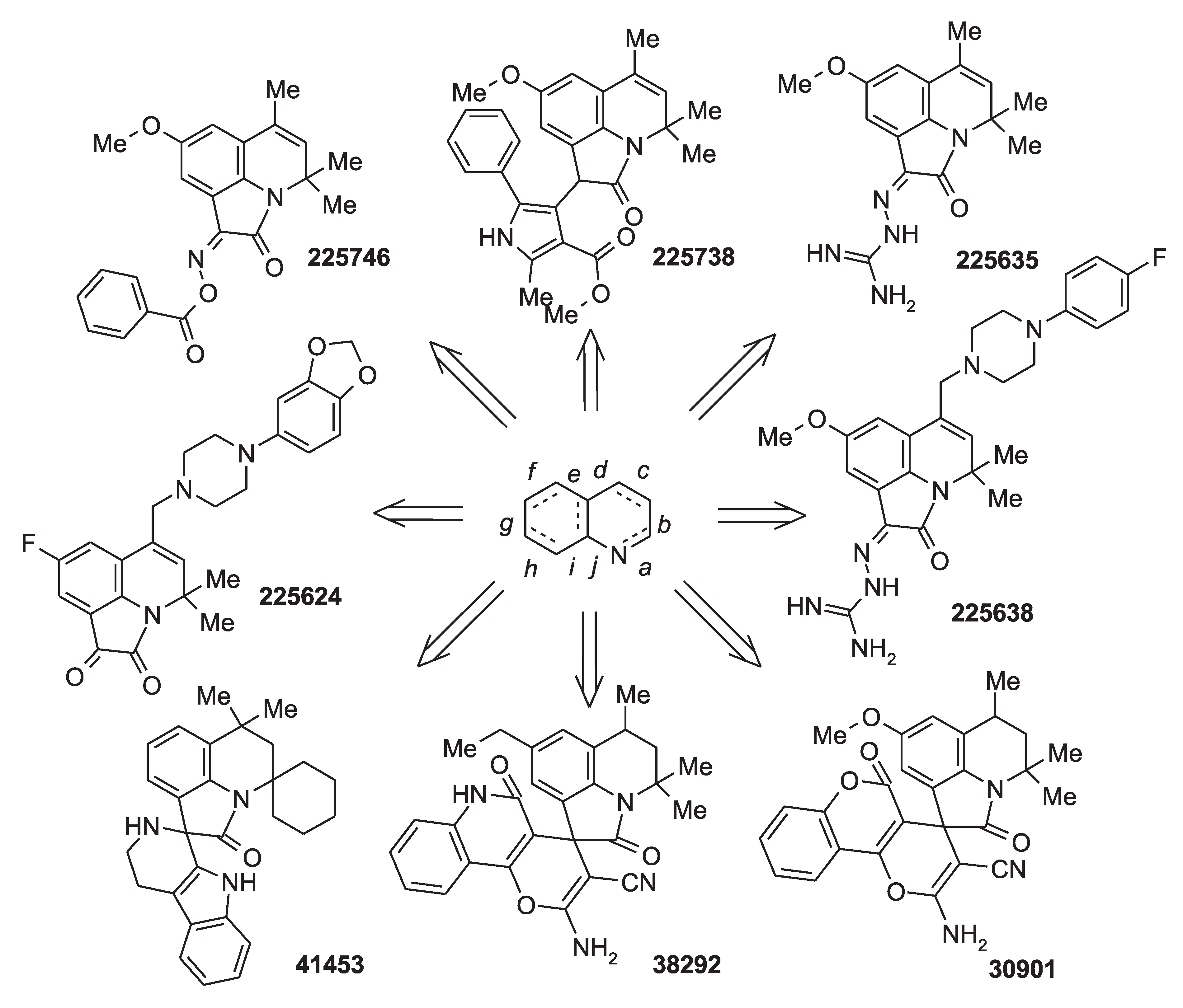
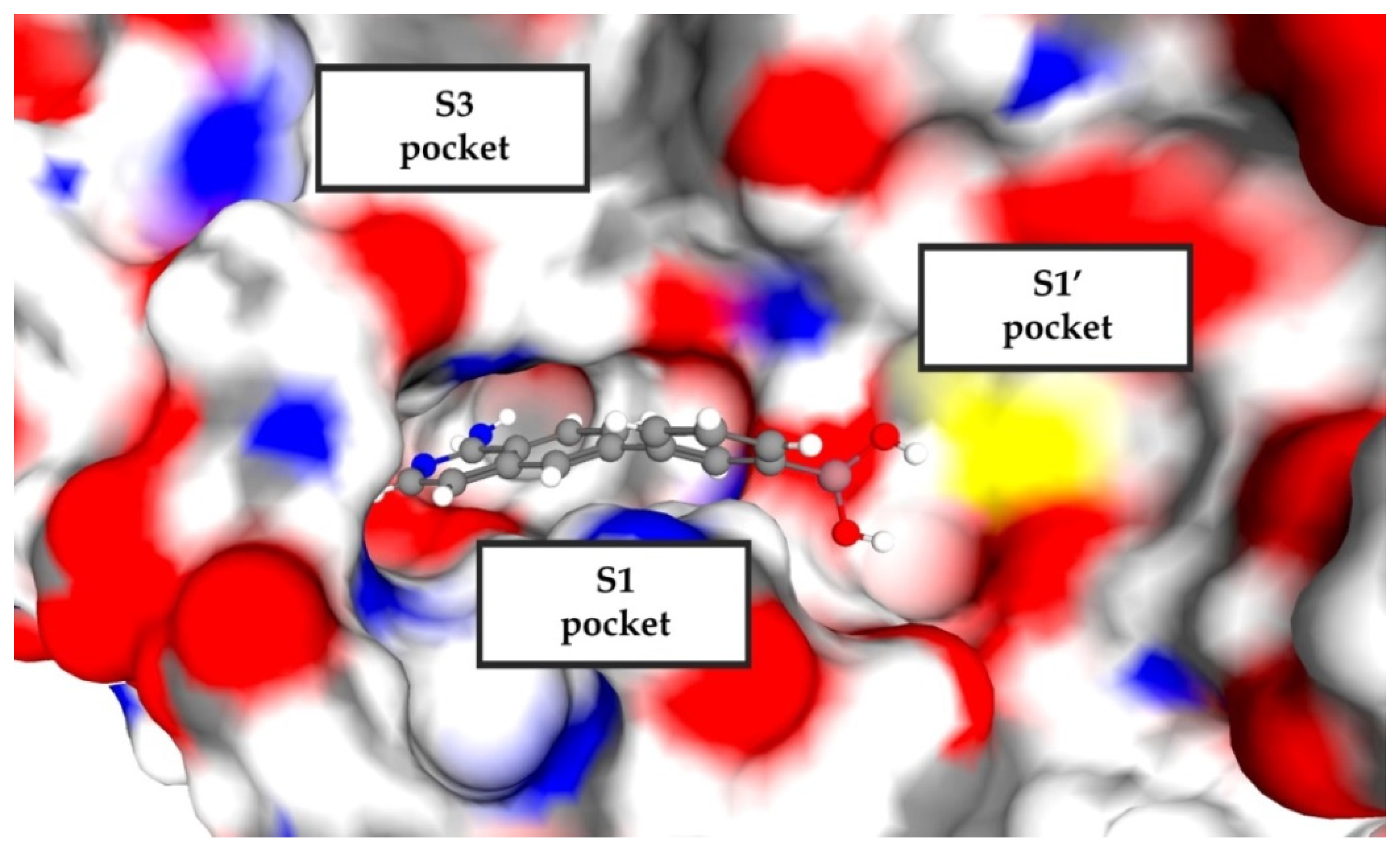
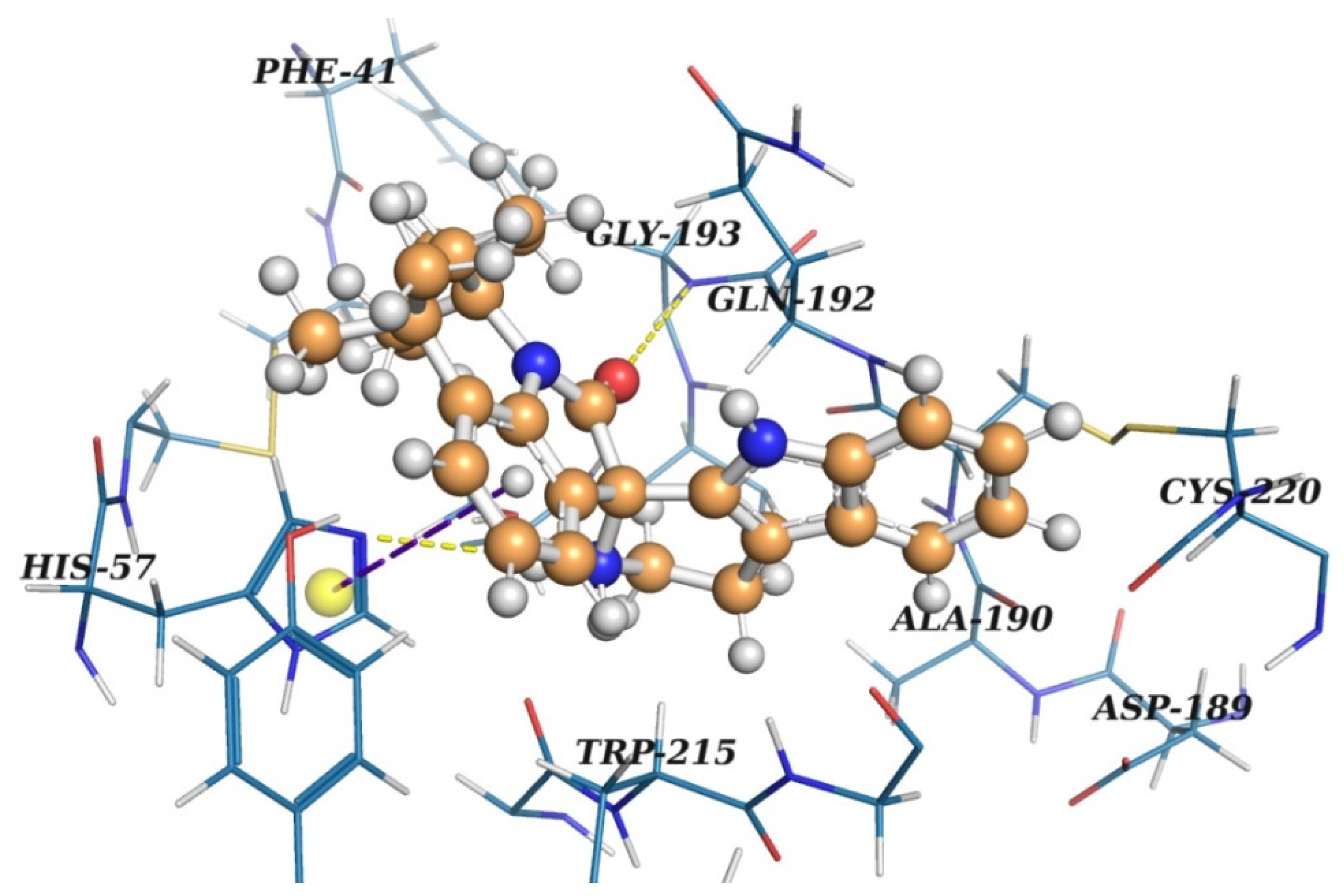
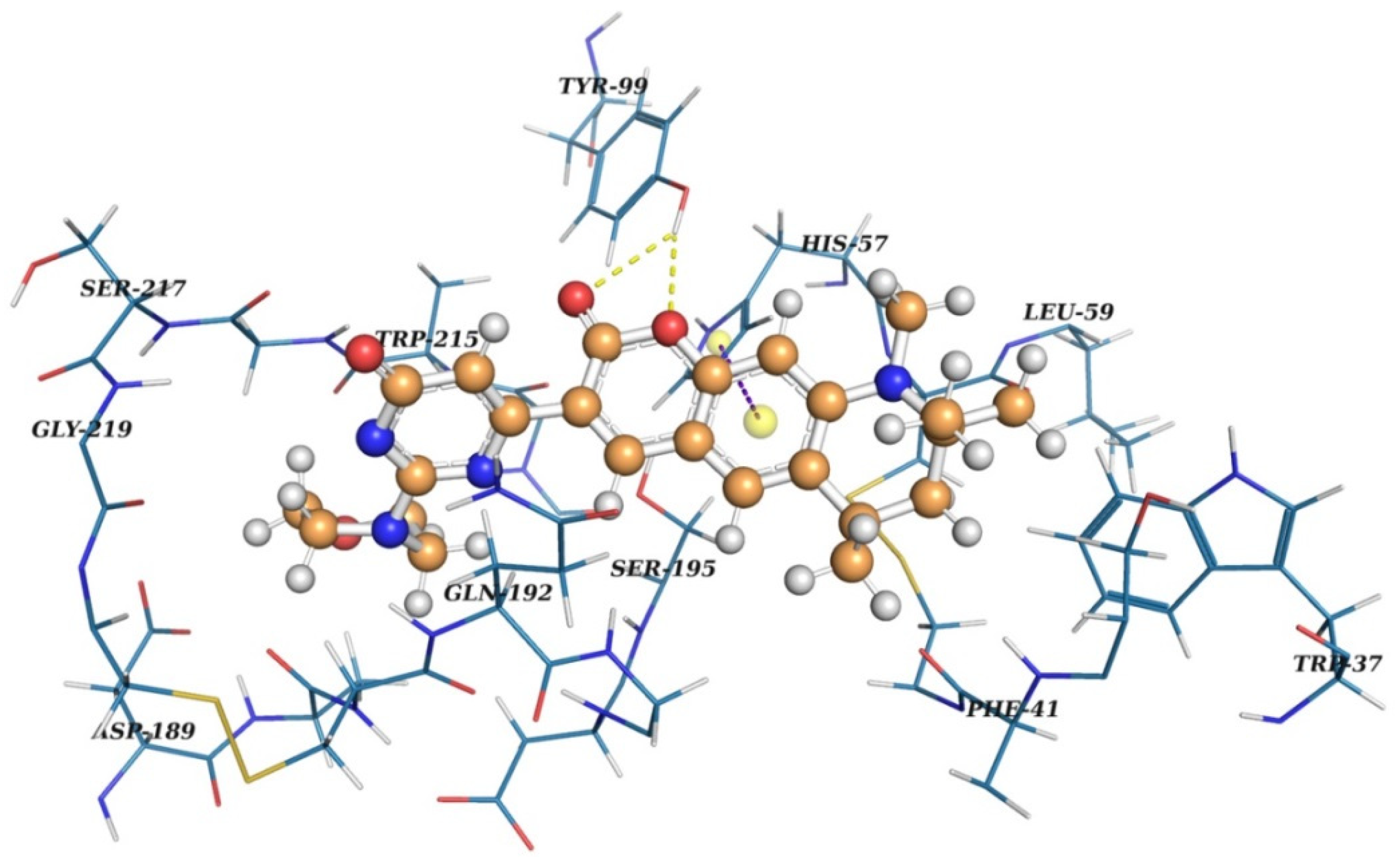
2.2. Virtual Screening
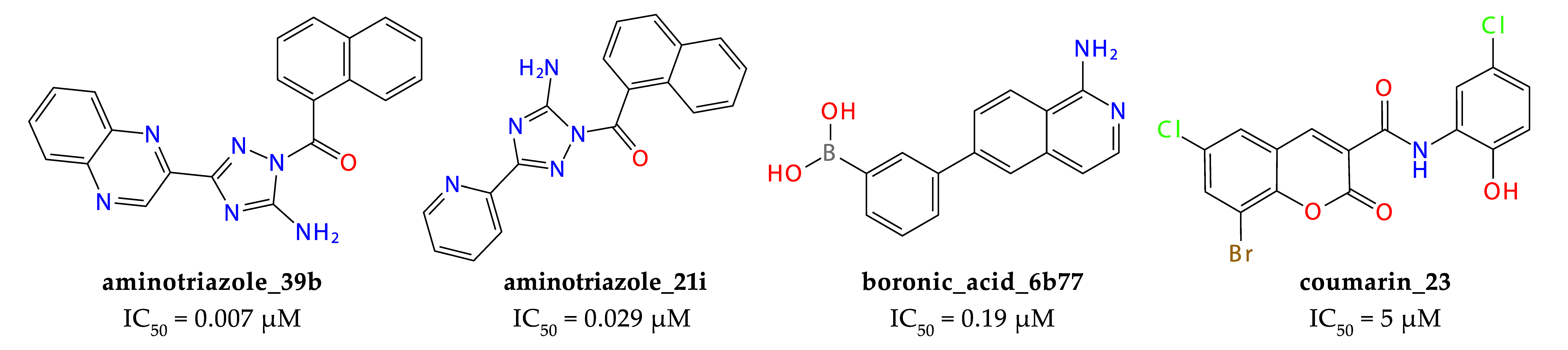
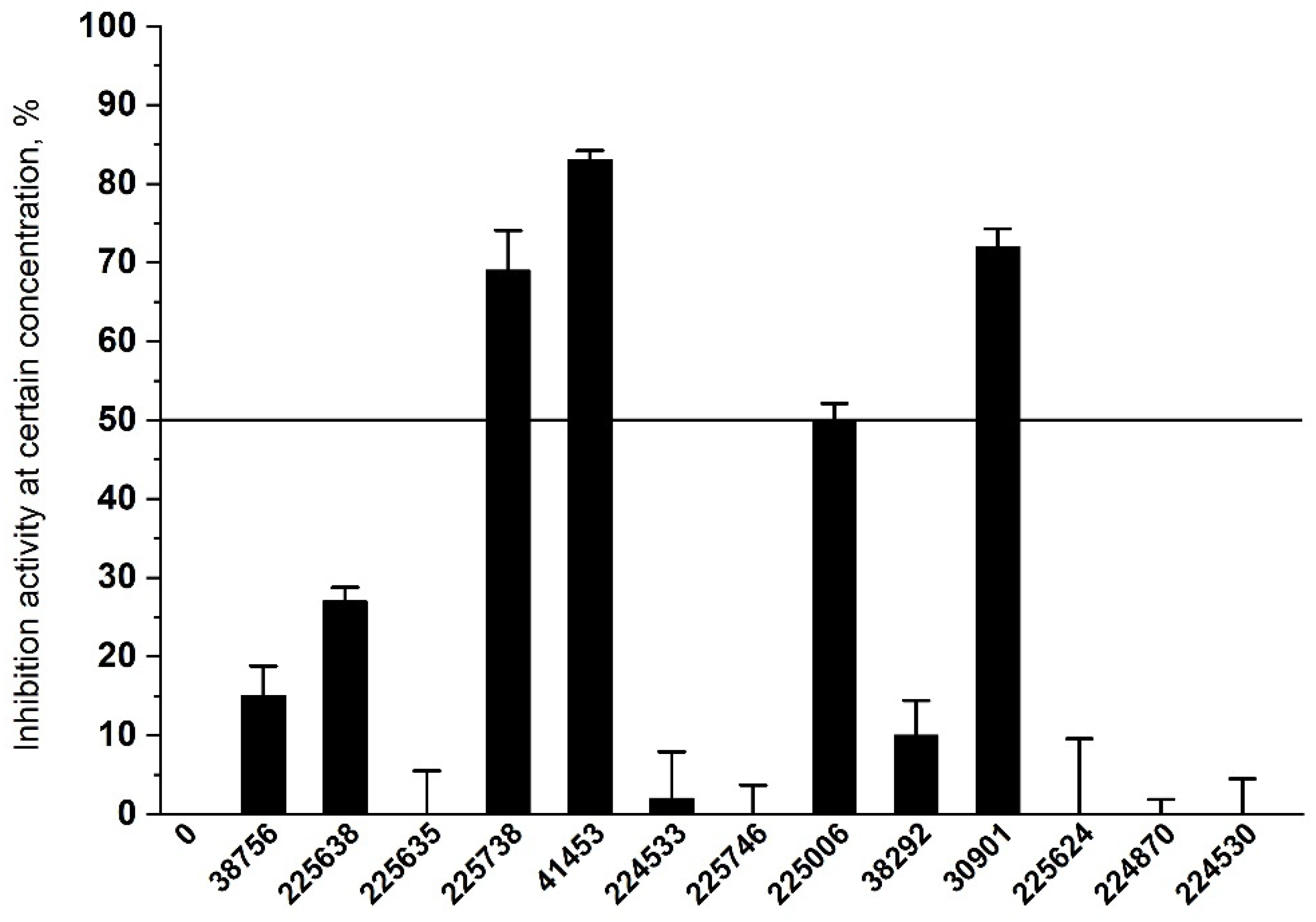
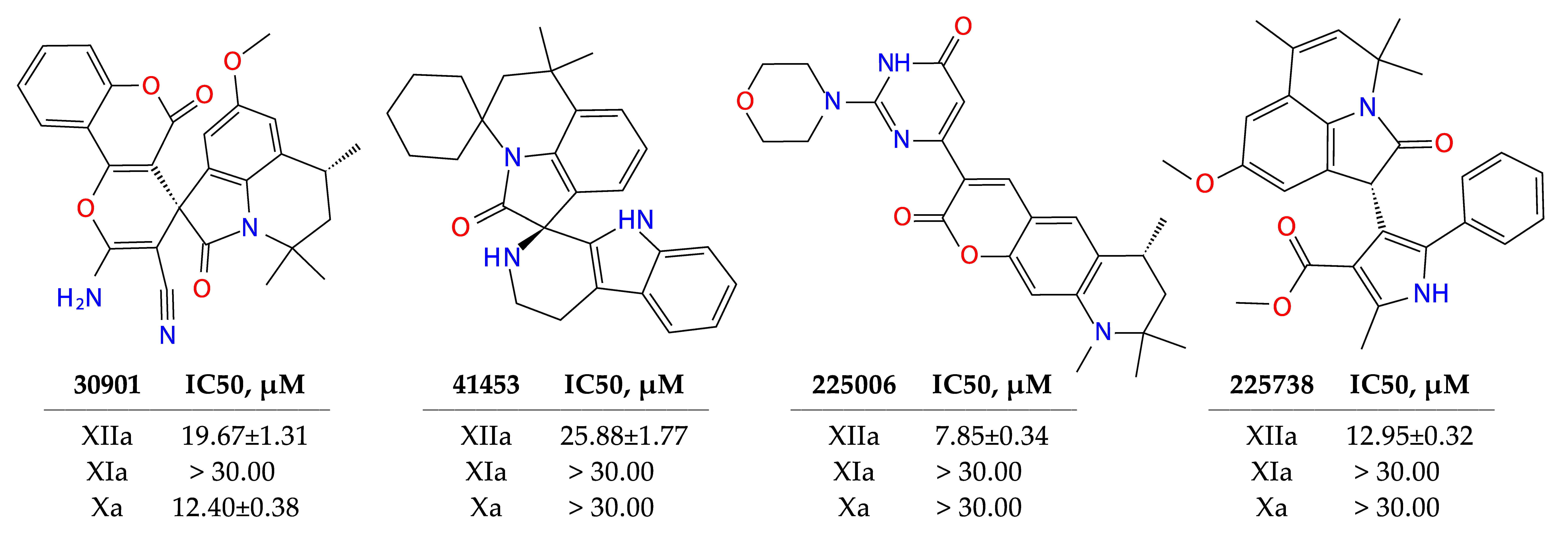
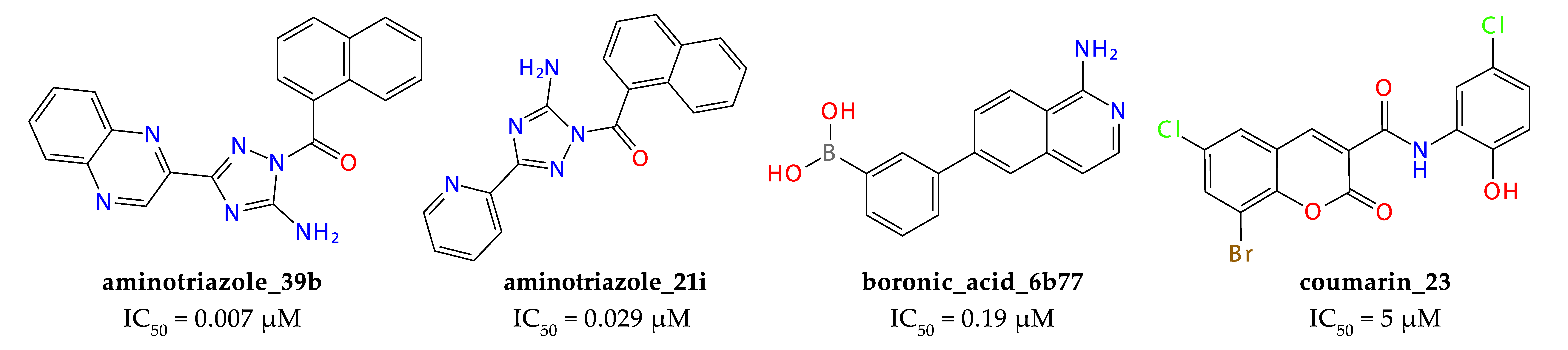
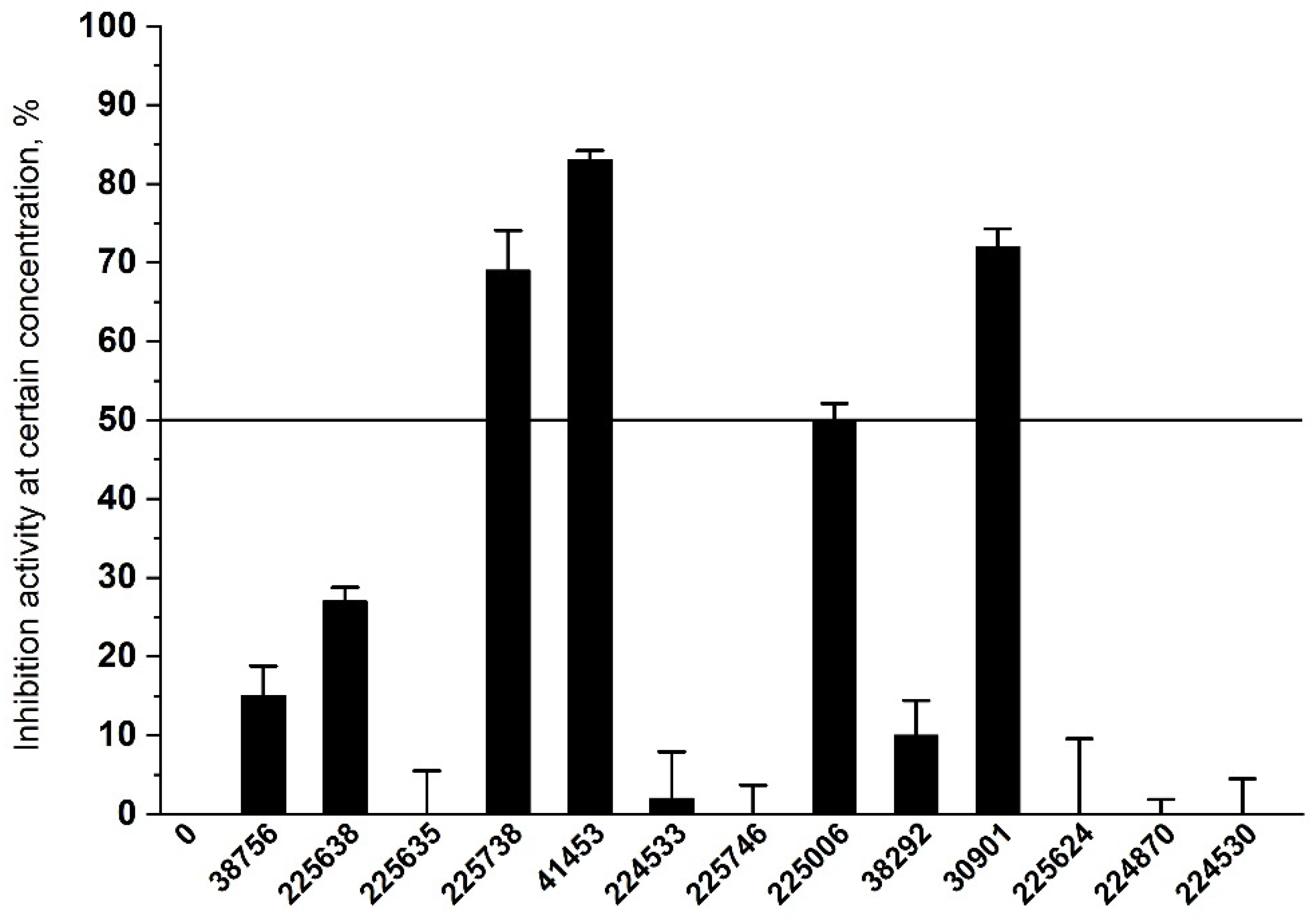
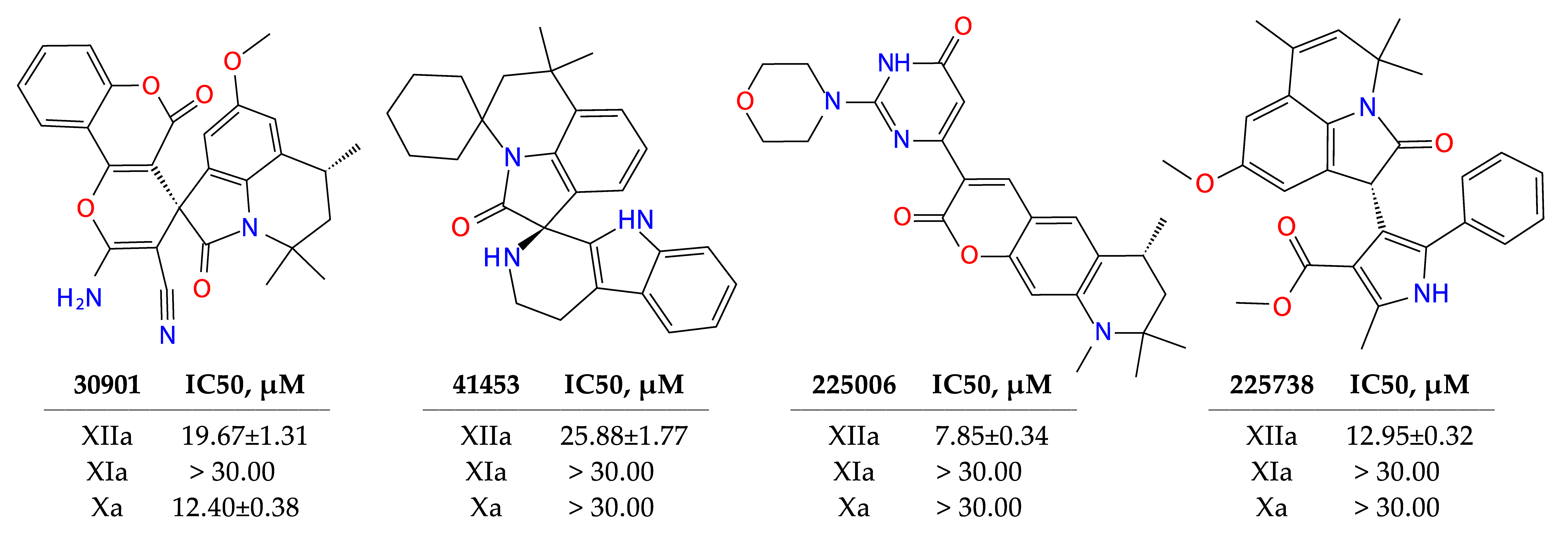
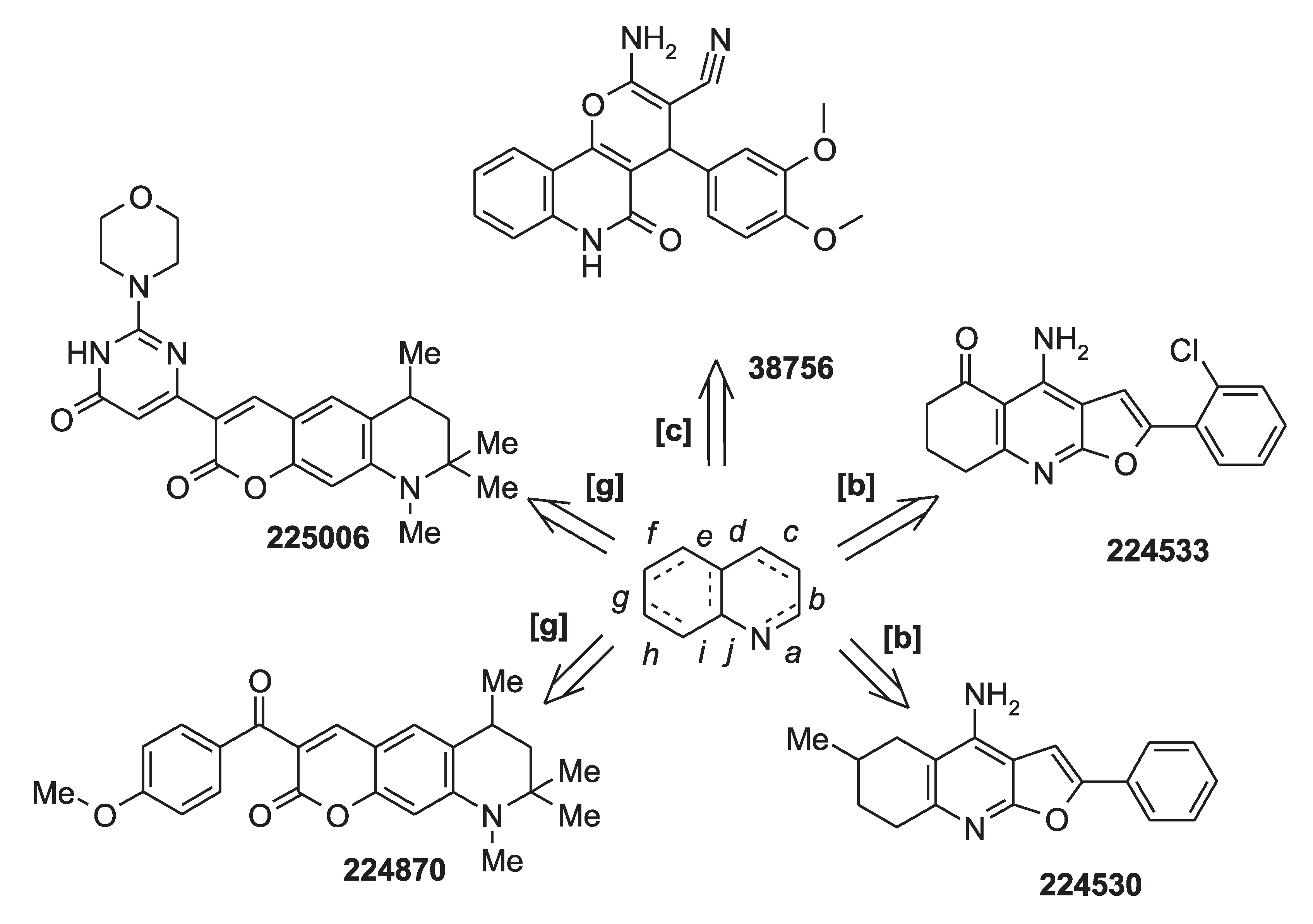
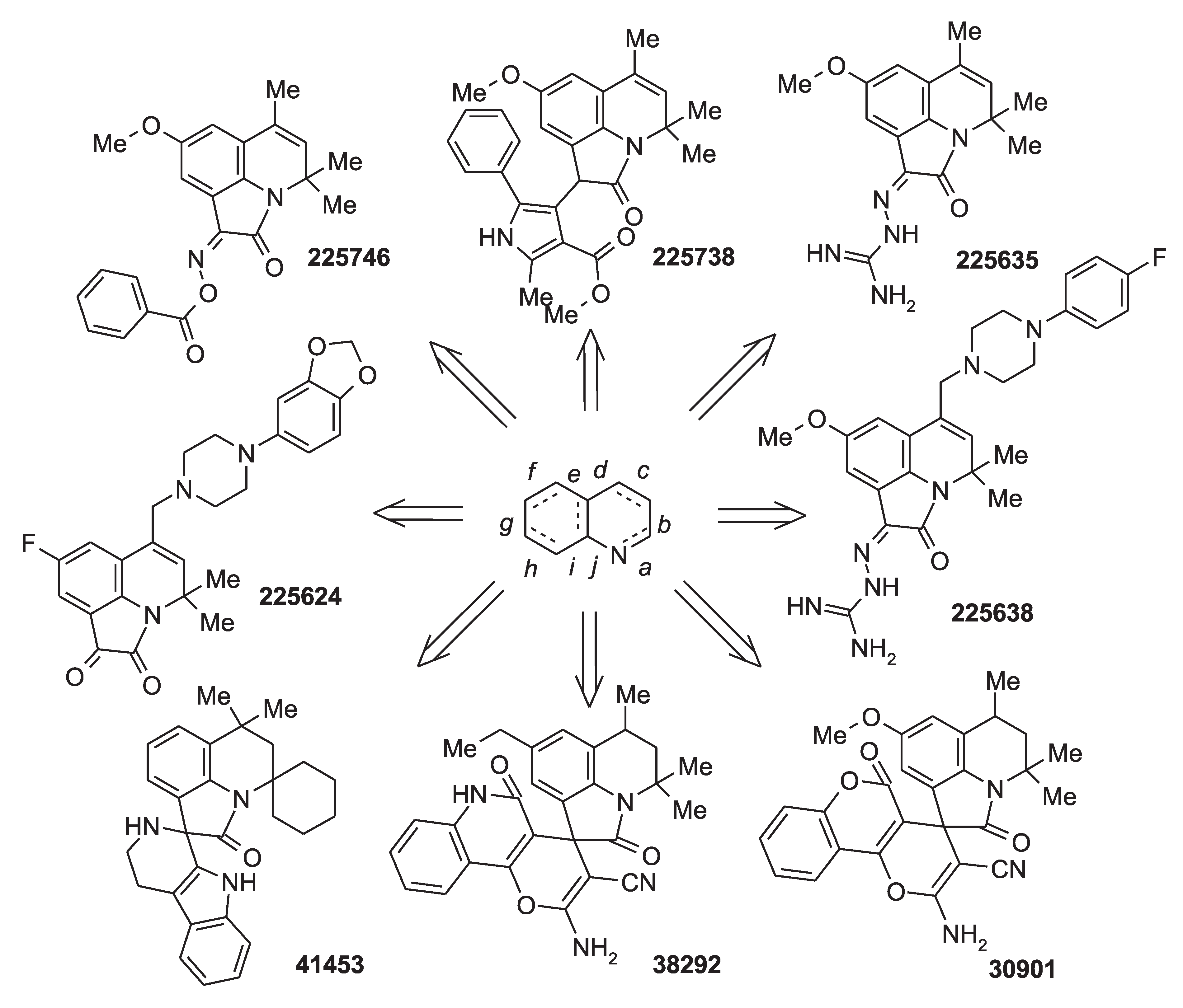
2.3. Results of Experimental Testing
3. Materials and Methods
3.1. Preparation of Protein and Ligands
3.2. Ligand Docking
3.3. Protein–Ligand Binding Enthalpy
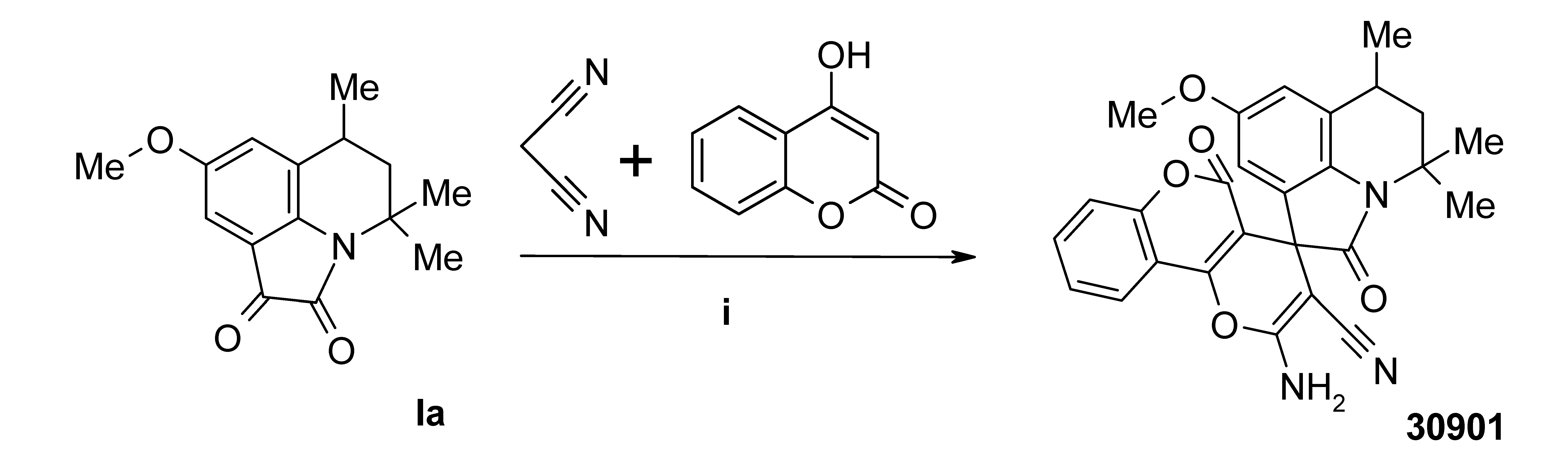
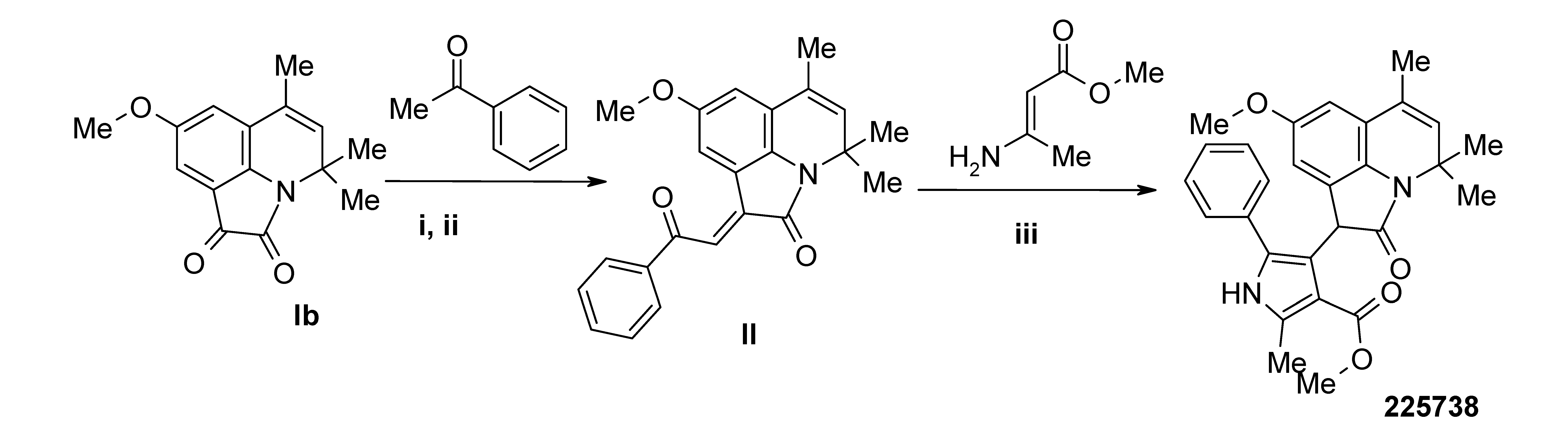
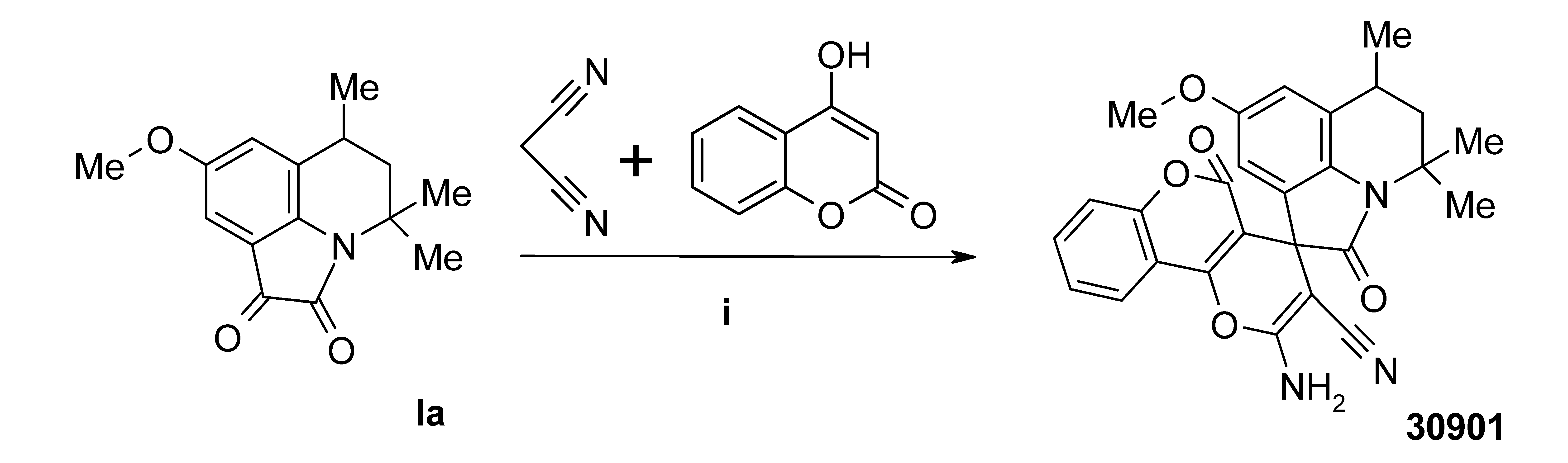
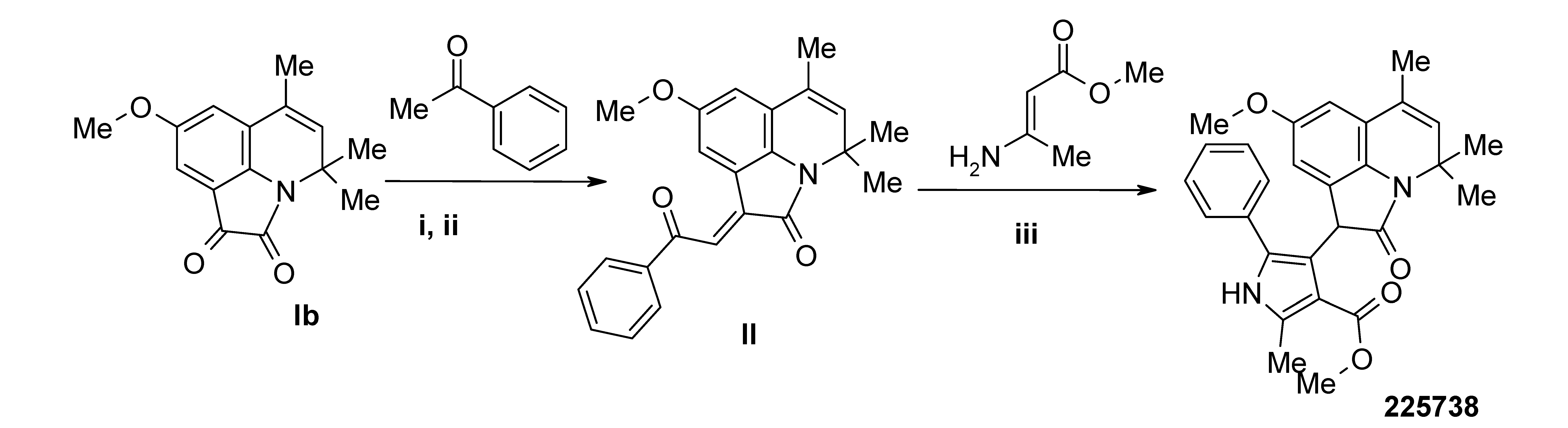
3.4. Synthesis
3.4.1. Instrumentation
3.4.2. Chemicals
3.5. In Vitro Assay
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Xu, D.; Xue, G.; Peng, B.; Feng, Z.; Hongling, L.; Gong, L. High-Throughput Docking and Molecular Dynamics Simulations towards the Identification of Potential Inhibitors against Human Coagulation Factor XIIa. Comput. Math. Methods Med. 2020, 2020, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Podoplelova, N.A.; Sulimov, V.B.; Tashchilova, A.S.; Ilin, I.S.; Panteleev, M.A.; Ledeneva, I.V.; Shikhaliev, K.S. Blood coagulation in the 21st cetrury: Existing knowledge, current strategies for treatment and perspective. Pediatr. Hematol. Immunopathol. 2020, 19, 139–157. (in Russian). [Google Scholar] [CrossRef]
- Kleinschnitz, C.; Stoll, G.; Bendszus, M.; Schuh, K.; Pauer, H.-U.; Burfeind, P.; Renneé, C.; Gailani, D.; Nieswandt, B.; Renneé, T. Targeting coagulation factor XII provides protection from pathological thrombosis in cerebral ischemia without interfering with hemostasis. J. Exp. Med. 2006, 203, 513–518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Müller, F.; Gailani, D.; Renné, T. Factor XI and XII as antithrombotic targets. Curr. Opin. Hematol. 2011, 18. [Google Scholar] [CrossRef] [Green Version]
- Hagedorn, I.; Schmidbauer, S.; Pleines, I.; Kleinschnitz, C.; Kronthaler, U.; Stoll, G.; Dickneite, G.; Nieswandt, B. Factor XIIa Inhibitor Recombinant Human Albumin Infestin-4 Abolishes Occlusive Arterial Thrombus Formation Without Affecting Bleeding. Circulation 2010, 121, 1510–1517. [Google Scholar] [CrossRef] [Green Version]
- Cheng, Q.; Tucker, E.I.; Pine, M.S.; Sisler, I.; Matafonov, A.; Sun, M.; White-Adams, T.C.; Smith, S.A.; Hanson, S.R.; McCarty, O.J.T.; et al. A role for factor XIIa–mediated factor XI activation in thrombus formation in vivo. Blood 2010, 116, 3981–3989. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Cai, T.-Q.; Castriota, G.; Zhou, Y.; Hoos, L.; Jochnowitz, N.; Loewrigkeit, C.; Cook, J.A.; Wickham, A.; Metzger, J.M.; et al. Factor XIIa inhibition by Infestin-4: In vitro mode of action and in vivo antithrombotic benefit. Thromb Haemost 2014, 111, 694–704. [Google Scholar] [CrossRef] [Green Version]
- Matafonov, A.; Leung, P.Y.; Gailani, A.E.; Grach, S.L.; Puy, C.; Cheng, Q.; Sun, M.; McCarty, O.J.T.; Tucker, E.I.; Kataoka, H.; et al. Factor XII inhibition reduces thrombus formation in a primate thrombosis model. Blood 2014, 123, 1739–1746. [Google Scholar] [CrossRef] [Green Version]
- Pathak, M.; Wilmann, P.; Awford, J.; Li, C.; Hamad, B.K.; Fischer, P.M.; Dreveny, I.; Dekker, L.V.; Emsley, J. Coagulation factor XII protease domain crystal structure. J. Thromb. Haemost. 2015, 13, 580–591. [Google Scholar] [CrossRef] [Green Version]
- Cool, D.E.; Edgell, C.J.; Louie, G.V.; Zoller, M.J.; Brayer, G.D.; MacGillivray, R.T. Characterization of human blood coagulation factor XII cDNA. Prediction of the primary structure of factor XII and the tertiary structure of beta-factor XIIa. J. Biol. Chem. 1985, 260, 13666–13676. [Google Scholar] [CrossRef]
- Dementiev, A.; Silva, A.; Yee, C.; Li, Z.; Flavin, M.T.; Sham, H.; Partridge, J.R. Structures of human plasma β–factor XIIa cocrystallized with potent inhibitors. Blood Adv. 2018, 2, 549–558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larsson, M.; Rayzman, V.; Nolte, M.W.; Nickel, K.F.; Björkqvist, J.; Jämsä, A.; Hardy, M.P.; Fries, M.; Schmidbauer, S.; Hedenqvist, P.; et al. A Factor XIIa Inhibitory Antibody Provides Thromboprotection in Extracorporeal Circulation Without Increasing Bleeding Risk. Sci. Transl. Med. 2014, 6, 222ra17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baeriswyl, V.; Calzavarini, S.; Chen, S.; Zorzi, A.; Bologna, L.; Angelillo-Scherrer, A.; Heinis, C. A Synthetic Factor XIIa Inhibitor Blocks Selectively Intrinsic Coagulation Initiation. ACS Chem. Biol. 2015, 10, 1861–1870. [Google Scholar] [CrossRef] [PubMed]
- Woodruff, R.S.; Xu, Y.; Layzer, J.; Wu, W.; Ogletree, M.L.; Sullenger, B.A. Inhibiting the intrinsic pathway of coagulation with a factor XII-targeting RNA aptamer. J. Thromb. Haemost. 2013, 11, 1364–1373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, T.-Q.; Wu, W.; Shin, M.K.; Xu, Y.; Jochnowitz, N.; Zhou, Y.; Hoos, L.; Bentley, R.; Strapps, W.; Thankappan, A.; et al. Factor XII full and partial null in rat confers robust antithrombotic efficacy with no bleeding. Blood Coagul. Fibrinolysis 2015, 26, 893–902. [Google Scholar] [CrossRef] [PubMed]
- Revenko, A.S.; Gao, D.; Crosby, J.R.; Bhattacharjee, G.; Zhao, C.; May, C.; Gailani, D.; Monia, B.P.; MacLeod, A.R. Selective depletion of plasma prekallikrein or coagulation factor XII inhibits thrombosis in mice without increased risk of bleeding. Blood 2011, 118, 5302–5311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davoine, C.; Bouckaert, C.; Fillet, M.; Pochet, L. Factor XII/XIIa inhibitors: Their discovery, development, and potential indications. Eur. J. Med. Chem. 2020, 208, 112753. [Google Scholar] [CrossRef]
- Bouckaert, C.; Serra, S.; Rondelet, G.; Dolušić, E.; Wouters, J.; Dogné, J.-M.; Frédérick, R.; Pochet, L. Synthesis, evaluation and structure-activity relationship of new 3-carboxamide coumarins as FXIIa inhibitors. Eur. J. Med. Chem. 2016, 110, 181–194. [Google Scholar] [CrossRef]
- Bouckaert, C.; Zhu, S.; Govers-Riemslag, J.W.P.; Depoorter, M.; Diamond, S.L.; Pochet, L. Discovery and assessment of water soluble coumarins as inhibitors of the coagulation contact pathway. Thromb. Res. 2017, 157, 126–133. [Google Scholar] [CrossRef]
- Chen, J.J.F.; Visco, D.P. Identifying novel factor XIIa inhibitors with PCA-GA-SVM developed vHTS models. Eur. J. Med. Chem. 2017, 140, 31–41. [Google Scholar] [CrossRef]
- Platte, S.; Korff, M.; Imberg, L.; Balicioglu, I.; Erbacher, C.; Will, J.M.; Daniliuc, C.G.; Karst, U.; Kalinin, D.V. Microscale Parallel Synthesis of Acylated Aminotriazoles Enabling the Development of Factor XIIa and Thrombin Inhibitors. ChemMedChem 2021, 16, 3672–3690. [Google Scholar] [CrossRef] [PubMed]
- Korff, M.; Imberg, L.; Will, J.M.; Bückreiß, N.; Kalinina, S.A.; Wenzel, B.M.; Kastner, G.A.; Daniliuc, C.G.; Barth, M.; Ovsepyan, R.A.; et al. Acylated 1H-1,2,4-Triazol-5-amines Targeting Human Coagulation Factor XIIa and Thrombin: Conventional and Microscale Synthesis, Anticoagulant Properties, and Mechanism of Action. J. Med. Chem. 2020, 63, 13159–13186. [Google Scholar] [CrossRef] [PubMed]
- Sulimov, A.V.; Kutov, D.C.; Oferkin, I.V.; Katkova, E.V.; Sulimov, V.B. Application of the docking program SOL for CSAR benchmark. J. Chem. Inf. Model. 2013, 53, 1946–1956. [Google Scholar] [CrossRef] [PubMed]
- Sulimov, V.B.; Ilin, I.S.; Kutov, D.C.; Sulimov, A. V Development of docking programs for Lomonosov supercomputer. J. Turkish Chem. Soc. Sect. A Chem. 2020, 7, 259–276. [Google Scholar] [CrossRef]
- Stewart, J.J. Optimization of parameters for semiempirical methods VI: More modifications to the NDDO approximations and re-optimization of parameters. J. Mol. Model. 2013, 19, 1–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klamt, A.; Schuurmann, G. COSMO: A new approach to dielectric screening in solvents with explicit expressions for the screening energy and its gradient. J. Chem. Soc. Perkin Trans. 2 1993, 799–805. [Google Scholar] [CrossRef]
- Stewart, J.J.P. Stewart Computational Chemistry. MOPAC2016. Available online: http://openmopac.net/MOPAC2016.html (accessed on 30 July 2020).
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [Green Version]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Veber, D.F.; Johnson, S.R.; Cheng, H.-Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef]
- Hughes, J.D.; Blagg, J.; Price, D.A.; Bailey, S.; Decrescenzo, G.A.; Devraj, R.V.; Ellsworth, E.; Fobian, Y.M.; Gibbs, M.E.; Gilles, R.W.; et al. Physiochemical drug properties associated with in vivo toxicological outcomes. Bioorg. Med. Chem. Lett. 2008, 18, 4872–4875. [Google Scholar] [CrossRef]
- Marvin Was Used for Drawing, Displaying and Characterizing Chemical Structures, Substructures and Reactions, Marvin 21.3.0. 2021. Available online: https://chemaxon.com/products/marvin (accessed on 9 January 2022).
- DeLano, W.L. The PyMOL Molecular Graphics System; De-Lano Scientific: San Carlos, CA, USA, 2002. [Google Scholar] [CrossRef] [Green Version]
- Salentin, S.; Schreiber, S.; Haupt, V.J.; Adasme, M.F.; Schroeder, M. PLIP: Fully automated protein-ligand interaction profiler. Nucleic Acids Res. 2015, 43, W443–W447. [Google Scholar] [CrossRef] [PubMed]
- Kutov, D.C.; Katkova, E.V.; Kondakova, O.A.; Sulimov, A.V.; Sulimov, V.B. Influence of the method of hydrogen atoms incorporation into the target protein on the protein-ligand binding energy. Bull. South Ural State Univ. Ser. Math. Model. Program. Comput. Softw. 2017, 10, 94–107. [Google Scholar] [CrossRef]
- Halgren, T.A. Merck molecular force field. J. Comput. Chem. 1996, 17, 490–641. [Google Scholar] [CrossRef]
- Sinauridze, E.I.; Romanov, A.N.; Gribkova, I.V.; Kondakova, O.A.; Surov, S.S.; Gorbatenko, A.S.; Butylin, A.A.; Monakov, M.Y.; Bogolyubov, A.A.; Kuznetsov, Y.V.; et al. New synthetic thrombin inhibitors: Molecular design and experimental verification. PLoS ONE 2011, 6, e19969. [Google Scholar] [CrossRef]
- Sulimov, V.B.; Katkova, E.V.; Oferkin, I.V.; Sulimov, A.V.; Romanov, A.N.; Roschin, A.I.; Beloglazova, I.B.; Plekhanova, O.S.; Tkachuk, V.A.; Sadovnichiy, V.A. Application of molecular modeling to urokinase inhibitors development. Biomed Res. Int. 2014, 2014, 625176. [Google Scholar] [CrossRef]
- Beloglazova, I.B.; Plekhanova, O.S.; Katkova, E.V.; Rysenkova, K.D.; Stambol’skii, D.V.; Sulimov, V.B.; Tkachuk, V.A. Molecular modeling as a new approach to the development of urokinase inhibitors. Bull. Exp. Biol. Med. 2015, 158, 700–704. [Google Scholar] [CrossRef]
- Sulimov, V.B.; Gribkova, I.V.; Kochugaeva, M.P.; Katkova, E.V.; Sulimov, A.V.; Kutov, D.C.; Shikhaliev, K.S.; Medvedeva, S.M.; Krysin, M.Y.; Sinauridze, E.I.; et al. Application of molecular modeling to development of new factor Xa inhibitors. Biomed Res. Int. 2015, 2015, 15. [Google Scholar] [CrossRef] [Green Version]
- Novichikhina, N.; Ilin, I.; Tashchilova, A.; Sulimov, A.; Kutov, D.; Ledenyova, I.; Krysin, M.; Shikhaliev, K.; Gantseva, A.; Gantseva, E.; et al. Synthesis, docking, and in vitro anticoagulant activity assay of hybrid derivatives of pyrrolo[3,2,1-ij]quinolin-2(1H)-one as new inhibitors of factor Xa and factor XIa. Molecules 2020, 25, 1889. [Google Scholar] [CrossRef] [Green Version]
- Ilin, I.S.; Lipets, E.N.; Sulimov, A.V.; Kutov, D.C.; Shikhaliev, K.S.; Potapov, A.Y.; Krysin, M.Y.; Zubkov, F.I.; Sapronova, L.V.; Ataullakhanov, F.I.; et al. New factor Xa inhibitors based on 1,2,3,4-tetrahydroquinoline developed by molecular modelling. J. Mol. Graph. Model. 2019, 89, 215–224. [Google Scholar] [CrossRef]
- Medvedeva, S.M.; Krysin, M.Y.; Zubkov, F.I.; Nikitina, E.V.; Shikhaliev, K.S. New Heterocyclic Systems Based on Substituted 3,4-Dihydro-1H-Spiro[Quinoline-2,1’-Cycloalkanes]. Chem. Heterocycl. Compd. 2014, 50, 1280–1290. [Google Scholar] [CrossRef]
- Potapov, A.Y.; Paponov, B.V.; Podoplelova, N.A.; Panteleev, M.A.; Potapov, M.A.; Ledenyova, I.V.; Stolpovskaya, N.V.; Shikhaliev, K.S. Synthesis of 2H-pyrano[3,2-g]quinolin-2-ones containing a pyrimidinone moiety and characterization of their anticoagulant activity via inhibition of blood coagulation factors Xa and XIa. Chem. Heterocycl. Compd. 2021, 57, 574–580. [Google Scholar] [CrossRef]
- Medvedeva, S.M.; Sabynin, A.L.; Shikhaliev, K.S. Efficient methods for the synthesis of spiroheterocyclic systems based on 4,4,6-trimethyl-4H-pyrrolo[3,2,1-ij]quinoline-1,2-diones. Russ. Chem. Bull. 2014, 63, 2693–2701. [Google Scholar] [CrossRef]
- Novichikhina, N.P.; Skoptsova, A.A.; Shestakov, A.S.; Potapov, A.Y.; Kosheleva, E.A.; Kozaderov, O.A.; Ledenyova, I.V.; Podoplelova, N.A.; Panteleev, M.A.; Shikhaliev, K.S. Synthesis and Anticoagulant Activity of New Ethylidene and Spiro Derivatives of Pyrrolo[3,2,1-ij]quinolin-2-ones. Russ. J. Org. Chem. 2020, 56, 1550–1556. [Google Scholar] [CrossRef]
- Lescheva, E.; Medvedeva, S.; Shikhaliev, K. Synthesis of 4,4,6-trimethyl-8-R-4H-pyrrolo [3,2,1-ij] quinoline-1,2-diones. Žurnal organìčnoï ta Farm. hìmìï 2014, 12, 15–20. [Google Scholar] [CrossRef] [Green Version]
- Galliford, C.V.; Scheidt, K.A. Pyrrolidinyl-Spirooxindole Natural Products as Inspirations for the Development of Potential Therapeutic Agents. Angew. Chemie Int. Ed. 2007, 46, 8748–8758. [Google Scholar] [CrossRef]
- Marti, C.; Carreira, E.M. Construction of Spiro[pyrrolidine-3,3′-oxindoles]—Recent Applications to the Synthesis of Oxindole Alkaloids. European J. Org. Chem. 2003, 2003, 2209–2219. [Google Scholar] [CrossRef]
- Williams, R.M.; Cox, R.J. Paraherquamides, Brevianamides, and Asperparalines: Laboratory Synthesis and Biosynthesis. An Interim Report. Acc. Chem. Res. 2003, 36, 127–139. [Google Scholar] [CrossRef]
- Alper, P.B.; Meyers, C.; Lerchner, A.; Siegel, D.R.; Carreira, E.M. Facile, novel methodology for the synthesis of spiro [pyrrolidin-3,3′-oxindoles]: Catalyzed ring expansion reactions of cyclopropanes by aldimines. Angew. Chemie Int. Ed. 1999, 38, 3186–3189. [Google Scholar] [CrossRef]
- Voevodin, V.V.; Antonov, A.S.; Nikitenko, D.A.; Shvets, P.A.; Sobolev, S.I.; Sidorov, I.Y.; Stefanov, K.S.; Voevodin, V.V.; Zhumatiy, S.A. Supercomputer Lomonosov-2: Large scale, deep monitoring and fine analytics for the user community. Supercomput. Front. Innov. 2019, 6, 4–11. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ID | Inhibition Activity at 30 µM, % | SOL, kcal/mol | ΔHbind, kcal/mol | ClogP | TPSA |
|---|---|---|---|---|---|
| 41453 | 83 | −7.03 | −43.8 | 5.06 | 48.1 |
| 30901 | 72 | −5.24 | −34.5 | 2.37 | 114.9 |
| 225738 | 69 | −5.11 | −37.9 | 4.66 | 71.6 |
| 225006 | 50 | −6.55 | −15.5 | 2.37 | 83.5 |
| 225638 | 27 | −5.39 | −70.0 | 2.61 | 110.3 |
| 38756 | 15 | −5.12 | −35.2 | 1.74 | 106.6 |
| 38292 | 10 | −5.36 | −36.6 | 3.30 | 108.5 |
| 224533 | 2 | −5.28 | −42.1 | 3.46 | 69.1 |
| 224870 | 0 | −6.52 | −30.4 | 4.54 | 55.8 |
| 225624 | 0 | −6.19 | −33.9 | 2.94 | 62.3 |
| 225635 | 0 | −5.01 | −69.0 | 1.30 | 103.8 |
| 224530 | 0 | −5.01 | −27.3 | 3.63 | 52.1 |
| 225746 | 0 | −5.44 | −38.4 | 4.22 | 68.2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tashchilova, A.; Podoplelova, N.; Sulimov, A.; Kutov, D.; Ilin, I.; Panteleev, M.; Shikhaliev, K.; Medvedeva, S.; Novichikhina, N.; Potapov, A.; et al. New Blood Coagulation Factor XIIa Inhibitors: Molecular Modeling, Synthesis, and Experimental Confirmation. Molecules 2022, 27, 1234. https://doi.org/10.3390/molecules27041234
Tashchilova A, Podoplelova N, Sulimov A, Kutov D, Ilin I, Panteleev M, Shikhaliev K, Medvedeva S, Novichikhina N, Potapov A, et al. New Blood Coagulation Factor XIIa Inhibitors: Molecular Modeling, Synthesis, and Experimental Confirmation. Molecules. 2022; 27(4):1234. https://doi.org/10.3390/molecules27041234
Chicago/Turabian StyleTashchilova, Anna, Nadezhda Podoplelova, Alexey Sulimov, Danil Kutov, Ivan Ilin, Mikhail Panteleev, Khidmet Shikhaliev, Svetlana Medvedeva, Nadezhda Novichikhina, Andrey Potapov, and et al. 2022. "New Blood Coagulation Factor XIIa Inhibitors: Molecular Modeling, Synthesis, and Experimental Confirmation" Molecules 27, no. 4: 1234. https://doi.org/10.3390/molecules27041234
APA StyleTashchilova, A., Podoplelova, N., Sulimov, A., Kutov, D., Ilin, I., Panteleev, M., Shikhaliev, K., Medvedeva, S., Novichikhina, N., Potapov, A., & Sulimov, V. (2022). New Blood Coagulation Factor XIIa Inhibitors: Molecular Modeling, Synthesis, and Experimental Confirmation. Molecules, 27(4), 1234. https://doi.org/10.3390/molecules27041234