
Carbonic Anhydrase Inhibition with Sulfonamides Incorporating Pyrazole- and Pyridazinecarboxamide Moieties Provides Examples of Isoform-Selective Inhibitors

 , ,
, ,  , , ,
, , ,  and
and 
Abstract
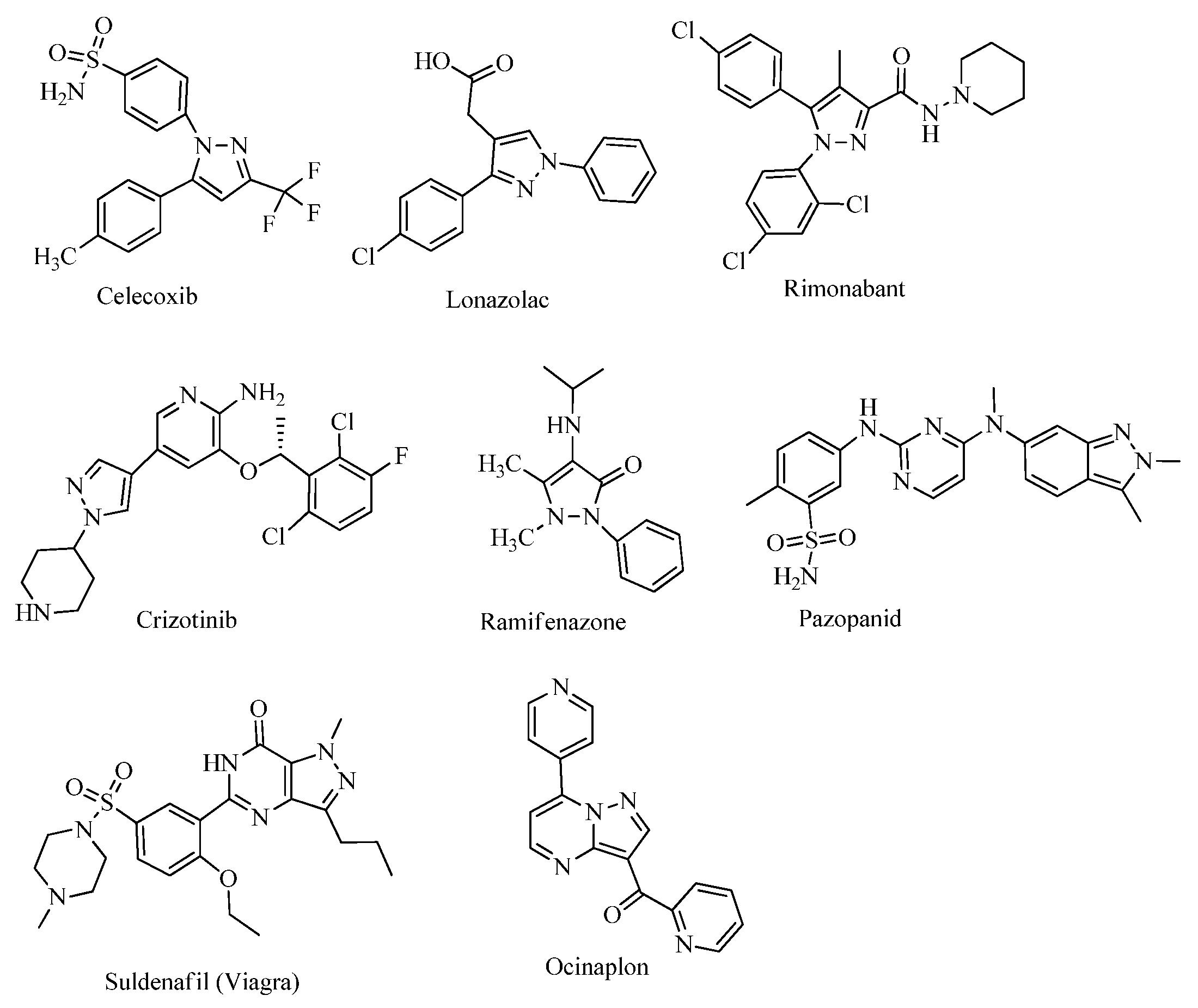
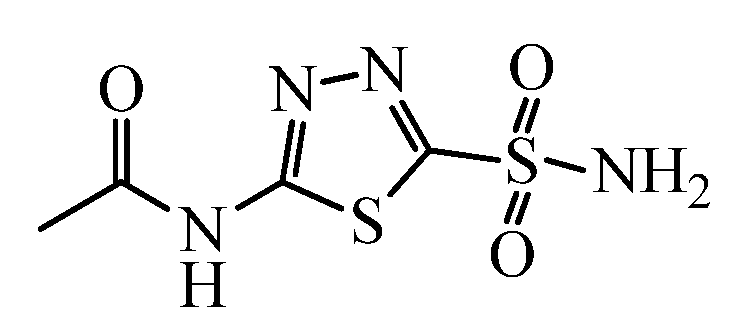
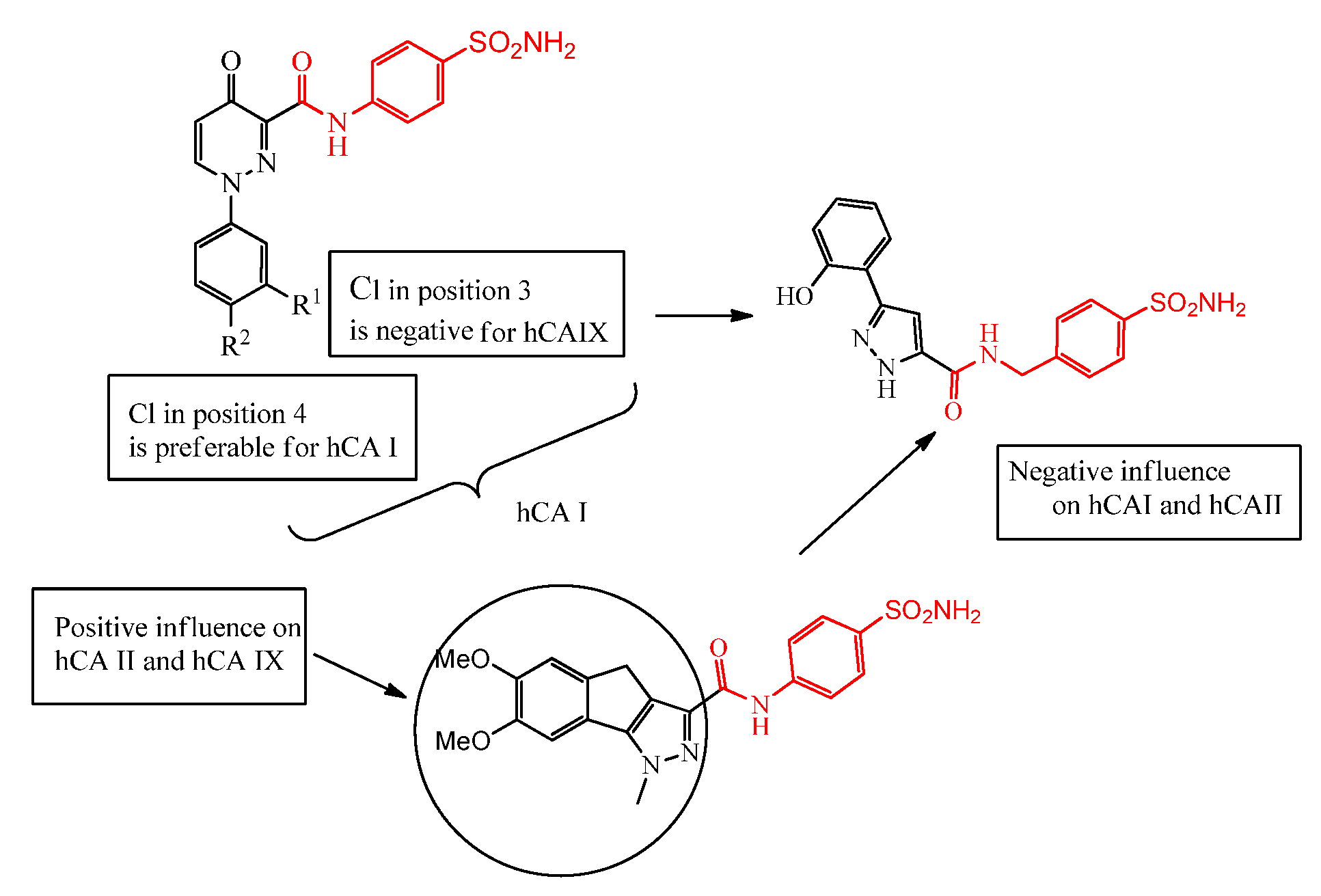
1. Introduction
2. Results
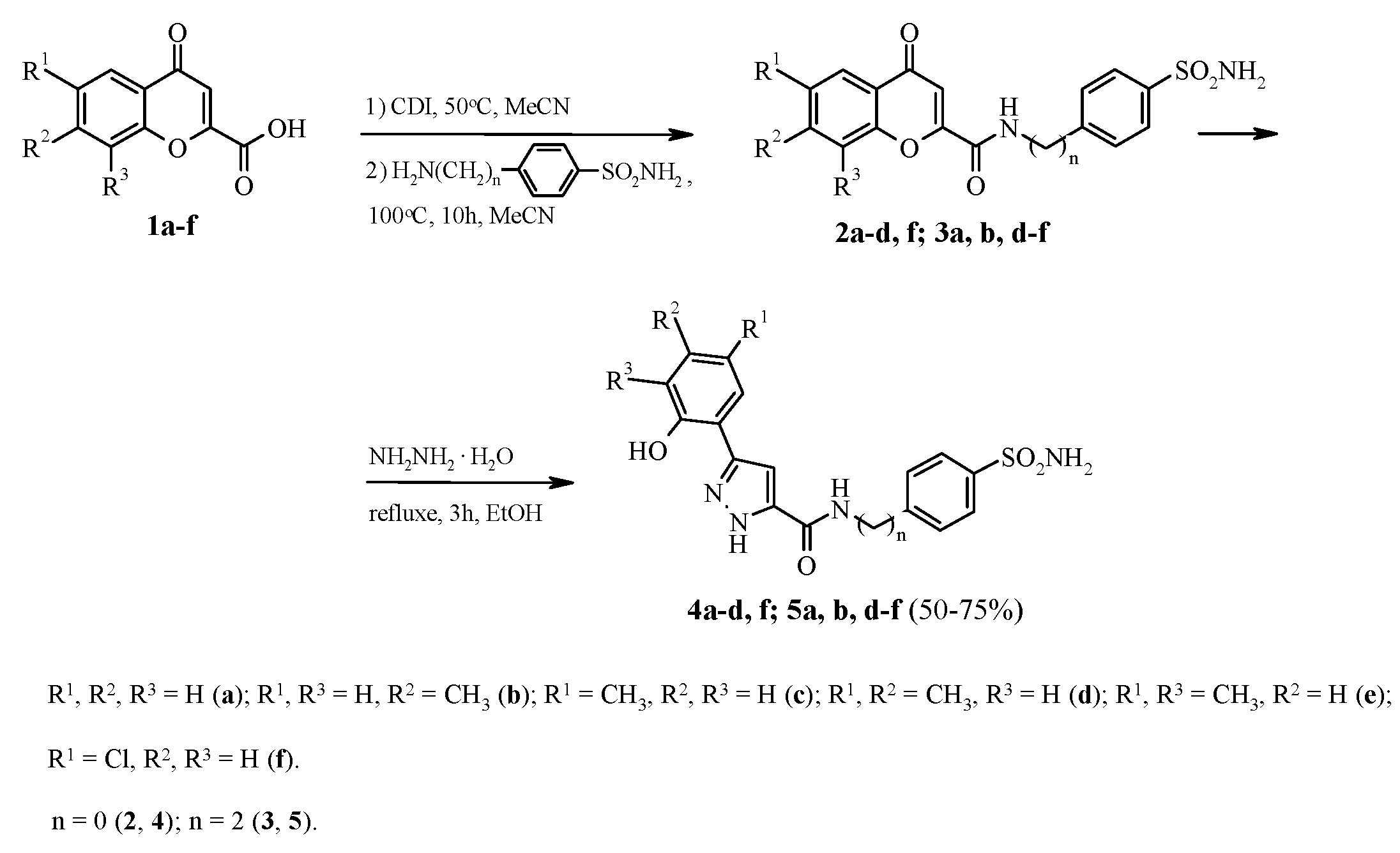
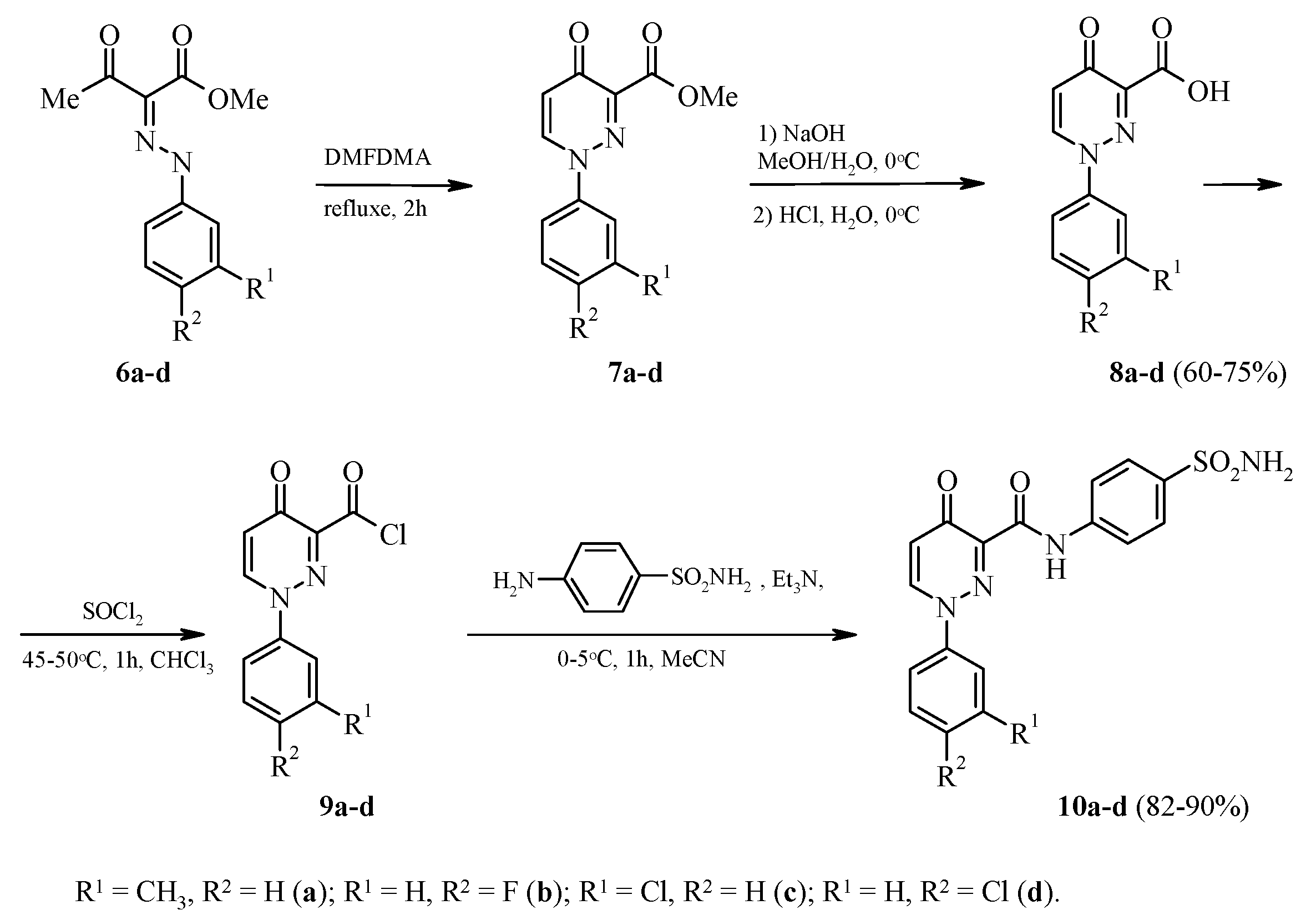
2.1. Chemistry
2.2. Biological Evaluation
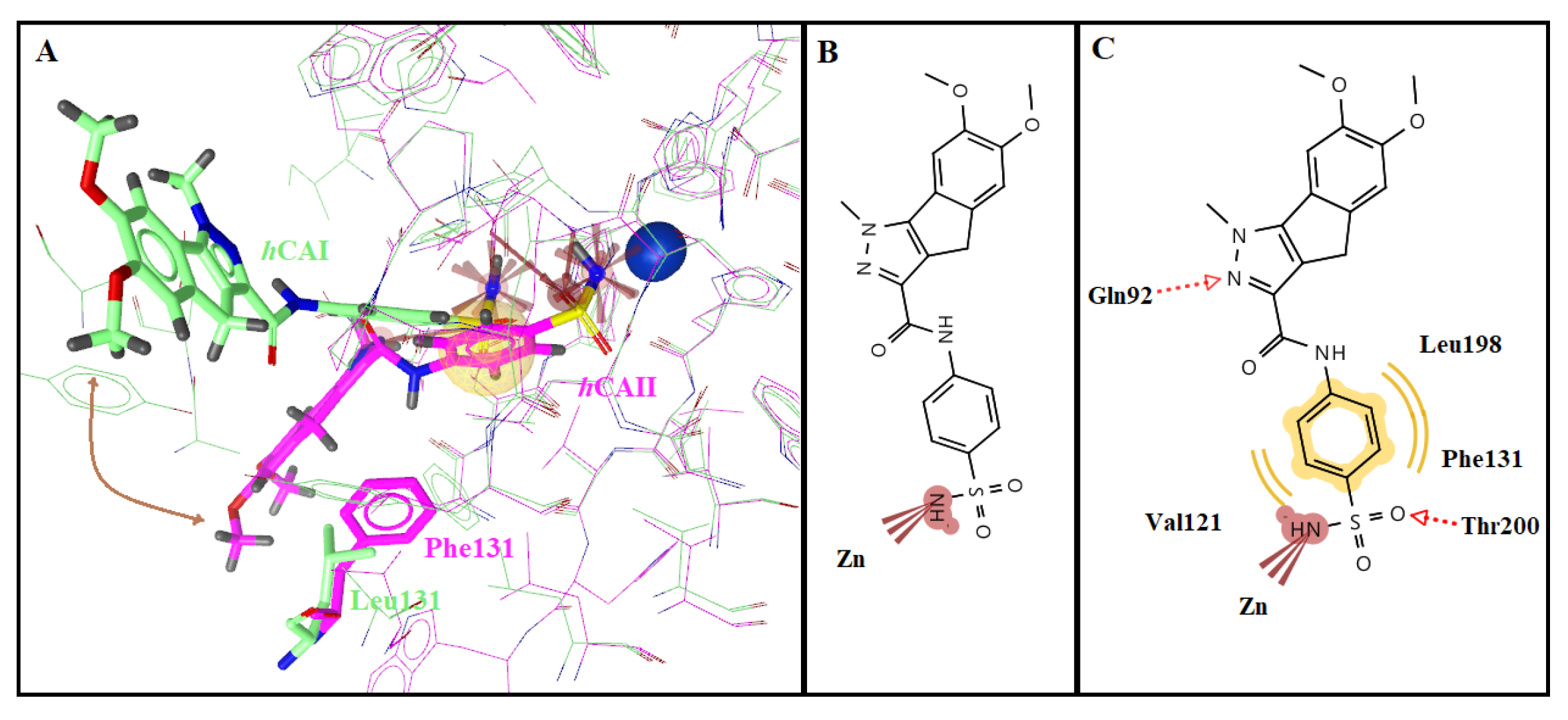
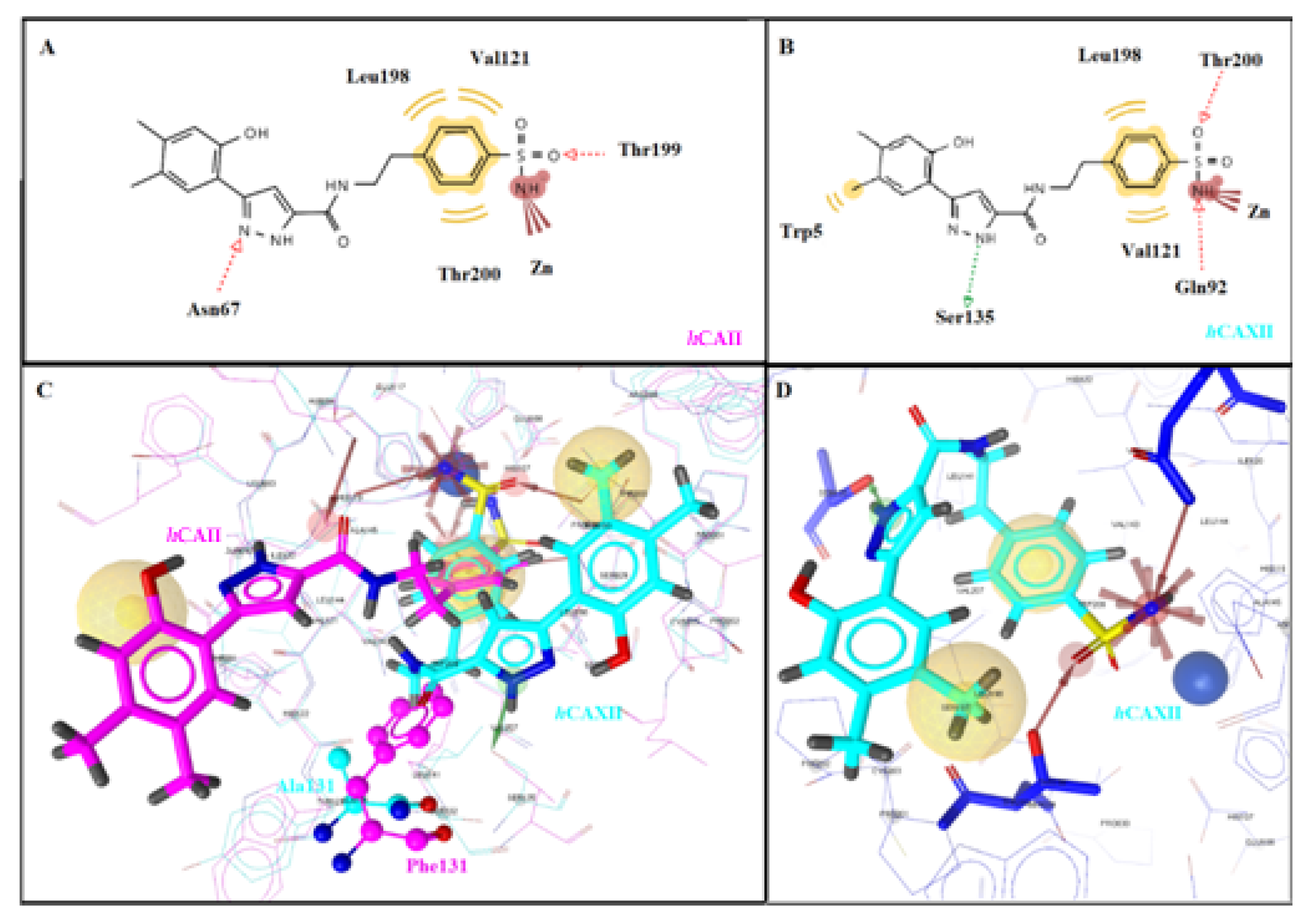
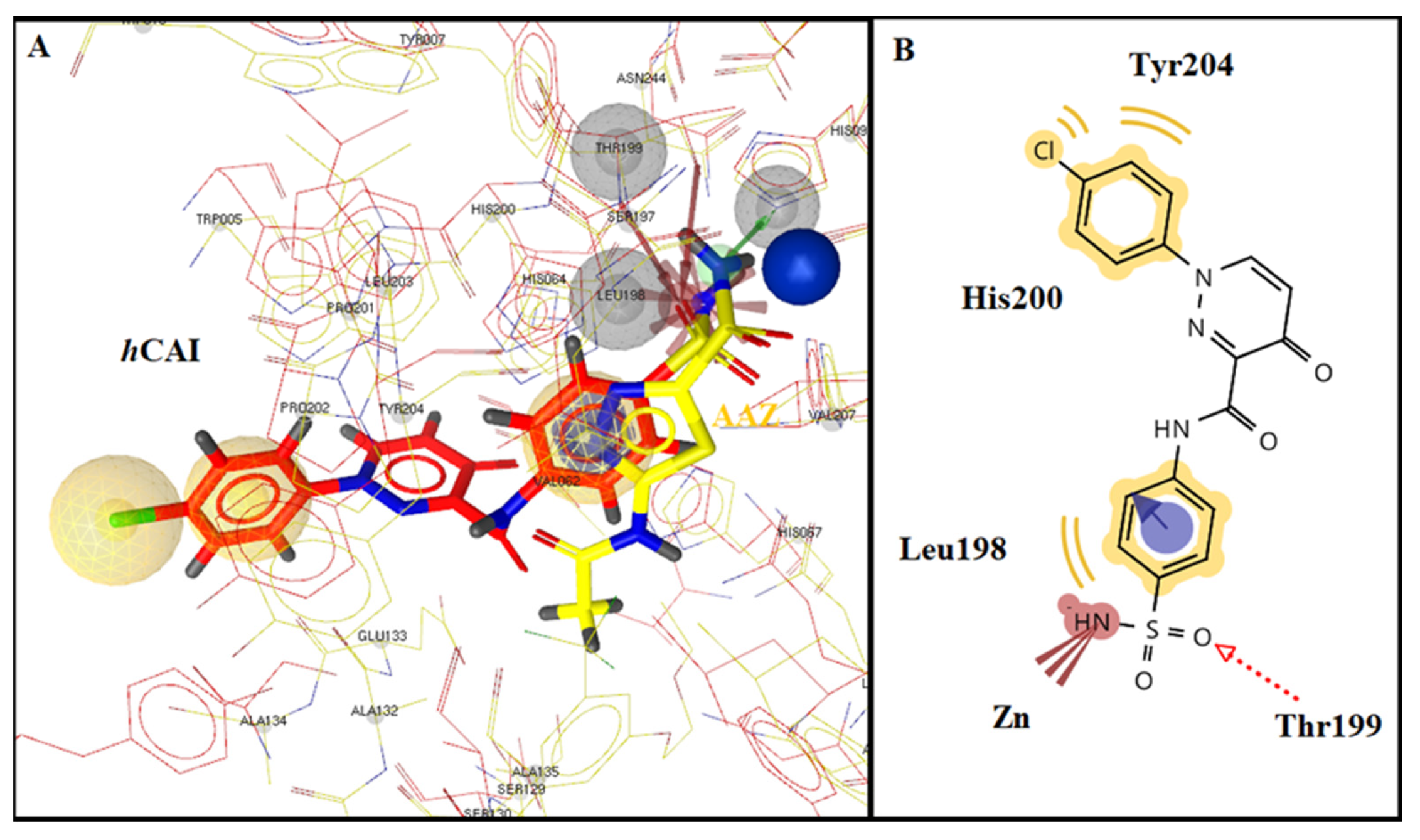
2.3. Molecular Docking Studies
3. Materials and Methods
3.1. General Procedure for the Synthesis of 3-(2-Hydroxyaryl)-1H-pyrazole-5-carboxamide 4a–d, f and 5a, b, d–f
3.2. General Procedure for the Synthesis of 4-Oxo-1-aryl-1,4-dihydropyridazine-3-carboxylic acid 8a–d
3.3. General Procedure for the Synthesis of 4-Oxo-1-aryl-1,4-dihydropyridazine-3-carbonyl chloride 9a–d
3.4. General Procedure for the Synthesis of N-[4-(aminosulfonyl)phenyl]-1-aryl-4-oxo-1,4-dihydropyridazine-3-carboxamide 10a–d
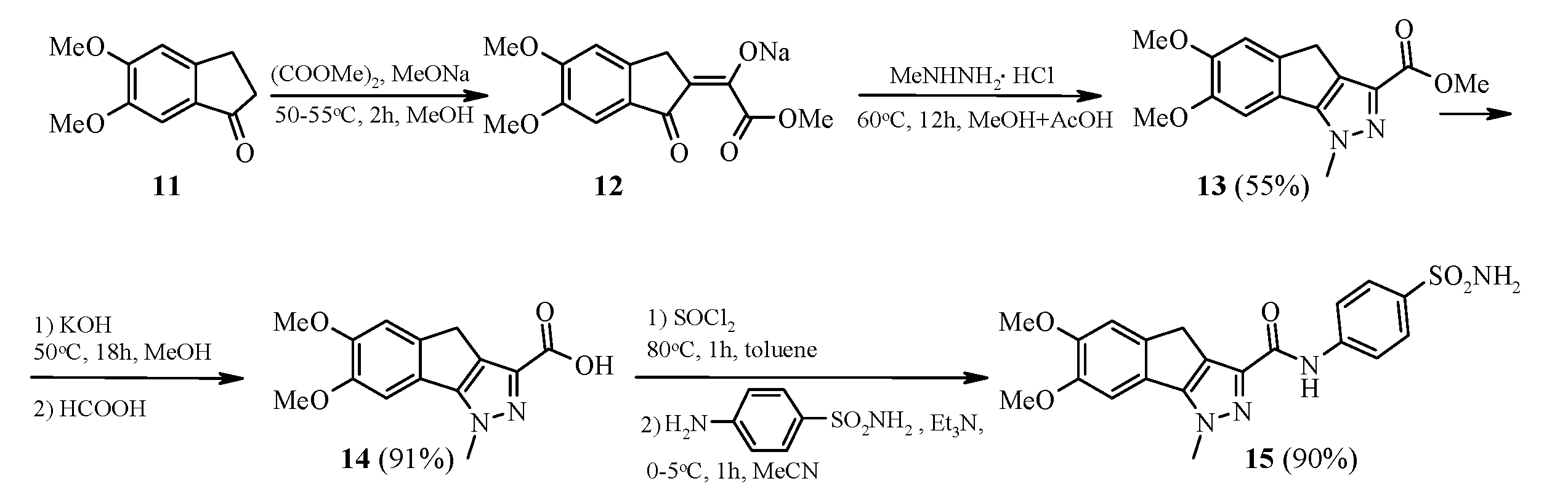
3.5. Synthesis of N-[4-(aminosulfonyl)phenyl]-6,7-dimethoxy-1-methyl-1,4-dihydroindeno[1,2-c]pyrazole-3-carboxamide 15
3.6. Molecular Docking Studies
3.7. CA Inhibition Assay
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Quade, B.N.; Parker, M.D.; Occhipinti, R. The therapeutic importance of acid-base balance. Biochem. Pharmacol. 2021, 183, 114278. [Google Scholar] [CrossRef] [PubMed]
- Supuran, C.T. Carbonic anhydrases: Novel therapeutic applications for inhibitors and activators. Nat. Rev. Drug Discov. 2008, 7, 168–181. [Google Scholar] [CrossRef]
- Supuran, C.T. Emerging role of carbonic anhydrase inhibitors. Clin. Sci. 2021, 135, 1233–1249. [Google Scholar] [CrossRef]
- Xu, Y.; Feng, L.; Jeffrey, P.D.; Shi, Y.; Morel, F.M. Structure and metal exchange in the cadmium carbonic anhydrase of marine diatoms. Nature 2008, 452, 56–61. [Google Scholar] [CrossRef] [PubMed]
- Capasso, C.; Supuran, C.T. An overview of the alpha-, beta- and gamma-carbonic anhydrases from bacteria: Can bacterial carbonic anhydrases shed new light on evolution of bacteria? J. Enzym. Inhib. Med. Chem. 2015, 30, 325–332. [Google Scholar] [CrossRef]
- Supuran, C.T.; Capasso, C. The eta-class carbonic anhydrases as drug targets for antimalarial agents. Expert. Opin. Ther. Targets 2015, 19, 551–563. [Google Scholar] [CrossRef] [PubMed]
- Akgul, O.; Di Cesare Mannelli, L.; Vullo, D.; Angeli, A.; Ghelardini, C.; Bartolucci, G.; Alfawaz Altamimi, A.S.; Scozzafava, A.; Supuran, C.T.; Carta, F. Discovery of novel nonsteroidal anti-inflammatory drugs and carbonic anhydrase inhibitors hybrids (NSAIDs-CAIs) for the management of rheumatoid arthritis. J. Med. Chem. 2018, 61, 4961–4977. [Google Scholar] [CrossRef]
- Bua, S.; Di Cesare Mannelli, L.; Vullo, D.; Ghelardini, C.; Bartolucci, G.; Scozzafava, A.; Supuran, C.T.; Carta, F. Design and synthesis of novel Nonsteroidal Anti-Inflammatory Drugs and Carbonic Anhydrase Inhibitors hybrids (NSAIDs-CAIs) for the treatment of rheumatoid arthritis. J. Med. Chem. 2017, 60, 1159–1170. [Google Scholar] [CrossRef]
- Lardner, A. The effects of extracellular pH on immune function. J. Leukoc. Biol. 2001, 69, 522–530. [Google Scholar]
- McDonald, P.C.; Chafe, S.C.; Brown, W.S.; Saberi, S.; Swayampakula, M.; Venkateswaran, G.; Nemirovsky, O.; Gillespie, J.A.; Karasinska, J.M.; Kalloger, S.E.; et al. Regulation of pH by Carbonic Anhydrase 9 Mediates Survival of Pancreatic Cancer Cells with Activated KRAS in Response to Hypoxia. Gastroenterology 2019, 157, 823–837. [Google Scholar] [CrossRef]
- Supuran, C.T. Carbonic anhydrase inhibitors as emerging agents for the treatment and imaging of hypoxic tumors. Expert Opin. Investig. Drugs 2018, 27, 963–970. [Google Scholar] [CrossRef] [PubMed]
- Supuran, C.T.; Winum, J.Y. Carbonic anhydrase IX inhibitors in cancer therapy: An update. Future Med. Chem. 2015, 7, 1407–1414. [Google Scholar] [CrossRef] [PubMed]
- Penning, T.D.; Talley, J.J.; Bertenshaw, S.R.; Carter, J.S.; Collins, P.W.; Doctor, S.; Graneto, M.J.; Lee, L.F.; Malecha, J.W.; Miyashiro, J.M.; et al. Synthesis and biological evaluation of the 1,5-diarylpyrazole class of cyclooxygenase-2 inhibitors: Identification of 4-[5-(4-methylphenyl)-3-(trifluoromethyl)-1H-pyrazol-1-yl]benze nesulfonamide (SC-58635, celecoxib). J. Med. Chem. 1997, 40, 1347–1365. [Google Scholar] [CrossRef]
- Fioravanti, R.; Bolasco, A.; Manna, F.; Rossi, F.; Orallo, F.; Ortuso, F.; Alcaro, S.; Cirilli, R. Synthesis and biological evaluation of N-substituted-3,5-diphenyl-2-pyrazoline derivatives as cyclooxygenase (COX-2) inhibitors. Eur. J. Med. Chem. 2010, 45, 6135–6138. [Google Scholar] [CrossRef]
- Riedel, R. Lonazolac-Ca = Calcium [3-(p-chlorophenyl)-1-phenylpyrazole-4[-acetate 1 Pharmacological properties of a new antiinflammatory/antirheumatic drug (author’s transl). Arzneim. Forsch. 1981, 31, 655–665. [Google Scholar]
- Isidro, M.L.; Cordido, F. Drug treatment of obesity: Established and emerging therapies. Mini-Rev. Med. Chem. 2009, 9, 664–673. [Google Scholar] [CrossRef] [PubMed]
- Devi, N.; Shankar, R.; Singh, V. 4-Formyl-Pyrazole-3-Carboxylate: A Useful Aldo-X Bifunctional Precursor for the Syntheses of Pyrazole-fused/Substituted Frameworks. J. Heterocycl. Chem. 2018, 55, 373–390. [Google Scholar] [CrossRef]
- Katz, R.J. Effects of zometapine, a structurally novel antidepressant, in an animal model of depression. Pharmacol. Biochem. Behav. 1984, 21, 487–490. [Google Scholar] [CrossRef]
- Lippa, A.; Czobor, P.; Stark, J.; Beer, B.; Kostakis, E.; Gravielle, M.; Bandyopadhyay, S.; Russek, S.J.; Gibbs, T.T.; Farb, D.H.; et al. Selective anxiolysis produced by ocinaplon, a GABAA receptor modulator. Proc. Natl. Acad. Sci. USA 2005, 102, 7380–7385. [Google Scholar] [CrossRef]
- Marinescu, M. Synthesis of Antimicrobial Benzimidazole-Pyrazole Compounds and Their Biological Activities. Antibiotics 2021, 10, 1002. [Google Scholar] [CrossRef]
- Çetin, A.; Bildirici, I. A study on synthesis and antimicrobial activity of 4-acyl-pyrazoles. J. Saudi Chem. Soc. 2018, 22, 279–296. [Google Scholar] [CrossRef]
- Muhammad, Z.A.; Alshehrei, F.; Zayed, M.E.M.; Farghaly, T.A.; Abdallah, M.A. Synthesis of Novel Bis-pyrazole Derivatives as Antimicrobial Agents. Mini Rev. Med. Chem. 2019, 19, 1276–1290. [Google Scholar] [CrossRef]
- Da Costa, L.; Scheers, E.; Coluccia, A.; Casulli, A.; Roche, M.; Di Giorgio, C.; Neyts, J.; Terme, T.; Cirilli, R.; La Regina, G.; et al. Structure-Based Drug Design of Potent Pyrazole Derivatives against Rhinovirus Replication. J. Med. Chem. 2018, 61, 8402–8416. [Google Scholar] [CrossRef]
- Corona, A.; Onnis, V.; Deplano, A.; Bianco, G.; Demurtas, M.; Distinto, S.; Cheng, Y.C.; Alcaro, S.; Esposito, F.; Tramontano, E. Design, synthesis and antiviral evaluation of novel heteroarylcarbothioamide derivatives as dual inhibitors of HIV-1 reverse transcriptase-associated RNase H and RDDP functions. Pathog. Dis. 2017, 75, ftx078. [Google Scholar] [CrossRef] [PubMed]
- Naim, M.J.; Alam, O.; Alam, M.J.; Hassan, M.Q.; Siddiqui, N.; Naidu, V.G.M.; Alam, M.I. Design, synthesis and molecular docking of thiazolidinedione based benzene sulphonamide derivatives containing pyrazole core as potential anti-diabetic agents. Bioorg. Chem. 2018, 76, 98–112. [Google Scholar] [CrossRef]
- Faidallah, H.M.; Al-Mohammadi, M.M.; Alamry, K.A.; Khan, K.A. Synthesis and biological evaluation of fluoropyrazolesulfonylurea and thiourea derivatives as possible antidiabetic agents. J. Enzyme Inhib. Med. Chem. 2016, 31, 157–163. [Google Scholar] [CrossRef]
- Li, X.; Yu, Y.; Tu, Z. Pyrazole Scaffold Synthesis, Functionalization, and Applications in Alzheimer’s Disease and Parkinson’s Disease Treatment (2011–2020). Molecules 2021, 26, 1202. [Google Scholar] [CrossRef]
- Grewal, A.S.; Sharma, S.K.; Pandita, D.; Lather, V. Synthesis, Docking and Evaluation of Novel Pyrazole Carboxamide Derivatives as Multifunctional Anti-Alzheimer’s Agents. J. Med. Chem. Toxicol. 2017, 2, 47–54. [Google Scholar]
- Meta, E.; Brullo, C.; Tonelli, M.; Franzblau, S.G.; Wang, Y.; Ma, R.; Baojie, W.; Orena, B.S.; Pasca, M.R.; Bruno, O. Pyrazole and imidazo[1,2-b]pyrazole Derivatives as New Potential Antituberculosis Agents. Med. Chem. 2019, 15, 17–27. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Gao, C.; Ren, Q.C.; Song, X.F.; Feng, L.S.; Lv, Z.S. Recent advances of pyrazole-containing derivatives as anti-tubercular agents. Eur. J. Med. Chem. 2017, 139, 429–440. [Google Scholar] [CrossRef]
- Bekhit, A.A.; Saudi, M.N.; Hassan, A.M.; Fahmy, S.M.; Ibrahim, T.M.; Ghareeb, D.; El-Seidy, A.M.; Al-Qallaf, S.M.; Habib, H.J.; Bekhit, A.E.A. Synthesis, molecular modeling and biological screening of some pyrazole derivatives as antileishmanial agents. Future Med. Chem. 2018, 10, 2325–2344. [Google Scholar] [CrossRef]
- Cagide, F.; Oliveira, C.; Reis, J.; Borges, F. Optimizing the synthetic route of chromone-2-carboxylic acids: A step forward to speed-up the discovery of chromone-based multitarget-directed ligands. Molecules 2019, 24, 4214. [Google Scholar] [CrossRef]
- Ruhemann, S.; Wragg, E. CXXV.—Condensation of phenols with esters of the acetylene series. Part VI. J. Chem. Soc. Trans. 1901, 79, 1185–1191. [Google Scholar] [CrossRef][Green Version]
- Golovchenko, A.V.; Solomyannyi, R.N.; Brovarets, V.S. Synthesis of C-heteryl-substituted aminomethylphosphonic acids derivatives. Russ. J. Gen. Chem. 2010, 80, 723–727. [Google Scholar] [CrossRef]
- Plescia, S.; Daidone, G.; Fabra, J.; Sprio, V. Studies on the synthesis of heterocyclic compounds. Part V. A novel synthesis of some pyridazin-4-(1H)one derivatives and their reaction with hydrazine. J. Het. Chem. 1981, 18, 333–334. [Google Scholar] [CrossRef]
- Wang, L.X.; Liu, X.; Xu, S.; Tang, Q.; Duan, Y.; Xiao, Z.; Zhi, J.; Jiang, L.; Zheng, P.; Zhu, W. Discovery of novel pyrrolo-pyridine/pyrimidine derivatives bearing pyridazinone moiety as c-Met kinase inhibitors. Eur. J. Med. Chem. 2017, 141, 538–551. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Kou, J.; Xiao, Z.; Tian, F.; Hu, J.; Zheng, P.; Zhu, W. Design, synthesis and biological evaluation of 6,7-disubstituted-4-phenoxyquinoline derivatives bearing pyridazinone moiety as c-met inhibitors. Molecules 2018, 23, 1543. [Google Scholar] [CrossRef] [PubMed]
- Altalbawy, F.M.A. Synthesis, in vitro antimicrobial and anticancer evaluation of some new pyridazines and polyfunctionally substituted heterocyclic compounds. Asian J. Chem. 2015, 27, 4361–4368. [Google Scholar] [CrossRef]
- Kunitomo, J.; Yoshikawa, M.; Fushimi, M.; Kawada, A.; Quinn, J.F.; Oki, H.; Kokubo, H.; Kondo, M.; Nakashima, K.; Kamiguchi, N.; et al. Discovery of 1-[2-fluoro-4-(1H-pyrazol-1-yl)phenyl]-5-methoxy-3-(1-phenyl-1H-pyrazol-5-yl)pyridazin-4(1H)-one (tak-063), a highly potent, selective, and orally active phosphodiesterase 10a (pde10a) inhibitor. J. Med. Chem. 2014, 57, 9627–9643. [Google Scholar] [CrossRef]
- Wang, L.; Xu, S.; Chen, X.; Liu, X.; Duan, Y.; Kong, D.; Zhao, D.; Zheng, P.; Tang, Q.; Zhu, W. Synthesis and bioevaluation study of novel N-methylpicolinamide and thienopyrimidine derivatives as selectivity c-Met kinase inhibitors. Bioorg. Med. Chem. 2018, 26, 245–256. [Google Scholar] [CrossRef]
- Pinna, G.A.; Pirisi, M.A.; Mussinu, J.-M.; Murineddu, G.; Loriga, G.; Pau, A.; Grella, G.E. Chromophore-modified bis-benzo[g]indole carboxamides: Synthesis and antiproliferative activity of bis-benzo[g]indazole-3-carboxamides and related dimers. Il Farmaco 2003, 58, 749–763. [Google Scholar] [CrossRef]
- Alterio, V.; Hilvo, M.; Di Fiore, A.; Supuran, C.T.; Pan, P.; Parkkila, S.; Scaloni, A.; Pastorek, J.; Patorekova, S.; Pedone, C.; et al. Crystal structure of the catalytic domain of the tumor associated human carbonic anhydrase IX. Proc. Natl. Acad. Sci. USA 2009, 106, 16233–16238. [Google Scholar] [CrossRef]
- Di Fiore, A.; Truppo, E.; Supuran, C.T.; Alterio, V.; Dathan, N.; Bootorabi, F.; Parkkila, S.; Monti, S.M.; De Simone, G. Crystal structure of the C183S/C217S mutant of human CA VII in complex with acetazolamide. Bioorg. Med. Chem. Lett. 2010, 20, 5023–5026. [Google Scholar] [CrossRef]
- Whittington, D.A.; Waheed, A.; Ulmasov, B.; Shah, G.N.; Grubb, J.H.; Sly, W.S.; Christianson, D.W. Crystal structure of the dimeric extracellular domain of human carbonic anhydrase XII, a bitopic membrane protein overexpressed in certain cancer tumor cells. Proc. Natl. Acad. Sci. USA 2001, 98, 9545–9550. [Google Scholar] [CrossRef] [PubMed]
- Supuran, C.T. Structure-based drug discovery of carbonic anhydrase inhibitors. J. Enzyme Inhib. Med. Chem. 2012, 27, 759–772. [Google Scholar] [CrossRef]
- Kanamori, K.; Roberts, J.D. Nitrogen-15 nuclear magnetic resonance study of benzenesulfonamide and cyanate binding to carbonic anhydrase. Biochemistry 1983, 22, 2658–2664. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. Autodock4 and AutoDockTools4: Automated docking with selective receptor flexiblity. J. Comput. Chem. 2009, 16, 2785–2791. [Google Scholar] [CrossRef] [PubMed]
- Available online: http://www.rcsb.org/ (accessed on 19 June 2021).
- Angeli, A.; Kartsev, V.; Petrou, A.; Pinteala, M.; Brovarets, V.; Slyvchuk, S.; Pilyo, S.; Geronikaki, A.; Supuran, C.T. Chromene-Containing Aromatic Sulfonamides with Carbonic Anhydrase Inhibitory Properties. Int. J. Mol. Sci. 2021, 22, 5082. [Google Scholar] [CrossRef]
- Khalifah, R.G. The carbon dioxide hydration activity of carbonic anhydrase. J. Biol. Chem. 1971, 246, 2561–2573. [Google Scholar] [CrossRef]
- Abdel-Aziz, A.A.; Angeli, A.; El-Azab, A.S.; Abu El-Enin, M.A.; Supuran, C.T. Synthesis and biological evaluation of cyclic imides incorporating benzenesulfonamide moieties as carbonic anhydrase I, II, IV and IX inhibitors. Bioorg. Med. Chem. 2017, 25, 1666–1671. [Google Scholar] [CrossRef]
- Gul, H.I.; Yamali, C.; Bulbuller, M.; Kirmizibayrak, P.B.; Gul, M.; Angeli, A.; Bua, S.; Supuran, C.T. Anticancer effects of new dibenzenesulfonamides by inducing apoptosis and autophagy pathways and their carbonic anhydrase inhibitory effects on hCA I, hCA II, hCA IX, hCA XII isoenzymes. Bioorg. Chem. 2018, 78, 290–297. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| KI (nM) 1 | ||||||||
|---|---|---|---|---|---|---|---|---|
| Com/ds | R1 | R2 | R3 | n | hCAI | hCAII | hCAIX | hCAXII |
| 4a | H | H | H | 0 | 3822 | 866.7 | 450.2 | 271.1 |
| 4b | H | H | Me | 0 | 93.3 | 308.6 | 78.9 | 372.5 |
| 4c | Me | H | H | 0 | 221.1 | 16.1 | 8.5 | 250.0 |
| 4f | Cl | H | H | 0 | 154.7 | 37.2 | 34.2 | 90.8 |
| 5a | H | H | H | 2 | 88.1 | 31.5 | 93.8 | 188.5 |
| 5b | H | H | Me | 2 | 344.6 | 16.2 | 16.1 | 329.5 |
| 5d | Me | Me | H | 2 | 218.5 | 25.2 | 63.5 | 61.3 |
| 5e | Me | H | Me | 2 | 71.4 | 16.2 | 52.3 | 191.2 |
| 5f | Cl | H | H | 2150 | 9.1 | 79.4 | 77.3 | |
| 10a | Me | H | - | - | 197.9 | 7.4 | 41.6 | 90.8 |
| 10b | H | F | - | - | 481.0 | 72.5 | 130.8 | 94.8 |
| 10c | Cl | H | - | - | 839.7 | 76.3 | 568.8 | 432.8 |
| 10d | H | Cl | - | - | 6.2 | 8.0 | 165.2 | 65.7 |
| 15 | OMe | OMe | - | - | 725.6 | 3.3 | 6.1 | 80.5 |
| AAZ | 250.0 | 12.1 | 25.8 | 5.7 | ||||
 | ||||||||
| No | R1 | R2 | R3 | n | hCA Isoform | Estimated Free Binding Energy (Kcal/mol) | Chelating the Zn (II) Ion | Residues Involved in H-Bond Interactions | Residues Involved in Hydrophobic Interactions |
|---|---|---|---|---|---|---|---|---|---|
| 5d | hCA I | −6.82 | Yes | Thr199 | Ile191, Val121, Leu198 | ||||
| Me | Me | H | 2 | hCA II | −8.17 | Yes | Thr199 | Val121, Leu198, Thr200 | |
| hCA IX | −6.84 | Yes | Thr200 | Val121, Trp209 | |||||
| hCA XII | −8.10 | Yes | Gln92, Ser135, Thr200 | Trp5, Val121, Leu198 | |||||
| hCA I | −7.16 | Yes | Thr199 | Ala132, Ala135, Leu198 | |||||
| 5e | Me | H | Me | 2 | hCA II | −8.82 | Yes | His94, Thr199 | Val121, Phe131, Thr200 |
| hCA IX | −7.71 | Yes | Thr199 | Val121, Leu198, Thr200 | |||||
| hCA XII | −6.21 | Yes | - | Val121, Leu198 | |||||
| hCA I | −12.38 | Yes | Thr199 | Leu198, His200, Tyr204 | |||||
| 10d | H | Cl | - | - | hCA II | −9.11 | Yes | Gln92, Thr199 | Val121, Phe131, Leu198 |
| hCA IX | −6.55 | Yes | Thr199 | Val121, Leu198 | |||||
| hCA XII | −7.02 | Yes | His94 | Val121, Ala131, Leu198 | |||||
| hCA I | −4.21 | Yes | - | - | |||||
| 15 | OMe | OMe | - | - | hCA II | −9.25 | Yes | Gln92, Thr200 | Val121, Phe131, Leu198 |
| hCA IX | −10.20 | Yes | Thr199, Thr200 | Val121, Val131, Leu198, Thr200 | |||||
| hCA XII | −7.11 | Yes | Thr200 | Val121, Leu198 | |||||
| hCA I | −8.28 | Yes | Gln92 | Leu198, Thr199, His200, Pro201, Trp209 | |||||
| AAZ | hCA II | −8.87 | Yes | Thr199, Thr200 | Val121, Phe131, Leu198, Trp209 | ||||
| hCA IX | −9.02 | Yes | Thr199, Thr200 | Val121, Val143, Val131, Leu198, Trp209 | |||||
| hCA XII | −9.14 | Yes | Thr199, Thr200 | Val121, Val143, Leu198, Trp209 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Angeli, A.; Kartsev, V.; Petrou, A.; Pinteala, M.; Brovarets, V.; Vydzhak, R.; Panchishin, S.; Geronikaki, A.; Supuran, C.T. Carbonic Anhydrase Inhibition with Sulfonamides Incorporating Pyrazole- and Pyridazinecarboxamide Moieties Provides Examples of Isoform-Selective Inhibitors. Molecules 2021, 26, 7023. https://doi.org/10.3390/molecules26227023
Angeli A, Kartsev V, Petrou A, Pinteala M, Brovarets V, Vydzhak R, Panchishin S, Geronikaki A, Supuran CT. Carbonic Anhydrase Inhibition with Sulfonamides Incorporating Pyrazole- and Pyridazinecarboxamide Moieties Provides Examples of Isoform-Selective Inhibitors. Molecules. 2021; 26(22):7023. https://doi.org/10.3390/molecules26227023
Chicago/Turabian StyleAngeli, Andrea, Victor Kartsev, Anthi Petrou, Mariana Pinteala, Volodymyr Brovarets, Roman Vydzhak, Svitlana Panchishin, Athina Geronikaki, and Claudiu T. Supuran. 2021. "Carbonic Anhydrase Inhibition with Sulfonamides Incorporating Pyrazole- and Pyridazinecarboxamide Moieties Provides Examples of Isoform-Selective Inhibitors" Molecules 26, no. 22: 7023. https://doi.org/10.3390/molecules26227023
APA StyleAngeli, A., Kartsev, V., Petrou, A., Pinteala, M., Brovarets, V., Vydzhak, R., Panchishin, S., Geronikaki, A., & Supuran, C. T. (2021). Carbonic Anhydrase Inhibition with Sulfonamides Incorporating Pyrazole- and Pyridazinecarboxamide Moieties Provides Examples of Isoform-Selective Inhibitors. Molecules, 26(22), 7023. https://doi.org/10.3390/molecules26227023