Intermolecular Interactions and Spectroscopic Signatures of the Hydrogen-Bonded System—n-Octanol in Experimental and Theoretical Studies

 , , , and
, , , and
Abstract
:1. Introduction
2. Materials and Methods
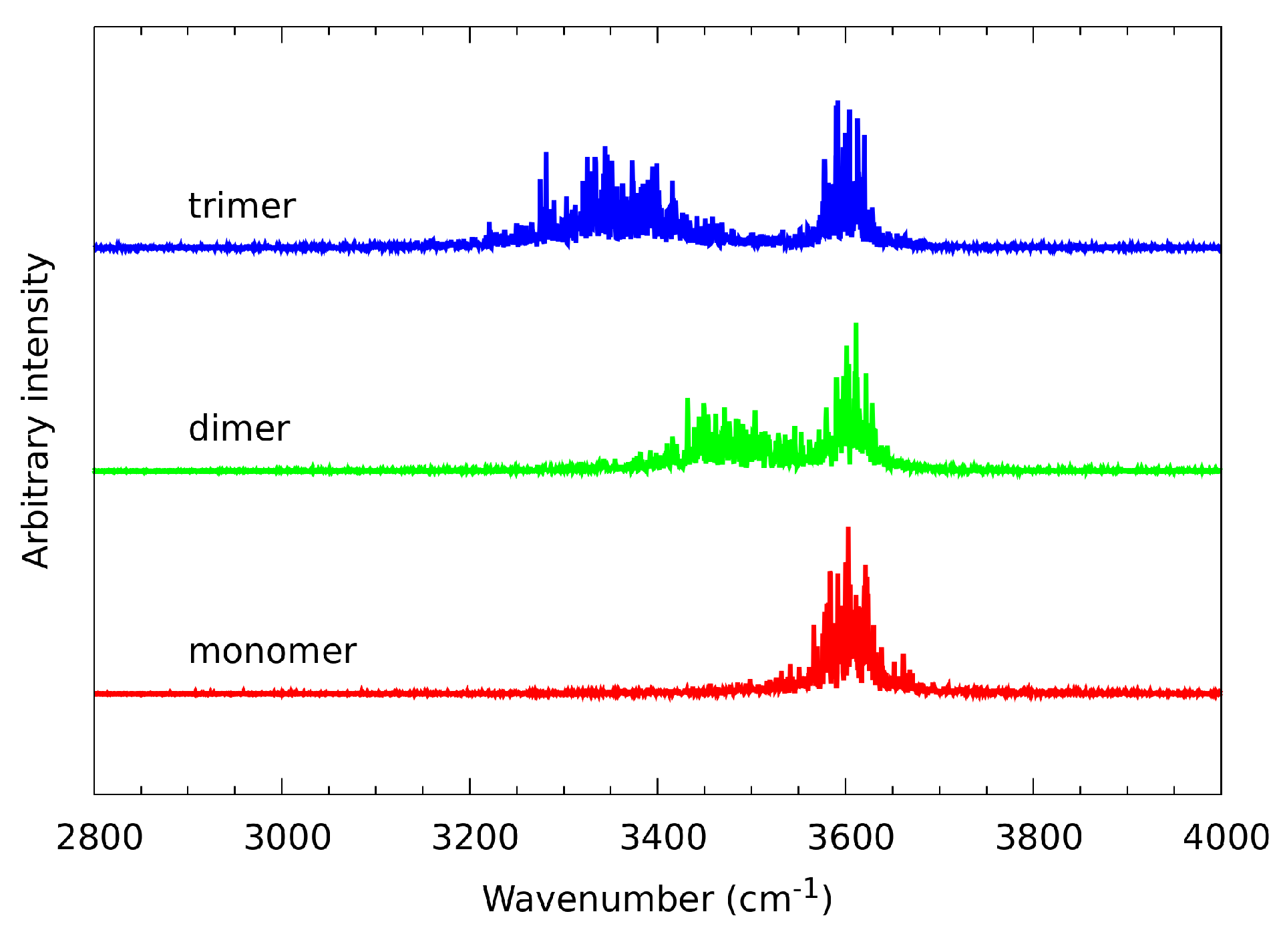
2.1. Experimental IR Spectra Measurements
2.2. Computational Methodology for the Static DFT and SAPT Models
2.2.1. Static Density Functional Theory (DFT)
2.2.2. Symmetry-Adapted Perturbation Theory (SAPT)
2.3. Computational Methodology for the Time-Evolution Study on the Basis of Classical and First-Principle Molecular Dynamics (FPMD)
2.3.1. Classical Molecular Dynamics (MD) Simulations
2.3.2. Car-Parrinello Molecular Dynamics (CPMD) in the Gas and Liquid Phases
2.3.3. Path Integral Molecular Dynamics (PIMD) in the Gas Phase
2.3.4. A Posteriori Inclusion of Quantum Effects
3. Results and Discussion
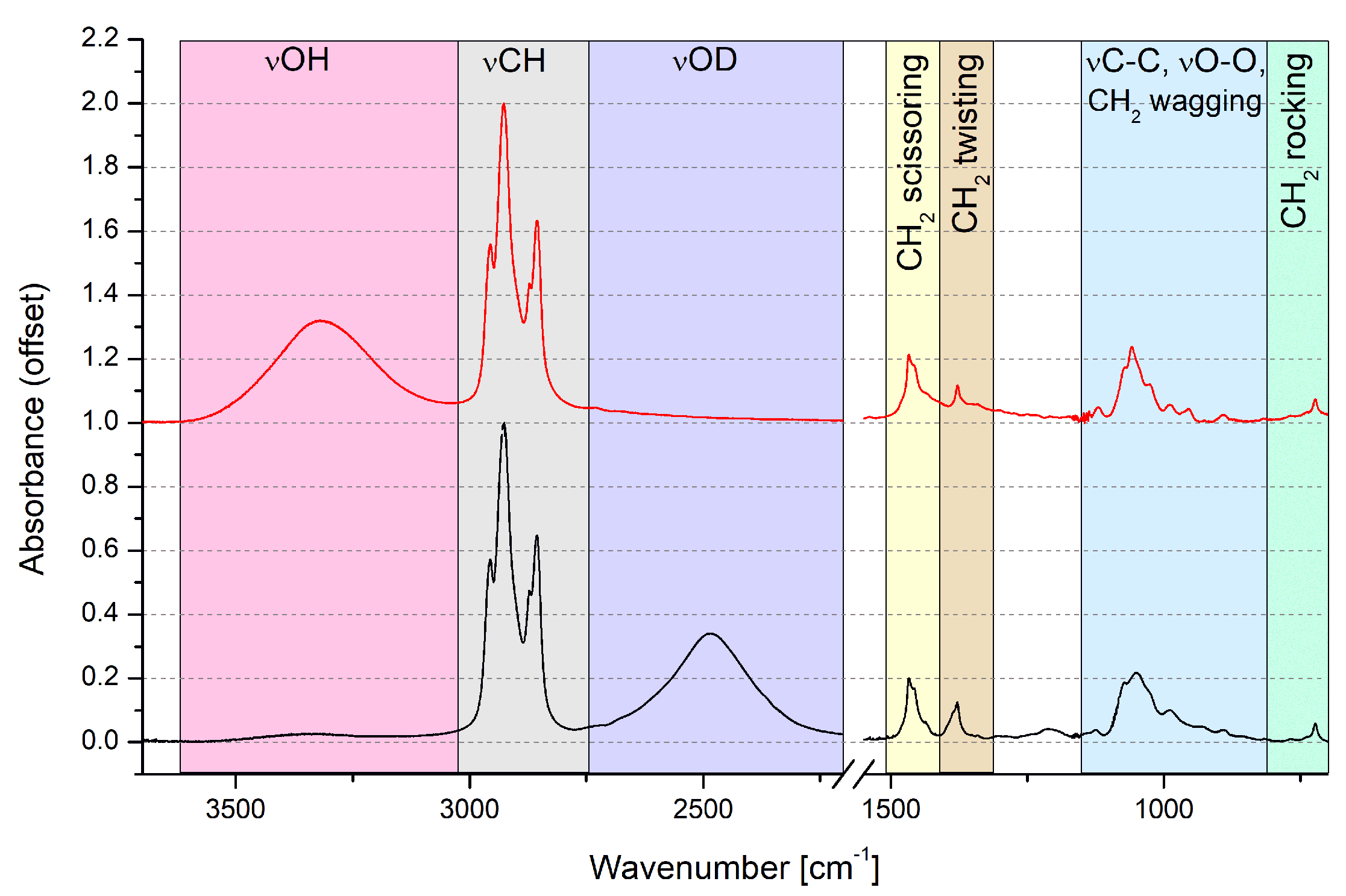
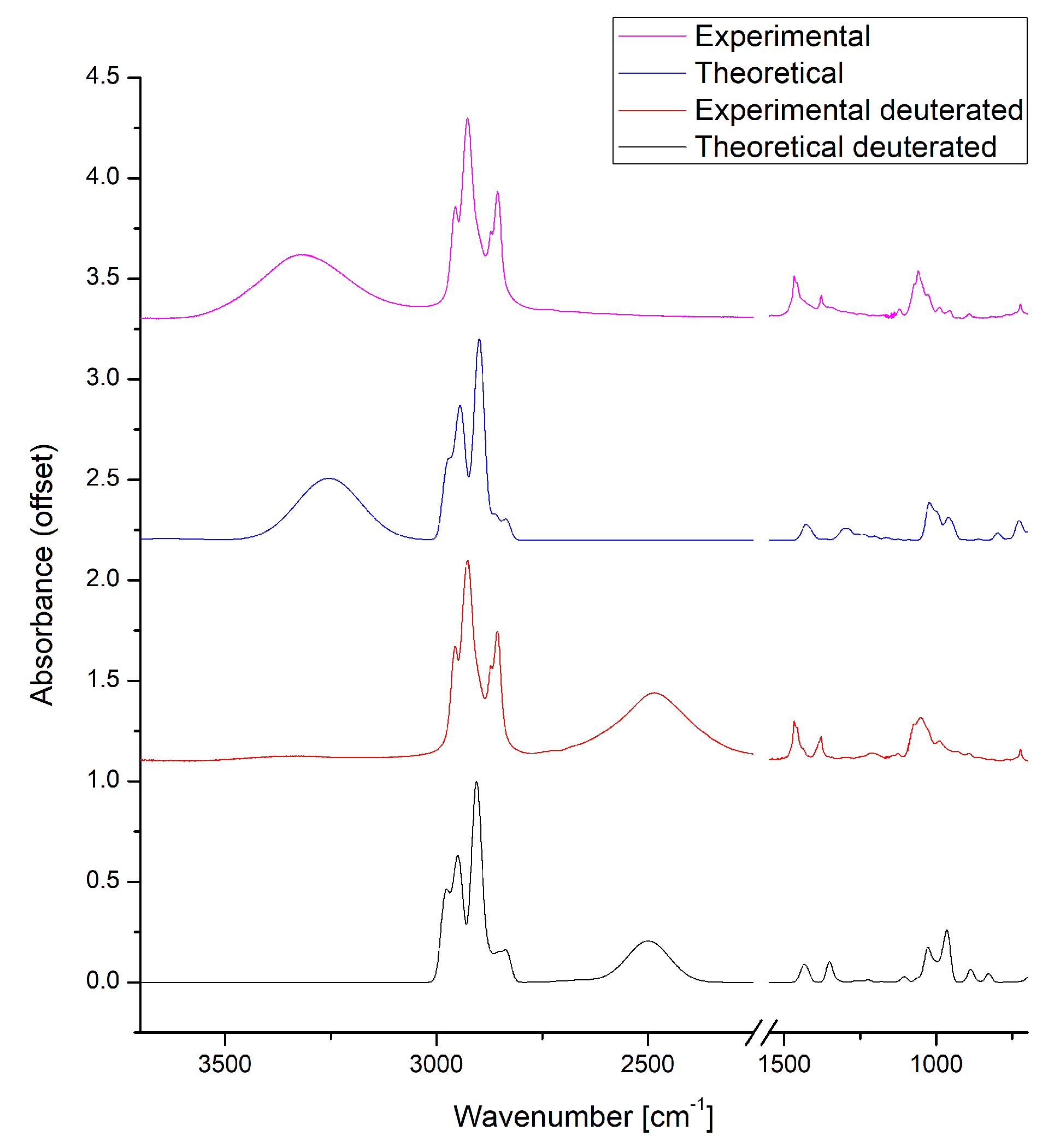
3.1. Experimental IR Data and Static Based DFT Models Describing Spectroscopic Properties of n-Octanol
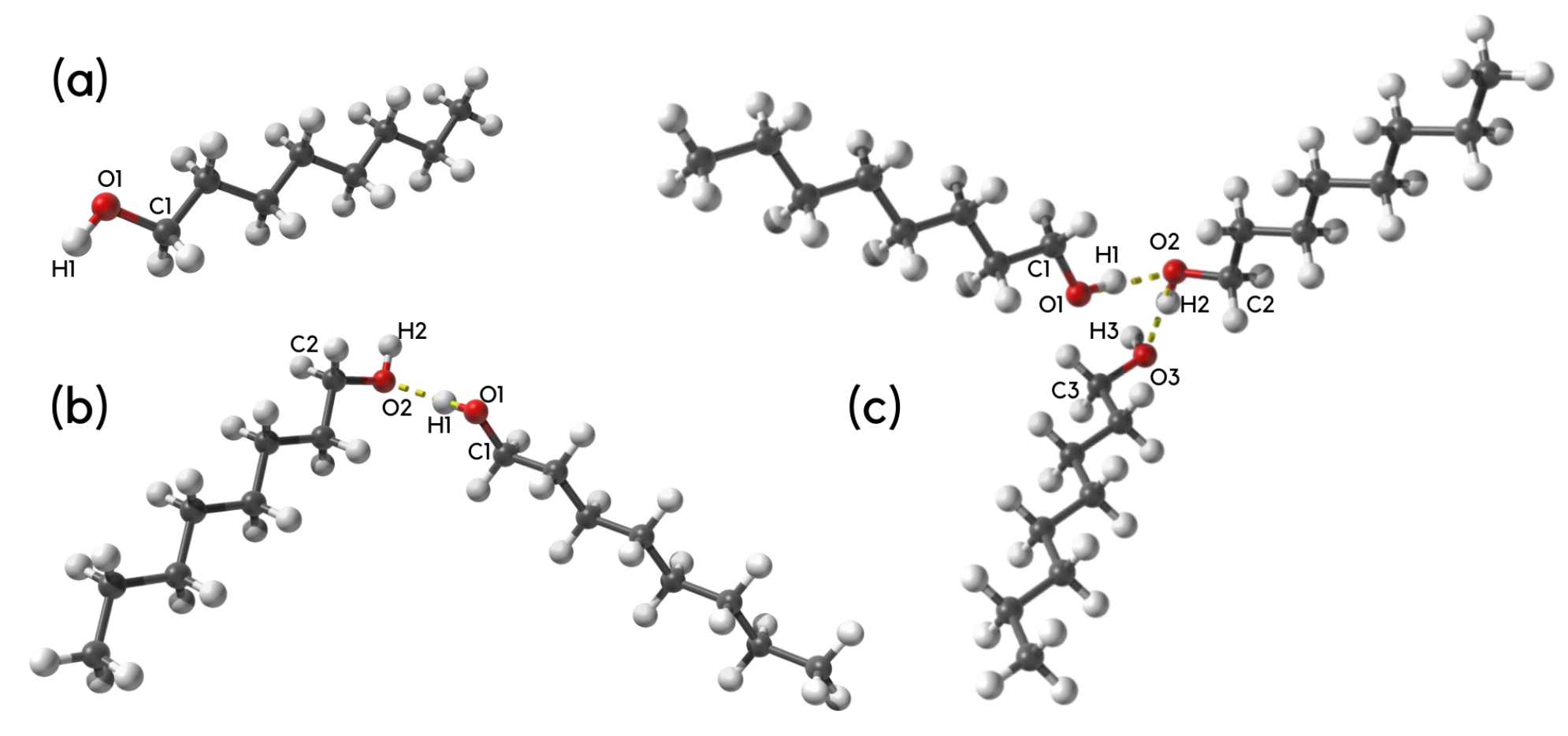
3.2. Molecular and Electronic Structure of n-Octanol Associates: A DFT and Atoms-in-MOLECULES Study
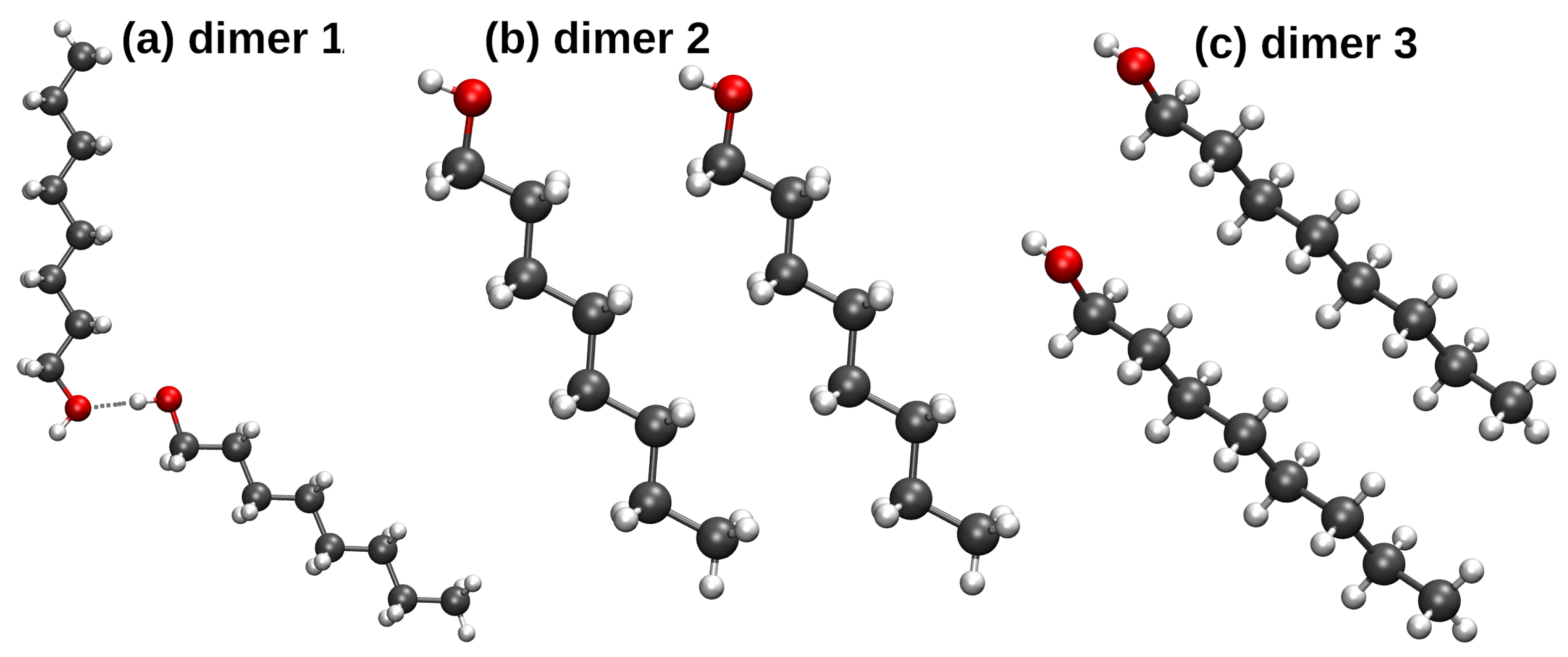
3.3. Origins of Interaction in n-Octanol Dimers—A Symmetry-Adapted Perturbation Theory Investigation
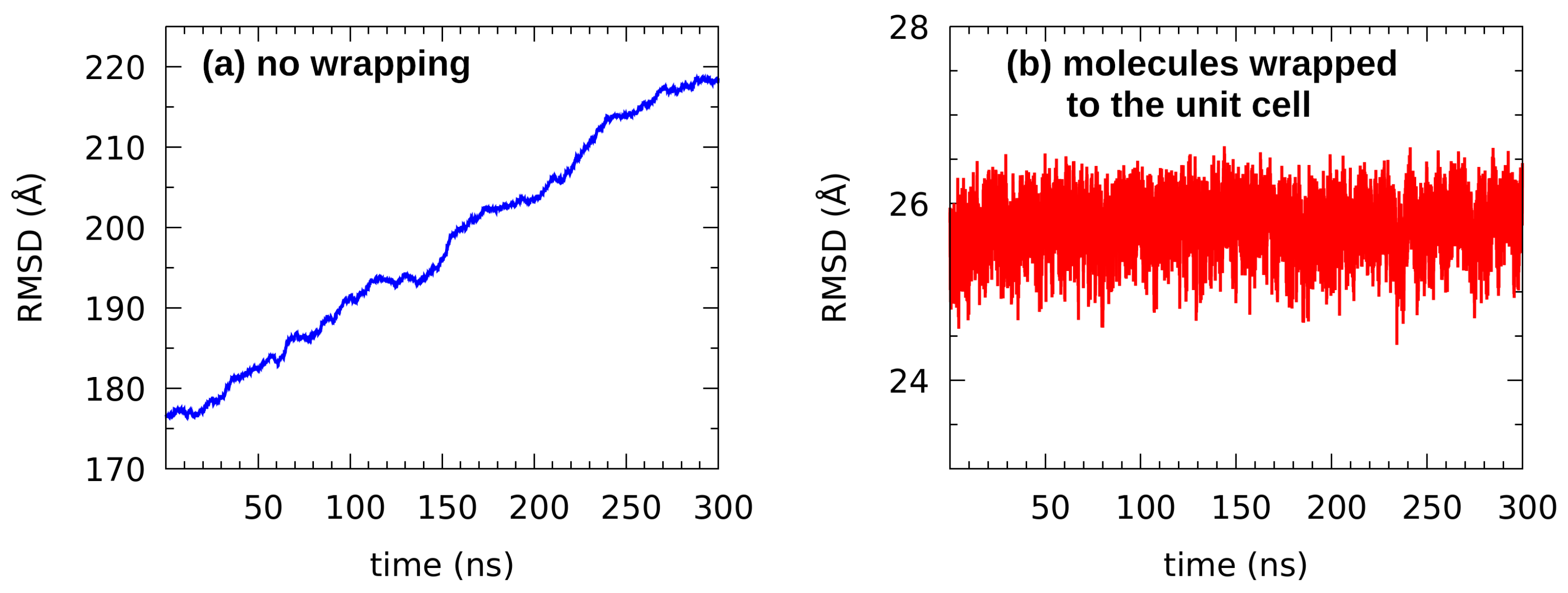
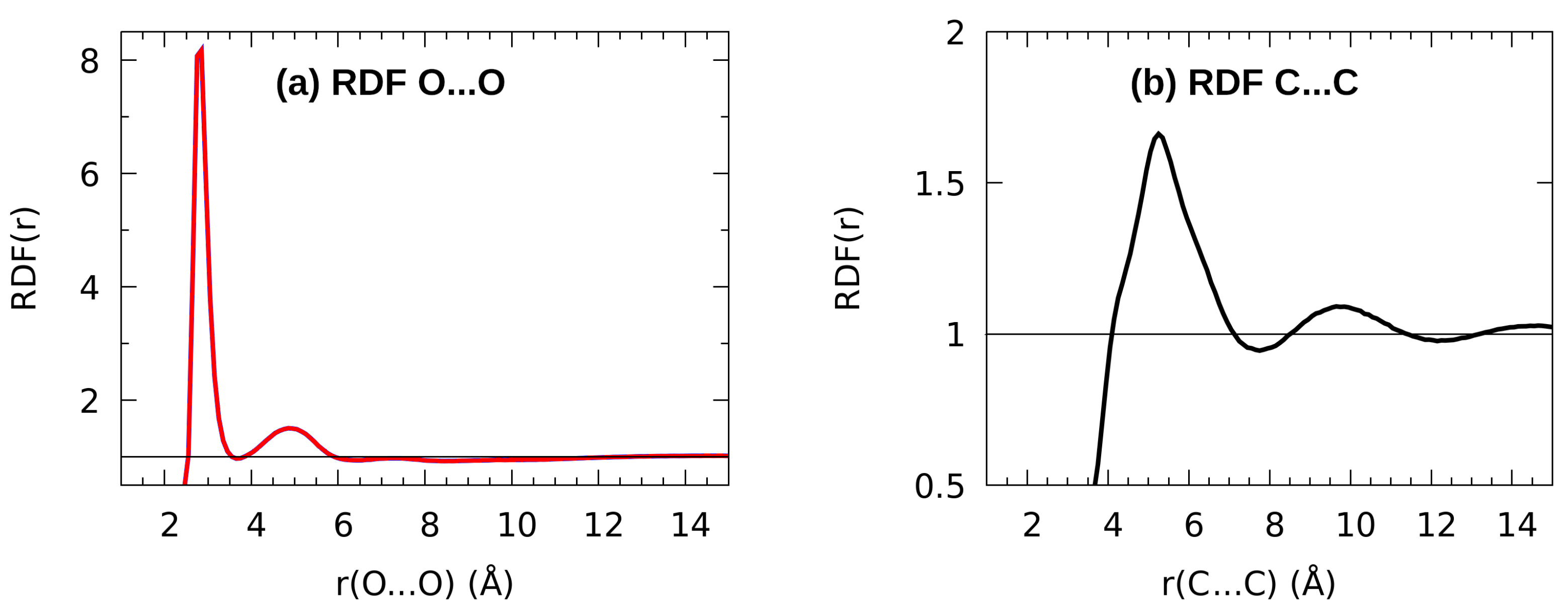
3.4. Classical Molecular Dynamics Study of Liquid n-Octanol
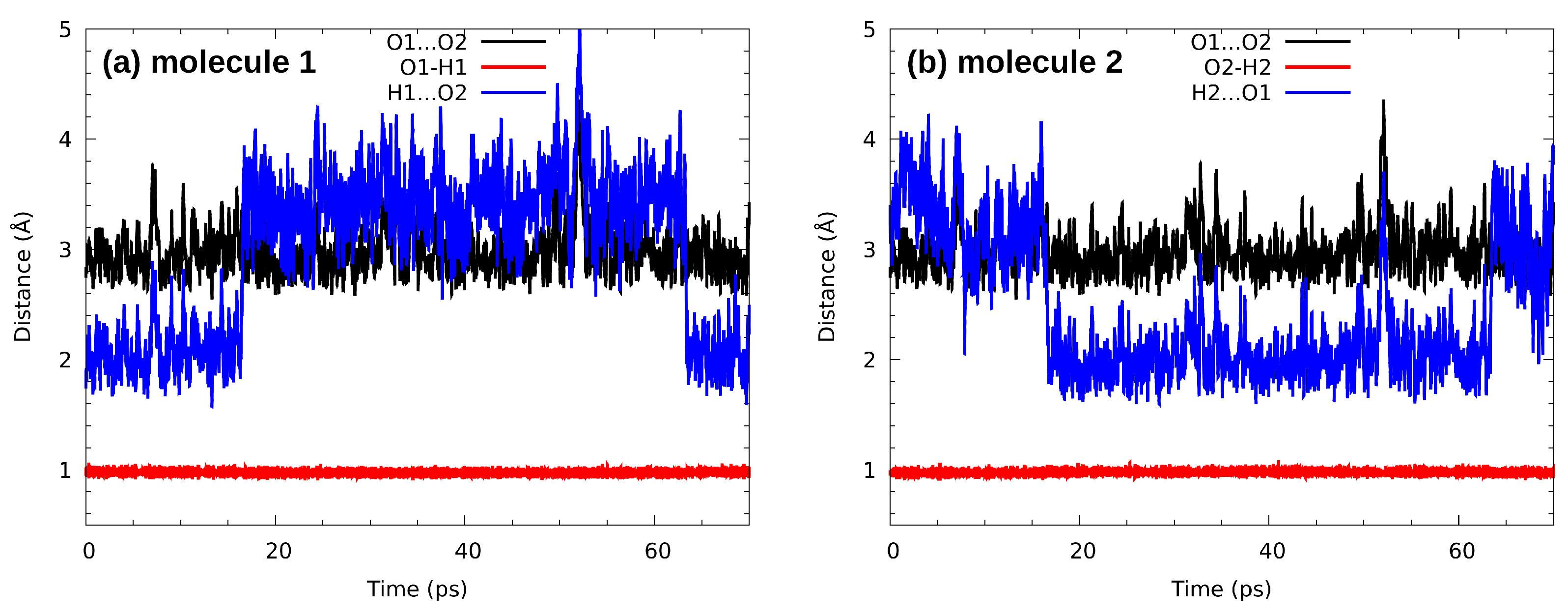
3.5. Car-Parrinello Molecular Dynamics Analysis of Intermolecular Hydrogen Bonding Network in n-Octanol
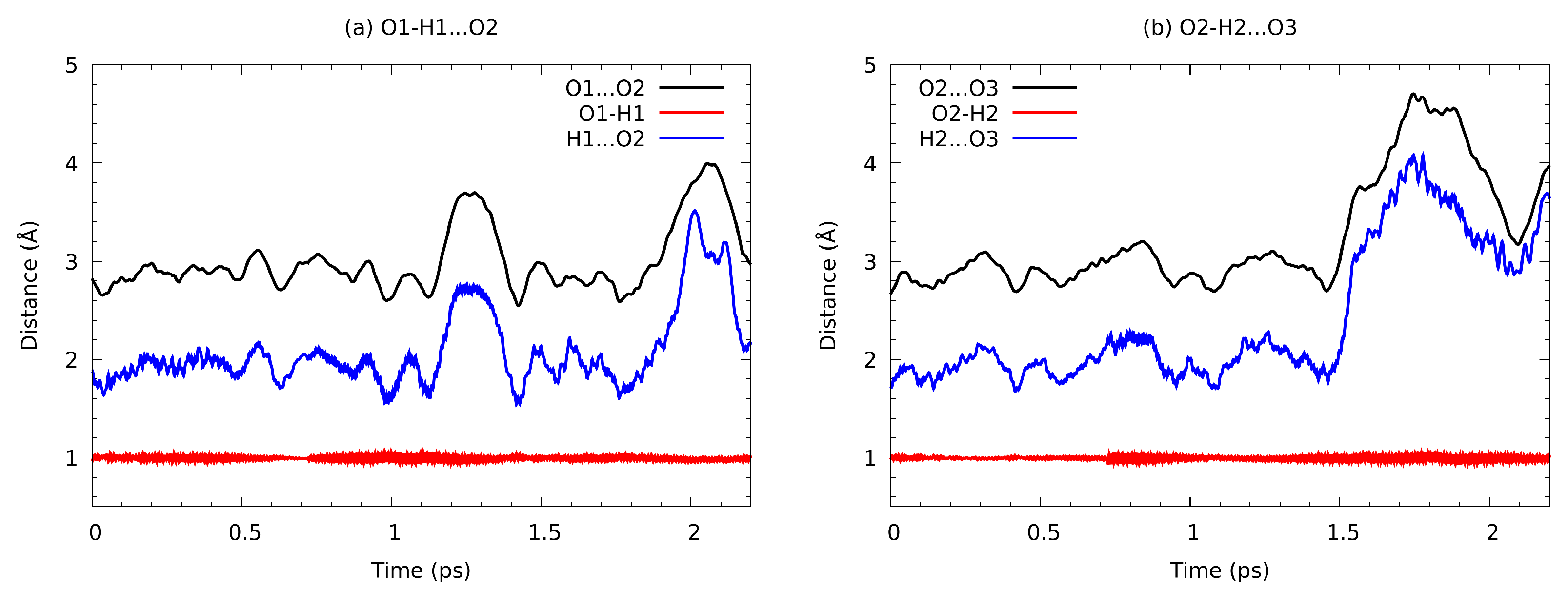
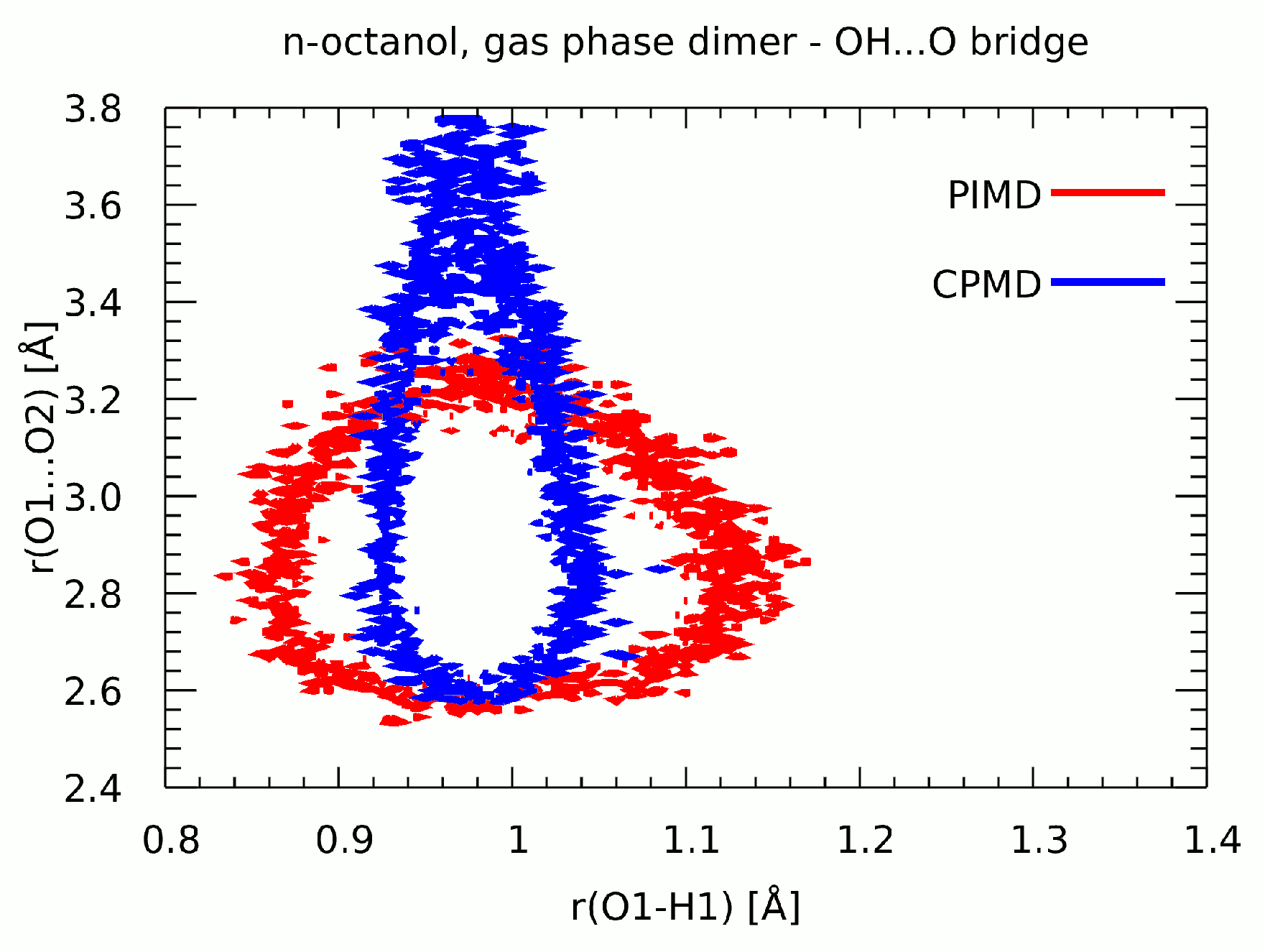
3.6. Path Integral Molecular Dynamics Results of the Intermolecular Hydrogen Bonding in the n-Octanol Dimer
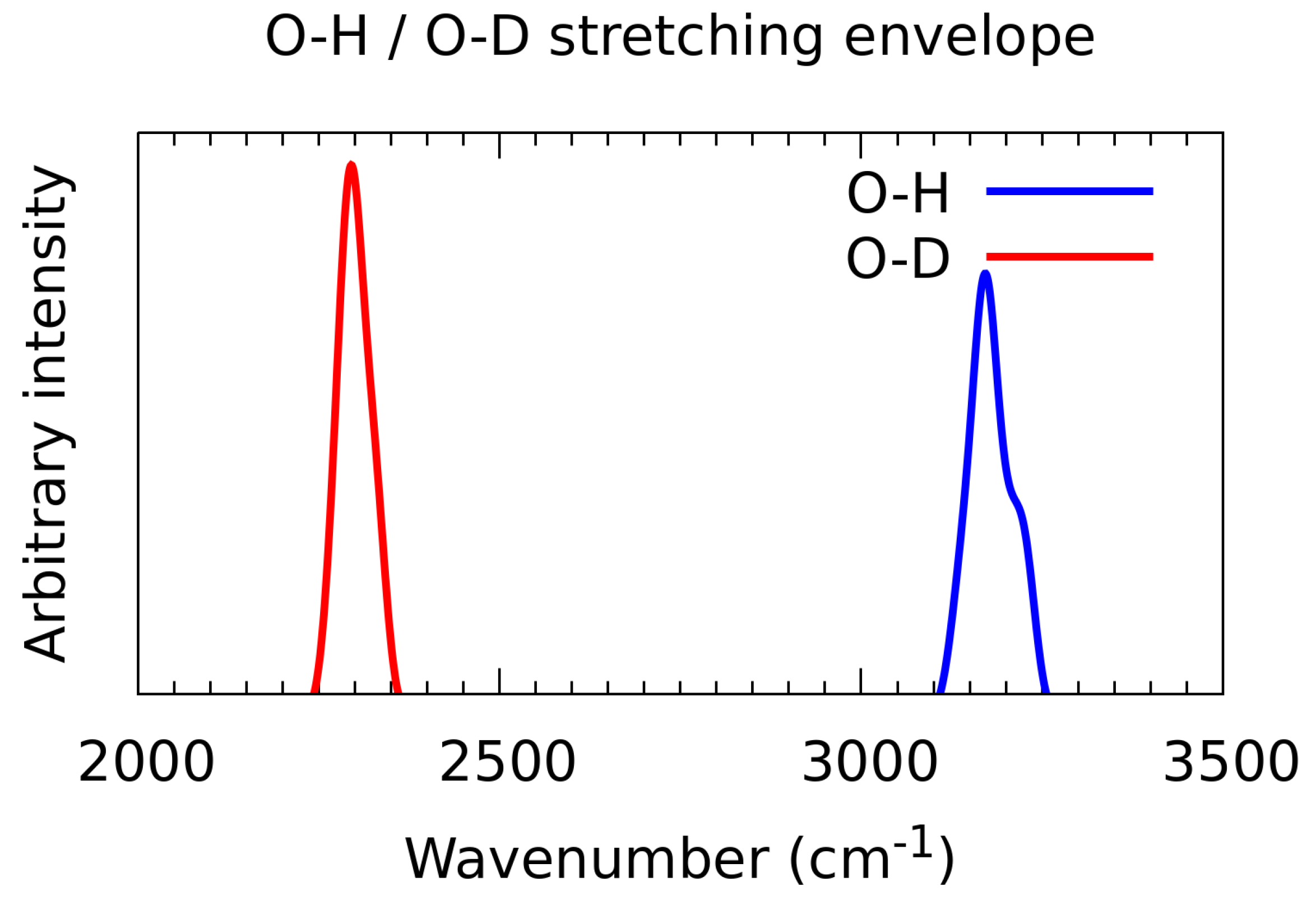
3.7. A Posteriori Inclusion of Quantum Corrections to the Nuclear Motion—Isotope Effect
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| FTIR | Fourier Transform Infrared Spectroscopy |
| DFT | Density Functional Theory |
| HB | hydrogen bond |
| PCM | Polarizable Continuum Model |
| AIM | Atoms In Molecules |
| MD | Molecular Dynamics |
| CPMD | Car-Parrinello Molecular Dynamics |
| PIMD | Path Integral Molecular Dynamics |
| NDE | Nonlinear Dielectric Effect |
| FPMD | First Principle Molecular Dynamics |
| CCDC | Cambridge Crystallographic Data Centre |
| PES | Potential Energy Surface |
| BCP | Bond Critical Point |
| RCP | Ring Critical Point |
| CCP | Cage Critical Point |
| SAPT | Symmetry-Adapted Perturbation Theory |
| BSSE | Basis Set Superposition Error |
| MP2 | Møller-Plesset second-order perturbation theory |
| PME | particle mesh Ewald |
| PBC | Periodic Boundary Conditions |
| RMSD | Root Mean Square Deviation |
| RDF | Radial Distribution Function |
References
- Kar, S.; Sanderson, H.; Roy, K.; Benfenati, E.; Leszczynski, J. Green Chemistry in the Synthesis of Pharmaceuticals. Chem. Rev. 2021, 4, 100174. [Google Scholar] [CrossRef] [PubMed]
- Crabtree, R.H. Homogeneous Transition Metal Catalysis of Acceptorless Dehydrogenative Alcohol Oxidation: Applications in Hydrogen Storage and to Heterocycle Synthesis. Chem. Rev. 2017, 117, 9228–9246. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Gong, Q.; Olson, D.H.; Li, J. Commensurate Adsorption of Hydrocarbons and Alcohols in Microporous Metal Organic Frameworks. Chem. Rev. 2012, 112, 836–868. [Google Scholar] [CrossRef]
- Bianchini, C.; Shen, P.K. Palladium-Based Electrocatalysts for Alcohol Oxidation in Half Cells and in Direct Alcohol Fuel Cells. Chem. Rev. 2009, 109, 4183–4206. [Google Scholar] [CrossRef]
- Ager, D.J.; Prakash, I.; Schaad, D.R. 1,2-Amino Alcohols and Their Heterocyclic Derivatives as Chiral Auxiliaries in Asymmetric Synthesis. Chem. Rev. 1996, 96, 835–876. [Google Scholar] [CrossRef] [PubMed]
- Falbe, J.; Bahrmann, H.; Lipps, W.; Mayer, D. Alcohols, Aliphatic. In Ullmann’s Encyclopedia of Industrial Chemistry; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2000. [Google Scholar] [CrossRef]
- Mattia, A. Saturated Acyclic Linear Primary Alcohols, Aldehydes and Acids; World Health Organization: Geneva, Switzerland, 1998. [Google Scholar]
- Akae, Y.; Sakurai, M.; Takata, T. Alcohol Moiety-Tethering Acylazides as Versatile Isocyanate Precursors for Polymerization Initiators and Monomers. Macromolecules 2021, 54, 8488–8494. [Google Scholar] [CrossRef]
- Jindal, A.; Vasudevan, S. Hydrogen Bonding in the Liquid State of Linear Alcohols: Molecular Dynamics and Thermodynamics. J. Phys. Chem. B 2020, 124, 3548–3555. [Google Scholar] [CrossRef]
- Chandra, A.K.; Sannigrahi, A.B. Hydrogen-Bonding Properties of Alcohols. J. Phys. Chem. 1965, 69, 2494–2499. [Google Scholar] [CrossRef]
- Dannhauser, W.; Bahe, L.W.; Lin, R.Y.; Flueckinger, A.F. Dielectric Constant of Hydrogen-Bonded Liquids. IV. Equilibrium and Relaxation Studies of Homologous Neo-Alcohols. J. Chem. Phys. 1965, 43, 257–266. [Google Scholar] [CrossRef]
- Fujii, A.; Sugawara, N.; Hsu, P.J.; Shimamori, T.; Li, Y.C.; Hamashima, T.; Kuo, J.L. Hydrogen bond network structures of protonated short-chain alcohol clusters. Phys. Chem. Chem. Phys. 2018, 20, 14971–14991. [Google Scholar] [CrossRef] [PubMed]
- Mazur, K.; Bonn, M.; Hunger, J. Hydrogen Bond Dynamics in Primary Alcohols: A Femtosecond Infrared Study. J. Phys. Chem. B 2015, 119, 1558–1566. [Google Scholar] [CrossRef] [PubMed]
- Sinton, C.M.; Krosser, B.I.; Walton, K.D.; Llinás, R.R. The effectiveness of different isomers of octanol as blockers of harmaline-induced tremor. Pflugers Arch. 1989, 414, 31–36. [Google Scholar] [CrossRef] [PubMed]
- Bushara, K.O.; Goldstein, S.R.; Grimes, G.J.; Burstein, A.H.; Hallett, M. Pilot trial of 1-octanol in essential tremor. Neurology 2004, 62, 122–124. [Google Scholar] [CrossRef] [PubMed]
- Vasylieva, A.; Doroshenko, I.; Pogorelov, V.; Sablinskas, V.; Balevicius, V. FTIR Spectra of n-pentanol and n-octanol in Liquid and Solid States. Ukr. J. Phys. 2015, 60, 723–727. [Google Scholar] [CrossRef]
- Chernolevska, Y.; Vaskivskyi, Y.; Pogorelov, V.; Doroshenko, I.; Doroshenko, O.; Sablinskas, V.; Balevicius, V. FTIR Spectra of n-pentanol and n-octanol in Liquid and Solid States. arXiv 2016, arXiv:1603.00630. [Google Scholar]
- Pocheć, M.; Małycha, K.; Orzechowski, K. Nonlinear Dielectric Effect in the Vicinity of Solid–Liquid Phase Transition of 1-Decanol. J. Phys. Chem. C 2019, 123, 16929–16933. [Google Scholar] [CrossRef]
- Pocheć, M.; Niu, H.; Ren, L.; Bai, S.; Orzechowski, K. Premelting Phenomena in n-Alcohols from Nonanol to Dodecanol. J. Phys. Chem. C 2020, 124, 21013–21017. [Google Scholar] [CrossRef]
- Pocheć, M.; Orzechowski, K.; Rutkowski, K. Indicators of premelting in 1-decanol and 1-nonanol studied by FTIR spectroscopy. Surf. Interfaces 2022, 28, 101676. [Google Scholar] [CrossRef]
- Desiraju, G.R. Hydrogen Bridges in Crystal Engineering: Interactions without Borders. Acc. Chem. Res. 2002, 35, 565–573. [Google Scholar] [CrossRef]
- Latimer, W.M.; Rodebush, W.H. Polarity and ionization from the standpoint of the Lewis theory of valence. J. Am. Chem. Soc. 1920, 42, 1419–1433. [Google Scholar] [CrossRef] [Green Version]
- Pauling, L. The Nature of the Chemical Bond; Cornell University Press: Ithaca, NY, USA, 1939. [Google Scholar]
- Pimentel, G.C.; McClellan, A.L. The Hydrogen Bond; W. H. Freeman and Company: San Fransisco, CA, USA, 1960. [Google Scholar]
- Huyskens, P.; Luck, W.; Zeegers-Huyskens, T. Intermolecular Forces: An Introduction to Modern Methods and Results; Springer: Berlin/Heidelberg, Germany, 1991. [Google Scholar]
- Jeffrey, G.A. An Introduction to Hydrogen Bonding; Oxford University Press: New York, NY, USA, 1997. [Google Scholar]
- Hobza, P.; Havlas, Z. Blue-Shifting Hydrogen Bonds. Chem. Rev. 2000, 100, 4253–4264. [Google Scholar] [CrossRef] [PubMed]
- Belkova, N.V.; Shubina, E.S.; Epstein, L.M. Diverse World of Unconventional Hydrogen Bonds. Acc. Chem. Res. 2005, 38, 624–631. [Google Scholar] [CrossRef] [PubMed]
- de Oliveira, B.G.; Ramos, M.N. Dihydrogen bonds and blue-shifting hydrogen bonds: A theoretical study of AHHCF3 and TH2 HCF3 model systems with A = Li or Na and T = Be or Mg. Int. J. Quantum Chem. 2009, 110, 307–316. [Google Scholar] [CrossRef]
- Arunan, E.; Desiraju, G.R.; Klein, R.A.; Sadlej, J.; Scheiner, S.; Alkorta, I.; Clary, D.C.; Crabtree, R.H.; Dannenberg, J.J.; Hobza, P.; et al. Definition of the hydrogen bond (IUPAC Recommendations 2011). Pure Appl. Chem. 2011, 83, 1637–1641. [Google Scholar] [CrossRef]
- Wulf, O.R.; Liddel, U.; Hendricks, S.B. The Effect of Ortho Substitution on the Absorption of the OH Group of Phenol in the Infrared1. J. Am. Chem. Soc. 1936, 58, 2287–2293. [Google Scholar] [CrossRef]
- Kollman, P.A.; Allen, L.C. Theory of the hydrogen bond. Chem. Rev. 1972, 72, 283–303. [Google Scholar] [CrossRef]
- Grabowski, S.J. What Is the Covalency of Hydrogen Bonding? Chem. Rev. 2011, 111, 2597–2625. [Google Scholar] [CrossRef]
- Grabowski, S.J. Hydrogen and halogen bonds are ruled by the same mechanisms. Phys. Chem. Chem. Phys. 2013, 15, 7249. [Google Scholar] [CrossRef]
- Cavallo, G.; Metrangolo, P.; Milani, R.; Pilati, T.; Priimagi, A.; Resnati, G.; Terraneo, G. The Halogen Bond. Chem. Rev. 2016, 116, 2478–2601. [Google Scholar] [CrossRef] [Green Version]
- Grabowski, S. New Type of Halogen Bond: Multivalent Halogen Interacting with π- and σ-Electrons. Molecules 2017, 22, 2150. [Google Scholar] [CrossRef] [Green Version]
- Kryachko, E.S.; Karpfen, A.; Remacle, F. Nonconventional Hydrogen Bonding between Clusters of Gold and Hydrogen Fluoride. J. Phys. Chem. A 2005, 109, 7309–7318. [Google Scholar] [CrossRef] [PubMed]
- Espinosa, E.; Molins, E.; Lecomte, C. Hydrogen bond strengths revealed by topological analyses of experimentally observed electron densities. Chem. Phys. Lett. 1998, 285, 170–173. [Google Scholar] [CrossRef]
- Sobczyk, L.; Grabowski, S.J.; Krygowski, T.M. Interrelation between H-Bond and Pi-Electron Delocalization. Chem. Rev. 2005, 105, 3513–3560. [Google Scholar] [CrossRef] [PubMed]
- Grabowski, S. Chapter 1. Hydrogen Bond—Definitions, Criteria of Existence and Various Types. In Understanding Hydrogen Bonds: Theoretical and Experimental Views; Royal Society of Chemistry: London, UK, 2020; pp. 1–40. [Google Scholar] [CrossRef]
- Szalewicz, K. Hydrogen Bond, Encyclopedia of Physical Science and Technology, 3rd ed.; Academic Press: Cambridge, MA, USA, 2001; pp. 505–538. [Google Scholar]
- Jaffé, H.H. Inter- and Intramolecular Hydrogen Bonds1. J. Am. Chem. Soc. 1957, 79, 2373–2375. [Google Scholar] [CrossRef]
- Clements, R.; Dean, R.L.; Singh, T.R.; Wood, J.L. Proton double minimum potentials in symmetric hydrogen bonds. J. Chem. Soc. D 1971, 18, 1125–1126. [Google Scholar] [CrossRef]
- Clements, R.; Dean, R.L.; Wood, J.L. Proton double minimum potentials in asymmetric hydrogen bonds. J. Chem. Soc. D 1971, 18, 1127–1129. [Google Scholar] [CrossRef]
- Fonseca Guerra, C.; Bickelhaupt, F.M.; Snijders, J.G.; Baerends, E.J. The Nature of the Hydrogen Bond in DNA Base Pairs: The Role of Charge Transfer and Resonance Assistance. Chem. Eur. J. 1999, 5, 3581–3594. [Google Scholar] [CrossRef]
- Murthy, A.S.N.; Rao, C.N.R. Spectroscopic Studies of the Hydrogen Bond. Appl. Spectrosc. Rev. 1968, 2, 69–191. [Google Scholar] [CrossRef]
- Fujii, A.; Patwari, G.; Ebata, T.; Mikami, N. Vibrational spectroscopic evidence of unconventional hydrogen bonds. Int. J. Mass Spectrom. 2002, 220, 289–312. [Google Scholar] [CrossRef]
- Cubero, E.; Orozco, M.; Hobza, P.; Luque, F.J. Hydrogen Bond versus Anti-Hydrogen Bond: A Comparative Analysis Based on the Electron Density Topology. J. Phys. Chem. A 1999, 103, 6394–6401. [Google Scholar] [CrossRef]
- Mikenda, W.; Steinböck, S. Stretching frequency vs. bond distance correlation of hydrogen bonds in solid hydrates: A generalized correlation function. J. Mol. Struct. 1996, 384, 159–163. [Google Scholar] [CrossRef]
- Novak, A. Hydrogen bonding in solids correlation of spectroscopic and crystallographic data. In Large Molecules; Springer: Berlin/Heidelberg, Germany, 1974; pp. 177–216. [Google Scholar] [CrossRef]
- Joseph, J.; Jemmis, E.D. Red-, Blue-, or No-Shift in Hydrogen Bonds: A Unified Explanation. J. Am. Chem. Soc. 2007, 129, 4620–4632. [Google Scholar] [CrossRef] [PubMed]
- Pogorelov, V.; Chernolevska, Y.; Vaskivskyi, Y.; Pettersson, L.G.; Doroshenko, I.; Sablinskas, V.; Balevicius, V.; Ceponkus, J.; Kovaleva, K.; Malevich, A.; et al. Structural transformations in bulk and matrix-isolated methanol from measured and computed infrared spectroscopy. J. Mol. Liq. 2016, 216, 53–58. [Google Scholar] [CrossRef]
- Buck, U.; Huisken, F. Infrared Spectroscopy of Size-Selected Water and Methanol Clusters. (Chem. Rev. 100, 3863–3890. Published on the Web Sept 23, 2000.). Chem. Rev. 2000, 101, 205–206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barlow, S.J.; Bondarenko, G.V.; Gorbaty, Y.E.; Yamaguchi, T.; Poliakoff, M. An IR Study of Hydrogen Bonding in Liquid and Supercritical Alcohols. J. Phys. Chem. A 2002, 106, 10452–10460. [Google Scholar] [CrossRef]
- Wu, X.; Chen, Y.; Yamaguchi, T. Hydrogen bonding in methanol studied by infrared spectroscopy. J. Mol. Spectrosc. 2007, 246, 187–191. [Google Scholar] [CrossRef]
- Larsen, R.W.; Suhm, M.A. The benefits of alternation and alkylation: Large amplitude hydrogen bond librational modes of alcohol trimers and tetramers. Phys. Chem. Chem. Phys. 2010, 12, 8152. [Google Scholar] [CrossRef]
- Hohenberg, P.; Kohn, W. Inhomogeneous Electron Gas. Phys. Rev. 1964, 136, B864–B871. [Google Scholar] [CrossRef] [Green Version]
- Kohn, W.; Sham, L.J. Self-Consistent Equations Including Exchange and Correlation Effects. Phys. Rev. 1965, 140, A1133–A1138. [Google Scholar] [CrossRef] [Green Version]
- Bader, R. Atoms in Molecules: A Quantum Theory; International Series of Monographs on Chemistry; Clarendon Press: Oxford, UK, 1994. [Google Scholar]
- Jeziorski, B.; Moszynski, R.; Szalewicz, K. Perturbation Theory Approach to Intermolecular Potential Energy Surfaces of van der Waals Complexes. Chem. Rev. 1994, 94, 1887–1930. [Google Scholar] [CrossRef]
- Car, R.; Parrinello, M. Unified Approach for Molecular Dynamics and Density-Functional Theory. Phys. Rev. Lett. 1985, 55, 2471–2474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marx, D.; Parrinello, M. The Effect of Quantum and Thermal Fluctuations on the Structure of the Floppy Molecule C2H3+. Science 1996, 271, 179–181. [Google Scholar] [CrossRef]
- Tuckerman, M.E.; Marx, D.; Klein, M.L.; Parrinello, M. Efficient and general algorithms for path integral Car-Parrinello molecular dynamics. J. Chem. Phys. 1996, 104, 5579–5588. [Google Scholar] [CrossRef]
- Denisov, G.S.; Mavri, J.; Sobczyk, L. Hydrogen Bonding-New Insights (Challenges and Advances in Computational Chemistry and Physics, 3); Grabowski, S.J., Ed.; Springer: Dordrecht, The Netherlands, 2006; pp. 377–416. [Google Scholar]
- CCDC Structural Database. Available online: https://www.ccdc.cam.ac.uk/ (accessed on 7 May 2021).
- Shallard-Brown, H.; Watkin, D.; Cowley, A. CCDC 263655: Experimental Crystal Structure Determination. 2005. Available online: https://doi.org/10.5517/CC8VC0G (accessed on 15 December 2021).
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dunning, T.H. Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen. J. Chem. Phys. 1989, 90, 1007–1023. [Google Scholar] [CrossRef]
- Kendall, R.A.; Dunning, T.H.; Harrison, R.J. Electron affinities of the first-row atoms revisited. Systematic basis sets and wave functions. J. Chem. Phys. 1992, 96, 6796–6806. [Google Scholar] [CrossRef] [Green Version]
- Cossi, M.; Barone, V.; Cammi, R.; Tomasi, J. Ab initio study of solvated molecules: A new implementation of the polarizable continuum model. Chem. Phys. Lett. 1996, 255, 327–335. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian˜16 Revision A.03; Gaussian Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Keith, T.A.; Gristmill, T. AIMAll; (Version 19.10.12); Software; Overland Park, KS, USA, 2019. [Google Scholar]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD—Visual Molecular Dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- OneAngstrom. SAMSON Platform for Molecular Design. Available online: https://www.samson-connect.net/ (accessed on 15 December 2021).
- Hohenstein, E.G.; Sherrill, C.D. Density fitting of intramonomer correlation effects in symmetry-adapted perturbation theory. J. Chem. Phys. 2010, 133, 014101. [Google Scholar] [CrossRef] [Green Version]
- Boys, S.; Bernardi, F. The calculation of small molecular interactions by the differences of separate total energies. Some procedures with reduced errors. Mol. Phys. 1970, 19, 553–566. [Google Scholar] [CrossRef]
- Parrish, R.M.; Burns, L.A.; Smith, D.G.A.; Simmonett, A.C.; DePrince, A.E.; Hohenstein, E.G.; Bozkaya, U.; Sokolov, A.Y.; Remigio, R.D.; Richard, R.M.; et al. Psi4 1.1: An Open-Source Electronic Structure Program Emphasizing Automation, Advanced Libraries, and Interoperability. J. Chem. Theory Comput. 2017, 13, 3185–3197. [Google Scholar] [CrossRef] [PubMed]
- Mercury—Crystal Structure Visualisation. Available online: http://www.ccdc.cam.ac.uk/Solutions/CSDSystem/Pages/Mercury.aspx (accessed on 15 December 2021).
- Wang, J.; Wang, W.; Kollman, P.A.; Case, D.A. Automatic atom type and bond type perception in molecular mechanical calculations. J. Mol. Graph. Model. 2006, 25, 247–260. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef] [PubMed]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N·log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef] [Green Version]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef] [Green Version]
- Heaps, C. LEAP: The Low Emissions Analysis Platform, Software version: 2020.1.52; Stockholm Environment Institute: Somerville, MA, USA, 2021; Available online: https://leap.sei.org (accessed on 15 December 2021).
- Salomon-Ferrer, R.; Case, D.A.; Walker, R.C. An overview of the Amber biomolecular simulation package. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2012, 3, 198–210. [Google Scholar] [CrossRef]
- Case, D.A.; Aktulga, H.M.; Belfon, K.; Ben-Shalom, I.; Brozell, S.R.; Cerutti, D.S.; Cheatham, T.E., III; Cruzeiro, V.W.D.; Darden, T.A.; Duke, R.E.; et al. Amber 2021; University of California: San Francisco, CA, USA, 2021. [Google Scholar]
- Adelman, S.A. Generalized Langevin equation approach for atom/solid-surface scattering: General formulation for classical scattering off harmonic solids. J. Chem. Phys. 1976, 64, 2375. [Google Scholar] [CrossRef]
- Feller, S.E.; Zhang, Y.; Pastor, R.W.; Brooks, B.R. Constant pressure molecular dynamics simulation: The Langevin piston method. J. Chem. Phys. 1995, 103, 4613–4621. [Google Scholar] [CrossRef]
- Ryckaert, J.P.; Ciccotti, G.; Berendsen, H.J. Numerical integration of the cartesian equations of motion of a system with constraints: Molecular dynamics of n-alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef] [Green Version]
- Troullier, N.; Martins, J.L. Efficient pseudopotentials for plane-wave calculations. Phys. Rev. B 1991, 43, 1993–2006. [Google Scholar] [CrossRef]
- Nosé, S. A unified formulation of the constant temperature molecular dynamics methods. J. Chem. Phys. 1984, 81, 511–519. [Google Scholar] [CrossRef] [Green Version]
- Hoover, W.G. Canonical dynamics: Equilibrium phase-space distributions. Phys. Rev. A 1985, 31, 1695–1697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grimme, S. Semiempirical GGA-type density functional constructed with a long-range dispersion correction. J. Comput. Chem. 2006, 27, 1787–1799. [Google Scholar] [CrossRef] [PubMed]
- CPMD ver. 4.3-4610. Copyright IBM Corp. (1990–2004). Copyright MPI für Festkoerperforschung Stuttgart (1997–2001). Available online: http://www.cpmd.org/ (accessed on 15 December 2021).
- Stare, J.; Mavri, J. Numerical solving of the vibrational time-independent Schrödinger equation in one and two dimensions using the variational method. Comput. Phys. Commun. 2002, 143, 222–240. [Google Scholar] [CrossRef]
- Kochen, A.; Limbach, H.H. Isotope Effects in Chemistry and Biology; CRC Press: New York, NY, USA, 2005. [Google Scholar]
- Pavon, J.A.; Fitzpatrick, P.F. Intrinsic Isotope Effects on Benzylic Hydroxylation by the Aromatic Amino Acid Hydroxylases: Evidence for Hydrogen Tunneling, Coupled Motion, and Similar Reactivities. J. Am. Chem. Soc. 2005, 127, 16414–16415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, T.; Kelley, C. Gnuplot 4.4: An Interactive Plotting Program. 2010. Available online: http://gnuplot.sourceforge.net/ (accessed on 15 December 2021).
- Petrus, J.; Cieślik-Boczula, K.; Czarnik-Matusewicz, B.; Jaszczyszyn, A.; Gasiorowski, K.; Świątek, P.; Malinka, W. Moving-window 2D correlation spectroscopy in studies of fluphenazine–DPPC dehydrated film as a function of temperature. J. Mol. Struct. 2010, 974, 192. [Google Scholar] [CrossRef]
- Zerbi, G.; Magni, R.; Gussoni, M.; Moritz, K.H.; Bigotto, A.; Dirlikov, S. Molecular mechanics for phase transition and melting of n-alkanes: A spectroscopic study of molecular mobility of solid n-nonadecane. J. Chem. Phys. 1981, 75, 3175–3194. [Google Scholar] [CrossRef]
- Zerbi, G.; Conti, G.; Minoni, G.; Pison, S.; Bigotto, A. Premelting phenomena in fatty acids: An infrared and Raman study. J. Phys. Chem. 1987, 91, 2386–2393. [Google Scholar] [CrossRef]
- Corsetti, S.; Rabl, T.; McGloin, D.; Kiefer, J. Intermediate phases during solid to liquid transitions in long-chain n-alkanes. Phys. Chem. Chem. Phys. 2017, 19, 13941–13950. [Google Scholar] [CrossRef] [Green Version]
- Doroshenko, I.; Pogorelov, V.; Sablinskas, V. Infrared Absorption Spectra of Monohydric Alcohols. Dataset Pap. Chem. 2013, 2013, 1329406. [Google Scholar] [CrossRef] [Green Version]
- Vaskivskyi, Y.; Chernolevska, Y.; Doroshenko, I.; Doroshenko, O.; Pogorelov, V. Cluster structure changes during melting of 1-decanol: FTIR study and DFT calculations. J. Mol. Liq. 2017, 247, 188–192. [Google Scholar] [CrossRef]
- Vasileva, A.; Golub, P.; Doroshenko, I.; Pogorelov, V.; Sablinskas, V.; Balevicius, V.; Ceponkus, J. FTIR Spectra of n-Octanol in Liquid and Solid States. Dataset Pap. Sci. 2014, 2014, 921308. [Google Scholar] [CrossRef]
- Golub, P.; Pogorelov, V.; Doroshenko, I. The structural peculiarities of liquid n-heptanol and n-octanol. J. Mol. Liq. 2012, 169, 80–86. [Google Scholar] [CrossRef]
- Quinan, J.R.; Wiberley, S.E. Infrared and Raman Spectra of Series of Deuterated Alcohols. Anal. Chem. 1954, 26, 1762–1764. [Google Scholar] [CrossRef]
- Wallberg, J.; Kjaergaard, H.G. Absolute fundamental and overtone OH and OD stretching intensities of alcohols. Spectrochim. Acta A Mol. Biomol. Spectr. 2019, 208, 315–324. [Google Scholar] [CrossRef]
- Koch, U.; Popelier, P.L.A. Characterization of C-H-O Hydrogen Bonds on the Basis of the Charge Density. J. Phys. Chem. 1995, 99, 9747–9754. [Google Scholar] [CrossRef]
- Haynes, W.M. (Ed.) CRC Handbook of Chemistry and Physics, 95th ed.; Taylor & Francis Ltd.: Boca Raton, FL, USA, 2014–2015; pp. 3–426. [Google Scholar]
- MacCallum, J.L.; Tieleman, D.P. Structures of Neat and Hydrated 1-Octanol from Computer Simulations. J. Am. Chem. Soc. 2002, 124, 15085–15093. [Google Scholar] [CrossRef]
- Bakker, H.J.; Rezus, Y.L.A.; Timmer, R.L.A. Molecular Reorientation of Liquid Water Studied with Femtosecond Midinfrared Spectroscopy. J. Phys. Chem. A 2008, 112, 11523–11534. [Google Scholar] [CrossRef]
- Legon, A.C.; Millen, D.J. Angular geometries and other properties of hydrogen-bonded dimers: A simple electrostatic interpretation of the success of the electron-pair model. Chem. Soc. Rev. 1987, 16, 467. [Google Scholar] [CrossRef]
- Howard, A.; Shallard-Brown, D.J.; Watkin, A.R. Cowley, n-Octanol. Acta Crystallogr. E 2005, 61, o213–o214. [Google Scholar]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Literature Assignment | Experimental | Theoretical (in n-Octanol Dimer) |
|---|---|---|
| [16,17,98,99,100,101] | ||
| stretching OH | 3319 | 3698, 3387 |
| stretching CH | 2955, 2926 | 3025, 3018, 2998, 2990, 2983 |
| 2871, 2855 | 2954, 2951, 2946, 2936, 2886 | |
| bending OH | 1420, 1120 | 1424, 1180 |
| stretching OD | 2490 | 2700, 2537 |
| scissoring CH | 1467 | 1462, 1451 |
| twisting CH | 1378 | 1252 |
| stretching C-O | 1058, 989, 954 | 1038, 1020, 1005 |
| and C-C | 975, 961 | |
| rocking CH | 722 | 706, 702, 701 |
| Atom | Atomic Charge [e] | |||||
|---|---|---|---|---|---|---|
| Monomer | Dimer | Trimer | ||||
| Gas Phase | PCM | Gas Phase | PCM | GasPhase | PCM | |
| qH1 | 0.5437 | 0.5423 | 0.5834 | 0.5845 | 0.5922 | 0.5944 |
| qO1 | −1.0562 | −1.0503 | −1.1092 | −1.1052 | −1.1217 | −1.1048 |
| qC1 | 0.4688 | 0.4625 | 0.4891 | 0.4839 | 0.4931 | 0.4909 |
| qH2 | 0.5560 | 0.5568 | 0.5982 | 0.5958 | ||
| qO2 | −1.0588 | −1.0553 | −1.1145 | −1.1077 | ||
| qC2 | 0.4457 | 0.4434 | 0.4702 | 0.4635 | ||
| qH3 | 0.5561 | 0.5592 | ||||
| qO3 | −1.0595 | −1.0582 | ||||
| qC3 | 0.4247 | 0.4366 | ||||
| Dimer | ||||
| Gas phase | PCM | |||
| BCP | ||||
| C1-O1 | 0.2585 | −0.5887 | 0.2564 | −0.5916 |
| O1-H1 | 0.3503 | −2.4583 | 0.3458 | −2.4082 |
| H1…O2 | 0.0310 | 0.0830 | 0.0351 | 0.0894 |
| O2-C2 | 0.2443 | −0.5164 | 0.2431 | −0.5165 |
| Trimer | ||||
| Gas phase | PCM | |||
| BCP | ||||
| C1-O1 | 0.2596 | −0.5915 | 0.2595 | −0.6138 |
| O1-H1 | 0.3425 | −2.4002 | 0.3407 | −2.3819 |
| H1…O2 | 0.0373 | 0.0882 | 0.0383 | 0.0897 |
| O2-C2 | 0.2522 | −0.5533 | 0.2495 | −0.5516 |
| O2-H2 | 0.3413 | −2.4018 | 0.3401 | −2.3802 |
| H2…O3 | 0.0384 | 0.0884 | 0.0392 | 0.0904 |
| O3-C3 | 0.2385 | −0.4912 | 0.2412 | −0.5044 |
| X-ray Structures | DFT-Optimized Structures | |||||
|---|---|---|---|---|---|---|
| SAPT Term | Dimer 1 | Dimer 2 | Dimer 3 | Dimer 1 | Dimer 2 | Dimer 3 |
| Electrostatics | −7.99 | −1.55 | −1.04 | −10.64 | −0.60 | −1.18 |
| Exchange | 9.44 | 3.55 | 4.60 | 12.37 | 1.64 | 3.54 |
| Induction | −2.56 | −0.22 | −0.42 | −3.85 | −0.14 | −0.40 |
| Dispersion | −3.27 | −5.21 | −7.93 | −3.76 | −3.10 | −6.59 |
| Total SAPT2 | −4.39 | −3.43 | −4.79 | −5.87 | −2.21 | −4.62 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pocheć, M.; Krupka, K.M.; Panek, J.J.; Orzechowski, K.; Jezierska, A. Intermolecular Interactions and Spectroscopic Signatures of the Hydrogen-Bonded System—n-Octanol in Experimental and Theoretical Studies. Molecules 2022, 27, 1225. https://doi.org/10.3390/molecules27041225
Pocheć M, Krupka KM, Panek JJ, Orzechowski K, Jezierska A. Intermolecular Interactions and Spectroscopic Signatures of the Hydrogen-Bonded System—n-Octanol in Experimental and Theoretical Studies. Molecules. 2022; 27(4):1225. https://doi.org/10.3390/molecules27041225
Chicago/Turabian StylePocheć, Michał, Katarzyna M. Krupka, Jarosław J. Panek, Kazimierz Orzechowski, and Aneta Jezierska. 2022. "Intermolecular Interactions and Spectroscopic Signatures of the Hydrogen-Bonded System—n-Octanol in Experimental and Theoretical Studies" Molecules 27, no. 4: 1225. https://doi.org/10.3390/molecules27041225
APA StylePocheć, M., Krupka, K. M., Panek, J. J., Orzechowski, K., & Jezierska, A. (2022). Intermolecular Interactions and Spectroscopic Signatures of the Hydrogen-Bonded System—n-Octanol in Experimental and Theoretical Studies. Molecules, 27(4), 1225. https://doi.org/10.3390/molecules27041225