


Electrochemical Characterization and Voltammetric Determination of Methylisothiazolinone on a Boron-Doped Diamond Electrode


Abstract
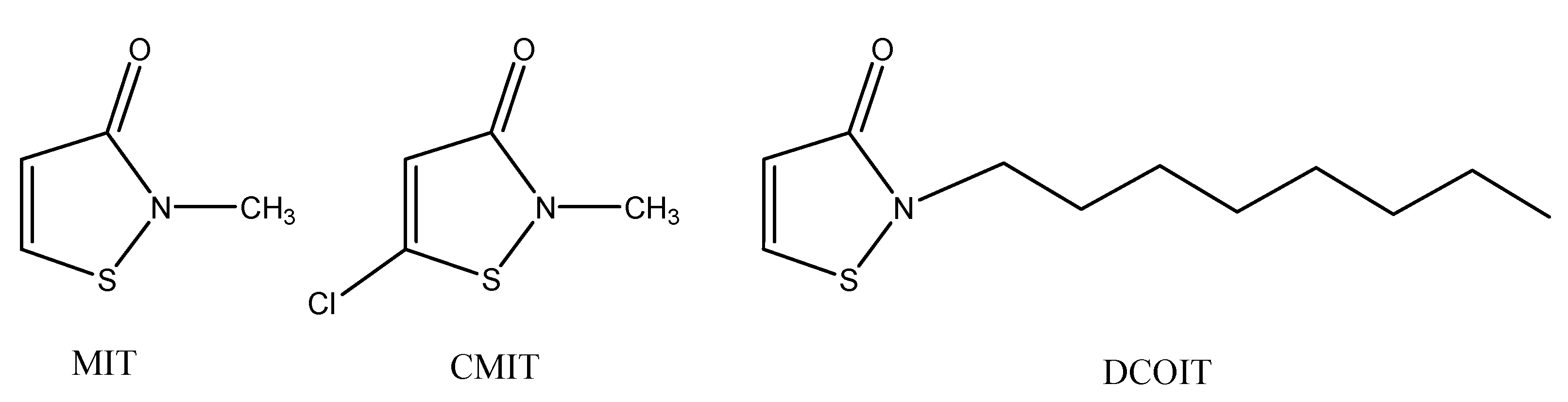
1. Introduction
2. Results and Discussion
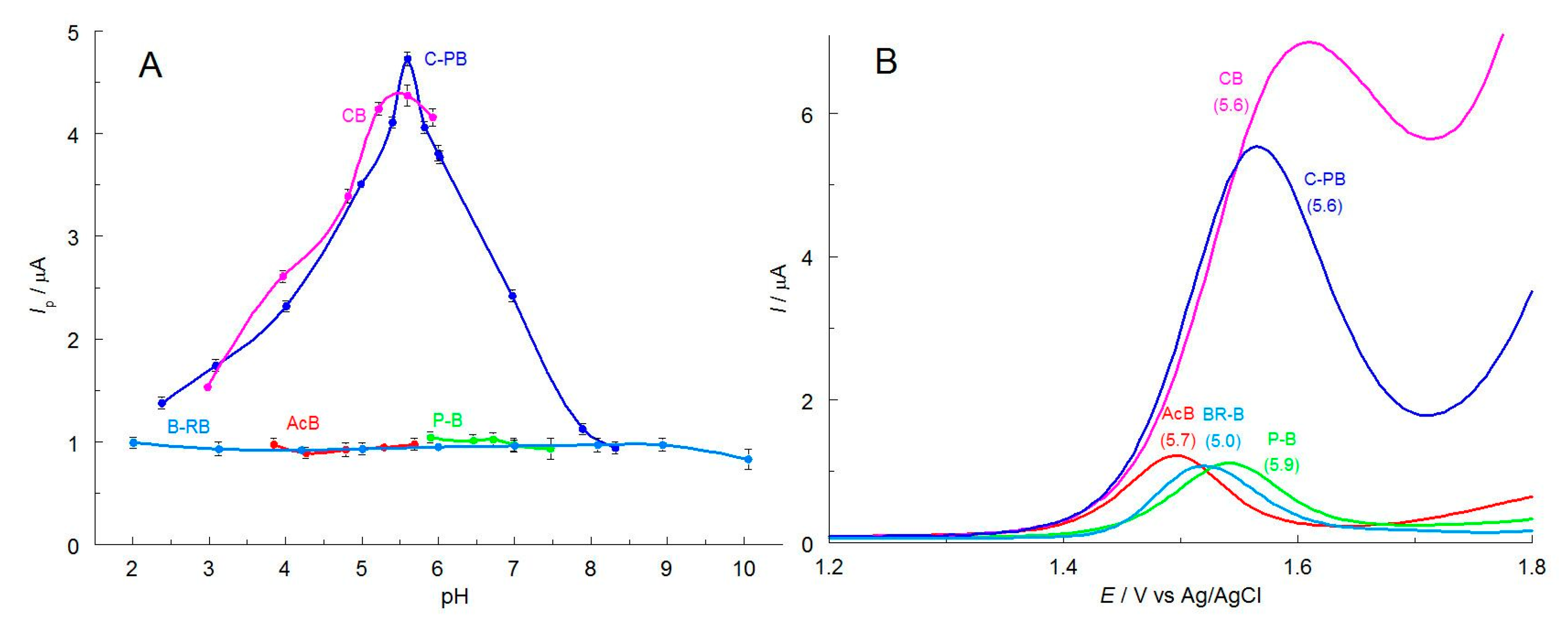
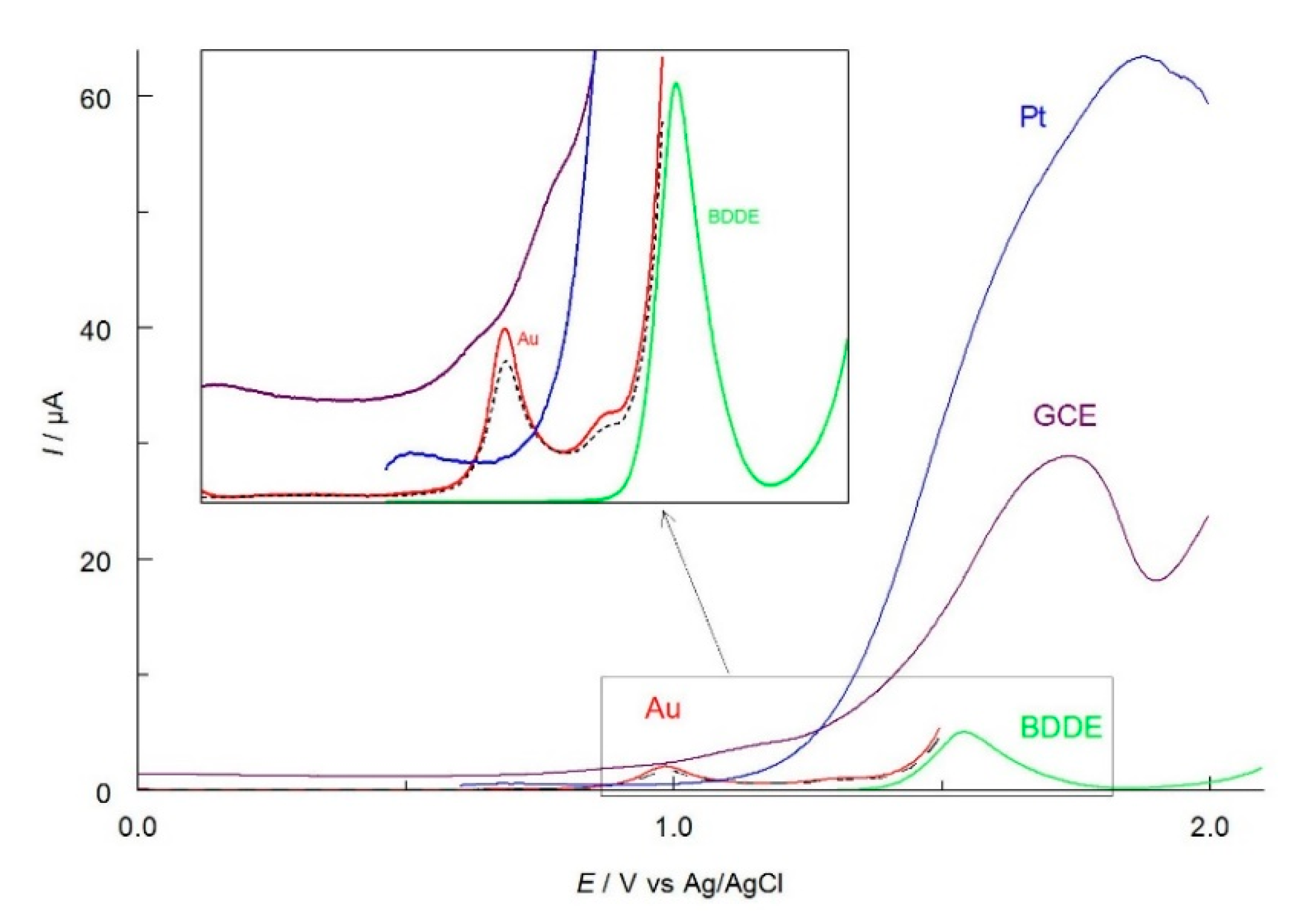
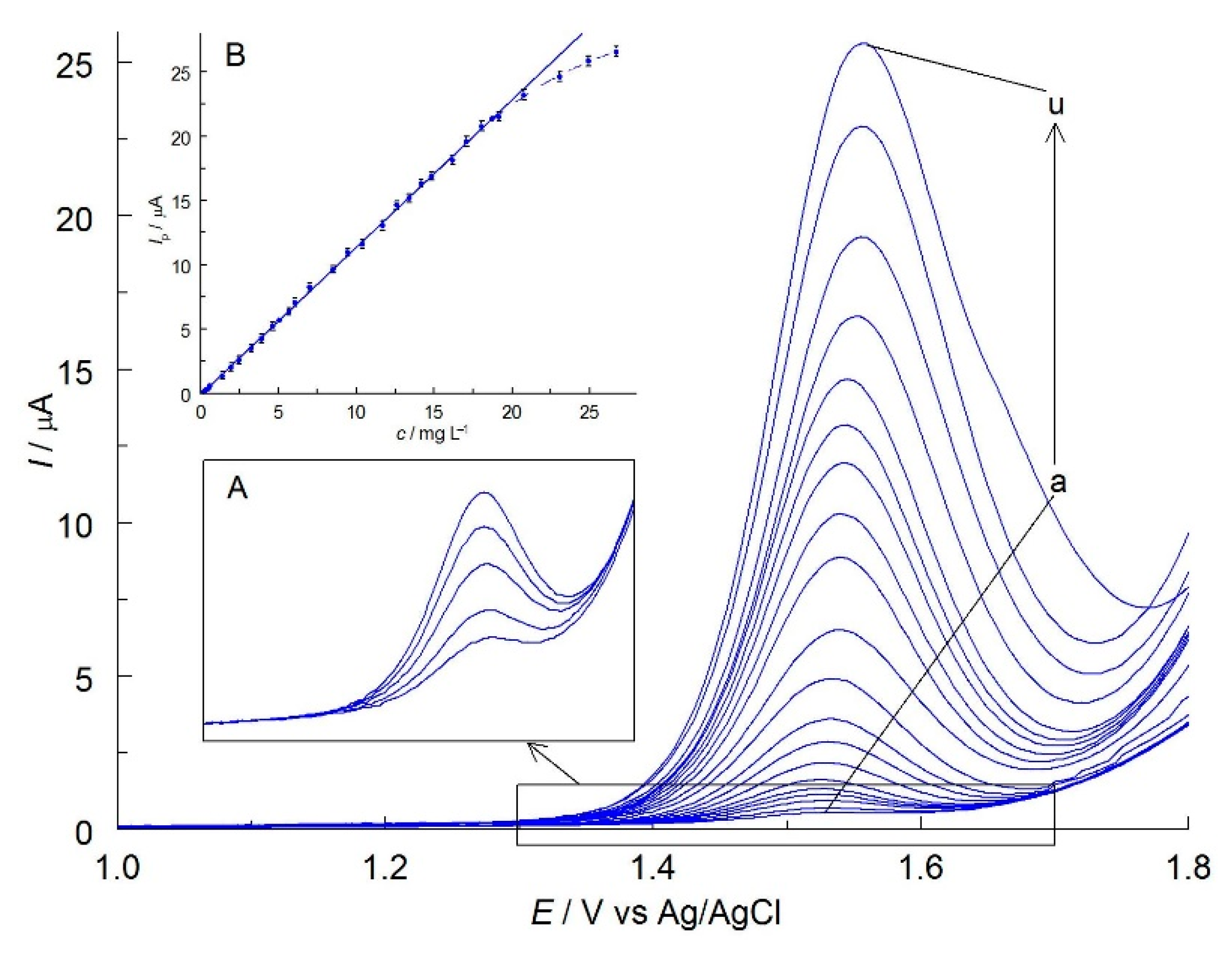
2.1. Selection of the Best Voltammetric Conditions
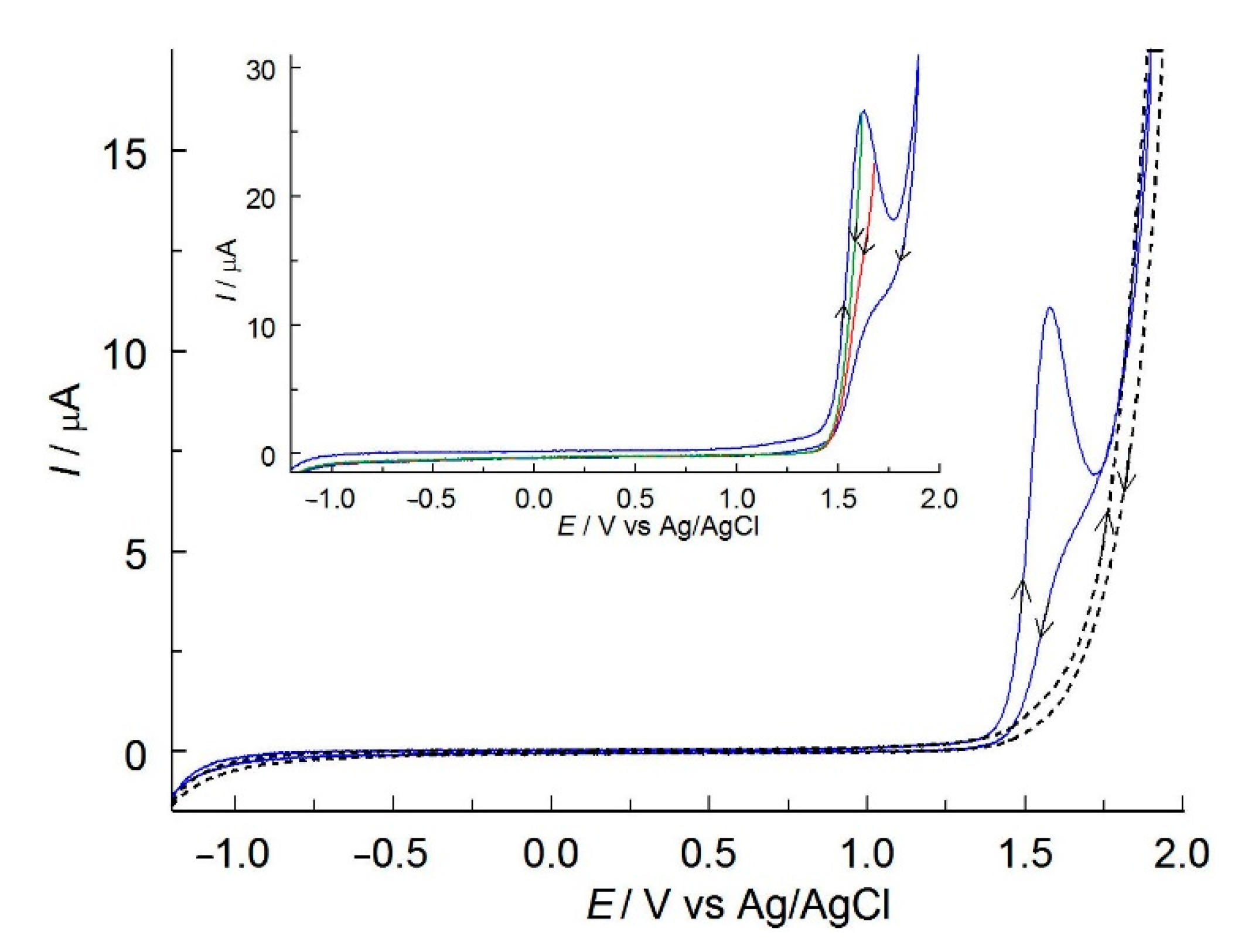
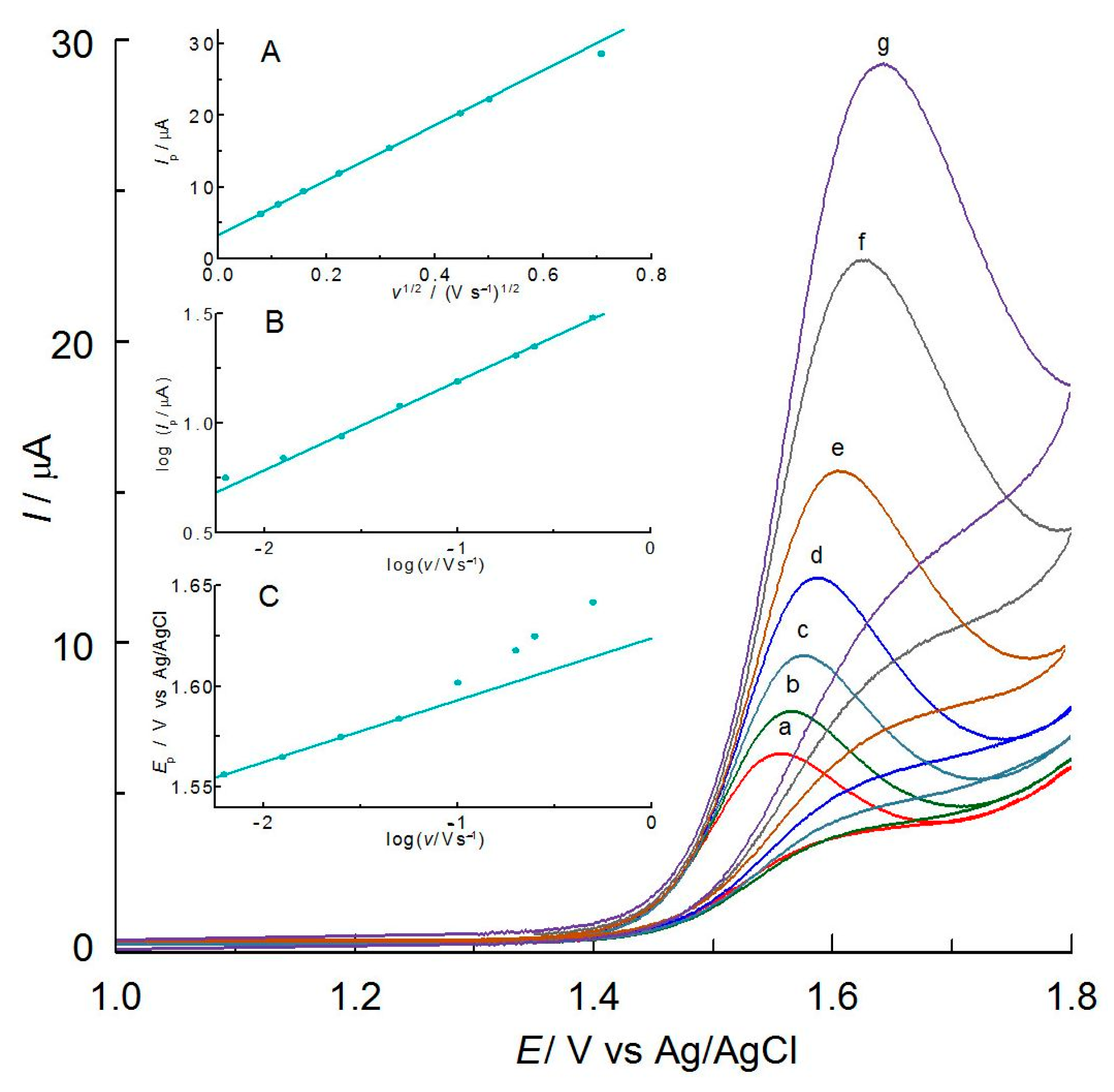
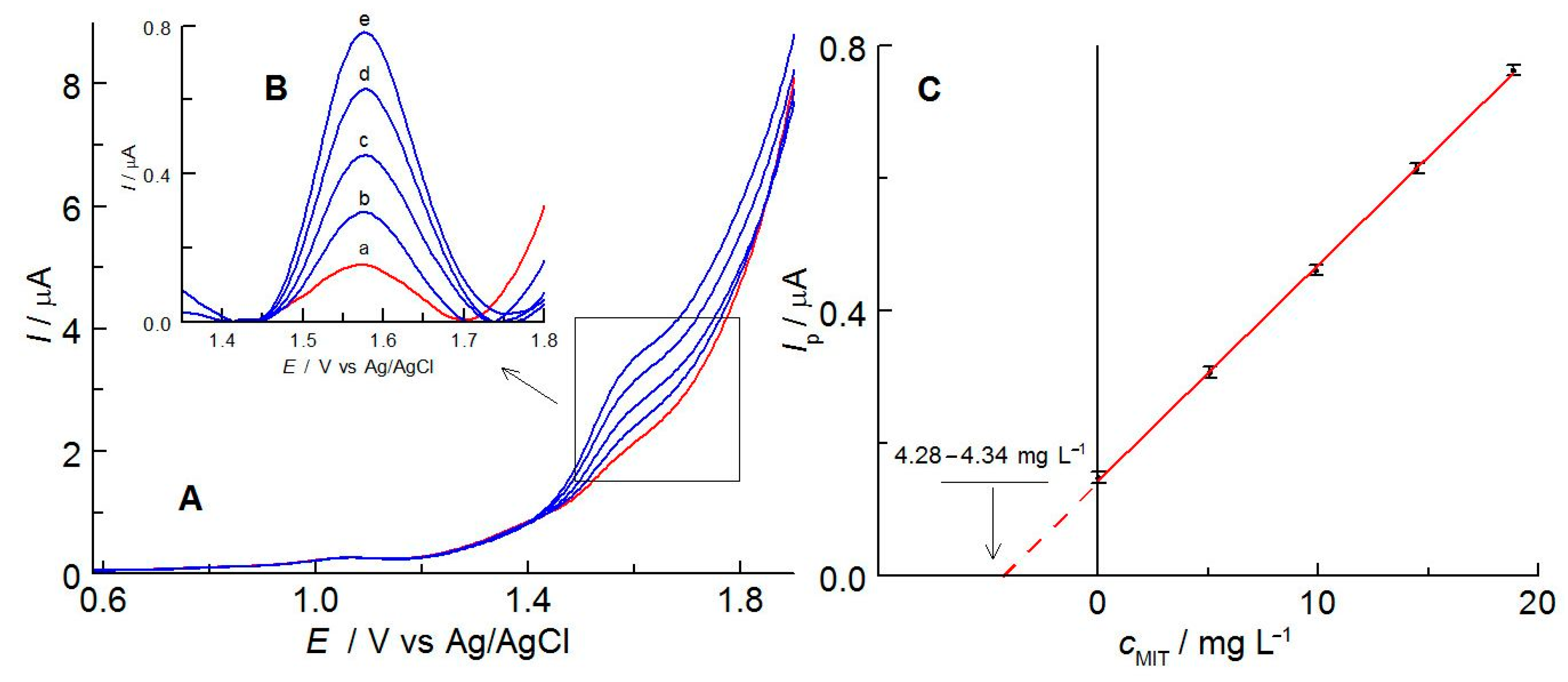
2.2. Electrochemical Properties of MIT in C-PB
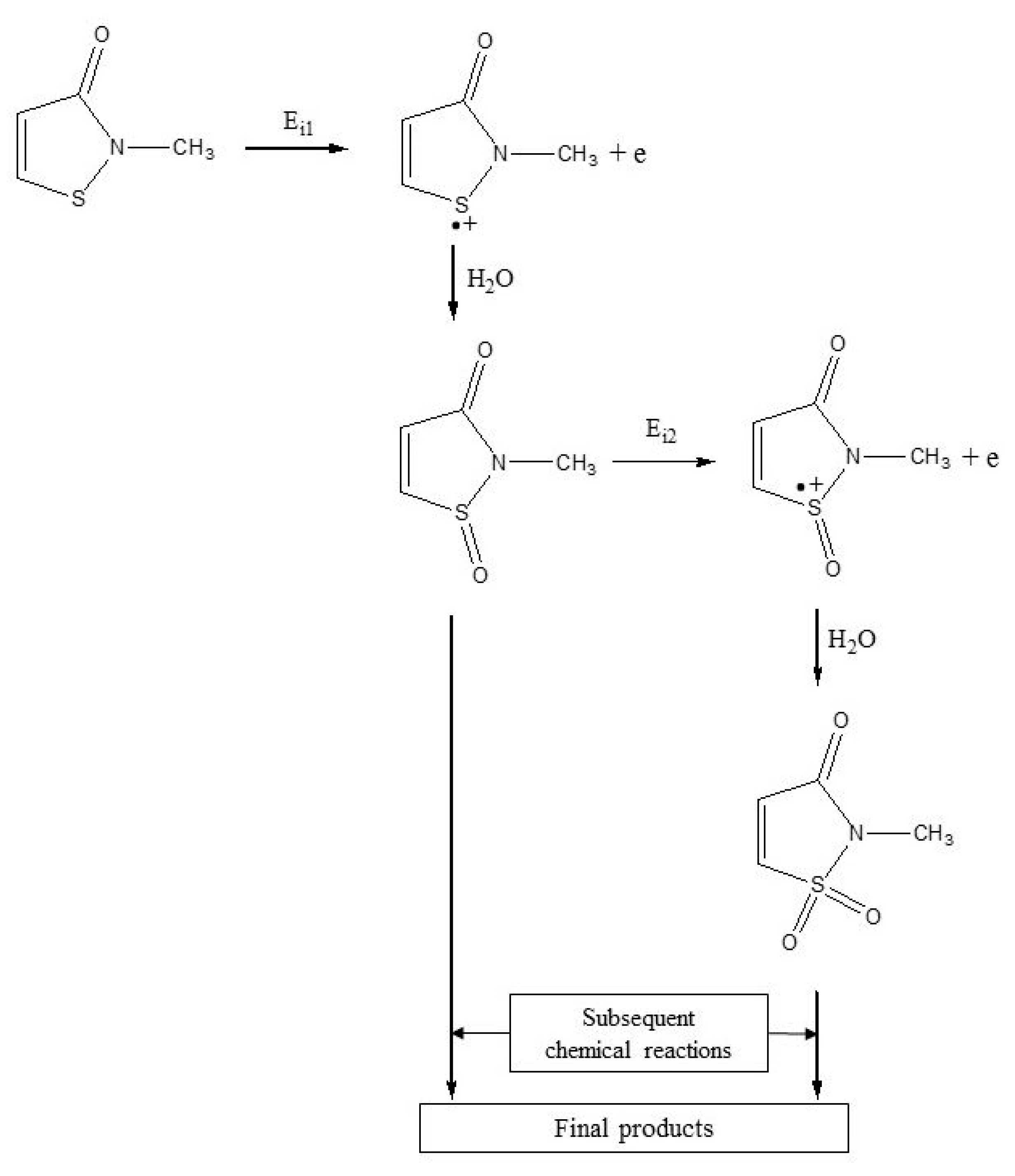
2.3. Mechanism
2.4. Validation of the MIT Determination Method
3. Materials and Methods
3.1. Reagents
3.2. Apparatus
3.3. Electrochemical Measurements
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Wittenberg, J.B.; Canas, B.J.; Zhou, W.; Wang, P.G.; Rua, D.; Krynitsky, A.J. Determination of methylisothiazolinone and methylchloroisothiazolinone in cosmetic products by ultra high performance liquid chromatography with tandem mass spectrometry. J. Sep. Sci. 2015, 38, 2983–2988. [Google Scholar] [CrossRef] [PubMed]
- Hefnawy, M.; Al-Majed, A.; Mohammed, M.; Al-Ghusn, A.; Al-Musallam, A.; Al-Sowidan, N.; Al-Hamid, M.; Al-Homoud, A. Fast and Sensitive Liquid Chromatography Method for Simultaneous Determination of Methylisothiazolinone, Salicylic Acid and Parabens in Cosmetic Products. Curr. Anal. Chem. 2017, 13, 430–438. [Google Scholar] [CrossRef]
- Alvarez-Rivera, G.; Dagnac, T.; Lores, M.; Garcia-Jares, C.; Sanchez-Prado, L.; Lamas, J.P.; Llompart, M. Determination of isothiazolinone preservatives in cosmetics and household products by matrix solid-phase dispersion followed by high-performance liquid chromatography-tandem mass spectrometry. J. Chromatogr. A 2012, 1270, 41–50. [Google Scholar] [CrossRef]
- Goodier, M.C.; Zang, L.Y.; Siegel, P.D.; Warshaw, E.M. Isothiazolinone Content of US Consumer Adhesives: Ultrahigh-Performance Liquid Chromatographic Mass Spectrometry Analysis. Dermatitis 2019, 30, 129–134. [Google Scholar] [CrossRef] [PubMed]
- Lundov, M.D.; Kolarik, B.; Bossi, R.; Gunnarsen, L.; Johansen, J.D. Emission of isothiazolinones from water-based paints. Environ. Sci. Technol. 2014, 48, 6989–6994. [Google Scholar] [CrossRef] [PubMed]
- Kahrilas, G.A.; Blotevogel, J.; Stewart, P.S.; Borch, T. Biocides in hydraulic fracturing fluids: A critical review of their usage, mobility, degradation, and toxicity. Environ. Sci. Technol. 2015, 49, 16–32. [Google Scholar] [CrossRef] [PubMed]
- Luz, G.V.S.; Sousa, B.A.S.M.; Guedes, A.V.; Barreto, C.C.; Brasil, L.M. Biocides used as additives to biodiesels and their risks to the environment and public health: A review. Molecules 2018, 23, 2698. [Google Scholar] [CrossRef] [PubMed]
- Isaksson, M.; Gruvberger, B.; Bruze, M. Patch testing with serial dilutions of various isothiazolinones in patients hypersensitive to methylchloroisothiazolinone/methylisothiazolinone. Contact Dermat. 2014, 70, 270–275. [Google Scholar] [CrossRef]
- Critchley, M.; Bentham, R. The efficacy of biocides and other chemical additives in cooling water systems in the control of amoebae. J. Appl. Microbiol. 2009, 106, 784–789. [Google Scholar] [CrossRef]
- Lin, Q.B.; Wang, T.J.; Song, H.; Li, B. Analysis of isothiazolinone biocides in paper for food packaging by ultra-high-performance liquid chromatography-tandem mass spectrometry. Food Addit. Contam.-Part A 2010, 27, 1775–1781. [Google Scholar] [CrossRef]
- Sukakul, T.; Kanchanapenkul, D.; Bunyavaree, M.; Limphoka, P.; Kumpangsin, T.; Boonchai, W. Methylchloroisothiazolinone and/or methylisothiazolinone in cosmetic products—A market survey. Contact Dermat. 2019, 80, 110–113. [Google Scholar] [CrossRef] [PubMed]
- Aerts, O.; Meert, H.; Goossens, A.; Janssens, S.; Lambert, J.; Apers, S. Methylisothiazolinone in selected consumer products in Belgium: Adding fuel to the fire? Contact Dermat. 2015, 73, 142–149. [Google Scholar] [CrossRef]
- Nielsen, H. Occupational exposdure to isothializolinones. A study based on a product register. Contact Dermatit. 1994, 31, 18–21. [Google Scholar] [CrossRef]
- Martinez-Mera, C.; González, M.A.; Hospital, M.; Turrión-Merino, L. Isothiazolinones in paint as a cause of airborne contact dermatitis in a patient with psoriasis receiving anti-interleukin-17 therapy. Contact Dermatit. 2019, 80, 328–329. [Google Scholar] [CrossRef] [PubMed]
- Vauhkala, A.R.; Pesonen, M.; Suomela, S.; Kuuliala, O.; Suuronen, K.; Aalto-Korte, K. Occupational contact allergy to methylchloroisothiazolinone/methylisothiazolinone and methylisothiazolinone. Contact Dermatit. 2015, 73, 150–156. [Google Scholar] [CrossRef] [PubMed]
- Todberg, T.; Opstrup, M.S.; Johansen, J.D.; Hald, M. Occupational facial contact dermatitis caused by methylchloroisothiazolinone/methylisothiazolinone in a stainless steel aerosol spray. Contact Dermatit. 2017, 77, 171–172. [Google Scholar] [CrossRef]
- Urwin, R.; Warburton, K.; Carder, M.; Turner, S.; Agius, R.; Wilkinson, S.M. Methylchloroisothiazolinone and methylisothiazolinone contact allergy: An occupational perspective. Contact Dermatit. 2015, 72, 381–386. [Google Scholar] [CrossRef]
- Castanedo-Tardana, M.P.; Zug, K.A. Methylisothiazolinone. Dermatitis 2013, 24, 2–6. [Google Scholar] [CrossRef]
- Commission Regulation (EU) 2016/1198 of 22 July 2016 Amending Annex to Regulation (EU) No. 1223/2009 of the European Parliament and of the Council on Cosmetic Products (Text with EEA Relevance). Off. J. L32016R1198, 2016. Available online: https://eur-lex.europa.eu/legal-content/EN/TXT/?uri=CELEX%3A32016R1198 (accessed on 13 December 2022).
- Commission Regulation (EU) 2017/1224 of 6 July 2017 Amending Annex V to Regulation (EC) No 1223/2009 of the European Parliament and of the Council on Cosmetic Products (Text with EEA Relevance). Off. J. 32017R1224, 2017. Available online: https://eur-lex.europa.eu/legal-content/EN/TXT/?toc=OJ:L:2017:174:TOC&uri=uriserv:OJ.L_.2017.174.01.0016.01.ENG (accessed on 13 December 2022).
- Baranowska, I.; Wojciechowska, I. The determination of preservatives in cosmetics and environmental waters by HPLC. Pol. J. Environ. Stud. 2013, 22, 1609–1625. [Google Scholar]
- Abad-Gil, L.; Lucas-Sánchez, S.; Gismera, M.J.; Sevilla, M.T.; Procopio, J.R. Determination of paraben-, isothiazolinone- and alcohol-type preservatives in personal care products by HPLC with dual (diode-array and fluorescence) detection. Microchem. J. 2021, 160, 105613. [Google Scholar] [CrossRef]
- Rastogi, S.C. Analytical control of preservative labelling on skin creams. Contact Dermatit. 2000, 43, 339–343. [Google Scholar] [CrossRef] [PubMed]
- Kawakami, T.; Isama, K.; Nishimura, T. Analysis of Isothiazolinones and Other Preservatives in Gel-Products Used for Cooling in Japan. J. Environ. Chem. 2012, 22, 205–211. [Google Scholar] [CrossRef][Green Version]
- Heo, J.J.; Kim, U.J.; Oh, J.E. Simultaneous quantitative analysis of four isothiazolinones and 3-iodo-2-propynyl butyl carbamate in hygienic consumer products. Environ. Eng. Res. 2019, 24, 137–143. [Google Scholar] [CrossRef]
- Bester, K.; Lamani, X. Determination of biocides as well as some biocide metabolites from facade run-off waters by solid phase extraction and high performance liquid chromatographic separation and tandem mass spectrometry detection. J. Chromatogr. A 2010, 1217, 5204–5214. [Google Scholar] [CrossRef]
- Speksnijder, P.; van Ravestijn, J.; de Voogt, P. Trace analysis of isothiazolinones in water samples by large-volume direct injection liquid chromatography tandem mass spectrometry. J. Chromatogr. A 2010, 1217, 5184–5189. [Google Scholar] [CrossRef]
- Rafoth, A.; Gabriel, S.; Sacher, F.; Brauch, H.J. Analysis of isothiazolinones in environmental waters by gas chromatography-mass spectrometry. J. Chromatogr. A 2007, 1164, 74–81. [Google Scholar] [CrossRef] [PubMed]
- Baranowska, I.; Wojciechowska, I.; Solarz, N.; Krutysza, E. Determination of preservatives in cosmetics, cleaning agents and pharmaceuticals using fast liquid chromatography. J. Chromatogr. Sci. 2014, 52, 88–94. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.; Wang, C.; Wang, X.; Ma, Q. Simultaneous determination of 21 preservatives in cosmetics by ultra performance liquid chromatography. Int. J. Cosmet. Sci. 2008, 30, 367–372. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.Y.; Lee, Y.J.; Kim, K.B. Analytical method development of methylisothiazolinone, a preservative, in rat plasma using LC-MS/MS. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2018, 1100–1101, 27–32. [Google Scholar] [CrossRef]
- Hoa, L.T.H.; Hung, V.T.N.; Trang, D.T.; Thu, T.N.H.; Le, D.C. Development and Validation of an HPLC Method for Simultaneous Assay of MCI and MI in Shampoos Containing Plant Extracts. Int. J. Anal. Chem. 2019, 2019, 1851796. [Google Scholar] [CrossRef]
- Abad-Gil, L.; Lucas-Sánchez, S.; Jesús Gismera, M.; Teresa Sevilla, M.; Procopio, J.R. HPLC method with electrochemical detection on gold electrode for simultaneous determination of different antimicrobial agents in cosmetics. Microchem. J. 2022, 182, 107881. [Google Scholar] [CrossRef]
- Abad-Gil, L.; Gismera, M.J.; Sevilla, M.T.; Procopio, J.R. Determination of methylisothiazolinone in waters. Comprehensive study about electrochemical behaviour on gold electrode and optimization of square-wave voltammetric methods. J. Electroanal. Chem. 2021, 880, 114831. [Google Scholar] [CrossRef]
- Abad-Gil, L.; Gismera, M.J.; Sevilla, M.T.; Procopio, J.R. Methylisothiazolinone response on disposable electrochemical platforms modified with carbon, nickel or gold-based nanomaterials. Microchim. Acta 2020, 187, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Fang, F.-G.; Tu, W.-J. Determination of isothiazolinones in cosmetics using differential pulse voltammetry on an ultramicroelectrode. Int. J. Pharm. Ther. 2014, 5, 207–212. [Google Scholar]
- Ruiz Montoya, M.; Giráldez, I.; Morales, E.; Estévez Brito, R.; Rodríguez Mellado, J.M. Electrochemical oxidation of isothiazolinone biocides and their interaction with cysteine. Electrochim. Acta 2020, 337, 135770. [Google Scholar] [CrossRef]
- Han, W.; Chen, Y.; Wang, L.; Sun, X.; Li, J. Mechanism and kinetics of electrochemical degradation of isothiazolin-ones using Ti/SnO2-Sb/PbO2 anode. Desalination 2011, 276, 82–88. [Google Scholar] [CrossRef]
- Wang, Y.; Chen, M.; Wang, C.; Meng, X.; Zhang, W.; Chen, Z.; Crittenden, J. Electrochemical degradation of methylisothiazolinone by using Ti/SnO2-Sb2O3/A, Β-PbO2 electrode: Kinetics, energy efficiency, oxidation mechanism and degradation pathway. Chem. Eng. J. 2019, 374, 626–636. [Google Scholar] [CrossRef]
- Kandavelu, V.; Yoshihara, S.; Kumaravel, M.; Murugananthan, M. Anodic oxidation of isothiazolin-3-ones in aqueous medium by using boron-doped diamond electrode. Diam. Relat. Mater. 2016, 69, 152–159. [Google Scholar] [CrossRef]
- Peng, L.; Liu, H.; Wang, W.L.; Xu, Z.B.; Ni, X.Y.; Wu, Y.H.; Wu, Q.Y.; Hu, H.Y. Degradation of methylisothiazolinone biocide using a carbon fiber felt-based flow-through electrode system (FES) via anodic oxidation. Chem. Eng. J. 2020, 384, 123239. [Google Scholar] [CrossRef]
- Scholz, F. Electroanalytical Methods. Guide to Experiments and Applications. Second Revised and Extended Edition; Springer: Berlin/Heidelberg, Germany, 2010. [Google Scholar]
- Gosser, D.K. Cyclic Voltammetry; Simulation and Analysis of Reaction Mechanisms; VCH Publishers, Inc.: New York, NY, USA, 1994. [Google Scholar]
- Michalkiewicz, S.; Skorupa, A.; Jakubczyk, M. Carbon materials in electroanalysis of preservatives: A review. Materials 2021, 14, 7630. [Google Scholar] [CrossRef]
- Oliveira, S.C.B.; Oliveira-Brett, A.M. Boron doped diamond electrode pre-treatments effect on the electrochemical oxidation of dsDNA, DNA bases, nucleotides, homopolynucleotides and biomarker 8-oxoguanine. J. Electroanal. Chem. 2010, 648, 60–66. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Tested Values | Optimal Value |
|---|---|---|
| Amplitude, dE/mV | 5–150 | 50 |
| Potential step, Es/mV | 2–13 | 5 |
| Pulse width/ms | 20–140 | 80 |
| Sample | MIT Taken/ mg L−1 | 1) MIT Found/ mg L−1 % (w/w) | 2)RSD/% (n = 5) | 3)R/% | |
|---|---|---|---|---|---|
| Control | 0.96 | 0.95 ± 0.02 | ‒ | 1.0 | 99.0 |
| Control | 4.98 | 4.96 ± 0.03 | ‒ | 0.5 | 99.5 |
| Control with Perwoll matrix | 0.96 | 5.17 ± 0.04 | 0.0126 ± 0.001 | 0.7 | 538.5 |
| Control with Perwoll matrix | 4.98 | 9.15 ± 0.04 | 0.0224 ± 0.001 | 0.4 | 183.7 |
| Perwoll | ‒ | 4.31 ± 0.03 | 0.0103 ± 0.0001 | 0.6 | ‒ |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jakubczyk, M.; Michalkiewicz, S.; Skorupa, A.; Krajcarz, K. Electrochemical Characterization and Voltammetric Determination of Methylisothiazolinone on a Boron-Doped Diamond Electrode. Molecules 2022, 27, 9013. https://doi.org/10.3390/molecules27249013
Jakubczyk M, Michalkiewicz S, Skorupa A, Krajcarz K. Electrochemical Characterization and Voltammetric Determination of Methylisothiazolinone on a Boron-Doped Diamond Electrode. Molecules. 2022; 27(24):9013. https://doi.org/10.3390/molecules27249013
Chicago/Turabian StyleJakubczyk, Magdalena, Slawomir Michalkiewicz, Agata Skorupa, and Kinga Krajcarz. 2022. "Electrochemical Characterization and Voltammetric Determination of Methylisothiazolinone on a Boron-Doped Diamond Electrode" Molecules 27, no. 24: 9013. https://doi.org/10.3390/molecules27249013
APA StyleJakubczyk, M., Michalkiewicz, S., Skorupa, A., & Krajcarz, K. (2022). Electrochemical Characterization and Voltammetric Determination of Methylisothiazolinone on a Boron-Doped Diamond Electrode. Molecules, 27(24), 9013. https://doi.org/10.3390/molecules27249013