

The Origin of Stereoselectivity in the Hydrogenation of Oximes Catalyzed by Iridium Complexes: A DFT Mechanistic Study

 and
and 
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
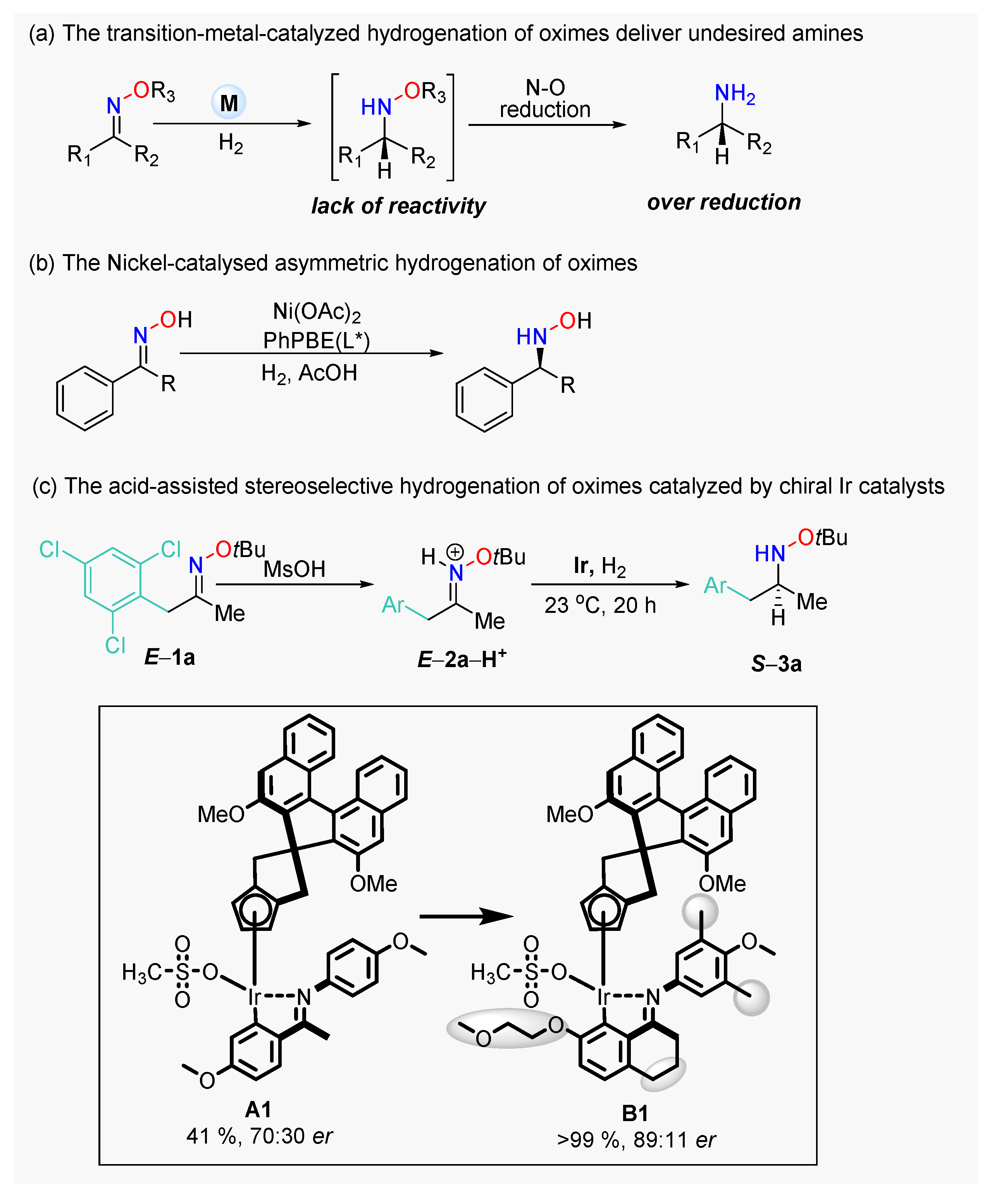
1. Introduction
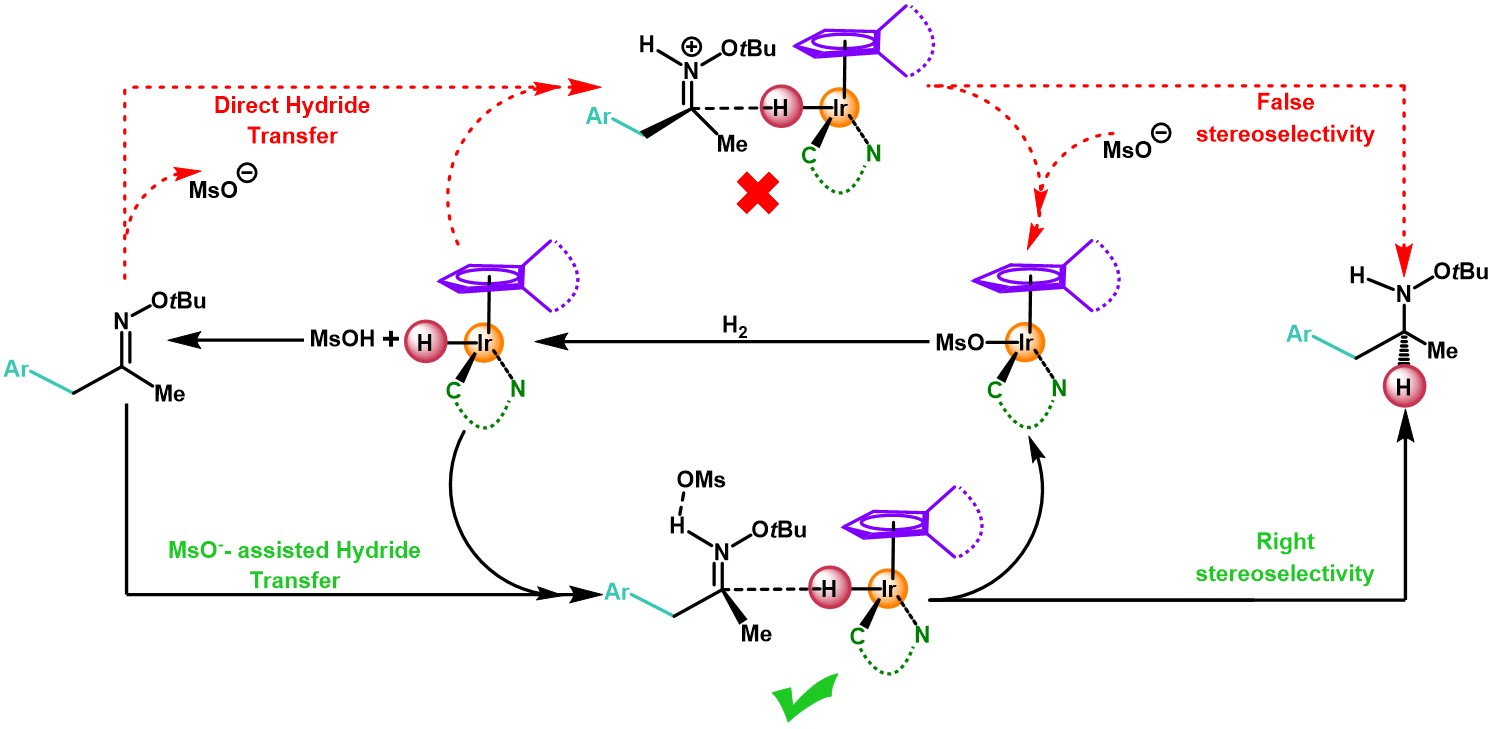
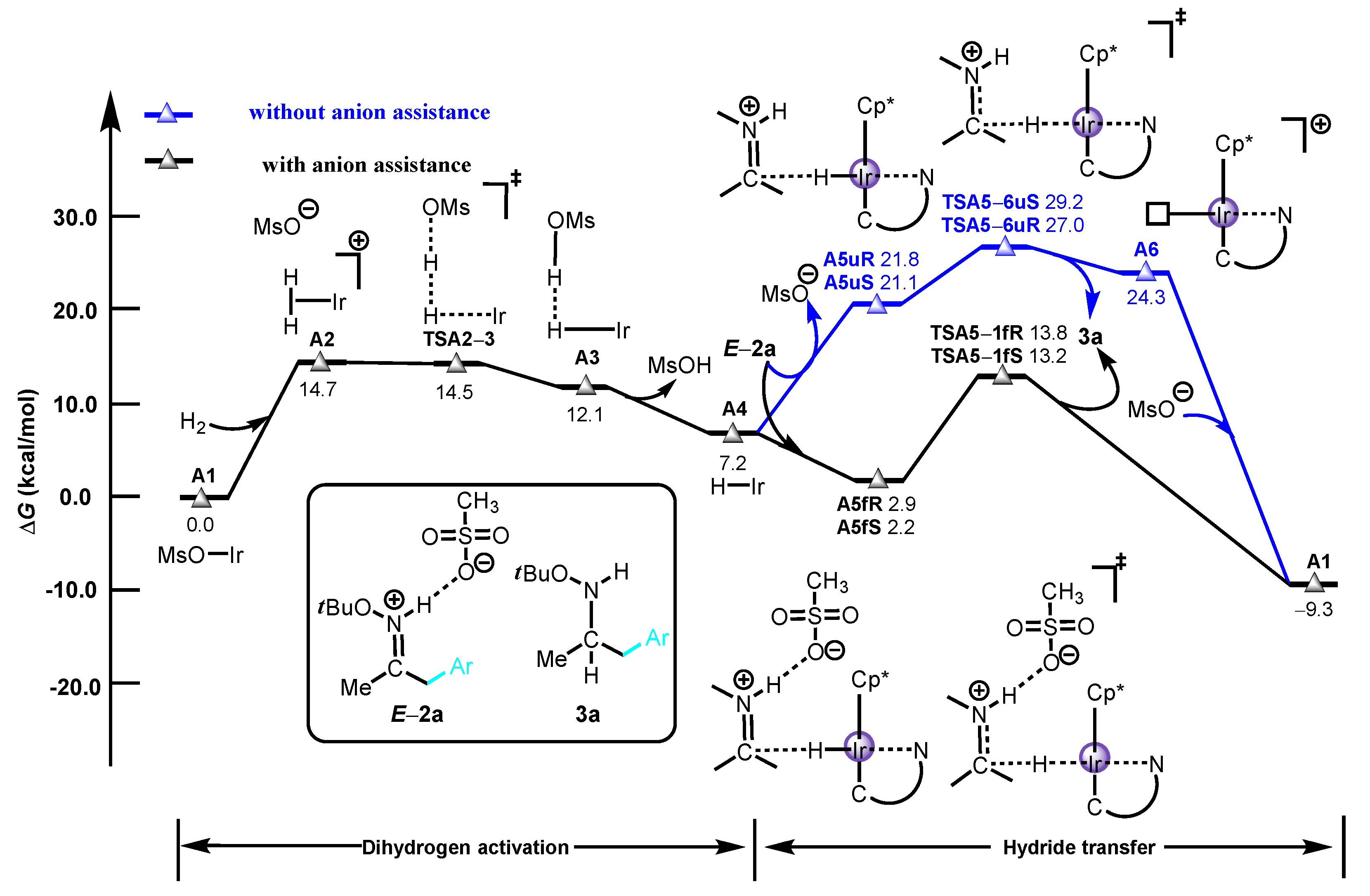
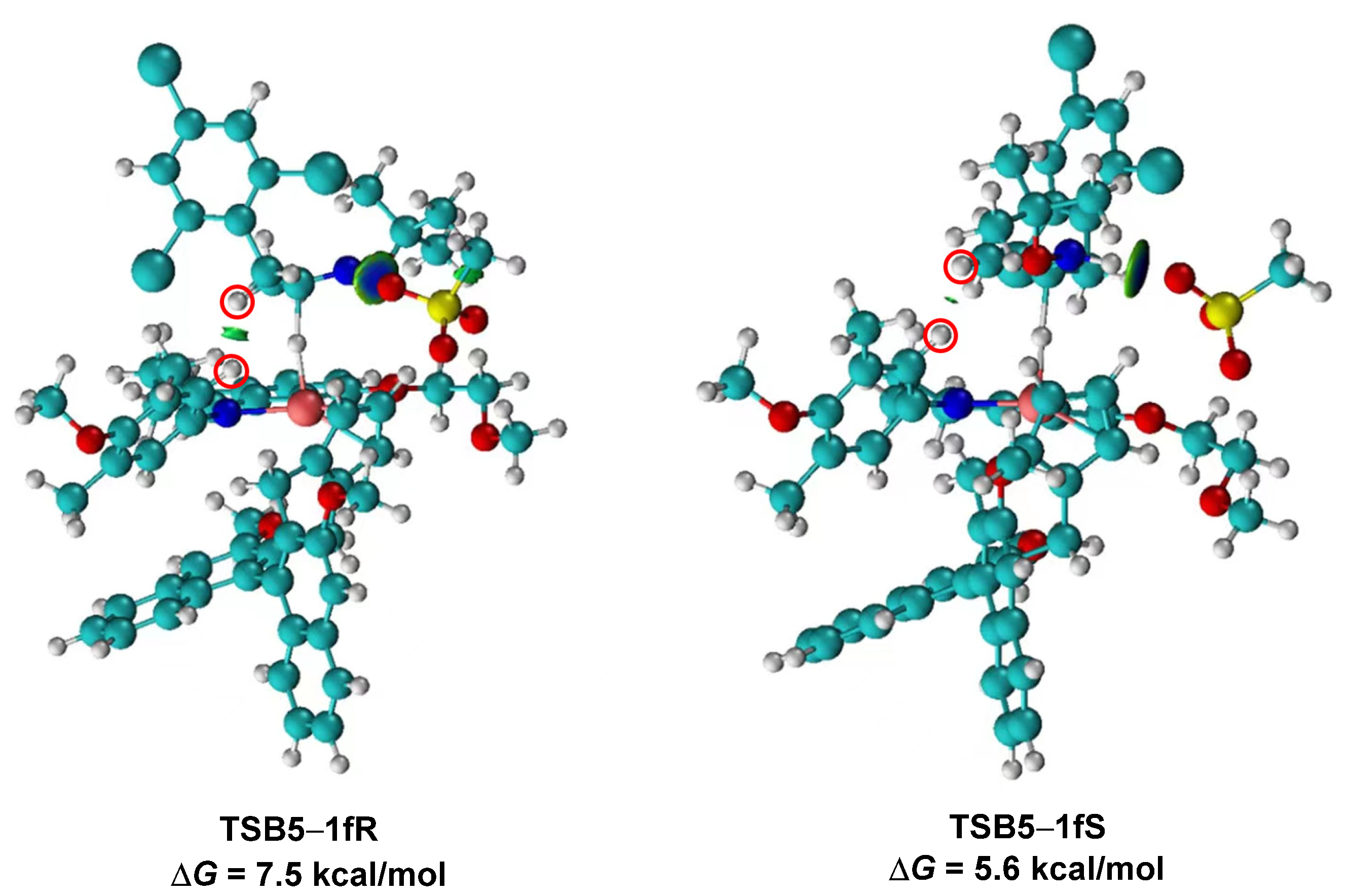
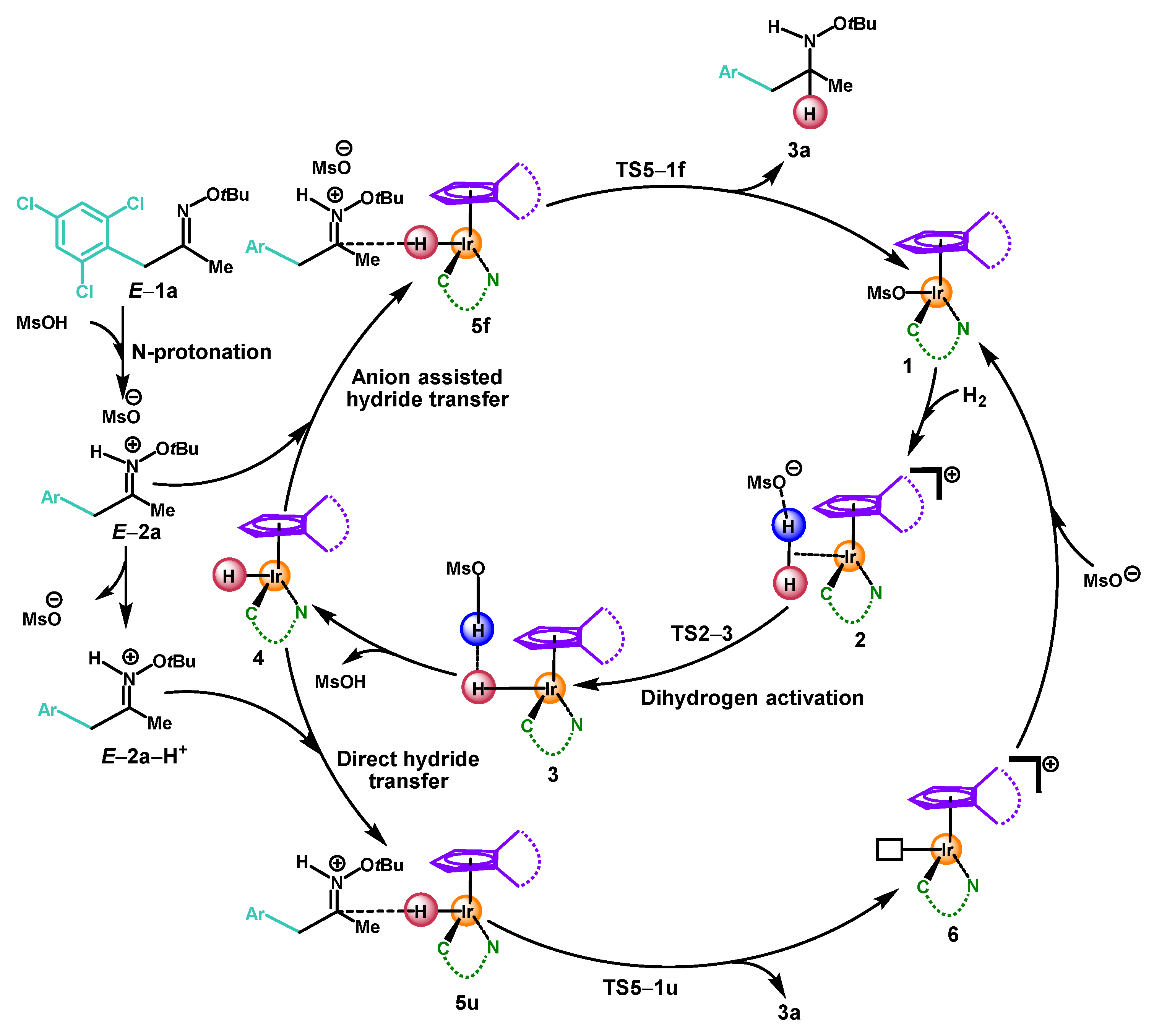
2. Results and Discussion
3. Computational Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Paudyal, M.P.; Adebesin, A.M.; Burt, S.R.; Ess, D.H.; Ma, Z.; Kürti, L.; Falck, J.R. Dirhodium-catalyzed c-h arene amination using hydroxylamines. Science 2016, 353, 1144–1147. [Google Scholar] [CrossRef] [PubMed]
- Ager, D.J.; de Vries, A.H.; de Vries, J.G. Asymmetric homogeneous hydrogenations at scale. Chem. Soc. Rev. 2012, 41, 3340–3380. [Google Scholar]
- Maj, A.M.; Suisse, I.; Agbossou-Niedercorn, F. Asymmetric hydrogenation of 2,3-dihydro-1H-inden-1-one oxime and derivatives. Tetrahedron Asymmetry 2016, 27, 268–273. [Google Scholar] [CrossRef]
- Li, B.; Liu, D.; Hu, Y.; Chen, J.; Zhang, Z.; Zhang, W. Nickel-catalyzed asymmetric hydrogenation of hydrazones. Eur. J. Org. Chem. 2021, 2021, 3421–3425. [Google Scholar] [CrossRef]
- Liu, D.; Li, B.; Chen, J.; Gridnev, I.D.; Yan, D.; Zhang, W. Ni-catalyzed asymmetric hydrogenation of n-aryl imino esters for the efficient synthesis of chiral alpha-aryl glycines. Nat. Commun. 2020, 11, 5935. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Chen, J.; Zhang, Z.; Gridnev, I.D.; Zhang, W. Nickel-catalyzed asymmetric hydrogenation of N-sulfonyl imines. Angew. Chem. Int. Ed. 2019, 58, 7329–7334. [Google Scholar] [CrossRef]
- Quan, M.; Wang, X.; Wu, L.; Gridnev, I.D.; Yang, G.; Zhang, W. Ni(ii)-catalyzed asymmetric alkenylations of ketimines. Nat. Commun. 2018, 9, 2258. [Google Scholar] [CrossRef]
- Mohr, J.; Oestreich, M. B(C6F5)3-catalyzed hydrogenation of oxime ethers without cleavage of the N-O bond. Angew. Chem. Int. Ed. 2014, 53, 13278–13281. [Google Scholar] [CrossRef]
- Li, B.; Chen, J.; Liu, D.; Gridnev, I.D.; Zhang, W. Nickel-catalysed asymmetric hydrogenation of oximes. Nat. Chem. 2022, 14, 920–927. [Google Scholar] [CrossRef]
- Mas-Rosello, J.; Smejkal, T.; Cramer, N. Iridium-catalyzed acid-assisted asymmetric hydrogenation of oximes to hydroxylamines. Science 2020, 368, 1098–1102. [Google Scholar] [CrossRef]
- Kozuch, S.; Shaik, S. How to conceptualize catalytic cycles? The energetic span model. Acc. Chem. Res. 2011, 44, 101–110. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.-w.; Zhang, L.; Pu, M.; Lei, M. Mechanistic understanding of base-catalyzed aldimine/ketoamine condensations: An old story and a new model. Asian J. Org. Chem. 2021, 10, 634–641. [Google Scholar] [CrossRef]
- Liao, G.; Wu, Y.-J.; Shi, B.-F. Noncovalent interaction in transition metal-catalyzed selective c-h activation. Acta Chim. Sinica 2020, 78, 289–298. [Google Scholar] [CrossRef]
- Jeffery, I.S. The Curtin-Hammett principle and the Winstein-Holness equation: New definition and recent extensions to classical concepts. J. Chem. Educ. 1986, 63, 42–48. [Google Scholar]
- Humphrey, W.; Dalke, A.; Schulten, K. Vmd: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Zhang, L.; Pu, M.; Lei, M. A phosphine-free Mn(i)-NNS catalyst for asymmetric transfer hydrogenation of acetophenone: A theoretical prediction. Dalton Trans. 2021, 50, 14738–14744. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Yue, X.; Li, L.; Li, Z.; Zhang, L.; Pu, M.; Yang, Z.; Wang, C.; Xiao, J.; Lei, M. Asymmetric induction with a chiral amine catalyzed by a ru-pnp pincer complex: Insight from theoretical investigation. Inorg. Chem. 2020, 59, 8404–8411. [Google Scholar] [CrossRef]
- Feng, R.; Xiao, A.; Zhang, X.; Tang, Y.; Lei, M. Origins of enantioselectivity in asymmetric ketone hydrogenation catalyzed by a Ruh2(binap)(cydn) complex: Insights from a computational study. Dalton Trans. 2013, 42, 2130–2145. [Google Scholar] [CrossRef]
- Li, L.; Pan, Y.; Lei, M. The enantioselectivity in asymmetric ketone hydrogenation catalyzed by Ruh2(diphosphine)(diamine) complexes: Insights from a 3D-QSSR and DFT study. Catal. Sci. Technol. 2016, 6, 4450–4457. [Google Scholar] [CrossRef]
- Xiao, M.; Yue, X.; Xu, R.; Tang, W.; Xue, D.; Li, C.; Lei, M.; Xiao, J.; Wang, C. Transition-metal-free hydrogen autotransfer: Diastereoselective n-alkylation of amines with racemic alcohols. Angew. Chem. Int. Ed. 2019, 58, 10528–10536. [Google Scholar] [CrossRef]
- Liu, Y.; Yue, X.; Luo, C.; Zhang, L.; Lei, M. Mechanisms of ketone/imine hydrogenation catalyzed by transition-metal complexes. Energy Environ. Mater. 2019, 2, 292–312. [Google Scholar] [CrossRef]
- Liu, C.; Zhang, L.; Li, L.; Lei, M. Theoretical design of a catalyst with both high activity and selectivity in C-H borylation. J. Org. Chem. 2021, 86, 16858–16866. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Zhao, Y.; Shi, X.; Tang, Y.; Yang, Z.; Pu, M.; Lei, M. A theoretical study on the hydrogenation of CO2 to methanol catalyzed by ruthenium pincer complexes. Dalton Trans. 2022, 51, 10020–10028. [Google Scholar] [CrossRef]
- Zhao, Y.; Zhang, L.; Tang, Y.; Pu, M.; Lei, M. A theoretical study of asymmetric ketone hydrogenation catalyzed by Mn complexes: From the catalytic mechanism to the catalyst design. Phys. Chem. Chem. Phys. 2022, 24, 13365–13375. [Google Scholar] [CrossRef] [PubMed]
- Gaussian, version 09, revision D.01; Gaussian Inc.: Pittsburgh, PA, USA, 2010.
- Chai, J.-D.; Head-Gordon, M. Long-range corrected hybrid density functionals with damped atom-atom dispersion corrections. Phys. Chem. Chem. Phys. 2008, 10, 6615–6620. [Google Scholar] [CrossRef]
- Hay, P.J.; Wadt, W.R. Ab initio effective core potentials for molecular calculations. Potentials for K to Au including the outermost core orbitals. J. Chem. Phys. 1985, 82, 299–310. [Google Scholar] [CrossRef]
- Ditchfield, R.; Hehre, W.J.; Pople, J.A. Self-consistent molecular-orbital methods. IX. An extended gaussian-type basis for molecular-orbital studies of organic molecules. J. Chem. Phys. 1971, 54, 724–728. [Google Scholar] [CrossRef]
- Hehre, W.J.; Ditchfield, R.; Pople, J.A. Self—Consistent molecular orbital methods. XII. Further extensions of gaussian—Type basis sets for use in molecular orbital studies of organic molecules. J. Chem. Phys. 1972, 56, 2257–2261. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef]
- Neese, F. Software update: The orca program system, version 4.0. WIREs Comput. Mol. Sci. 2018, 8, e1327. [Google Scholar] [CrossRef]
- Neese, F.; Wennmohs, F.; Becker, U.; Riplinger, C. The orca quantum chemistry program package. J. Chem. Phys. 2020, 152, 224108. [Google Scholar] [CrossRef] [PubMed]
- Mardirossian, N.; Head-Gordon, M. ωB97m-V: A combinatorially optimized, range-separated hybrid, meta-GGA density functional with VV10 nonlocal correlation. J. Chem. Phys. 2016, 144, 214110. [Google Scholar] [CrossRef] [PubMed]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal solvation model based on solute electron density and on a continuum model of the solvent defined by the bulk dielectric constant and atomic surface tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [Google Scholar] [CrossRef] [PubMed]
- Hratchian, H.P.; Schlegel, H.B. Accurate reaction paths using a Hessian based predictor-corrector integrator. J. Chem. Phys. 2004, 120, 9918–9924. [Google Scholar] [CrossRef]
- Grimme, S. Supramolecular binding thermodynamics by dispersion-corrected density functional theory. Chem. Eur. J. 2012, 18, 9955–9964. [Google Scholar] [CrossRef]
- Lu, T.; Chen, Q. Shermo: A general code for calculating molecular thermochemistry properties. Comput. Theor. Chem. 2021, 1200, 113249. [Google Scholar] [CrossRef]
- Bryantsev, V.S.; Diallo, M.S.; Goddard, W.A. Calculation of solvation free energies of charged solutes using mixed cluster/continuum models. J. Phys. Chem. B 2008, 112, 9709–9719. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef]
- Lu, T.; Chen, Q. Independent gradient model based on Hirshfeld partition: A new method for visual study of interactions in chemical systems. J. Comput. Chem. 2022, 43, 539–555. [Google Scholar] [CrossRef]
- Stephens, P.J.; Devlin, F.J.; Chabalowski, C.F.; Frisch, M.J. Ab initio calculation of vibrational absorption and circular dichroism spectra using density functional force fields. J. Phys. Chem. 1994, 98, 11623–11627. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [PubMed]
- Adamo, C.; Barone, V. Toward reliable density functional methods without adjustable parameters: The PBE0 model. J. Chem. Phys. 1999, 110, 6158–6170. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ali, Q.; Chen, Y.; Zhang, R.; Li, Z.; Tang, Y.; Pu, M.; Lei, M. The Origin of Stereoselectivity in the Hydrogenation of Oximes Catalyzed by Iridium Complexes: A DFT Mechanistic Study. Molecules 2022, 27, 8349. https://doi.org/10.3390/molecules27238349
Ali Q, Chen Y, Zhang R, Li Z, Tang Y, Pu M, Lei M. The Origin of Stereoselectivity in the Hydrogenation of Oximes Catalyzed by Iridium Complexes: A DFT Mechanistic Study. Molecules. 2022; 27(23):8349. https://doi.org/10.3390/molecules27238349
Chicago/Turabian StyleAli, Qaim, Yongyong Chen, Ruixue Zhang, Zhewei Li, Yanhui Tang, Min Pu, and Ming Lei. 2022. "The Origin of Stereoselectivity in the Hydrogenation of Oximes Catalyzed by Iridium Complexes: A DFT Mechanistic Study" Molecules 27, no. 23: 8349. https://doi.org/10.3390/molecules27238349
APA StyleAli, Q., Chen, Y., Zhang, R., Li, Z., Tang, Y., Pu, M., & Lei, M. (2022). The Origin of Stereoselectivity in the Hydrogenation of Oximes Catalyzed by Iridium Complexes: A DFT Mechanistic Study. Molecules, 27(23), 8349. https://doi.org/10.3390/molecules27238349