
Synthesis and Evaluation of NF-κB Inhibitory Activity of Mollugin Derivatives

 ,
,
Abstract

1. Introduction
2. Results and Discussion
2.1. Chemistry
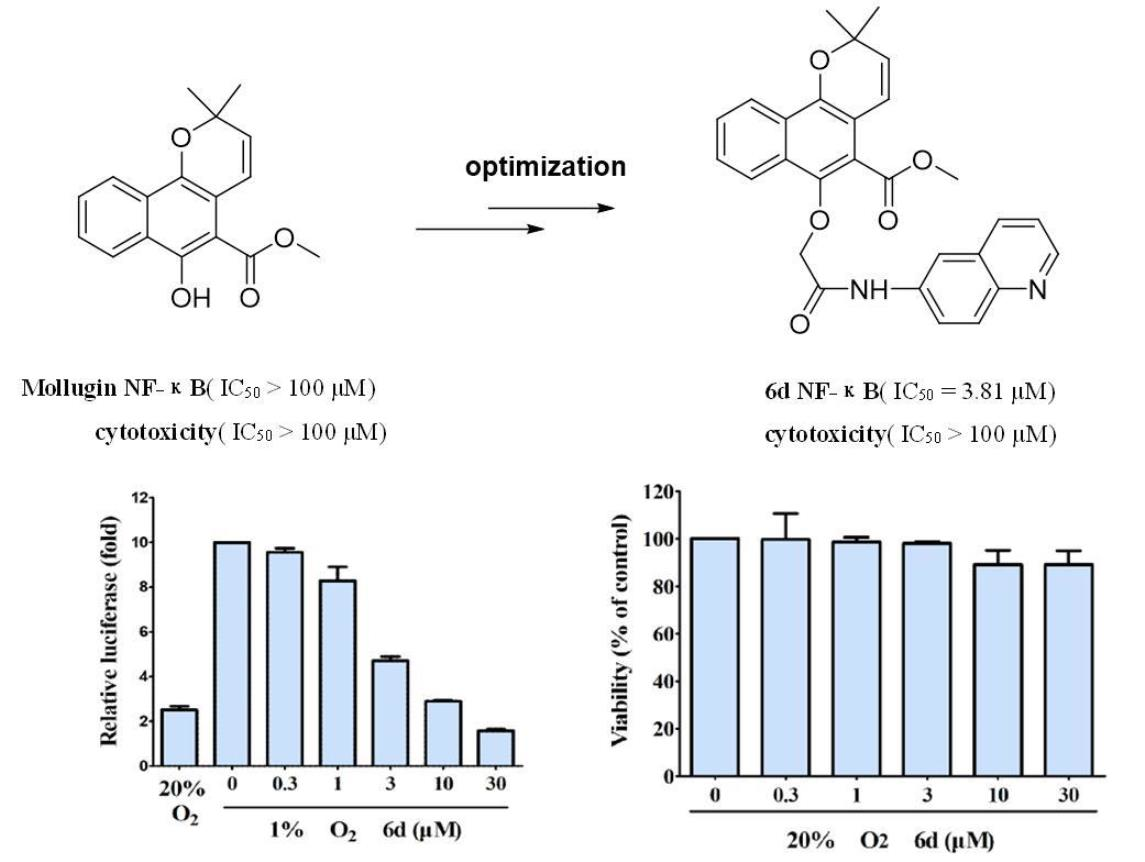
2.2. Evaluation of Biological Activities
2.2.1. Luciferase Reporter and MTT Analysis
2.2.2. Western Blot Analysis
2.2.3. Anti-Inflammatory Activity
2.3. Prediction of ADMET Properties
3. Materials and Methods
3.1. Experimental Compounds
3.1.1. General Procedures
3.1.2. Procedure for the Synthesis of the Mollugin 2
3.1.3. General Procedure for the Synthesis of the Intermediate 3a–i
3.1.4. General Procedure for the Synthesis of the Intermediate 5a–k
3.1.5. General Procedure for the Synthesis of the Intermediate 7a–b
3.1.6. General Procedure for the Preparation of Target Compounds 4a–i
Methyl 2,2-dimethyl-6-(2-oxo-2-(phenylamino)ethoxy)-2H-benzo [h] chromene-5-carboxyate (4a)
Methyl 6-(2-((4-chlorophenyl)amino)-2-oxoethoxy)-2,2-dimethyl-2H-benzo [h] chromene-5-carboxylate (4b)
Methyl 6-(2-((4-bromophenyl)amino)-2-oxoethoxy)-2,2-dimethyl-2H-Benzo [h] chromene-5-carboxylate (4c)
Methyl 6-(2-((4-methoxyphenyl)amino)-2-oxoethoxy)-2,2-dimethyl-2H-benzo [h] chromene-5-carboxylate (4d)
Methyl 2,2-dimethyl-6-(2-oxo-2-(p-tolylamino)ethoxy)-2H-benzo [h] chromene-5-carboxylate (4e)
Methyl 6-(2-((4-fluorophenyl)amino)-2-oxoethoxy)-2,2-dimethyl-2H-benzo [h] chromene-5-carboxylate (4f)
Methyl 2,2-dimethyl-6-(2-oxo-2-(o-tolylamino)ethoxy)-2H-Benzo [h] chromene-5-carboxylate (4g)
Methyl 2,2-dimethyl-6-(2-oxo-2-(m-tolylamino)ethoxy)-2H-benzo [h] chromene-5-carboxylate (4h)
Methyl 6-(2-((4-(cyanomethyl)phenyl)amino)-2-oxoethoxy)-2,2-dimethyl-2H-benzo [h] chromene-5-carboxylate (4i)
3.1.7. General Procedure for the Preparation of Target Compounds 6a–k
Methyl 6-(2-((1-benzylpiperidin-4-yl)amino)-2-oxoethoxy)-2,2-dimethyl-2H-benzo [h] chromene-5-carboxylate (6a)
Methyl 2,2-dimethyl-6-(2-((3-methylpyridin-4-yl)amino)-2-oxoethoxy)-2H-benzo [h] chromene-5-carboxylate (6b)
Methyl 6-(2-((4-chlorobenzyl)amino)-2-oxoethoxy)-2,2-dimethyl-2H-benzo [h] chromene-5-carboxylate (6c)
Methyl 2,2-dimethyl-6-(2-oxo-2-(quinolin-6-ylamino)ethoxy)-2H-benzo [h] Chromene-5-carboxylate (6d)
Methyl 2,2-dimethyl-6-(2-oxo-2-(pyridin-4-ylamino)ethoxy)-2H-benzo [h] chromene-5-carboxylate (6e)
Methyl 6-(2-(cyclopropylamino)-2-oxoethoxy)-2,2-dimethyl-2H-benzo [h] chromene-5-carboxylate (6f)
Methyl 6-(2-(benzhydrylamino)-2-oxoethoxy)-2,2-Dimethyl-2H-benzo [h] chromene-5-carboxylate (6g)
Methyl 2,2-dimethyl-6-(2-oxo-2-(pyrazin-2-ylamino)ethoxy)-2H-benzo [h] chromene-5-carboxylate (6h)
Methyl 2,2-dimethyl-6-(2-((6-methylpyridin-3-yl)amino)-2-oxoethoxy)-2H-benzo [h] chromene-5-carboxylate (6i)
Methyl 2,2-dimethyl-6-(2-oxo-2-(quinolin-8-ylamino)ethoxy)-2H-benzo [h] chromene-5-carboxylate (6j)
Methyl 2,2-dimethyl-6-(2-oxo-2-(quinolin-2-ylamino)ethoxy)-2H-benzo [h] chromene-5-carboxylate (6k)
3.1.8. General Procedure for the Preparation of Target Compounds 8a–c
Methyl 6-(2-bromoethoxy)-2,2-dimethyl-2H-benzo [h] chromene-5-carboxylate (8a)
Methyl 2,2-dimethyl-6-(2-(phenylamino)ethoxy)-2H-benzo[h]chromene-5-carboxylate (8b)
Methyl6-(2-((4-fluorophenyl)amino)ethoxy)-2,2-dimethyl-2H-benzo [h] chromene-5-carboxylate (8c)
3.2. Biological Evaluation
3.2.1. Cell Culture
3.2.2. Antibodies and Reagents
3.2.3. Plasmids, Transfections, and Luciferase Reporter Assay
3.2.4. Measurement of Cell Viability by MTT Assay
3.2.5. Western Blot Assay
3.3. Animals
Evaluation of Anti-Inflammatory Activity in Vivo
3.4. Prediction of ADMET Properties
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Serhan, C.N.; Savill, J. Resolution of inflammation: The beginning programs the end. Nat. Immunol. 2005, 6, 1191–1197. [Google Scholar] [CrossRef] [PubMed]
- Silva, L.M.C.M.; Lima, V.; Holanda, M.L.; Pinheiro, P.G.; Rodrigues, J.A.G.; Lima, M.E.P.; Benevides, N.M.B. Antinociceptive and anti-inflammatory activities of lectin from marine red alga Pterocladiella capillacea. Biol. Pharm. Bull. 2010, 33, 830–835. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Ma, S.; Zhang, T.-Y.; Wei, Z.-Y.; Wang, H.-M.; Guo, F.-Y.; Zheng, C.-J.; Piao, H.-R. Synthesis and biological evaluation of ursolic acid derivatives containing an aminoguanidine moiety. Med. Chem. Res. 2019, 28, 959–973. [Google Scholar] [CrossRef]
- Tanaka, T.; Narazaki, M.; Kishimoto, T. IL-6 in inflammation, immunity, and disease. Cold Spring Harb. Perspect. Biol. 2014, 6, 1–16. [Google Scholar] [CrossRef]
- Verma, S.; Jesus, P.D.; Chanda, S.K.; Verma, I.M. SNW1, a Novel Transcriptional Regulator of the NF-κB Pathway. Mol. Cell. Biol. 2019, 39, 415–418. [Google Scholar] [CrossRef]
- Lawrence, T. The nuclear factor NF-kappaB pathway in inflammation. Cold Spring Harb. Perspect. Biol. 2009, 1, 1–10. [Google Scholar] [CrossRef]
- Karin, M.; Ben-Neriah, Y. Phosphorylation meets ubiquitination: The control of NF-[kappa]B activity. Annu. Rev. Immunol. 2000, 18, 621–663. [Google Scholar] [CrossRef]
- Gilmore, T.D.; Herscovitch, M. Inhibitors of NF-kappaB signaling: 785 and counting. Oncogene 2006, 25, 6887–6899. [Google Scholar] [CrossRef]
- Lu, Y.; Liu, R.; Sun, C.; Pan, Y. An effective high-speed countercurrent chromatographic method for preparative isolation and purification of mollugin directly from the ethanol extract of the Chinese medicinal plant Rubia cordifolia. J. Sep. Sci. 2007, 30, 1313–1317. [Google Scholar] [CrossRef]
- Kim, K.J.; Lee, J.S.; Kwak, M.K.; Choi, H.G.; Yong, C.S.; Kim, J.A.; Lee, Y.R.; Lyoo, W.S.; Park, Y. Anti-inflammatory action of mollugin and its synthetic derivatives in HT-29 human colonic epithelial cells is mediated through inhibition of NF-kappaB activation. Eur. J. Pharmacol. 2009, 622, 52–57. [Google Scholar] [CrossRef]
- Kim, S.M.; Park, H.S.; Woo, H.J.; Woo, M.H.; Yang, C.H.; Kim, Y.H. Mollugin induces apoptosis in human Jurkat T cells through endoplasmic reticulum stress-mediated activation of JNK and caspase-12 and subsequent activation of mitochondria-dependent caspase cascade regulated by Bcl-xL. Toxicol. Appl. Pharmacol. 2009, 241, 210–220. [Google Scholar] [CrossRef] [PubMed]
- Jeong, G.-S.; Lee, D.-S.; Kim, D.-C.; Jahng, Y.; Son, J.-K.; Lee, S.-H.; Kim, Y.-C. Neuroprotective and anti-inflammatory effects of mollugin via up-regulation of heme oxygenase-1 in mouse hippocampal and microglial cells. Eur. J. Pharmacol. 2011, 654, 226–234. [Google Scholar] [CrossRef] [PubMed]
- Nishino, H.; Nakajima, Y.; Kakubari, Y.; Asami, N.; Deguchi, J.; Nugroho, A.E.; Hirasawa, Y.; Kaneda, T.; Kawasaki, Y.; Goda, Y.; et al. Syntheses and anti-inflammatory activity of azamollugin derivatives. Bioorganic Med. Chem. Lett. 2016, 26, 524–525. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhou, S.; Zhou, J.; Wang, D.; Zhou, T. Regulation of NF-κB/MAPK signaling pathway attenuates the acute lung inflammation in Klebsiella pneumonia rats by mollugin treatment. Microb. Pathog. 2019, 132, 369–373. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.; Han, D.-Z.; Jin, G.-Z. Study on the chemical constituents and anticancer activity from the roots of Rubia cordifolia L. Chin. J. Hosp. Pharm. 2012, 32, 1126–1128. [Google Scholar]
- Han, S.H.; Lee, K.S.; Park, E.H.; Park, J.D.; Kang, S.S.; Woo, S.Y.; Hong, K.B.; Kim, D.R.; Eun, B.R.; Cho, J.H. Preparation of Mollugin Derivatives for the Treatment of Inflammatory Bowel Disease. KR 10-2017-0122970, 7 November 2017. [Google Scholar]
- Ghosh, S.; May, M.J.; Kopp, E.B. NF-κB and Rel proteins: Evolutionarily conserved mediators of immune responses. Annu. Rev. Immunol. 1998, 16, 225–260. [Google Scholar] [CrossRef]
- Guo, Z.; Song, X.; Zhao, L.M.; Piao, M.G.; Quan, J.; Piao, H.R.; Jin, C.H. Synthesis and biological evaluation of novel benzo[c] [1,2,5]thiadiazol-5-yl and thieno [3,2-c]-pyridin-2-yl imidazole derivatives as ALK5 inhibitors. Bioorganic Med. Chem. Lett. 2019, 29, 2070–2075. [Google Scholar] [CrossRef]
- Liu, Y.M.; Feng, Y.D.; Lu, X.; Nie, J.B.; Li, W.; Wang, L.N.; Tian, L.J.; Liu, Q.H. Isosteroidal alkaloids as potent dual-binding site inhibitors of both acetylcholinesterase and butyrylcholinesterase from the bulbs of Fritillaria walujewii. Eur. J. Med. Chem. 2017, 137, 280–291. [Google Scholar] [CrossRef]
- Zhang, L.-H.; Zhang, Z.-H.; Li, M.-Y.; Wei, Z.-Y.; Jin, X.-J.; Piao, H.-R. Synthesis and evaluation of the HIF-1α inhibitory activities of novel ursolic acid tetrazole derivatives. Bioorganic Med. Chem. Lett. 2019, 29, 1440–1445. [Google Scholar] [CrossRef]
- Yang, A.; Li, M.Y.; Zhang, Z.H.; Wang, J.Y.; Xing, Y.; Ri, M.; Cheng, H.; Guang, H.; Lian, X.; Hong, L.; et al. Erianin regulates programmed cell death ligand 1 expression and enhances cytotoxic T lymphocyte activity. J. Ethnopharmacol. 2021, 273, 113598. [Google Scholar] [CrossRef]
- Wang, Z.; Li, M.Y.; Mi, C.; Wang, K.S.; Ma, J.; Jin, X. Mollugin Has an Anti-Cancer Therapeutic Effect by Inhibiting TNF-α-Induced NF-κB Activation. Int. J. Mol. Sci. 2017, 18, 1619. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | ||
|---|---|---|
| Compound | IC50 (μM) | |
| NF-κB a | Cytotoxicity b | |
| Mollugin | >100 | >100 |
| 4a | >100 | >100 |
| 4b | >100 | >100 |
| 4c | >100 | >100 |
| 4d | 88.25 | >100 |
| 4e | 55.12 | >100 |
| 4f | 18.53 | >100 |
| 4g | >100 | >100 |
| 4h | >100 | >100 |
| 4i | 22.93 | >100 |
 | ||
|---|---|---|
| Compound | IC50 (μM) | |
| NF-κB a | Cytotoxicity b | |
| 6a | 49.28 | >100 |
| 6b | 41.91 | >100 |
| 6c | 23.09 | >100 |
| 6d | 3.81 | >100 |
| 6e | >100 | >100 |
| 6f | >100 | >100 |
| 6g | >100 | >100 |
| 6h | 4.93 | >100 |
| 6i | >100 | >100 |
| 6j | 18.57 | >100 |
| 6k | >100 | >100 |
 | ||
|---|---|---|
| Compound | IC50 (μM) | |
| NF-κB a | Cytotoxicity b | |
| 8a | >100 | >100 |
| 8b | >100 | >100 |
| 8c | >100 | >100 |
| Dose (mg/kg) | Number of Mice | Edema Mean ± SD (mg) | Inhibition Rate (%) | |
|---|---|---|---|---|
| DMSO | 100 | 6 | 6.5333 ± 0.1795 | - |
| ibuprfen | 100 | 6 | 3.3167 ± 0.5550 *** | 49.75 |
| mesalazine | 100 | 6 | 3.2167 ± 1.1067 *** | 51.26 |
| mollugin | 100 | 6 | 3.0333 ± 0.9655 *** | 54.04 |
| 4a | 100 | 6 | 4.0500 ± 0.6188 *** | 38.64 |
| 4b | 100 | 6 | 2.9167 ± 0.8029 *** | 55.81 |
| 4c | 100 | 6 | 3.3500 ± 1.0579 *** | 49.24 |
| 4d | 100 | 6 | 2.5167 ± 0.6890 *** | 61.87 |
| 4e | 100 | 6 | 3.8667 ± 1.2023 *** | 41.41 |
| 4f | 100 | 6 | 1.1167 ± 0.2544 *** | 83.08 |
| 4g | 100 | 6 | 3.8000 ± 0.5066 *** | 42.42 |
| 4h | 100 | 6 | 2.4833 ± 1.5464 *** | 62.37 |
| 4i | 100 | 6 | 2.7000 ± 0.4619 *** | 59.09 |
| 6a | 100 | 6 | 2.8667 ± 1.2645 *** | 56.57 |
| 6b | 100 | 6 | 2.4333 ± 0.1795 *** | 63.13 |
| 6c | 100 | 6 | 1.7667 ± 0.3037 *** | 73.23 |
| 6d | 100 | 6 | 3.1000 ± 0.4899 *** | 76.77 |
| 6e | 100 | 6 | 2.2500 ± 0.3403 *** | 65.91 |
| 6f | 100 | 6 | 2.8167 ± 0.6962 *** | 57.32 |
| 6g | 100 | 6 | 2.7333 ± 0.4819 *** | 58.59 |
| 6h | 100 | 6 | 1.8000 ± 0.6880 *** | 72.73 |
| 6i | 100 | 6 | 3.7667 ± 1.1571 *** | 42.93 |
| 6j | 100 | 6 | 1.6333 ± 0.4384 *** | 75.25 |
| 6k | 100 | 6 | 2.1833 ± 0.6414 *** | 66.92 |
| 8a | 100 | 6 | 2.8167 ± 0.9370 *** | 57.32 |
| 8b | 100 | 6 | 1.5500 ± 0.3594 *** | 76.52 |
| 8c | 100 | 6 | 2.3833 ± 0.7104 *** | 63.89 |
| Time (h) | Dose (mg/kg) | Number of Mice | Inhibition(%) | ||
|---|---|---|---|---|---|
| 4f | 6d | Ibuprfen | |||
| 1 | 100 | 6 | 27.53% *** | 25.76% *** | 22.98% *** |
| 2 | 100 | 6 | 37.12% *** | 33.84% *** | 33.33% *** |
| 3 | 100 | 6 | 46.46% *** | 41.92% *** | 44.95% *** |
| 4 | 100 | 6 | 58.08% *** | 52.53% *** | 47.47% *** |
| 6 | 100 | 6 | 77.78% *** | 67.68% *** | 44.44% *** |
| 9 | 100 | 6 | 43.94% *** | 37.63% *** | 34.60% *** |
| 12 | 100 | 6 | 35.35% *** | 31.82% *** | 19.95% ** |
| 24 | 100 | 6 | 26.01% *** | 25.25% *** | 14.65% * |
| Time (h) | Dose (mg/kg) | Number of Mice | Inhibition(%) | ||
|---|---|---|---|---|---|
| 4f | 6d | Ibuprfen | |||
| 6 | 100 | 6 | 74.32% *** | 62.39% *** | 43.32% *** |
| 6 | 50 | 6 | 53.08% *** | 46.32% *** | 32.68% ** |
| 6 | 25 | 6 | 46.46% ** | 41.92% ** | 28.95% * |
| Compound | Absorption a | AS b | BBB c | CYP2D6 d | PPB e |
|---|---|---|---|---|---|
| 4f | 0 | −5.805 | 2 | FALSE | TRUE |
| 6d | 0 | −6.028 | 2 | FALSE | TRUE |
| mollugin | 0 | −4.383 | 2 | FALSE | TRUE |
 |  |  |
|---|---|---|
| Compound | R | Yield |
| Mollugin | — | — |
| 4a | H | 62% |
| 4b | 4-Cl | 70% |
| 4c | 4-Br | 65% |
| 4d | 4-OCH3 | 70% |
| 4e | 4-CH3 | 51% |
| 4f | 4-F | 55% |
| 4g | 2-CH3 | 67% |
| 4h | 3-CH3 | 72% |
| 4i | 4-CH2CN | 69% |
| 6a |  | 53% |
| 6b |  | 57% |
| 6c |  | 64% |
| 6d |  | 60% |
| 6e |  | 58% |
| 6f |  | 72% |
| 6g |  | 74% |
| 6h |  | 64% |
| 6i |  | 56% |
| 6j |  | 48% |
| 6k |  | 54% |
| 8a | Br | 80% |
| 8b | C6H5NH- | 77% |
| 8c | 4-FC6H5NH- | 69% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, L.-H.; Li, M.-Y.; Wang, D.-Y.; Jin, X.-J.; Chen, F.-E.; Piao, H.-R. Synthesis and Evaluation of NF-κB Inhibitory Activity of Mollugin Derivatives. Molecules 2022, 27, 7925. https://doi.org/10.3390/molecules27227925
Zhang L-H, Li M-Y, Wang D-Y, Jin X-J, Chen F-E, Piao H-R. Synthesis and Evaluation of NF-κB Inhibitory Activity of Mollugin Derivatives. Molecules. 2022; 27(22):7925. https://doi.org/10.3390/molecules27227925
Chicago/Turabian StyleZhang, Lin-Hao, Ming-Yue Li, Da-Yuan Wang, Xue-Jun Jin, Fen-Er Chen, and Hu-Ri Piao. 2022. "Synthesis and Evaluation of NF-κB Inhibitory Activity of Mollugin Derivatives" Molecules 27, no. 22: 7925. https://doi.org/10.3390/molecules27227925
APA StyleZhang, L.-H., Li, M.-Y., Wang, D.-Y., Jin, X.-J., Chen, F.-E., & Piao, H.-R. (2022). Synthesis and Evaluation of NF-κB Inhibitory Activity of Mollugin Derivatives. Molecules, 27(22), 7925. https://doi.org/10.3390/molecules27227925