
Methylglyoxal in the Brain: From Glycolytic Metabolite to Signalling Molecule


{kind=link}
{kind=link}
{kind=link}
Abstract
Highlights
- ●
- MGO may be essential for glycometabolism and bioenergetics in homeostasis and neural development;
- ●
- MGO may be an essential molecule in the regulation of neural homeostasis (redox homeostasis, lipid metabolism homeostasis, energy homeostasis, protein steady-state, epigenetic mechanisms, and neurotransmitters);
- ●
- Glycolysis is a source of protein homeostasis destruction. MGO formation as a by-product of glycolysis drives damage to the proteome.
Abstract
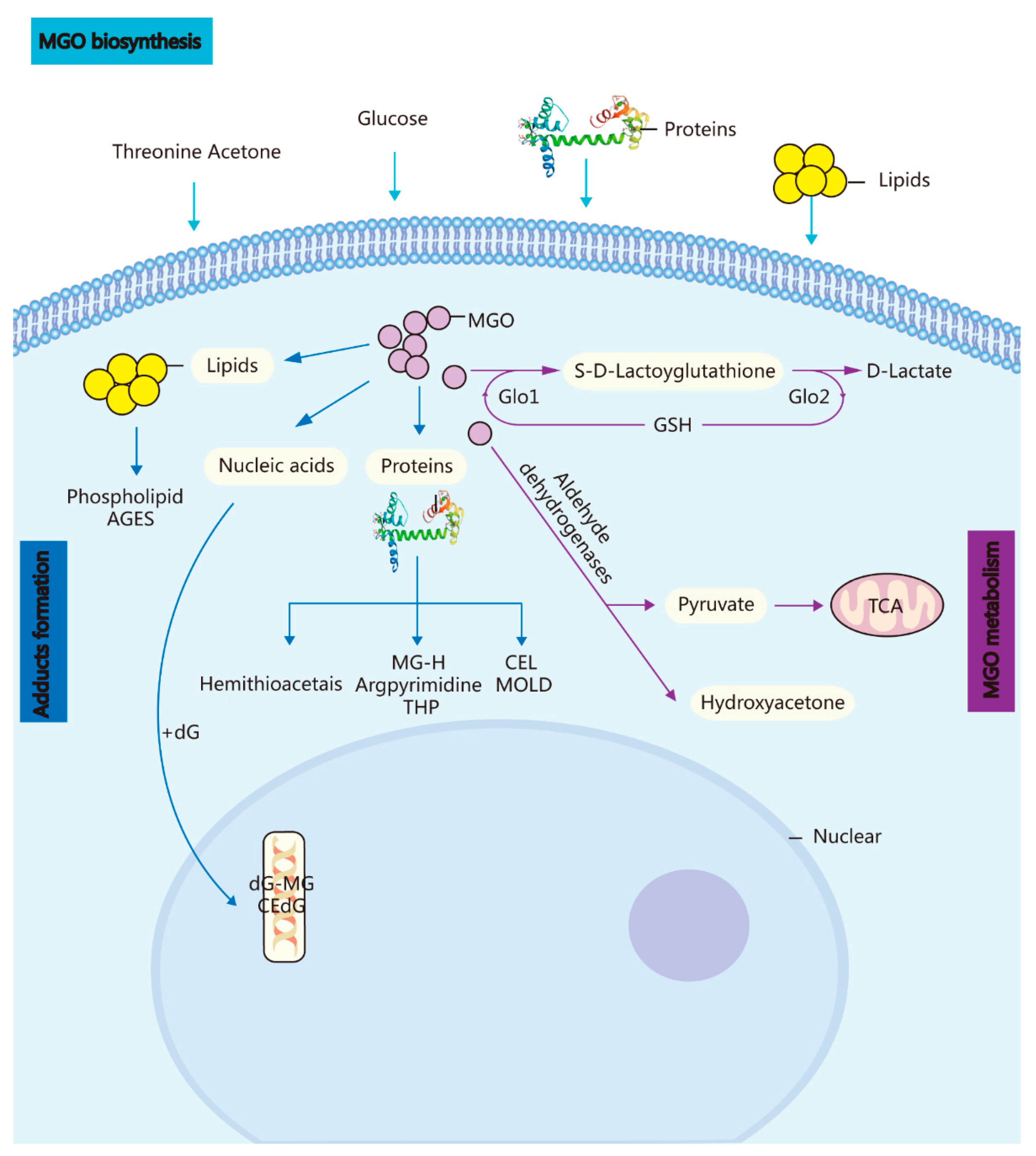
1. Methylglyoxal (A Metabolic Side-Product) and the GLO System In Vivo
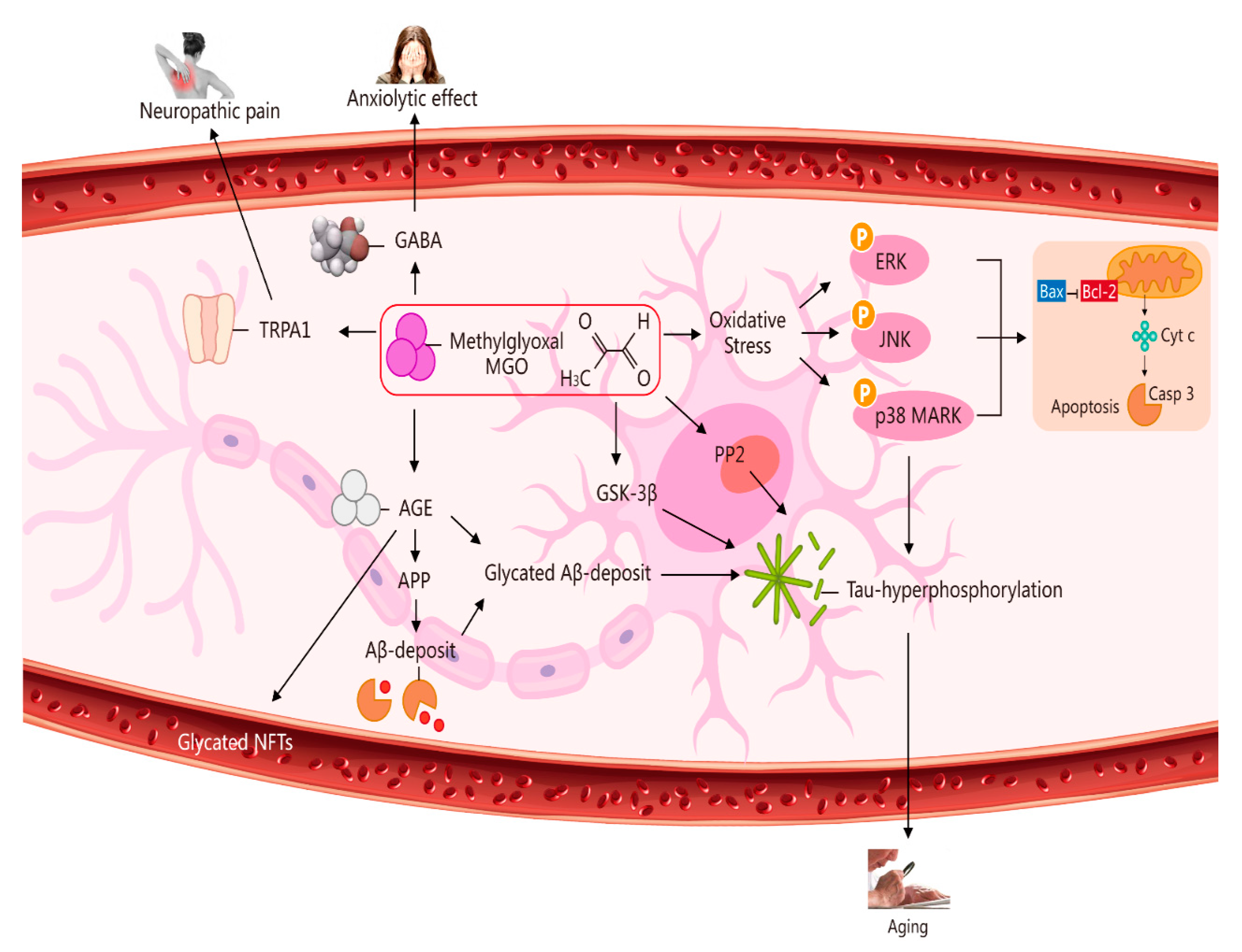
2. Potential Role of MGO in Ageing via Its Induction of Oxidative Stress, Neuropathic Pain, an Anxiolytic Effect, and Apoptosis [7]
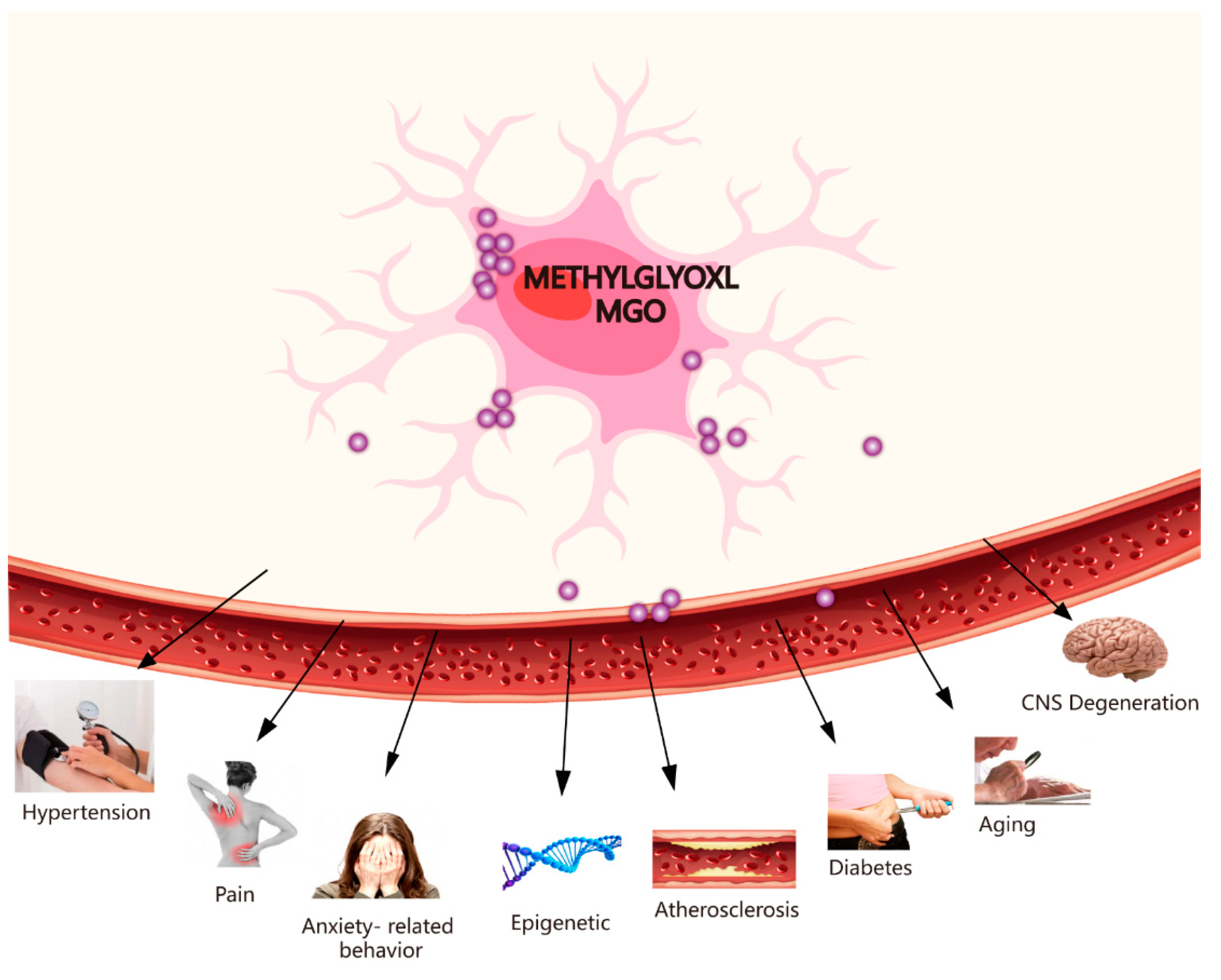
3. Toxicity of MGO and Some Diseases (e.g., CNS Degenerative Diseases, Pain, Hypertension, Ageing, Epigenetics, Diabetes-Related Neurological Complications, and Anxiety-Related Behaviour)
3.1. MGO-GLO System and CNS Degeneration
3.2. MGO and Diabetes-Relevant Neurological Complications
3.3. MGO and Inflammation
3.4. MGO and Anxiety-Related Behaviour
3.5. MGO and Neuropathic Pain
3.6. Other Mental Disorders and the MGO-Glo System
3.7. MGO and Atherosclerosis, Hypertension, Ageing, and Epigenetics
4. MGO Signalling Pathway: Role and Mechanism in Seizures
5. Potential Role of Small Molecules Such as MGO and GLO1 in Neuroprotection and Relevant Diseases
6. MGO and Homeostasis
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kalapos, M.P. The tandem of free radicals and methylglyoxal. Chem. Biol. Interact. 2008, 171, 251–271. [Google Scholar] [CrossRef] [PubMed]
- Thornalley, P.J.; Rabbani, N. Glyoxalase in tumourigenesis and multidrug resistance. Semin Cell Dev. Biol. 2011, 22, 318–325. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Chen, X. Glyoxalase II, a detoxifying enzyme of glycolysis byproduct methylglyoxal and a target of p63 and p73, is a pro-survival factor of the p53 family. J. Biol. Chem. 2006, 281, 26702–26713. [Google Scholar] [CrossRef] [PubMed]
- Shin, M.J.; Kim, D.W.; Lee, Y.P.; Ahn, E.H.; Jo, H.S.; Kim, D.S.; Kwon, O.S.; Kang, T.C.; Cho, Y.J.; Park, J.; et al. Tat-glyoxalase protein inhibits against ischemic neuronal cell damage and ameliorates ischemic injury. Free Radic. Biol. Med. 2014, 67, 195–210. [Google Scholar] [CrossRef]
- Bilova, T.; Paudel, G.; Shilyaev, N.; Schmidt, R.; Brauch, D.; Tarakhovskaya, E.; Milrud, S.; Smolikova, G.; Tissier, A.; Vogt, T.; et al. Global proteomic analysis of advanced glycation end products in the Arabidopsis proteome provides evidence for age-related glycation Hotspots. J. Biol. Chem. 2017, 22, 15758–15776. [Google Scholar] [CrossRef]
- Hasanuzzaman, M.; Hossain, M.A.; Fujita, M. Exogenous selenium pretreatment protects rapeseed seedlings from cadmium-induced oxidative stress by upregulating antioxidant defense and methylglyoxal detoxification systems. Biol. Trace Elem. Res. 2012, 149, 248–261. [Google Scholar] [CrossRef]
- Li, H.; Tang, Z.; Chu, P.; Song, Y.; Yang, Y.; Sun, B.; Niu, M.; Qaed, E.; Shopit, A.; Han, G.; et al. Neuroprotective effect of phosphocreatine on oxidative stress and mitochondrial dysfunction induced apoptosis in vitro and in vivo: Involvement of dual PI3K/Akt and Nrf2/HO-1 pathways. Free Radic. Biol. Med. 2018, 120, 228–238. [Google Scholar] [CrossRef]
- Rabbani, N.; Thornalley, P.J. Measurement of methylglyoxal by stable isotopic dilution analysis LC-MS/MS with corroborative prediction in physiological samples. Nat. Protoc. 2014, 9, 1969–1979. [Google Scholar] [CrossRef]
- Currais, A.; Maher, P. Functional consequences of age-dependent changes in glutathione status in the brain. Antioxid. Redox. Signal. 2012, 19, 813–822. [Google Scholar] [CrossRef]
- Volkenhoff, A.; Weiler, A.; Letzel, M.; Stehling, M.; Klämbt, C.; Schirmeier, S. Glial glycolysis is essential for neuronal survival in drosophila. Cell Metab. 2015, 22, 437–447. [Google Scholar] [CrossRef]
- Sada, N.; Lee, S.; Katsu, T.; Otsuki, T.; Inoue, T. Epilepsy treatment. Targeting LDH enzymes with a stiripentol analog to treat epilepsy. Science 2015, 347, 1362–1367. [Google Scholar] [CrossRef] [PubMed]
- Sotelo-Hitschfeld, T.; Niemeyer, M.I.; Mächler, P.; Ruminot, I.; Lerchundi, R.; Wyss, M.T.; Stobart, J.; Fernández-Moncada, I.; Valdebenito, R.; Garrido-Gerter, P.; et al. Channel-mediated lactate release by K(+)-stimulated astrocytes. J. Neurosci. 2015, 35, 4168–4178. [Google Scholar] [CrossRef] [PubMed]
- Pellerin, L.; Magistretti, P.J. Sweet sixteen for ANLS. J. Cereb. Blood Flow Metab. 2012, 32, 1152–1166. [Google Scholar] [CrossRef] [PubMed]
- Barros, L.F. Metabolic signaling by lactate in the brain. Trends Neurosci. 2013, 36, 396–404. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Song, M.; Yu, S.; Gao, P.; Yu, Y.; Wang, H.; Huang, L. Advanced glycation endproducts alter functions and promote apoptosis in endothelial progenitor cells through receptor for advanced glycation endproducts mediate overpression of cell oxidant stress. Mol. Cell Biochem. 2010, 335, 137–146. [Google Scholar] [CrossRef] [PubMed]
- Kaji, Y.; Amano, S.; Usui, T.; Oshika, T.; Yamashiro, K.; Ishida, S.; Suzuki, K.; Tanaka, S.; Adamis, A.P.; Nagai, R.; et al. Expression and function of receptors for advanced glycation end products in bovine corneal endothelial cells. Investig. Ophthalmol. Vis Sci. 2003, 44, 521–528. [Google Scholar] [CrossRef] [PubMed]
- Pieroh, P.; Wagner, D.-C.; Alessandri, B.; Nazari, M.D.; Ehrlich, A.; Ghadban, C.; Hobusch, C.; Birkenmeier, G.; Dehghani, F. Comparative Examination of Temporal Glyoxalase 1 Variations Following Perforant Pathway Transection, Excitotoxicity, and Controlled Cortical Impact Injury. Neurotox. Res. 2018, 33, 412–421. [Google Scholar] [CrossRef]
- Hovatta, I.; Hovatta, I.; Tennant, R.S.; Helton, R.; Marr, R.A.; Singer, O.; Redwine, J.M.; Ellison, J.A.; Schadt, E.E.; Verma, I.M.; et al. Glyoxalase 1 and glutathione reductase 1 regulate anxiety in mice. Nature 2005, 438, 662–666. [Google Scholar] [CrossRef]
- Distler, M.G.; Plant, L.D.; Sokoloff, G.; Hawk, A.J.; Aneas, I.; Wuenschell, G.E.; Termini, J.; Meredith, S.C.; Nobrega, M.A.; Palmer, A.A. Glyoxalase 1 increases anxiety by reducing GABAA receptor agonist methylglyoxal. J. Clin. Investig. 2012, 122, 2306–2315. [Google Scholar] [CrossRef]
- Hambsch, B.; Chen, B.-G.; Brenndörfer, J.; Meyer, M.; Avrabos, C.; Maccarrone, G.; Liu, R.H.; Eder, M.; Turck, C.W.; Landgraf, R. Methylglyoxal-mediated anxiolysis involves increased protein modification and elevated expression of glyoxalase 1 in the brain. J. Neurochem. 2010, 113, 1240–1251. [Google Scholar] [CrossRef]
- Thornalley, P.J. Pharmacology of methylglyoxal: Formation, modification of proteins and nucleic acids, and enzymatic detoxification—A role in pathogenesis and antiproliferative chemotherapy. Gen. Pharmacol. 1996, 27, 565–573. [Google Scholar] [CrossRef]
- Tye, K.M.; Deisseroth, K. Optogenetic investigation of neural circuits underlying brain disease in animal models. Nat. Rev. Neurosci. 2012, 13, 251–266. [Google Scholar] [CrossRef] [PubMed]
- Janak, P.H.; Tye, K.M. From circuits to behaviour in the amygdala. Nature 2015, 517, 284–292. [Google Scholar] [CrossRef] [PubMed]
- Heldt, S.; Ressler, K. Localized injections of midazolam into the amygdala and hippocampus induce differential changes in anxiolytic-like motor activity in mice. Behav. Pharmacol. 2006, 17, 349–356. [Google Scholar] [CrossRef]
- Vyas, A.; Chattarji, S. Modulation of different states of anxiety-like behavior by chronic stress. Behav. Neurosci. 2004, 118, 1450–1454. [Google Scholar] [CrossRef]
- Richarme, G.; Liu, C.; Mihoub, M.; Abdallah, J.; Leger, N.T.; Joly, N.; Liebart, J.-C.; Jurkunas, U.V.; Nadal, M.; Bouloc, P.; et al. Guanine glycation repair by DJ-1/Park7 and its bacterial homologs. Science 2017, 57, 208–211. [Google Scholar] [CrossRef]
- Sreejayan, N.; Yang, X.; Palanichamy, K.; Dolence, K.; Ren, J. Antioxidant properties of argpyrimidine. Eur. J. Pharmacol. 2008, 593, 30–35. [Google Scholar] [CrossRef]
- Jo-Watanabe, A.; Ohse, T.; Nishimatsu, H.; Takahashi, M.; Ikeda, Y.; Wada, T.; Shirakawa, J.; Nagai, R.; Miyata, T.; Nagano, T.; et al. Glyoxalase I reduces glycative and oxidative stress and prevents age-related endothelial dysfunction through modulation of endothelial nitric oxide synthase phosphorylation. Aging Cell 2014, 13, 519–528. [Google Scholar] [CrossRef]
- Li, W.; Maloney, R.E.; Circu, M.L.; Alexander, J.S.; Aw, T.Y. Acute carbonyl stress induces occludin glycation and brain microvascular endothelial barrier dysfunction: Role for glutathione-dependent metabolism of methylglyoxal. Free Radic. Biol. Med. 2013, 54, 51–61. [Google Scholar] [CrossRef]
- Miyazawa, N.; Abe, M.; Souma, T.; Tanemoto, M.; Abe, T.; Nakayama, M.; Ito, S. Methylglyoxal augments intracellular oxidative stress in human aortic endothelial cells. Free Radic. Res. 2010, 44, 101–107. [Google Scholar] [CrossRef]
- Lin, M.T.; Beal, M.F. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 2006, 443, 787–795. [Google Scholar] [CrossRef] [PubMed]
- Praticò, D. Oxidative stress hypothesis in Alzheimer’s disease: A reappraisal. Trends Pharmacol. Sci. 2008, 29, 609–615. [Google Scholar] [CrossRef] [PubMed]
- Grillo, M.A.; Colombatto, S. Advanced glycation end-products (AGEs): Involvement in aging and in neurodegenerative diseases. Amino Acids. 2008, 35, 29–36. [Google Scholar] [CrossRef] [PubMed]
- Jack, M.; Wright, D. Role of advanced glycation endproducts and glyoxalase I in diabetic peripheral sensory neuropathy. Transl. Res. 2012, 159, 355–365. [Google Scholar] [CrossRef]
- Beeri, M.S.; Moshier, E.; Schmeidler, J.; Godbold, J.; Uribarri, J.; Reddy, S.; Sano, M.; Grossman, H.T.; Weijing Cai, W.; Vlassara, H.; et al. Serum concentration of an inflammatory glycotoxin, methylglyoxal, is associated with increased cognitive decline in elderly individuals. Mech. Ageing Dev. 2011, 132, 583–587. [Google Scholar] [CrossRef] [PubMed]
- Sousa Silva, M.; Gomes, R.A.; Ferreira, A.E.N.; Ponces, F.A.; Cordeiro, C. The glyoxalase pathway: The first hundred years... and beyond. Biochem. J. 2013, 453, 1–15. [Google Scholar] [CrossRef]
- Xue, M.; Rabbani, N.; Momiji, H.; Imbasi, P.; Anwar, M.M.; Kitteringham, N.; Park, B.K.; Souma, T.; Moriguchi, T.; Yamamoto, M.; et al. Transcriptional control of glyoxalase 1 by Nrf2 provides a stress-responsive defence against dicarbonyl glycation. Biochem. J. 2012, 443, 213–222. [Google Scholar] [CrossRef]
- Eberhardt, M.J.; Filipovic, M.R.; Leffler, A.; Roche, J.D.L.; Kistner, K.; Fischer, M.J.; Fleming, T.; Zimmermann, K.; Ivanovic-Burmazovic, I.; Nawroth, P.P.; et al. Methylglyoxal activates nociceptors through transient receptor ootential channel A1 (TRPA1). A possible mechanism of metabolic neuropathies. J. Biol. Chem. 2012, 287, 28291–28306. [Google Scholar] [CrossRef]
- Chen, K.; Maley, J.; Yu, P.H. Potential inplications of endogenous aldehydes in beta-amyloid misfolding, oligomerization and fibrillogenesis. J. Neurochem. 2006, 99, 1413–1424. [Google Scholar] [CrossRef]
- Schipper, H.M. Apolipoprotein E: Implications for AD neurobiology, epidemiology and risk assessment. Neurobiol. Aging 2011, 32, 778–790. [Google Scholar] [CrossRef]
- Bu, G. Apolipoprotein e and its receptors in Alzheimer’s disease: Pathways, pathogenesis and therapy. Nat. Rev. Neurosci. 2009, 10, 333–344. [Google Scholar] [CrossRef] [PubMed]
- Kok, E.; Haikonen, S.; Luoto, T.; Huhtala, H.; Goebeler, S.; Haapasalo, H.; Karhunen, P.J. Apolipoprotein E-dependent accumulation of alzheimer disease-related lesions begins in middle age. Ann. Neurol. 2009, 65, 650–657. [Google Scholar] [PubMed]
- Polvikoski, T.; Sulkava, R.; Haltia, M.; Kainulainen, K.; Vuorio, A.; Verkkoniemi, A.; Niinistö, L.; Halonen, P.; Kontula, K. Apolipoprotein E, dementia, and cortical deposition of beta-amyloid protein. N. Engl. J. Med. 1995, 333, 1242–1247. [Google Scholar] [CrossRef] [PubMed]
- Rabbani, N.; Thornalley, P.J. Methylglyoxal, glyoxalase 1 and the dicarbonyl proteome. Amino Acids. 2012, 42, 1133–1142. [Google Scholar] [PubMed]
- Wautier, J.L.; Guillausseau, P.J. Advanced glycation end products, their receptors and diabetic angiopathy. Diabetes Metab. 2001, 27, 535–542. [Google Scholar] [CrossRef]
- Münch, G.; Westcott, B.; Menini, T.; Gugliucci, A. Advanced glycation end products and their pathogenic roles in neurological disorders. Amino Acids. 2012, 42, 1221–1236. [Google Scholar] [CrossRef]
- Lüth, H.J.; Ogunlade, V.; Kuhla, B.; Kientsch-Engel, R.; Stahl, P.; Webster, J.; Arendt, T.; Münch, G. Age- and stage-dependent accumulation of advanced glycation end products in intracellular deposits in normal and Alzheimer’s disease brains. Cereb. Cortex. 2005, 15, 211–220. [Google Scholar] [CrossRef]
- Boeck, K.; Schmidt, A.; Ogunlade, V.; Arendt, T.; Münch, G.; Lüth, H.-J. Age- and stage-dependent glyoxalase I expression and its activity in normal and Alzheimer’s disease brains. Neurobiol. Aging 2007, 28, 29–41. [Google Scholar]
- Lee, J.Y.; Song, J.; Kwon, K.; Jang, S.; Kim, C.; Baek, K.; Kim, J.; Park, C. Human DJ-1 and its homologs are novel glyoxalases. Hum. Mol. Genet. 2012, 21, 3215–3225. [Google Scholar] [CrossRef]
- Vicente, M.H.; Szego, É.M.; Oliveira, L.M.A.; Breda, C.; Darendelioglu, E.; de Oliveira, R.M.; Ferreira, D.G.; Gomes, M.A.; Rott, R.; Oliveira, M.; et al. Glycation potentiates α-synuclein-associated neurodegeneration in synucleinopathies. Brain 2017, 140, 1399–1419. [Google Scholar] [CrossRef]
- Mostafa, A.A.; Randell, E.W.; Vasdev, S.C.; Gill, V.D.; Han, Y.; Gadag, V.; Ahmed, A.; Raouf, A.A.; Said, H.E. Plasma protein advanced glycation end products, carboxymethyl cysteine, and carboxyethyl cysteine, are elevated and related to nephropathy in patients with diabetes. Mol. Cell Biochem. 2007, 302, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Biosa, A.; Outeiro, T.F.; Bubacco, L.; Bisaglia, M. Diabetes Mellitus as a Risk Factor for Parkinson’s Disease: A Molecular Point of View. Mol. Neurobiol. 2018, 55, 8754–8763. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.; Cancino, G.I.; Zahr, S.K.; Guskjolen, A.; Voronova, A.; Gallagher, D.; Frankland, P.W.; Kaplan, D.R.; Miller, F.D. A Glo1-Methylglyoxal Pathway that Is Perturbed in Maternal Diabetes Regulates Embryonic and Adult Neural Stem Cell Pools in Murine Offspring. Cell Rep. 2016, 17, 1022–1036. [Google Scholar] [CrossRef]
- Khan, M.A.; Schultz, S.; Othman, A.; Fleming, T.; Lebrón-Galán, R.; Rades, D.; Clemente, D.; Nawroth, P.P.; Schwaninger, M. Hyperglycemia in Stroke Impairs Polarization of Monocytes/Macrophages to a Protective Noninflammatory Cell Type. J. Neurosci. 2016, 36, 9313–9325. [Google Scholar] [CrossRef]
- Morcos, M.; Du, X.; Pfisterer, F.; Hutter, H.; Sayed, A.A.R.; Thornalley, P.; Ahmed, N.; Baynes, J.; Thorpe, S.; Kukudov, G. Glyoxalase-1 prevents mitochondrial protein modification and enhances lifespan in Caenorhabditis elegans. Aging Cell 2008, 7, 260–269. [Google Scholar] [CrossRef] [PubMed]
- Fleming, T.H.; Theilen, T.-M.; Masania, J.; Marius Wunderle, M.; Jamshid Karimi, J.; Vittas, S.; Bernauer, R.; Bierhaus, A.; Rabbani, N.; Thornalley, P.L.; et al. Aging-dependent reduction in glyoxalase 1 delays wound healing. Gerontology 2013, 59, 427–437. [Google Scholar] [CrossRef]
- Ikeda, Y.; Inagi, R.; Miyata, T.; Nagai, R.; Arai, M.; Miyashita, M.; Itokawa, M.; Fujita, T.; Nangaku, M. Glyoxalase I retards renal senescence. Am. J. Pathol. 2011, 179, 2810–2821. [Google Scholar] [CrossRef]
- Gu, Q.; Wang, B.; Zhang, X.F.; Ma, Y.P.; Liu, J.D.; Wang, X.Z. Contribution of receptor for advanced glycation end products to vasculature-protecting effects of exercise training in aged rats. Eur. J. Pharmacol. 2014, 741, 186–194. [Google Scholar] [CrossRef]
- Xue, M.; Rabbani, N.; Thornalley, P.J. Glyoxalase in ageing. Semin. Cell Dev. Biol. 2011, 22, 293–301. [Google Scholar] [CrossRef]
- Kollmannsberger, L.K.; Gassen, N.C.; Bultmann, A.; Hartmann, J.; Weber, P.; Schmidt, M.V.; Rein, T. Increased glyoxalase-1 levels in Fkbp5 knockout mice caused by glyoxalase-1 gene duplication. G3 (Bethesda) 2013, 3, 1311–1313. [Google Scholar] [CrossRef]
- Reiner-Benaim, A.; Yekutieli, D.; Letwin, N.E.; Elmer, G.I.; Lee, N.H.; Kafkafi, N.; Benjamini, Y. Associating quantitative behavioral traits with gene expression in the brain: Searching for diamonds in the hay. Bioinformatics 2007, 23, 2239–2246. [Google Scholar] [CrossRef] [PubMed]
- Loos, M.; Sluis, S.V.D.; Bochdanovits, Z.; Zutphen, I.J.V.; Pattij, T.; Stiedl, O.; Neuro-BSIK Mouse Phenomics Consortium; Smit, A.B.; Spijker, S. Activity and impulsive action are controlled by different genetic and environmental factors. Genes Brain Behav. 2009, 8, 817–828. [Google Scholar] [CrossRef] [PubMed]
- Distler, M.G.; Palmer, A.A. Role of Glyoxalase 1 (Glo1) and methylglyoxal (MG) in behavior: Recent advances and mechanistic insights. Front Genet. 2012, 3, 250. [Google Scholar] [CrossRef] [PubMed]
- Jack, M.M.; Ryals, J.M.; Wright, D.E. Characterisation of glyoxalase I in a streptozocin-induced mouse model of diabetes with painful and insensate neuropathy. Diabetologia 2011, 54, 2174–2182. [Google Scholar] [CrossRef]
- Bierhaus, A.; Fleming, T.; Stoyanov, S.; Leffler, A.; Babes, A.; Neacsu, C.; Sauer, S.K.; Eberhardt, M.; Schnölzer, M.; Lasitschka, F. Methylglyoxal mo.dification of Na(v)1.8 facilitates nociceptive neuron firing and causes hyperalgesia in diabetic neuropathy. Nat. Med. 2012, 18, 926–933. [Google Scholar] [CrossRef]
- Zhang, Y.; Filiou, M.D.; Reckow, S.; Gormanns, P.; Maccarrone, G.; Kessler, M.S.; Frank, E.; Hambsch, B.; Holsboer, F.; Landgraf, R.; et al. Proteomic and metabolomic profiling of a trait anxiety mouse model implicate affected pathways. Mol. Cell. Proteom. 2011, 10, M111.008110. [Google Scholar] [CrossRef]
- Shinohara, M.; Thornalley, P.J.; Giardino, I.; Beisswenger, P.; Thorpe, S.R.; Onorato, J.; Brownlee, M. Overexpression of glyoxalase-I in bovine endothelial cells inhibits intracellular advanced glycation endproduct formation and prevents hyperglycemia-induced increases in macromolecular endocytosis. J. Clin. Investig. 1998, 101, 1142–1147. [Google Scholar] [CrossRef]
- Duran-Jimenez, B.; Dobler, D.; Moffatt, S.; Rabbani, N.; Charles, H.; Streuli, C.H.; Thornalley, P.J.; Tomlinson, D.R.; Gardiner, N.J. Advanced glycation end products in extracellular matrix proteins contribute to the failure of sensory nerve regeneration in diabetes. Diabetes 2009, 58, 2893–2903. [Google Scholar] [CrossRef]
- Rehnström, K.; Ylisaukko-Oja, T.; Vanhala, R.; Wendt, L.V.; Peltonen, L.; Hovatta, I. No association between common variants in glyoxalase 1 and autism spectrum disorders. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2008, 147B, 124–127. [Google Scholar] [CrossRef]
- Silverman, J.L.; Yang, M.; Lord, C.; Crawley, J.N. Behavioural phenotyping assays for mouse models of autism. Nat. Rev. Neurosci. 2010, 11, 490–502. [Google Scholar] [CrossRef]
- Toyosima, M.; Maekawa, M.; Toyota, T.; Iwayama, Y.; Arai, M.; Ichikawa, T.; Miyashita, M.; Arinami, T.; Itokawa, M.; Yoshikawa, T. Schizophrenia with the 22q11.2 deletion and additional genetic defects: Case history. Br. J. Psychiatry 2011, 199, 245–246. [Google Scholar] [CrossRef] [PubMed]
- Arai, M.; Yuzawa, H.; Nohara, I.; Ohnishi, T.; Obata, N.; Iwayama, Y.; Haga, S.; Toyota, T.; Ujike, H.; Arai, M.; et al. Enhanced carbonyl stress in a subpopulation of schizophrenia. Arch. Gen. Psychiatry 2010, 67, 589–597. [Google Scholar] [CrossRef] [PubMed]
- Saremi, A.; Howell, S.; Schwenke, D.C.; Bahn, G.; Beisswenger, P.J.; Reaven, P.D.; VADT Investigators. Advanced Glycation End Products, Oxidation Products, and the Extent of Atherosclerosis During the VA Diabetes Trial and Follow-up Study. Diabetes Care 2017, 40, 591–598. [Google Scholar] [CrossRef] [PubMed]
- Tao, H.; Zhou, X.; Zhao, B.; Li, K. Conflicting Effects of Methylglyoxal and Potential Significance of miRNAs for Seizure Treatment. Front. Mol. Neurosci. 2018, 5, 11–70. [Google Scholar] [CrossRef] [PubMed]
- Distler, M.G.; Gorfinkle, N.; Ligia, A.; Papale, L.A.; Gerald, E.; Wuenschell, G.E.; Termini, J.; Escayg, A.; Winawer, M.R.; Abraham, A.; et al. Glyoxalase 1 and its substrate methylglyoxal are novel regulators of seizure susceptibility. Epilepsia 2013, 54, 649–657. [Google Scholar] [CrossRef]
- Macdonald, R.L.; Kang, J.Q.; Gallagher, M.J. Mutations in GABAA receptor subunits associated with genetic epilepsies. J. Physiol. 2010, 588, 1861–1869. [Google Scholar] [CrossRef]
- Lv, Q.; Gu, C.; Chen, C. Venlafaxine protects methylglyoxal-induced apoptosis in the cultured human brain microvascular endothelial cells. Neurosci. Lett. 2014, 569, 99–103. [Google Scholar] [CrossRef]
- Scott, G.F.; Nguyen, A.Q.; Cherry, B.H.; Roger, A.; Hollrah, R.A.; Salinas, I.; Arthur, G.W., Jr.; Myoung-Gwi, R.; Mallet, R.T. Featured Article: Pyruvate preserves antiglycation defenses in porcine brain after cardiac arrest. Exp. Biol. Med. 2017, 242, 1095–1103. [Google Scholar] [CrossRef]
- Thornalley, P.J. Glyoxalase I -Structure, function and a critical role in the enzymatic defence against glycation. Biochem. Soc. Trans. 2003, 31, 1343–1348. [Google Scholar] [CrossRef]
- Wang, B.; Yee Aw, T.; Stokes, K.Y. N-acetylcysteine attenuates systemic platelet activation and cerebral vessel thrombosis in diabetes. Redox. Biol. 2018, 14, 218–228. [Google Scholar] [CrossRef]
- Hipkiss, A.R. Aging risk factors and Parkinson’s disease: Contrasting roles of common dietary constituents. Neurobiol. Aging 2014, 35, 1469–1472. [Google Scholar] [CrossRef] [PubMed]
- Williams, M.E.; Bolton, W.K.; Khalifah, R.G.; Degenhardt, T.P.; Schotzinger, R.J.; McGill, J.B. Effects of pyridoxamine in combined phase 2 studies of patients with type 1 and type 2 diabetes and overt nephropathy. Am. J. Nephrol. 2007, 27, 605–614. [Google Scholar] [CrossRef] [PubMed]
- Kanazawa, I.; Yamamoto, M.; Yamaguchi, T.; Sugimoto, T. Effects of Metformin and Pioglitazone on Serum Pentosidine Levels in Type 2 Diabetes Mellitus. Exp. Clin. Endocrinol. Diabetes 2011, 119, 362–365. [Google Scholar] [CrossRef] [PubMed]
- Engelen, L.; Lund, S.S.; Ferreira, I.; Tarnow, L.; Parving, H.H.; Gram, J.; Winther, K.; Pedersen, O.; Teerlink, T.; Barto, R.; et al. Improved glycemic control induced by both metformin and repaglinide is associated with a reduction in blood levels of 3-deoxyglucosone in nonobese patients with type 2 diabetes. Eur. J. Endocrinol. 2011, 164, 371–379. [Google Scholar] [CrossRef] [PubMed]
- Desai, K.; Wu, L. Methylglyoxal and advanced glycation endproducts: New therapeutic horizons? Recent Pat. Cardiovasc. Drug Discov. 2007, 2, 89–99. [Google Scholar] [CrossRef] [PubMed]
- Yan, S.F.; Ramasamy, R.; Schmidt, A.M. Soluble RAGE: Therapy and biomarker in unraveling the RAGE axis in chronic disease and aging. Biochem. Pharmacol. 2010, 79, 1379–1386. [Google Scholar] [CrossRef]
- Baynes, J.W.; Thorpe, S.R. Glycoxidation and lipoxidation in atherogenesis. Free Radic. Biol. Med. 2000, 28, 1708–1716. [Google Scholar] [CrossRef]
- Gomes, R.A.; Silva, M.S.; Miranda, V.H.; Ferreira, A.E.N.; Cordeiro, C.A.A.; Freire, A.P. Protein glycation in Saccharomyces cerevisiae. Argpyrimidine formation and methylglyoxal catabolism. FEBS J. 2005, 272, 4521–4531. [Google Scholar] [CrossRef]
- Yao, D.; Brownlee, M. Hyperglycemia-induced reactive oxygen species increase expression of the receptor for advanced glycation end products (RAGE) and RAGE ligands. Diabetes 2010, 59, 249–255. [Google Scholar] [CrossRef]
- Kuhla, B.; Boeck, K.; Lüth, H.J.; Schmidt, A.; Weigle, B.; Schmitz, M.; Ogunlade, V.; Münch, G.; Arendt, T. Age-dependent changes of glyoxalase I expression in human brain. Neurobiol. Aging 2006, 27, 815–822. [Google Scholar] [CrossRef]
- Antognelli, C.; Trapani, E.; Delle Monache, S.; Perrelli, A.; Daga, M.; Pizzimenti, S.; Barrera, G.; Cassoni, P.; Angelucci, A.; Trabalzini, L.; et al. KRIT1 loss-of-function induces a chronic Nrf2-mediated adaptivehomeostasisthat sensitizes cells to oxidative stress: Implication for Cerebral Cavernous Malformation disease. Free Radic. Biol. Med. 2018, 115, 202–218. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Hu, F.; Wu, J.; Zhang, S. Cannabidiol attenuates OGD/R-induced damage by enhancing mitochondrial bioenergetics and modulating glucose metabolism via pentose-phosphate pathway in hippocampal neurons. Redox. Biol. 2017, 11, 577–585. [Google Scholar] [CrossRef] [PubMed]
- Barkley-Levenson, A.M.; Lagarda, F.A.; Palmer, A.A. Glyoxalase 1 (GLO1) Inhibition or Genetic Overexpression Does Not Alter Ethanol’s Locomotor Effects: Implications for GLO1 as a Therapeutic Target in Alcohol Use Disorders. Alcohol. Clin. Exp. Res. 2018, 42, 869–878. [Google Scholar] [CrossRef]
- Jiang, B.; Le, L.; Liu, H.; Xu, L.; He, C.; Hu, K.; Peng, Y.; Xiao, P. Marein protects against methylglyoxal-induced apoptosis by activating the AMPK pathway in PC12cells. Free Radic. Res. 2016, 50, 1173–1187. [Google Scholar] [CrossRef] [PubMed]
- Santarius, T.; Bignell, G.R.; Greenman, C.D.; Widaa, S.; Chen, L.; Mahoney, C.L.; Butler, A.; Edkins, S.; Waris, S.; Thornalley, P.J.; et al. GLO1-A novel amplified gene in human cancer. Genes Chromosomes Cancer 2010, 49, 711–725. [Google Scholar] [CrossRef] [PubMed]
- Qi, W.; Keenan, H.A.; Li, Q.; Ishikado, A.; Kannt, A.; Sadowski, T.; Yorek, M.A.; Wu, I.-H.; Lockhart, S.; Coppey, L.J.; et al. Pyruvate kinase M2 activation may protect against the progression of diabetic glomerular pathology and mitochondrial dysfunction. Nat. Med. 2017, 23, 753–762. [Google Scholar] [CrossRef]
- Chaudhuri, J.; Bains, Y.; Guha, S.; Kahn, A.; Hall, D.; Bose, N.; Gugliucci, A.; Kapahi, P. The Role of Advanced Glycation End Products in Aging and Metabolic Diseases: Bridging Association and Causality. Cell Metab. 2018, 28, 337–352. [Google Scholar] [CrossRef]
- Galligan, J.J.; Wepy, J.A.; Streeter, M.D.; Kingsley, P.J.; Mitchener, M.M.; Wauchope, O.R.; Beavers, W.N.; Rose, K.L.; Wang, T.; Spiegel, D.A.; et al. Methylglyoxal-derived post Transl. ational arginine modifications are abundant histone marks. Proc. Natl. Acad. Sci. USA 2018, 115, 9228–9233. [Google Scholar] [CrossRef]
- Bollong, M.J.; Lee, G.; Coukos, J.S.; Yun, H.; Zambaldo, C.; Chang, J.W.; Chin, E.N.; Ahmad, I.; Chatterjee, A.K.; Lairson, L.L.; et al. A metabolite-derived protein modification integrates glycolysis with KEAP1-NRF2 signalling. Nature 2018, 562, 600–604. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, Z.; Zhang, W.; Lu, H.; Cai, S. Methylglyoxal in the Brain: From Glycolytic Metabolite to Signalling Molecule. Molecules 2022, 27, 7905. https://doi.org/10.3390/molecules27227905
Yang Z, Zhang W, Lu H, Cai S. Methylglyoxal in the Brain: From Glycolytic Metabolite to Signalling Molecule. Molecules. 2022; 27(22):7905. https://doi.org/10.3390/molecules27227905
Chicago/Turabian StyleYang, Zeyong, Wangping Zhang, Han Lu, and Shu Cai. 2022. "Methylglyoxal in the Brain: From Glycolytic Metabolite to Signalling Molecule" Molecules 27, no. 22: 7905. https://doi.org/10.3390/molecules27227905
APA StyleYang, Z., Zhang, W., Lu, H., & Cai, S. (2022). Methylglyoxal in the Brain: From Glycolytic Metabolite to Signalling Molecule. Molecules, 27(22), 7905. https://doi.org/10.3390/molecules27227905