
Promising Oxygen- and Nitrogen-Rich Azidonitramino Ether Plasticizers for Energetic Materials

 , ,
, ,
Abstract
1. Introduction
2. Results and Discussion
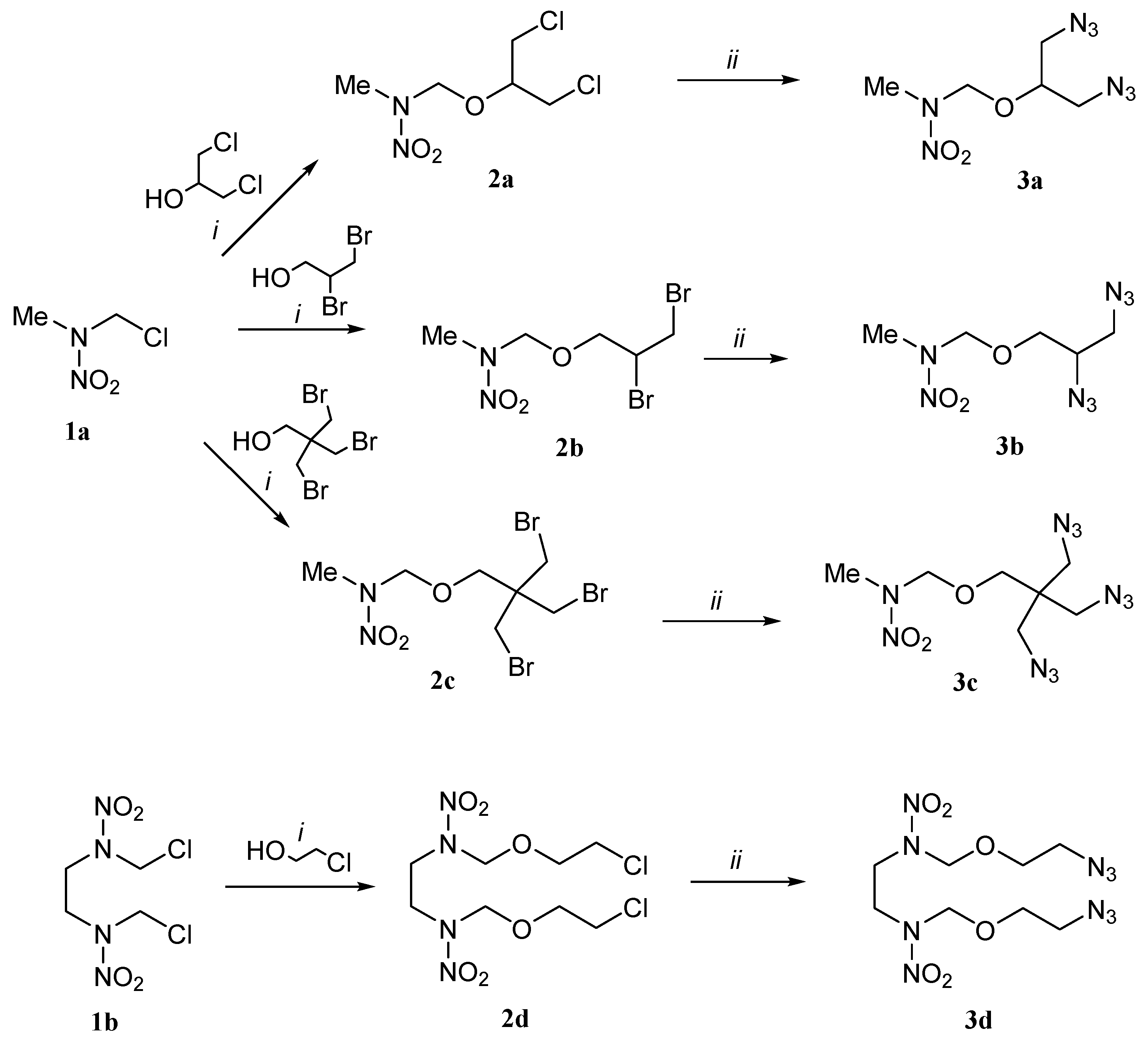
2.1. Synthesis
2.2. Sensitivity Measurements
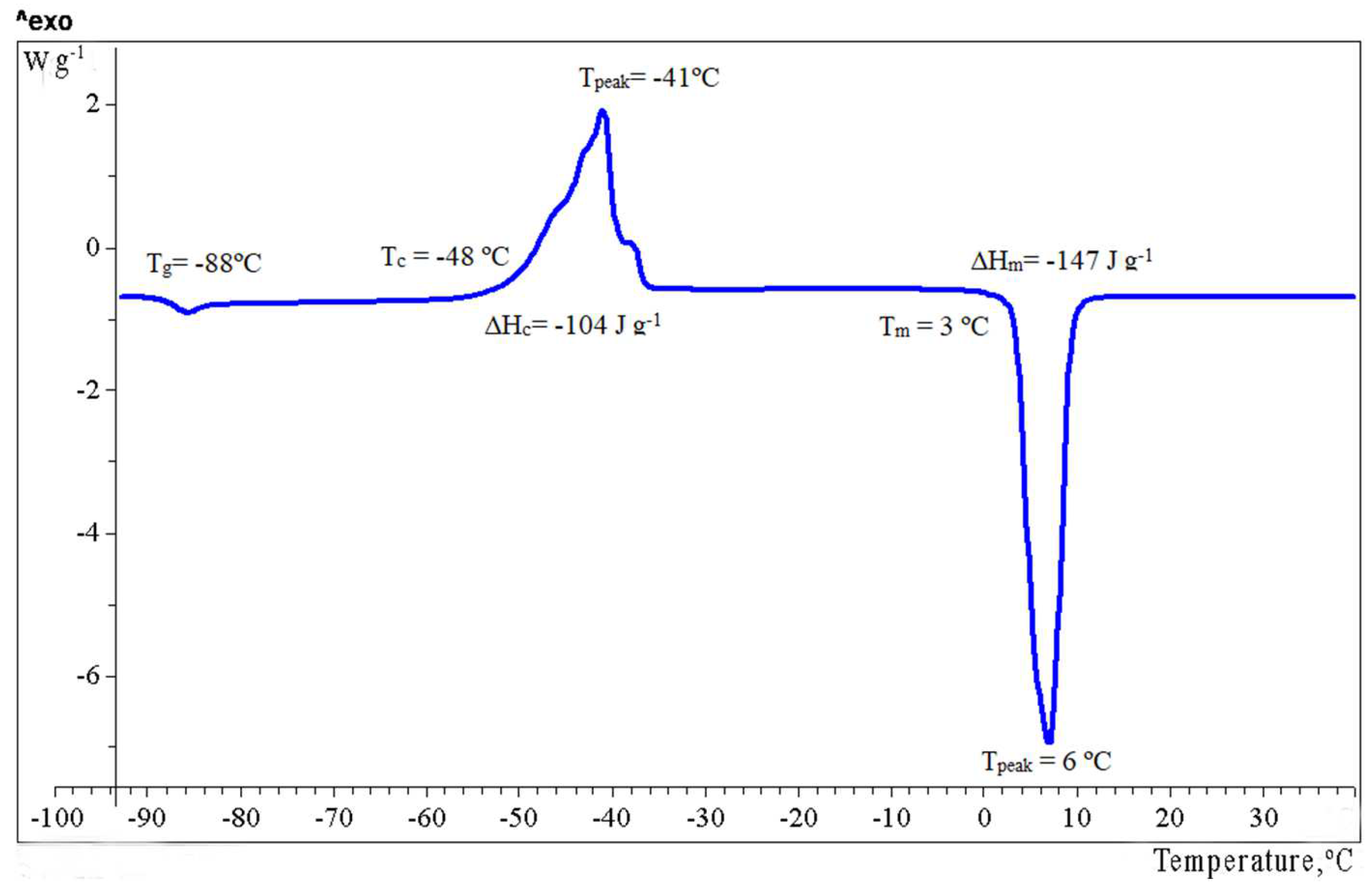
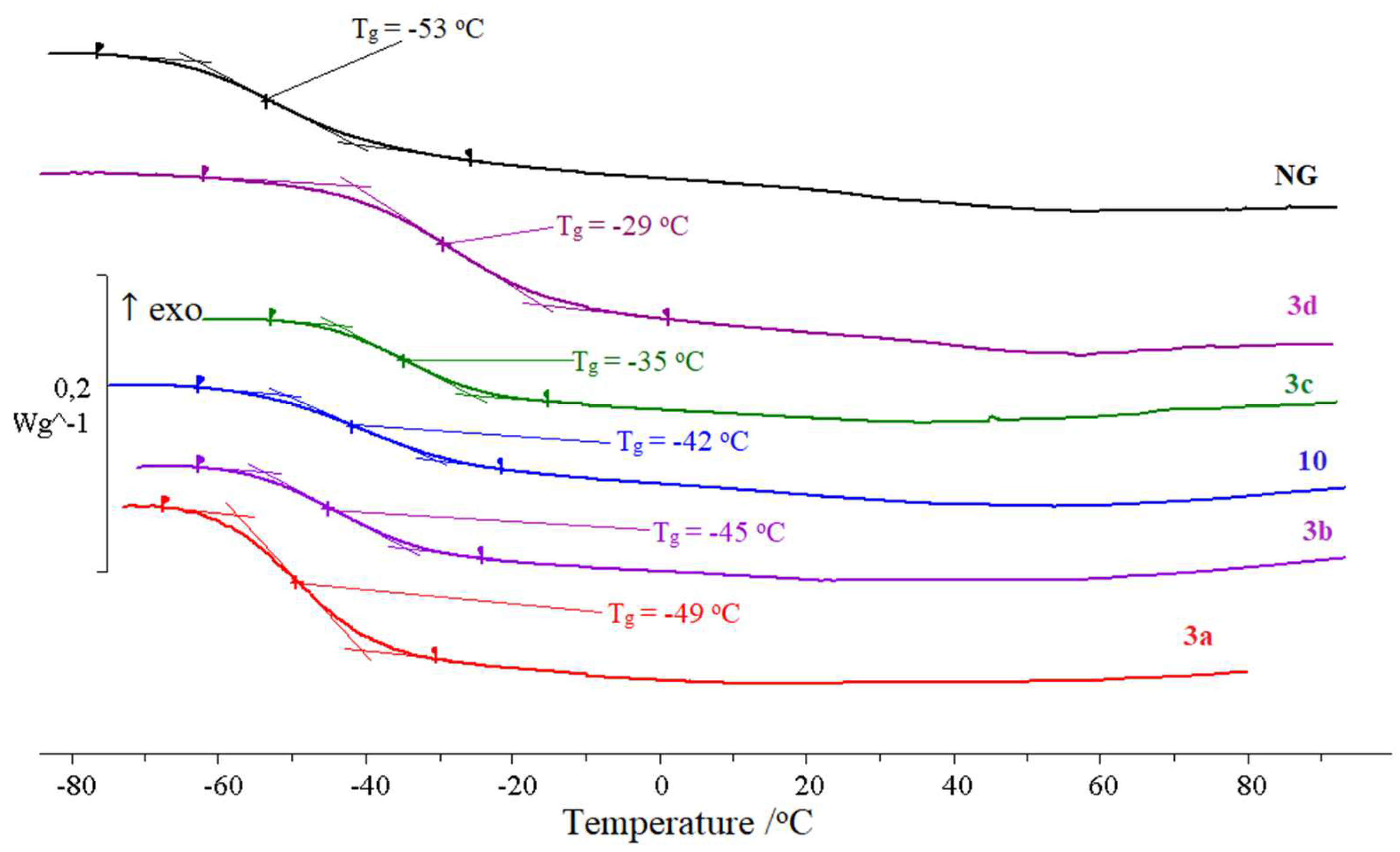
2.3. Relaxation and Phase Transitions
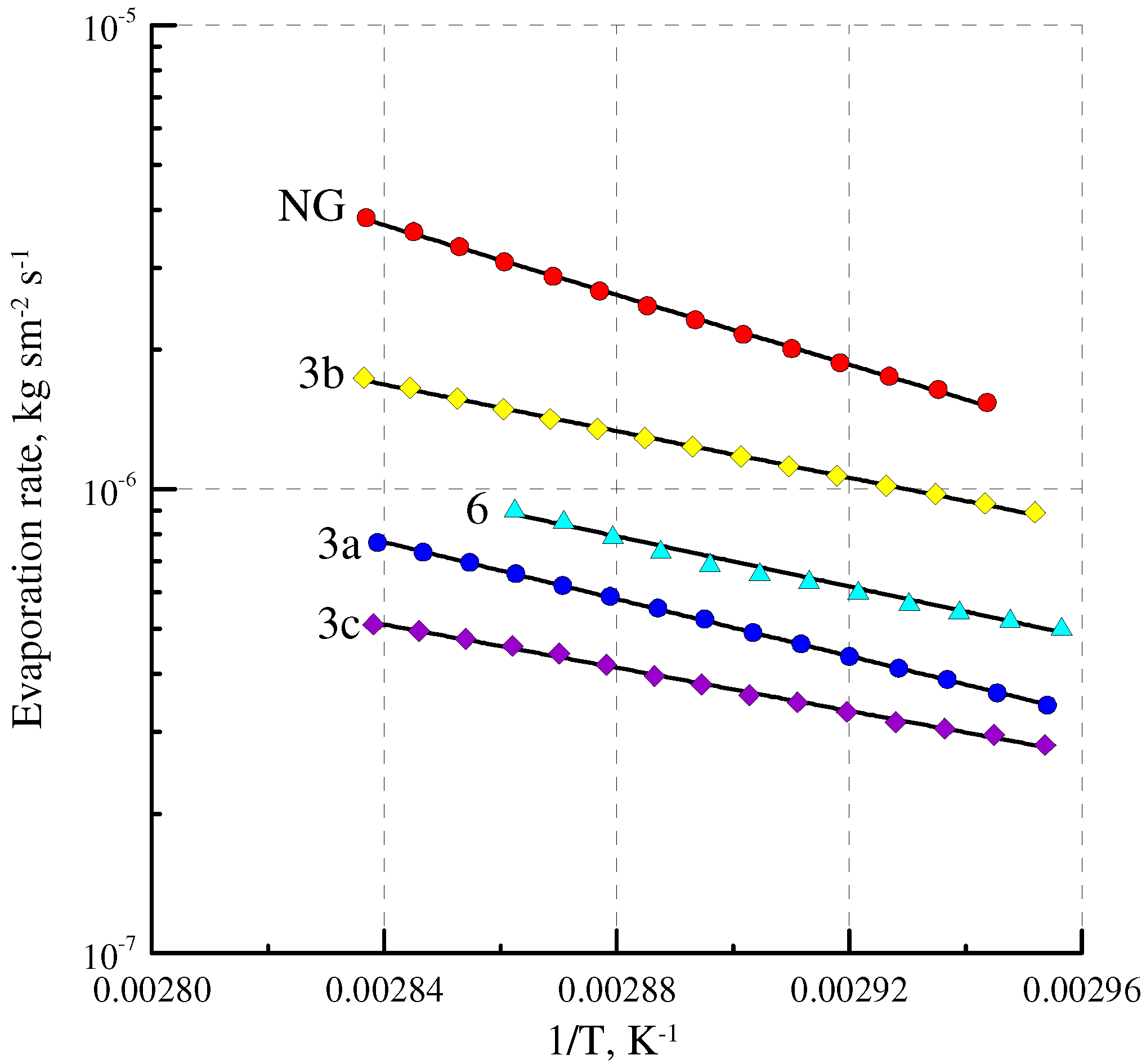
2.4. Volatility
2.5. Thermal Analysis
2.6. Combustion
2.7. Plasticizing Ability
2.8. Physical and Energy Properties
3. Materials and Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
Sample Availability
References
- Modyachin, F.P.; Tikhonova, N.A. Components and Combustion Products of Pyrotechnic Compositions; Polymers and Oligomers; KSTU: Kazan, Russia, 2008; Volume 2. (In Russian) [Google Scholar]
- DeLuca, L.T.; Shimada, T.; Sinditskii, V.; Calabro, M. Chemical Rocket Propulsion—A Comprehensive Survey of Energetic Materials; Springer International Publishing: Cham, Switzerland, 2017. [Google Scholar] [CrossRef]
- Paraskos, A.J. Energetic Polymers: Synthesis and Applications. In Energetic Materials; Shukla, M., Boddu, V., Steevens, J., Damavarapu, R., Leszczynski, J., Eds.; Springer: Cham, Switzerland, 2017; Chapter 4; pp. 91–134. [Google Scholar] [CrossRef]
- Gayathri, S.; Reshmi, S. Nitrato Functionalized Polymers for High Energy Propellants and Explosives: Recent Advances. Polym. Adv. Technol. 2017, 28, 1539–1550. [Google Scholar] [CrossRef]
- Kizhnyaev, V.N.; Golobokova, T.V.; Pokatilov, F.A.; Vereshchagin, L.I.; Estrin, Y.I. Synthesis of energetic triazole- and tetrazole-containing oligomers and polymers. Chem. Heterocycl. Comp. 2017, 53, 682–692. [Google Scholar] [CrossRef]
- Jarosz, T.; Stolarczyk, A.; Wawrzkiewicz-Jalowiecka, A.; Pawlus, K.; Miszczyszyn, K. Glycidyl Azide Polymer and its Derivatives-Versatile Binders for Explosives and Pyrotechnics: Tutorial Review of Recent Progress. Molecules 2019, 24, 4475. [Google Scholar] [CrossRef]
- Cheng, T. Review of novel energetic polymers and binders—High energy propellant ingredients for the new space race. Des. Monomers Polym. 2019, 22, 54–65. [Google Scholar] [CrossRef]
- Zinoviev, V.M.; Kutsenko, G.V.; Ermilov, A.S.; Boldavnin, I.I. High-Energy Plasticizers of Solid Rocket and Gun Propellants. Perm STU Publishing House: Russia, 2010. (In Russian) [Google Scholar]
- Ang, H.G.; Pisharath, S. Energetic Polymers: Binders and Plasticizers for Enhancing Performance, 1st ed.; WILEY-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2012. [Google Scholar]
- Kumari, D.; Balakshe, R.; Banerjee, S.; Singh, H. Energetic Plasticizers for Gun & Rocket Propellants. Rev. J. Chem. 2012, 2, 240–262. [Google Scholar] [CrossRef]
- Zhou, J.; Zhang, J.L.; Wang, B.Z.; Qiu, L.L.; Xu, R.Q.; Sheremetev, A.B. Recent Synthetic Efforts towards High Energy Density Materials: How to Design High-Performance Energetic Structures? FirePhysChem 2022, 2, 83–139. [Google Scholar] [CrossRef]
- Barshtein, R.S.; Kirilovich, V.I.; Nosovsky, Y.E. Plasticizers for Polymers; Khimiya: Moscow, Russia, 1982. (In Russian) [Google Scholar]
- Witucki, E.F.; Wilson, E.R.; Flanagan, J.E.; Frankel, M.B. Synthesis of Novel Energetic Compounds. J. Chem. Eng. Data 1983, 28, 285–286. [Google Scholar] [CrossRef]
- Gareev, G.A.; Kirillova, L.P.; Cherkashina, N.A.; Shul’gina, V.M.; Kozlova, L.M.; Vereshchagin, L.I. Synthesis of ethers of 2-nitro-2-aza-1-propanol. Zh. Org. Khim. 1990, 26, 71–75. [Google Scholar]
- Tartakovsky, V.A.; Ermakov, A.S.; Strelenko, Y.A.; Vinogradov, D.B.; Serkov, S.A. Synthesis of N-nitrooxazolidines and N-nitrotetrahydro-1,3-oxazines from N-(2-hydroxyalkyl)- and N-(3-hydroxyalkyl)sulfamates. Russ. Chem. Bull. 2001, 50, 911–913, Translation of Izv. Akad. Nauk Ser. Khim. 2001, 872–874. [Google Scholar] [CrossRef]
- Miroshnichenko, E.A.; Kon’kova, T.S.; Pashchenko, L.L.; Matyushin, Y.N.; Inozemtsev, Y.O.; Tartakovskii, V.A. Energy characteristics of nitrooxazolidines and their radicals. Russ. Chem. Bull. 2016, 65, 1876–1878. [Google Scholar] [CrossRef]
- Vinogradov, D.B.; Bulatov, P.V.; Petrov, E.Y.; Tartakovsky, V.A. New access to azido-substituted alkylnitramines. Mendeleev Commun. 2021, 31, 795–796. [Google Scholar] [CrossRef]
- Sinditskii, V.P.; Smirnova, A.D.; Serushkin, V.V.; Aleksandrova, N.S.; Sheremetev, A.B. Furazan-fused azacyclic nitramines: Influence of structural features on the combustion and the thermolysis. ChemistrySelect 2020, 5, 3868–13877. [Google Scholar] [CrossRef]
- Gribov, P.S.; Mikhailova, M.V.; Kon’kova, T.S.; Matyushin, Y.N.; Sheremetev, A.B. Bis(propargyl)nitramine: Synthesis and reactivity. Mendeleev Commun. 2022, 32, 218–220. [Google Scholar] [CrossRef]
- Gribov, P.S.; Getmanova, A.D.; Sheremetev, A.B. Nitrolysis of N-alkylurotropinium Salts. Chem. Heterocycl. Comp. 2022, 58, 345–348. [Google Scholar] [CrossRef]
- Lipilin, D.L.; Sheremetev, A.B. 3-(Nitramino)acrylates: Synthesis and reactivity. Russ. Chem. Bull. 2022, 71, 2006–2011. [Google Scholar] [CrossRef]
- Gribov, P.S.; Suponitsky, K.Y.; Sheremetev, A.B. Efficient Synthesis of N-(Chloromethyl)nitramines via TiCl4-Catalyzed Chlorodeacetoxylation. New J. Chem. 2022, 46, 17548–17553. [Google Scholar] [CrossRef]
- Valiulin, R.A.; Kutateladze, A.G. 2,6,7-Trithiabicyclo [2.2.2]octanes as Promising Photolabile Tags for Combinatorial Encoding. J. Org. Chem. 2008, 73, 335–338. [Google Scholar] [CrossRef]
- Frohlich, T.; Hahn, F.; Belmudes, L.; Leidenberger, M.; Friedrich, O.; Kappes, B.; Coute, Y.; Marschall, M.; Tsogoeva, S.B. Synthesis of Artemisinin-Derived Dimers, Trimers and Dendrimers: Investigation of Their Antimalarial and Antiviral Activities Including Putative Mechanisms of Action. Chem. Eur. J. 2018, 24, 8103–8113. [Google Scholar] [CrossRef]
- Ikeda, T. Poly(ionic liquid)s with branched side chains: Polymer design for breaking the conventional record of ionic conductivity. Polym. Chem. 2021, 12, 711–718. [Google Scholar] [CrossRef]
- Brase, S.; Banert, K. (Eds.) Organic Azides: Syntheses and Applications; John Wiley & Sons Ltd.: Hoboken, NJ, USA, 2010. [Google Scholar]
- Stanovnik, B. Application of organic azides in the synthesis of heterocyclic systems. Adv. Heterocycl. Chem. 2020, 130, 145–194. [Google Scholar] [CrossRef]
- Sivaguru, P.; Ning, Y.; Bi, X. New Strategies for the Synthesis of Aliphatic Azides. Chem. Rev. 2021, 121, 4253–4307. [Google Scholar] [CrossRef]
- Grieco, P.A. Organic Synthesis in Water; Springer: New York, NY, USA, 1998. [Google Scholar] [CrossRef]
- Chanda, A.; Fokin, V.V. Organic Synthesis “On Water”. Chem. Rev. 2009, 109, 725–748. [Google Scholar] [CrossRef]
- Butler, R.N.; Coyne, A.G. Organic synthesis reactions on-water at the organic–liquid water interface. Org. Biomol. Chem. 2016, 14, 9945–9960. [Google Scholar] [CrossRef]
- Kitanosono, T.; Masuda, K.; Xu, P.; Kobayashi, S. Catalytic Organic Reactions in Water toward Sustainable Society. Chem. Rev. 2018, 118, 679–746. [Google Scholar] [CrossRef] [PubMed]
- Yufit, S.S. Mechanism of Phase Transfer Catalysis; Nauka: Moscow, Russia, 1984. (In Russian) [Google Scholar]
- Xue, J.; Wang, W.; Xu, Y.; Han, S.; Liu, F.; Zhang, D.; Shang, B. A Preparation Method of 1,5-Diazido-3-Nitro Aza-Pentane. CN Patent 104262194, 2015. [Google Scholar]
- Manser, G.E.; Fletcher, R.W. Nitramine Oxetanes and Polyethers Formed Therefrom. U.S. Patent 4707540, 1986. [Google Scholar]
- Gaur, P.; Dev, S.; Kumar, S.; Kumar, M.; Vargeese, A.A.; Soni, P.; Siril, P.F.; Ghosh, S. Dendritic Polynitrato Energetic Motifs: Development and Exploration of Physicochemical Behavior through Theoretical and Experimental Approach. ACS Omega 2017, 2, 8227–8233. [Google Scholar] [CrossRef]
- Golobokova, T.V.; Proidakov, A.G.; Vereshchagin, L.I.; Kizhnyaev, V.N. Epichlorohydrin as a Precursor of Functionally Substituted 1,2,3-Triazoles and Tetrazoles. Russ. J. Org. Chem. 2019, 55, 186–192, Translation of Zh. Org. Khim. 2019, 55, 234–241. [Google Scholar] [CrossRef]
- Moini, N.; Zohuriaan-Mehr, M.J.; Kabiri, K.; Khonakdar, H.A. “Click” on SAP: Superabsorbent polymer surface modification via CuAAC reaction toward antibacterial activity and improved swollen gel strength. Appl. Surf. Sci. 2019, 487, 1131–1144. [Google Scholar] [CrossRef]
- Witucki, E.F.; Frankel, M.B. Synthesis of novel energetic aliphatic compounds. J. Chem. Eng. Data 1979, 24, 247–249. [Google Scholar] [CrossRef]
- Guo, S.; Su, T.; Feng, L.; Wang, X.; Zhang, L.; Zhang, C. Synthesis of 1,3-diazido-2-nitryloxypropane. Hanneng Cailiao 2003, 11, 149–152. [Google Scholar]
- Muravyev, N.V.; Meerov, D.B.; Monogarov, K.A.; Melnikov, I.N.; Kosareva, E.K.; Fershtat, L.L.; Sheremetev, A.B.; Dalinger, I.L.; Fomenkov, I.V.; Pivkina, A.N. Sensitivity of Energetic Materials: Evidence of Thermodynamic Factor on a Large Array of CHNOFCl Compounds. Chem. Eng. J. 2021, 421, 129804. [Google Scholar] [CrossRef]
- Orlenko, L.P. (Ed.) Physics of Explosion, 3rd ed.; Fismatlit: Moscow, Russia, 2002; Volume 1. (In Russian) [Google Scholar]
- Braak, E.C. The melting points of stable-form nitrate ester crystals. J. Energ. Mater. 1990, 8, 21–39. [Google Scholar] [CrossRef]
- Ou, Y.; Chen, B.; Yan, H.; Jia, H.; Li, J.; Dong, S. Development of energetic additives for propellants in China. J. Propuls. Power 1995, 11, 838–847. [Google Scholar] [CrossRef]
- Price, D.M.; Hawkins, M. Vapour pressures of hydroxybenzophenone UV Absorbers. Thermochim. Acta 1999, 329, 73–76. [Google Scholar] [CrossRef]
- Price, D.M. Vapor pressure determination by termogravimetry. Thermochim. Acta 2001, 367/368, 253–262. [Google Scholar] [CrossRef]
- Suceska, M.; Musanic, S.M.; Houra, I.F. Kinetics and enthalpy of nitroglycerin evaporation from double base propellants by isothermal thermogravimetry. Termochim. Acta 2010, 510, 9–16. [Google Scholar] [CrossRef]
- Phang, P.; Dollimore, D.; Evans, S.J. A comparative method for developing vapor pressure curves based on evaporation data obtained from a simultaneous TG–DTA unit. Thermochim. Acta 2002, 392/393, 119–125. [Google Scholar] [CrossRef]
- Egorshev, V.Y.; Sinditskii, V.P.; Berezin, M.V. Study on combustion of liquid and gelatinized glycidyl azide oligomers. In New Trends in Research of Energetic Materials (NTREM) (2004), Proceedings of the 7th Seminar, Pardubice, Czech Republic, 22–24 April 2004; University of Pardubice Czech Republic: Pardubice, Czech Republic, 2004; Volume 1, pp. 100–115. [Google Scholar]
- Scriven, E. (Ed.) Azides and Nitrenes: Reactivity and Utility; Academic Press Inc.: Cambridge, MA, USA, 1984. [Google Scholar]
- Manelis, G.B.; Nazin, G.M.; Rubsov, Y.I.; Strunin, V.A. Thermal Decomposition and Combustion of Explosives and Propellants; CRC Press: Boca Raton, FL, USA, 2003. [Google Scholar]
- Stepanov, R.S.; Kruglyakova, L.A. Thermal decomposition of polyfunctional azidocompounds. In Proceedings of the Fraunhofer Institut Fur Chemische Technologie, International Annual Conference, Karlsruhe, FRG; Fraunhofer-Institut für Chemische Technologie (ICT): Pfinztal, Germany, 1999; paper-47. [Google Scholar]
- Sinditskii, V.P.; Smirnova, A.D.; Serushkin, V.V.; Yudin, N.V.; Vatsadze, I.A.; Dalinger, I.L.; Kiselev, V.G.; Sheremetev, A.B. Nitroderivatives of N-pyrazolyltetrazoles: Thermal decomposition and combustion. Thermochim. Acta 2021, 698, 178876. [Google Scholar] [CrossRef]
- Sheremetev, A.B.; Mel’nikova, S.F.; Kokareva, E.S.; Nekrutenko, R.E.; Strizhenko, K.V.; Suponitsky, K.Y.; Pham, T.D.; Pivkina, A.N.; Sinditskii, V.P. Nitroxy- and azidomethyl azofurazans as advanced energetic materials. Def. Technol. 2022, 18, 1369–1381. [Google Scholar] [CrossRef]
- Kissinger, H.E. Reaction kinetics in differential thermal analysis. Anal. Chem. 1957, 29, 1702–1706. [Google Scholar] [CrossRef]
- Chen, J.K.; Brill, T.B. Thermal Decomposition of Energetic Materials 54. Kinetics and Near-Surface Products of Azide Polymers AMMO, BAMO, and GAP in Simulated Combustion. Combust. Flame 1991, 87, 157–168. [Google Scholar] [CrossRef]
- Recommendations on the Transport of Dangerous Goods, Manual of Tests and Criteria, 4th ed.; ST/SG/AC.10/11/Rev. 4; United Nations: New York, NY, USA; Geneva, Switzerland, 2003.
- Andreev, K.K. Thermal Decomposition and Combustion of Explosives; No. FTD-HT-23-1329-68, Foreign Technology Division, Wright-Patterson Air Force Base, Ohio (English Translation); Nauka: Moscow, Russia, 1969. [Google Scholar]
- Sergeev, V.V.; Kozhukh, M.S. Combustion of 1,3-diazidopropanol-2. Combust. Explos. Shock Waves 1975, 11, 343–349, Translation of Fiz. Gor. Vzryva 1975, 11, 403–412. . [Google Scholar] [CrossRef]
- Sinditskii, V.P.; Fogelzang, A.E.; Egorshev, V.Y.; Serushkin, V.V.; Kolesov, V.I. Effect of molecular structure on combustion of polynitrogen energetic materials. In Solid Propellant Chemistry, Combustion, and Motor Interior Ballistics, Progress in Astronautics and Aeronautics; AIAA: Restone, VA, USA, 2000; pp. 99–128. [Google Scholar] [CrossRef]
- Sinditskii, V.P. On the nature of the burning rate-controlling reaction of energetic materials for the gas-phase. Combust. Explos. Shock Waves 2007, 43, 297–308. [Google Scholar] [CrossRef]
- Lotmentsev, Y.M.; Kondakova, N.N.; Bakeshko, A.V.; Kozeev, A.M.; Sheremetev, A.B. 3-Alkyl-4-nitrofurazans–plasticizers for polymers. Chem. Heterocycl. Comp. 2017, 53, 740–745. [Google Scholar] [CrossRef]
- Davenas, A. Solid Rocket Propulsion Technology; Pergamon Press: Oxford, UK, 1993; pp. 477–485. [Google Scholar]
- Zimerman, G.A. Newey Embrittlement of propellants containing nitrate ester plasticizers. In Proceedings of the AIAA/SAE/ASME 18th Joint Propulsion Conference, Cleveland, OH, USA, 21–23 June 1982. [Google Scholar]
- Brun, I.; Longevialle, Y.; Rat, M. Etude de la cristallisation de diffkrents esters nitriques. Propellants Explos. Pyrotech. 1991, 16, 182–187. [Google Scholar] [CrossRef]
- Malkin, A.Y.; Chalykh, A.E. Diffusion and Viscosity of Polymers; Khimiya: Moscow, Russia, 1979. (In Russian) [Google Scholar]
- Tretyakova, V.D.; Lotmentsev, Y.M.; Kondakova, N.N.; Pleshakov, D.V. Research of thermodynamic compatibility of energetically active plasticizers with polyetherurethane and divinylnitrile rubbers. Adv. Chem. Chem. Technol. 2008, 22, 69–72. (In Russian) [Google Scholar]
- Bhowmik, D.; Sadavarte, V.S.; Shrikant, M.; Pande, S.M.; Saraswat, B.S. An energetic binder for the formulation of advanced solid rocket propellants. Cent. Eur. J. Energ. Mater. 2015, 12, 145–158. [Google Scholar]
- Prisacariu, C. Polyurethane Elastomers: From Morphology to Mechanical Aspects; Springer: Vienna, Austria, 2011. [Google Scholar]
- Lempert, D.B.; Kazakov, A.I.; Sheremetev, A.B. Comparative ballistic efficiency of solid composite propellants: Which plasticizer/polymer combination is the energetically preferred binder? Mendeleev Commun. 2022, 32, 601–603. [Google Scholar] [CrossRef]
- Lysien, K.; Stolarczyk, A.; Jarosz, T. Solid propellant formulations: A review of recent progress and utilized components. Materials 2021, 14, 6657. [Google Scholar] [CrossRef]
- Benson, S.W.; Cruickshank, F.R.; Golden, D.M.; Haugen, G.R.; O’Neal, H.E.; Rodgers, A.S.; Shaw, R.; Walsh, R. Additivity rules for the estimation of thermochemical properties. Chem. Rev. 1969, 69, 279–324. [Google Scholar] [CrossRef]
- Lebedev, Y.A.; Miroshnichenko, E.A.; Knobel, Y.K. Thermochemistry of Nitro Compounds; Nauka: Moscow, Russia, 1970. (In Russian) [Google Scholar]
- Holmes, J.L.; Aubry, C. Group additivity values for estimating the enthalpy of formation of organic compounds: An update and reappraisal. 2. C,H,N,O,S, and halogens. J. Phys. Chem. A 2012, 116, 7196–7209. [Google Scholar] [CrossRef]
- Naef, R.; Acree, W.E., Jr. Revision and Extension of a Generally Applicable Group-Additivity Method for the Calculation of the Standard Heat of Combustion and Formation of Organic Molecules. Molecules 2021, 26, 6101. [Google Scholar] [CrossRef]
- Belov, G.B. Thermodynamic analysis of combustion products at high temperature and pressure. Propellants Explos. Pyrotech. 1998, 23, 86–89. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | IS a, % | Observed Effects |
|---|---|---|
| NG | 100 | Very loud sound |
| 13 | 100 | Very loud sound, carbon deposits on a striker and an anvil |
| 3c | 88 | Loud sound, carbon deposits on a striker and an anvil |
| 3a | 72 | Loud sound, carbon deposits on a striker and an anvil, smoke |
| 3b | 52 | Loud sound |
| 10 | 36 | Loud sound |
| 3d | 12 | A weak sound |
| Sample | Glass Transition | Crystallization | Melting | ||||
|---|---|---|---|---|---|---|---|
| Tg, a °C | ∆Cp, b J g−1 K−1 | Tc, c °C | Tpeak, d °C | ∆Hc, e J g−1 | Tm, f °C | ∆Hm, g J g−1 | |
| 3d | −61 | 0.75 | −7 | 10 | 44 | +21 | 42 |
| 6 | −88 | 0.87 | −48 | −41 | 104 | +3 i | 147 |
| 13 | −94 | 1.00 | Does not crystallize | ||||
| 3a | −81 | 0.67 | |||||
| 10 | −74 | 0.65 | |||||
| 3b | −81 | 0.87 | |||||
| 3c | −72 | 0.73 | |||||
| NG | −70 | 0.70 | |||||
| NGh | −70 | 0.70 | −26 | −16 | 75 | −1 | 86 |
| Sample | Molecular Weight, g mol−1 | Evaporation Rate G, kg·sec−1·m−2 × 106 | Relative Volatility Coefficient Frel |
|---|---|---|---|
| NG | 227 | 1.07 | 1 |
| DNDEG | 196 | 1.70 | 1.48 |
| 10 | 228 | 0.849 | 0.80 |
| 3b | 218 | 0.738 | 0.68 |
| 6 | 200 | 0.394 | 0.35 |
| 3c | 299 | 0.224 | 0.24 |
| 3a | 218 | 0.237 | 0.22 |
| Sample | DSC | TGA | |||
|---|---|---|---|---|---|
| Tonset, a °C | Tpeak, b °C | ∆Hd, c J g−1 (kJ mol−1) | Tonset/Tend, d °C | Δm, e % | |
| 3a | 213 | 243 | 816 (175) | 207/250 | 87 |
| 3b | 201 | 244 | 1463 (319) | 164/220 | 85 |
| 3c | 223 | 254 | 2017 (603) | 220/245 | 78 |
| 3d | 226 | 257 | 2413 (840) | 221/250 | 100 |
| 6 | 211 | 241 f | 1442 (288) | 165/200 | 98 |
| 10 | 211 | 243 | 666 (152) | 163/220 | 99 |
| 13 | 185 | 187 | - | 93/155 | 100 |
| NG | 192 | 198 | - | 140–173 | 100 |
| Sample | logA a | Ea, b kJ mol−1 | k × 1014 (at 298 K), c s−1 | k/kNG d | ƞ × 10−3, e % | SADT, f °C | Tign, g °C |
|---|---|---|---|---|---|---|---|
| 3a | 14.3 | 162.0 | 0.75 | 0.20 | 0.47 | 109 | 237 |
| 3c | 15.1 | 172.6 | 0.07 | 0.02 | 0.04 | 116 | 234 |
| 6 | 14.4 | 161.5 | 1.10 | 0.29 | 0.69 | 106 | 226 |
| GAP | 14.1 | 164.8 | 0.16 | 0.04 | 0.10 | ||
| NG | 15.4 | 164.4 | 3.76 | 1.00 | 2.37 | ||
| NC | 16.9 | 175.8 | 1.21 | 0.32 | 0.76 | 185–205 [58] |
| Sample | Burning Rate rb = Apn | rb 2, a мм/c | rb 10, b мм/c | ||
|---|---|---|---|---|---|
| A c | n d | ∆p, e MПa | |||
| 3df | 1.39 | 0.85 | 3.5–12 | 3.4 | 9.1 |
| NC (12% N) | 1.99 | 0.75 | 1–12 | 3.3 | 11.2 |
| 3cf | 1.16 | 1.08 | 9–12.6 | - | 13.8 |
| 3bf | 1.81 | 0.98 | 1–12 | 3.3 | 17.3 |
| 10f | 1.81 | 0.98 | 2–10 | 3.3 | 17.3 |
| NGf | 7.42 | 0.68 | 1–12 | 11.8 | 35.5 |
| Plasticizer | Tg, oC | ∆Cp, * J g−1 K−1 |
|---|---|---|
| NG | −53 | 0.35 |
| 10 | −42 | 0.28 |
| 3a | −49 | 0.45 |
| 3b | −45 | 0.35 |
| 3c | −35 | 0.29 |
| 3d | −29 | 0.46 |
| Sample | Formula Mw | α a | N, b % | d419, c g cm−3 | ΔHf0(l), d kJ mol−1 (kJ g−1) | Tf, e K |
|---|---|---|---|---|---|---|
| EtAENA | C4H9N5O2 159.15 | 0.160 | 44.01 | 1.320 | +204.8 (+1.29) | 1652 |
| BuAENA | C6H13N5O2 187.20 | 0.110 | 37.41 | 1.211 | +150.5 (+0.80) | 1377 |
| DANPE (6) | C4H8N8O2 200.16 | 0.167 | 55.98 | 1.330 | +539.2 (+2.69) | 2275 |
| 3a | C5H10N8O3 230.19 | 0.200 | 48.68 | 1.313 | +443.1 (+1.92) | 1940 |
| 3b | C5H10N8O3 230.19 | 0.200 | 48.68 | 1.319 | +451.4 (+1.96) | 1880 |
| 3c | C7H13N11O3 299.26 | 0.146 | 51.49 | 1.312 | +691.8 (+2.31) | 1720 |
| 3d | C8H16N10O6 348.28 | 0.250 | 40.22 | 1.366 | +351.1 (+1.01) | 1640 |
| 10 | C6H12N8O2 228.22 | 0.111 | 49.10 | 1.262 | +453.5 (+1.99) | 1770 |
| 13 | C3H5N7O3 187.12 | 0.353 | 52.40 | 1.379 | +432.2 (+2.31) | 2469 |
| NG | C3H5N3O9 227.09 | 1.059 | 18.50 | 1.593 | −370.3 (−1.63) | 3233 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vinogradov, D.B.; Bulatov, P.V.; Petrov, E.Y.; Gribov, P.S.; Kondakova, N.N.; Il’icheva, N.N.; Stepanova, E.R.; Denisyuk, A.P.; Sizov, V.A.; Sinditskii, V.P.; et al. Promising Oxygen- and Nitrogen-Rich Azidonitramino Ether Plasticizers for Energetic Materials. Molecules 2022, 27, 7749. https://doi.org/10.3390/molecules27227749
Vinogradov DB, Bulatov PV, Petrov EY, Gribov PS, Kondakova NN, Il’icheva NN, Stepanova ER, Denisyuk AP, Sizov VA, Sinditskii VP, et al. Promising Oxygen- and Nitrogen-Rich Azidonitramino Ether Plasticizers for Energetic Materials. Molecules. 2022; 27(22):7749. https://doi.org/10.3390/molecules27227749
Chicago/Turabian StyleVinogradov, Dmitry B., Pavel V. Bulatov, Evgeny Yu. Petrov, Pavel S. Gribov, Natalia N. Kondakova, Natalia N. Il’icheva, Evgenia R. Stepanova, Anatoly P. Denisyuk, Vladimir A. Sizov, Valery P. Sinditskii, and et al. 2022. "Promising Oxygen- and Nitrogen-Rich Azidonitramino Ether Plasticizers for Energetic Materials" Molecules 27, no. 22: 7749. https://doi.org/10.3390/molecules27227749
APA StyleVinogradov, D. B., Bulatov, P. V., Petrov, E. Y., Gribov, P. S., Kondakova, N. N., Il’icheva, N. N., Stepanova, E. R., Denisyuk, A. P., Sizov, V. A., Sinditskii, V. P., & Sheremetev, A. B. (2022). Promising Oxygen- and Nitrogen-Rich Azidonitramino Ether Plasticizers for Energetic Materials. Molecules, 27(22), 7749. https://doi.org/10.3390/molecules27227749