

Synthesis, Self-Assembly in Crystalline Phase and Anti-Tumor Activity of 2-(2-/4-Hydroxybenzylidene)thiazolo[3,2-a]pyrimidines
 , ,
, ,
Abstract
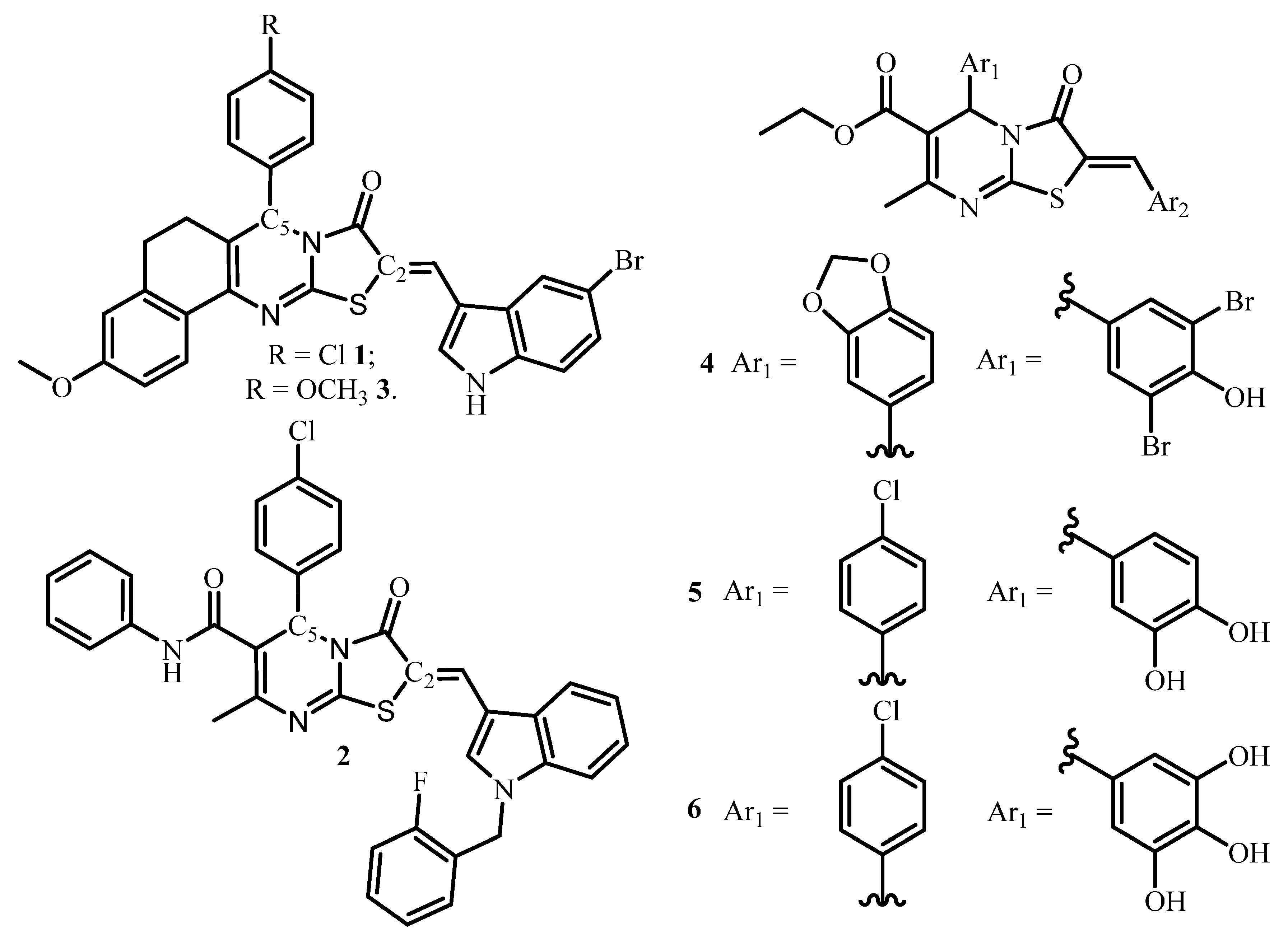
1. Introduction
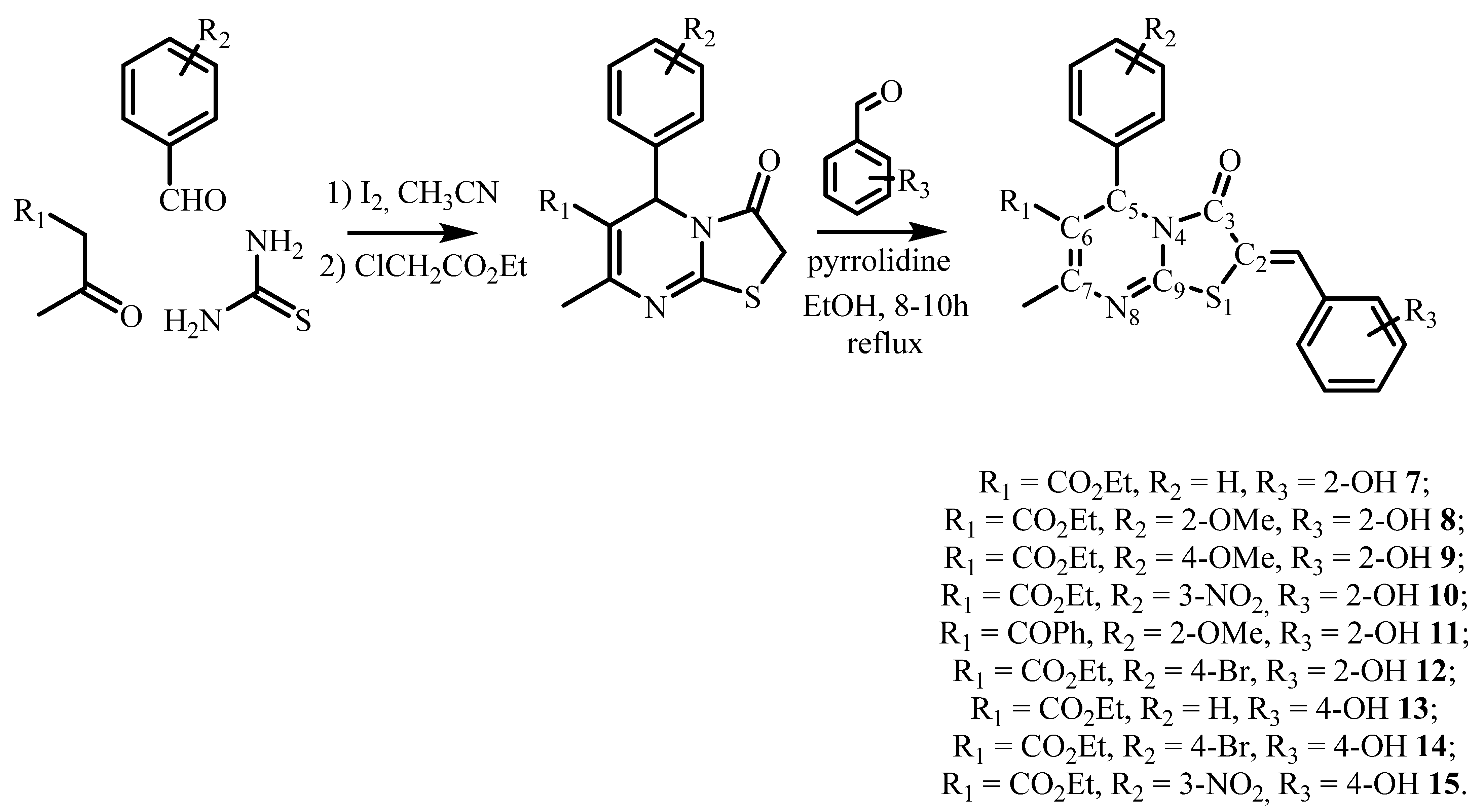
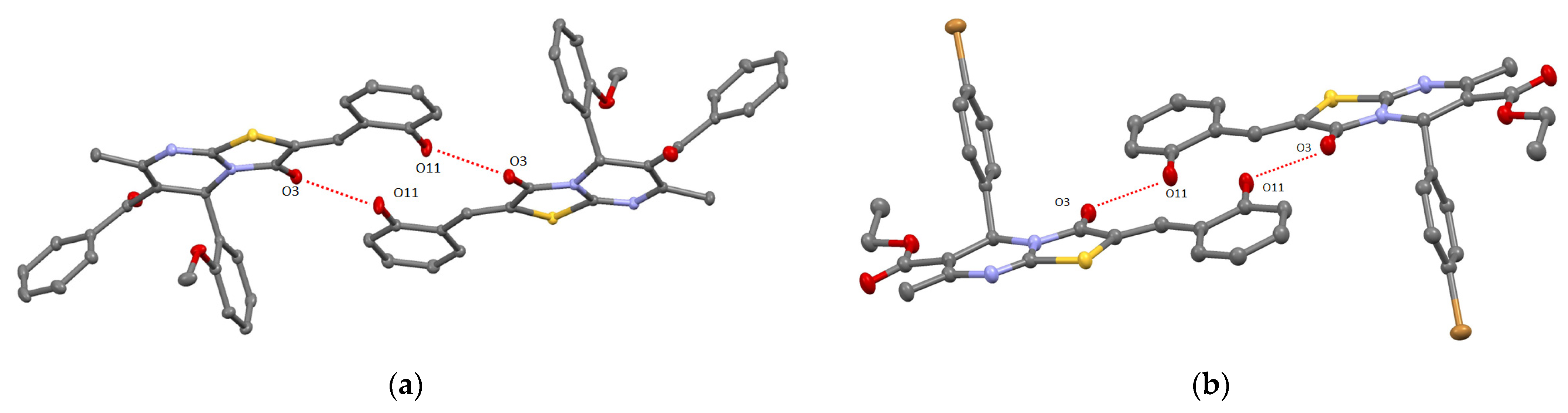
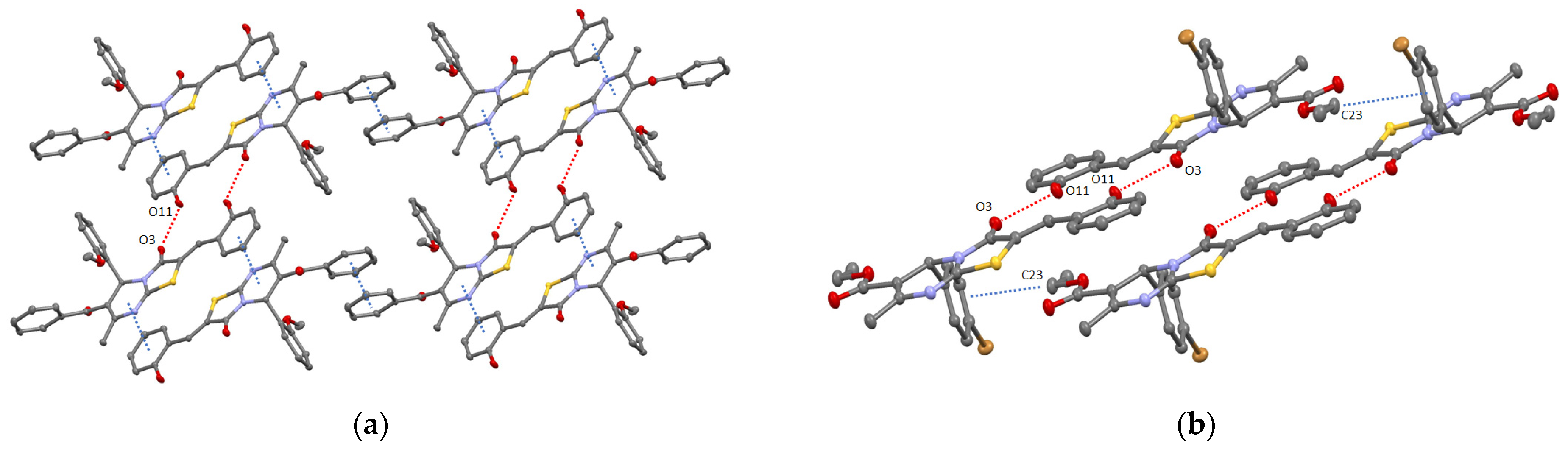
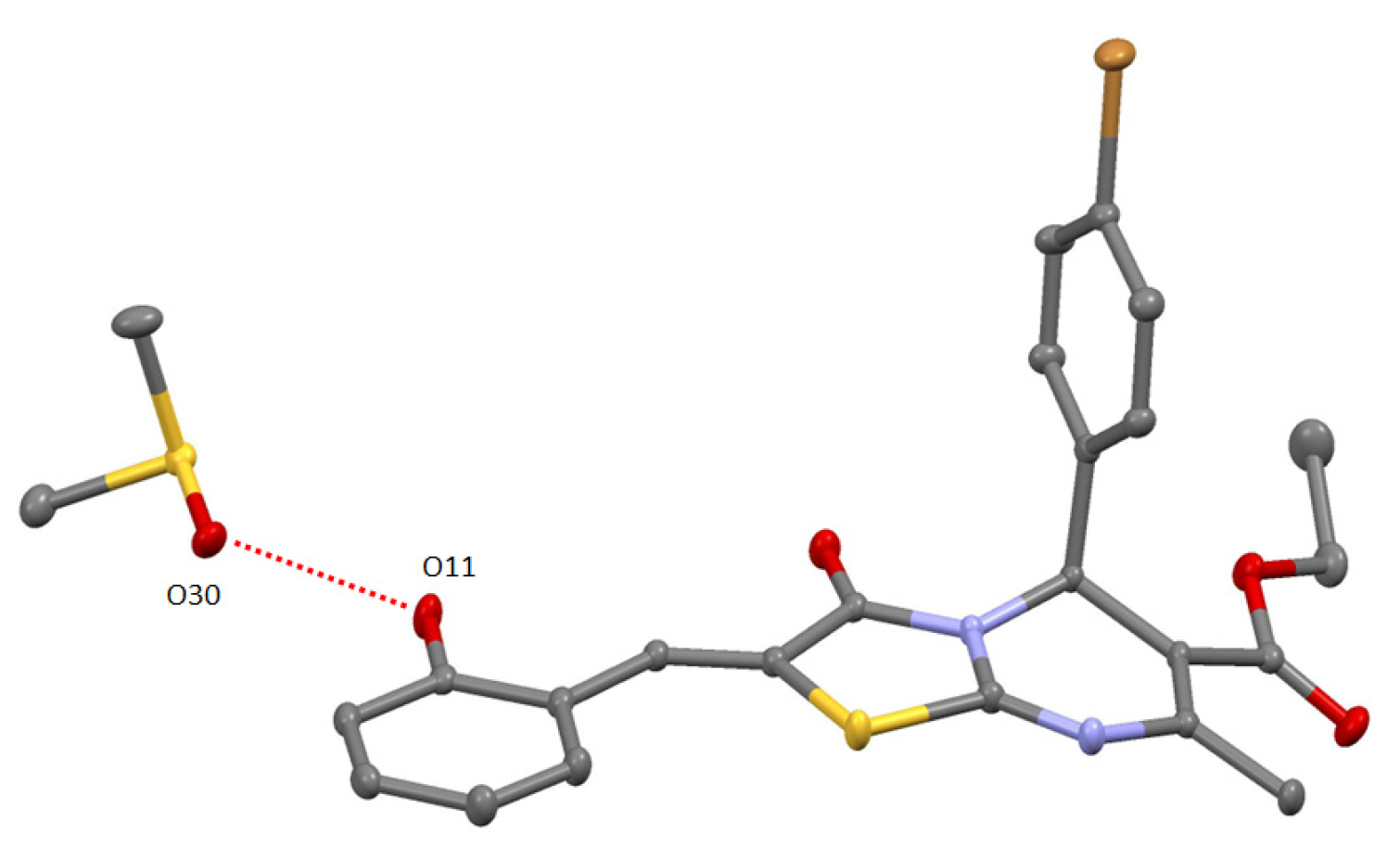
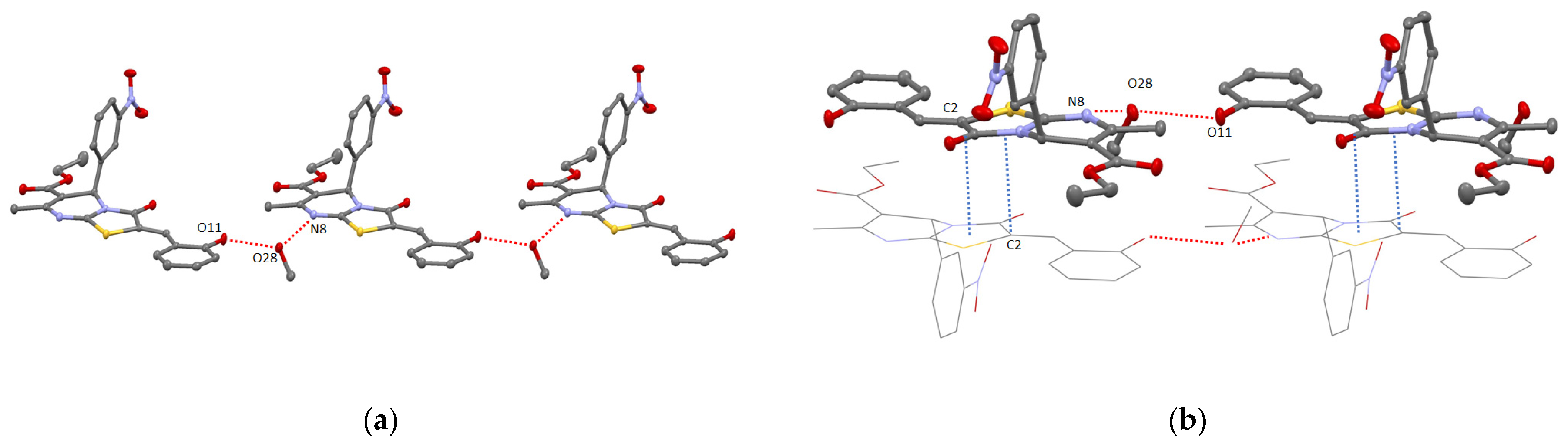
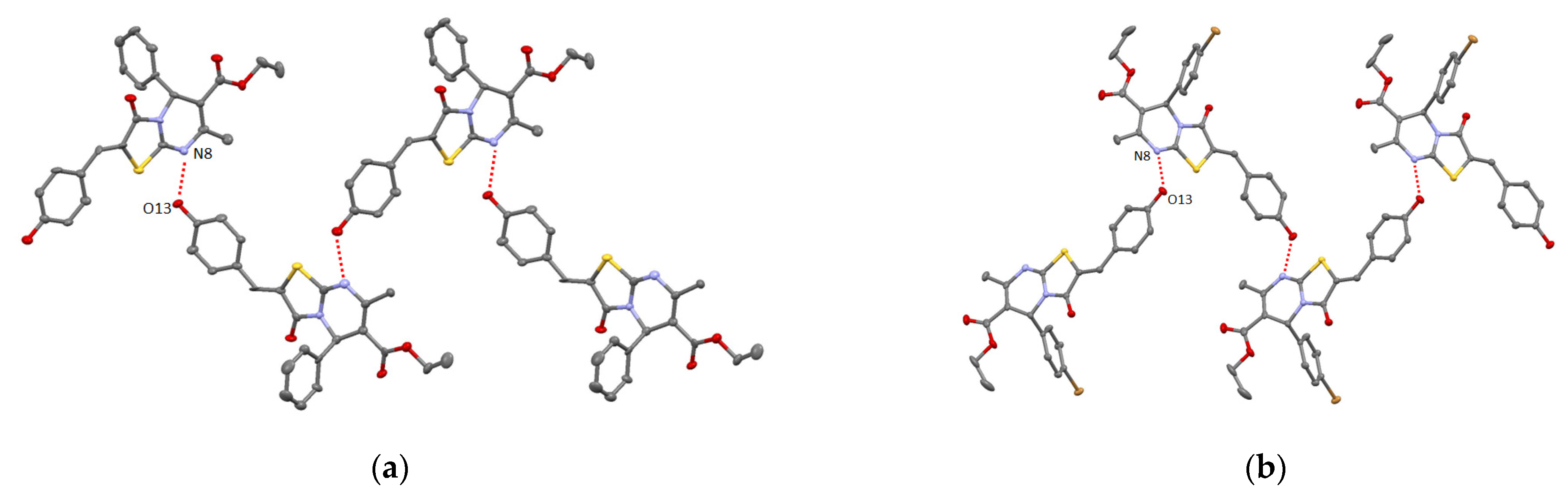
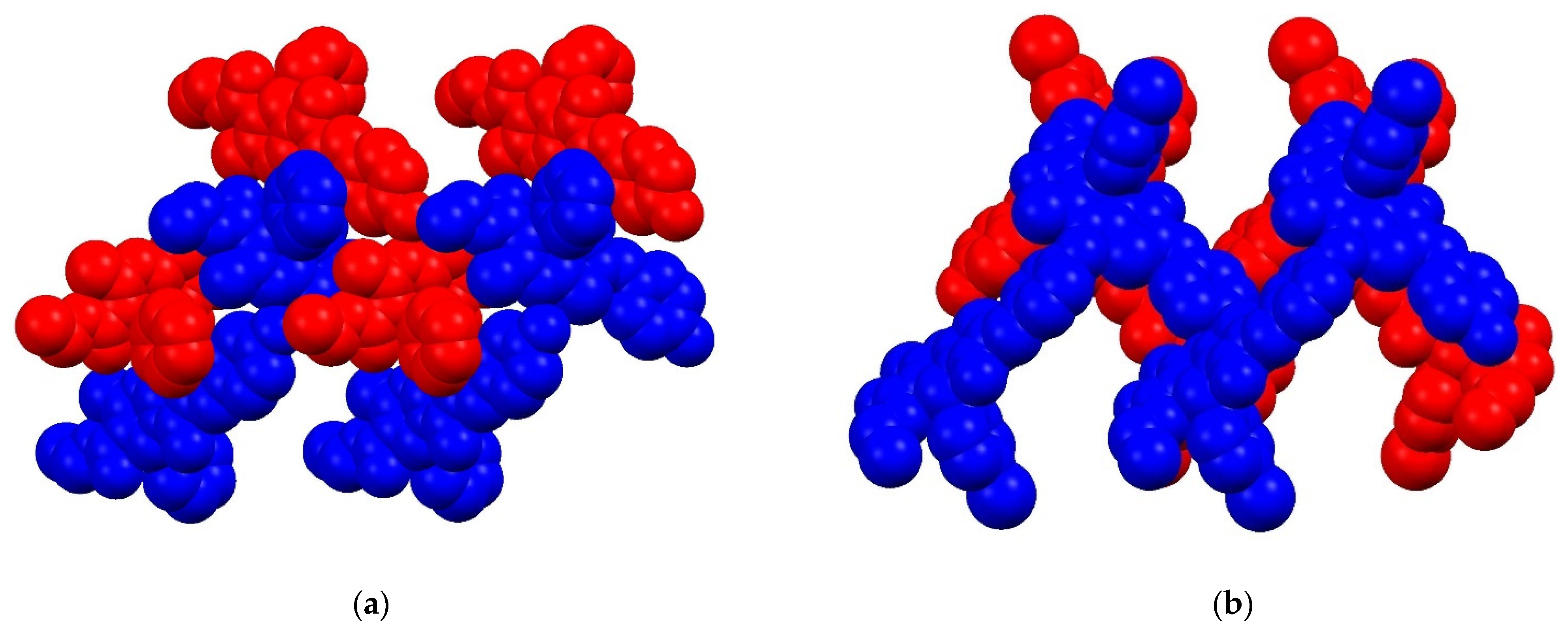
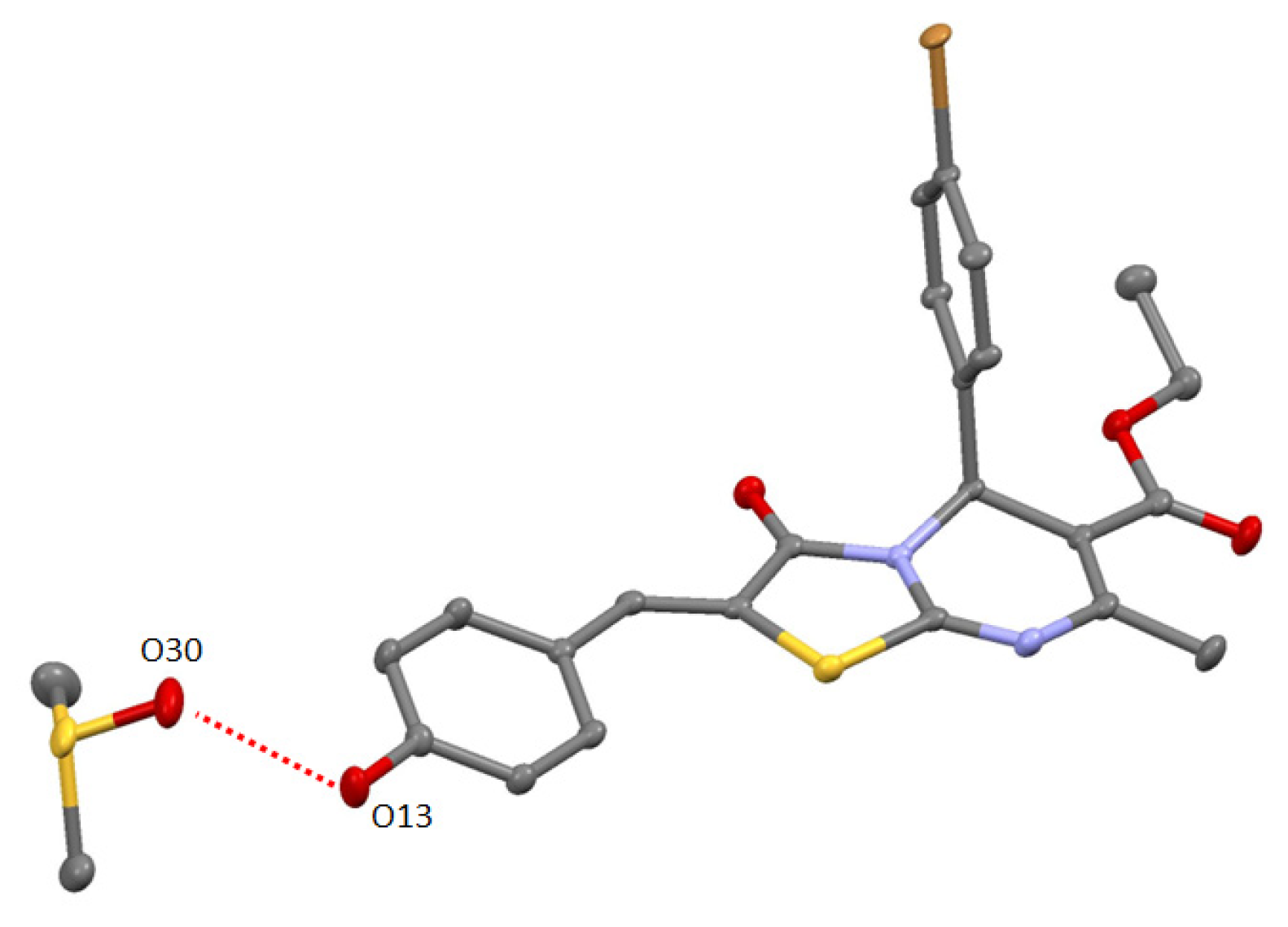
2. Results and Discussion
3. Materials and Methods
3.1. Synthesis and Characterisation
3.1.1. General Method for Compounds 10–15 Preparation
3.1.2. Crystallization Conditions
3.1.3. Single Crystal X-ray Diffraction
3.2. Biological Study
3.2.1. Cells and Materials
3.2.2. MTT Assay
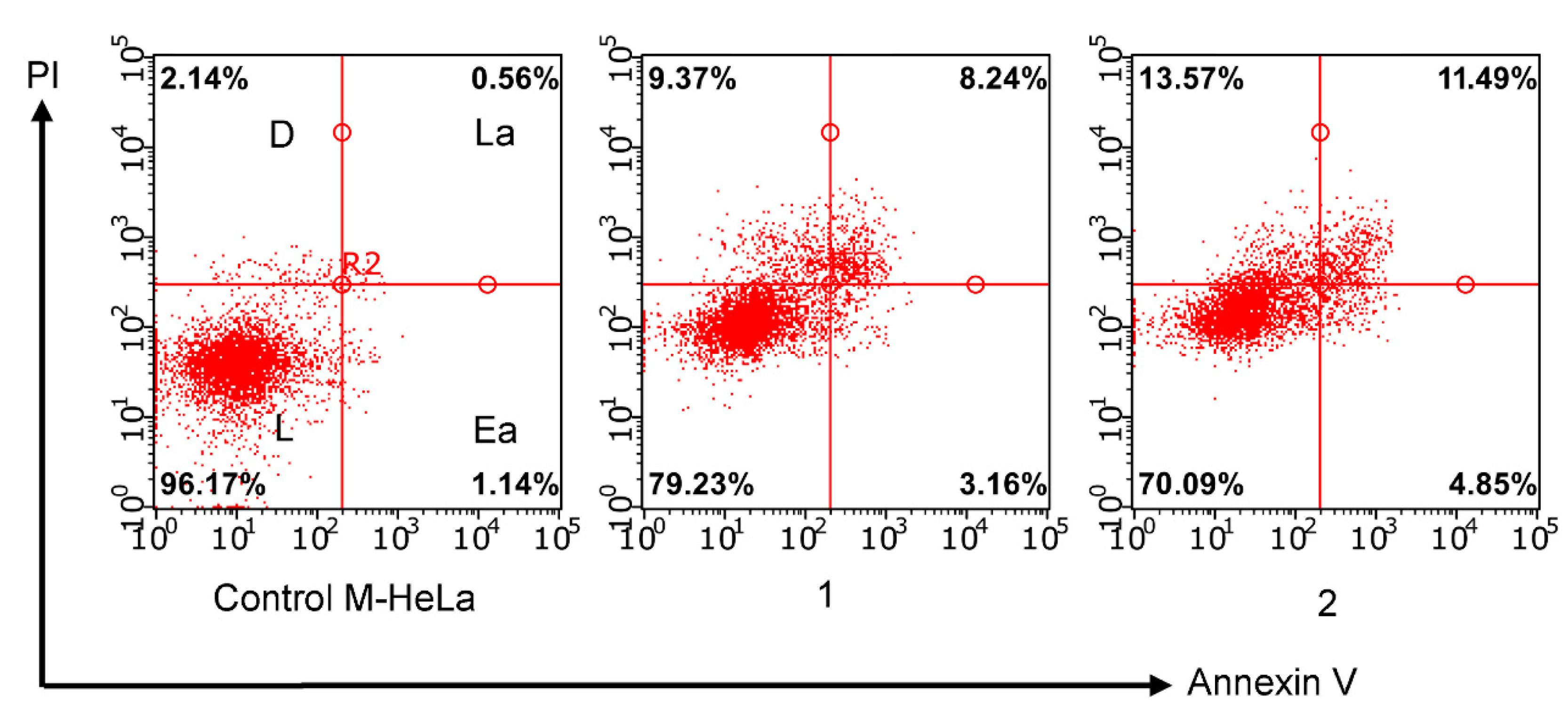
3.2.3. Induction of Apoptotic Effects by Test Compounds
Flow Cytometry Assay
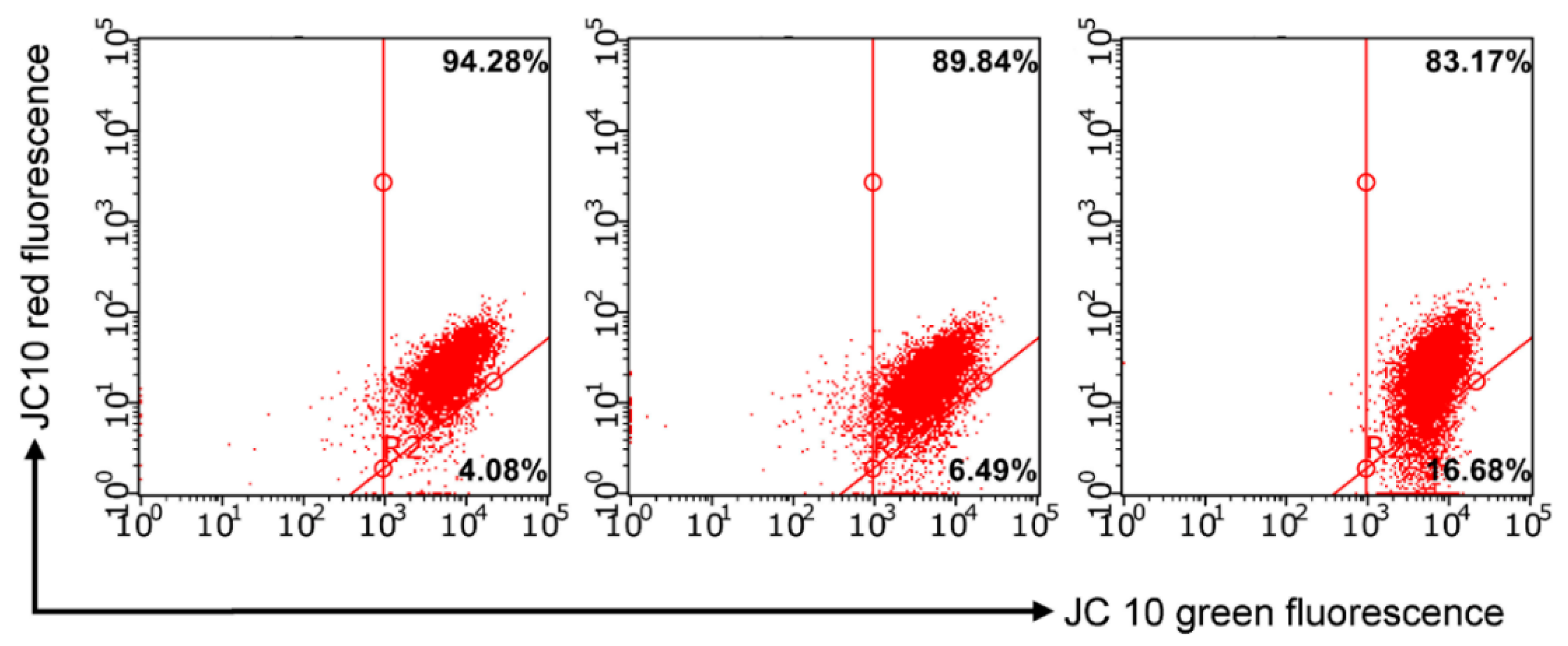
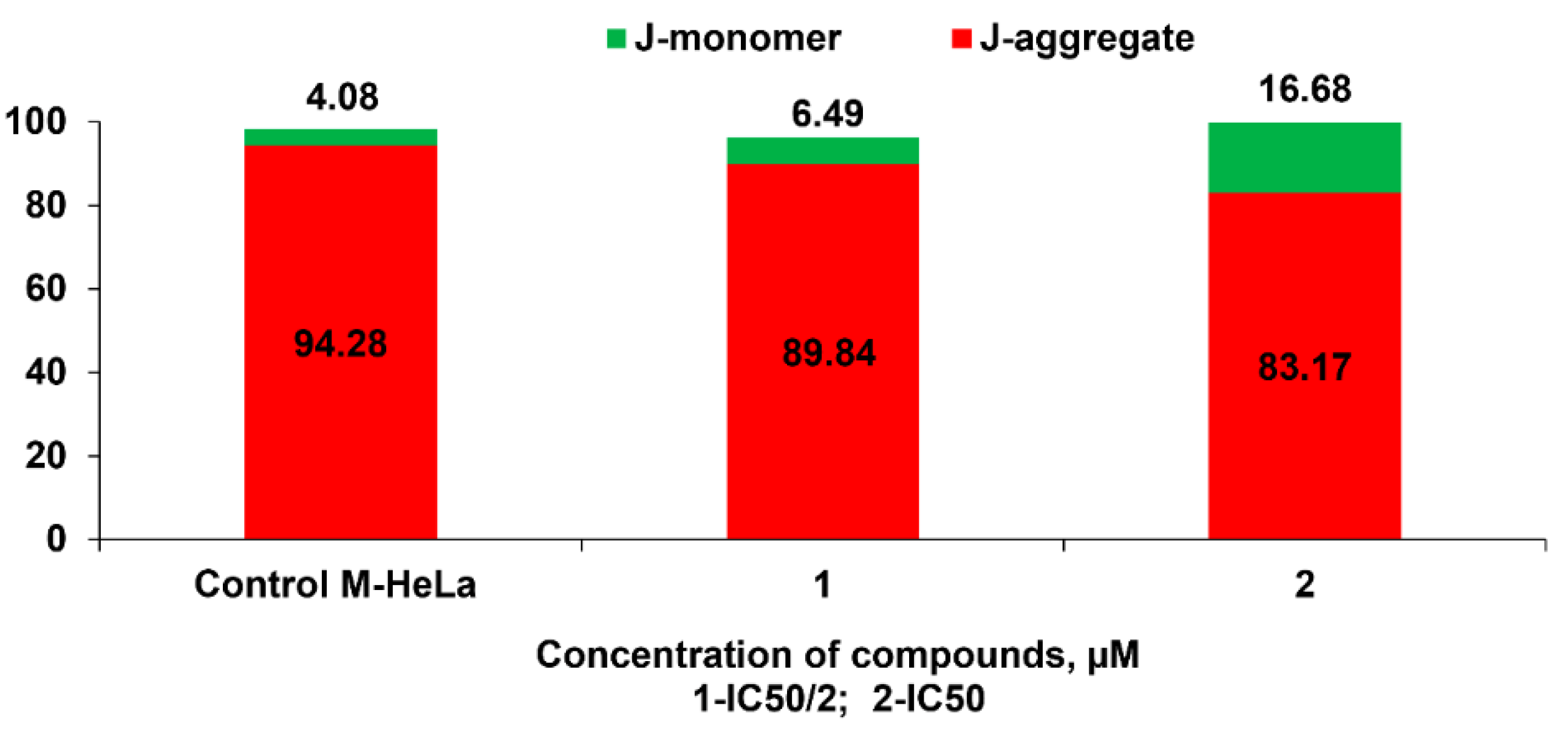
3.2.4. Detection of Intracellular ROS
3.2.5. Multiplex Analysis of Markers DNA Damage/Genotoxicity
Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Venkatesh, T.; Upendranath, K.; Nayaka, Y.A. Development of electrochemical and optoelectronic performance of new 7-{[1H-indol-3-ylmethylidene]amino}-4-methyl-2H-chromen-2-one dye. J. Solid State Electrochem. 2021, 25, 1237–1244. [Google Scholar] [CrossRef]
- Sukanya, S.H.; Venkatesh, T.; Rao, S.A.; Joy, M.N. Efficient L-Proline catalyzed synthesis of some new (4-substituted-phenyl)-1,5-dihydro-2H-pyrimido[4,5-d][1,3]thiazolo[3,2-a]pyrimidine-2,4(3H)-diones bearing thiazolopyrimidine derivatives and evaluation of their pharmacological activities. J. Mol. Struct. 2022, 1247, 131324. [Google Scholar] [CrossRef]
- Nagaraju, P.; Reddy, P.N.; Padmaja, P.; Ugale, V.G. Microwave-Assisted Synthesis of Thiazole/Benzothiazole Fused Pyranopyrimidine Derivatives and Evaluation of their Biological Activity. Lett. Org. Chem. 2021, 18, 49–57. [Google Scholar] [CrossRef]
- El-Shahat, M.; Salama, M.; El-Farargy, A.F.; Ali, M.M.; Ahmed, D.M. Effective pharmacophore for CDC25 phosphatases ezymeinhibitors: Newly synthesized bromothiazolopyrimidine derivatives. Mini Rev. Med. Chem. 2021, 21, 118–131. [Google Scholar] [CrossRef] [PubMed]
- Nagaraju, P.; Reddy, P.N.; Padmaja, P.; Ugale, V.G. Synthesis, Antiproliferative Activity and Molecular Docking of New Thiazole/Benzothiazole Fused Pyranopyrimidine Derivatives. Lett. Org. Chem. 2020, 17, 951–958. [Google Scholar] [CrossRef]
- Mohamed, S.F.; Abbas, E.M.; Khalaf, H.S.; Farghaly, T.A.; Abd El-Shafy, D.N. Triazolopyrimidines and thiazolopyrimidines: Synthesis, anti-HSV-1, cytotoxicity and mechanism of action. Mini Rev. Med. Chem. 2018, 18, 794–802. [Google Scholar] [CrossRef]
- Abdel-Hafez, N.A.; Mohamed, S.F.; El-Hag, F.A.; Hawas, U.W.; Awad, H.M. Synthesis and cytotoxicity evaluation of some new pyrimidinethione and thiazolopyrimidine derivatives linked to N-propylpiperidone. Der Pharma Chem. 2016, 8, 1–10. [Google Scholar]
- Keshari, K.A.; Singh, K.A.; Saha, S. Bridgehead nitrogen thiazolo [3, 2-a] pyrimidine: A privileged structural framework in drug discovery. Mini Rev. Med. Chem. 2017, 17, 1488–1499. [Google Scholar] [CrossRef]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Gali, R.; Banothu, J.; Porika, M.; Velpula, R.; Hnamte, S.; Bavantula, R.; Abbagani, S.; Busi, S. Indolylmethylene benzo[h]thiazolo[2, 3-b]quinazolinones: Synthesis, characterization and evaluation of anticancer and antimicrobial activities. Bioorg. Med. Chem. Lett. 2014, 24, 4239–4242. [Google Scholar] [CrossRef]
- Studzińska, R.; Kołodziejska, R.; Redka, M.; Modzelewska-Banachiewicz, B.; Augustyńska, B. Lipophilicity study of thiazolo[3, 2-a]pyrimidine derivatives as potential bioactive agents. J. Braz. Chem. Soc. 2016, 27, 1587–1593. [Google Scholar] [CrossRef]
- Zhou, B.; Li, X.; Li, Y.; Xu, Y.; Zhang, Z.; Zhou, M.; Zhang, X.; Liu, Z.; Zhou, J.; Cao, C.; et al. Discovery and Development of Thiazolo[3, 2-a]pyrimidinone Derivatives as General Inhibitors of Bcl-2 Family Proteins. ChemMedChem 2011, 6, 904–921. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Ding, X.; Chen, T.; Chen, L.; Liu, F.; Jia, X.; Luo, X.; Shen, X.; Chen, K.; Jiang, H.; et al. Design, synthesis, and interaction study of quinazoline-2(1H)-thione derivatives as novel potential Bcl-xL inhibitors. J. Med. Chem. 2010, 53, 3465–3479. [Google Scholar] [CrossRef] [PubMed]
- Kolb, S.; Mondésert, O.; Goddard, M.L.; Jullien, D.; Villoutreix, B.O.; Ducommun, B.; Garbay, C.; Braud, E. Development of novel thiazolopyrimidines as CDC25B phosphatase inhibitors. ChemMedChem Chem. Enabling Drug Discov. 2009, 4, 633–648. [Google Scholar] [CrossRef]
- Jin, C.H.; Jun, K.Y.; Lee, E.; Kim, S.; Kwon, Y.; Kim, K.; Na, Y. Ethyl 2-(benzylidene)-7-methyl-3-oxo-2,3-dihydro-5H-thiazolo[3, 2-a]pyrimidine-6-carboxylate analogues as a new scaffold for protein kinase casein kinase 2 inhibitor. Bioorg. Med. Chem. 2014, 22, 4553–4565. [Google Scholar] [CrossRef]
- Agarkov, A.S.; Gabitova, E.R.; Galieva, F.B.; Ovsyannikov, A.S.; Voloshina, A.D.; Shiryaev, A.K.; Litvinov, I.A.; Solovieva, S.E.; Antipin, I.S. Structure and Biological Properties of 2-Phenylhydrazone Derivatives of Thiazolopyrimidines. In Doklady Chemistry; Pleiades Publishing: New York, NY, USA, 2022; Volume 503, pp. 45–50. [Google Scholar]
- Agarkov, A.S.; Litvinov, I.A.; Gabitova, E.R.; Ovsyannikov, A.S.; Dorovatovskii, P.V.; Shiryaev, A.K.; Solovieva, S.E.; Antipin, I.S. Crystalline State Hydrogen Bonding of 2-(2-Hydroxybenzylidene)Thiazolo[3,2-a]Pyrimidines: A Way to Non-Centrosymmetric Crystals. Crystals 2022, 12, 494. [Google Scholar] [CrossRef]
- Lashmanova, E.A.; Rybakov, V.B.; Shiryaev, A.K. Synthesis of adamantylated pyrimidines using the Biginelli reaction. Synthesis 2016, 48, 3965–3970. [Google Scholar]
- Shiryaev, A.K.; Baranovskaya, N.S.; Eremin, M.S. Synthesis of 5H-thiazolo[3,2-a]pyrimidines. Chem. Heterocycl. Compd. 2012, 48, 1662–1667. [Google Scholar] [CrossRef]
- Shiryaev, A.K.; Kolesnikova, N.G.; Kuznetsova, N.M.; Lashmanova, E.A. Alkylation of tetrahydropyrimidine-2-thions with ethyl chloroacetate. Chem. Heterocycl. Compd. 2013, 49, 1812–1817. [Google Scholar]
- Lashmanova, E.A.; Kirdyashkina, A.I.; Slepukhin, P.A.; Shiryaev, A.K. Oxidation of thiazolo[3,2-a]pyrimidin-3(2H)-ones with DMSO and Lawesson’s reagent. Tetrahedron Lett. 2018, 59, 1099–1103. [Google Scholar] [CrossRef]
- Lashmanova, E.A.; Agarkov, A.S.; Rybakov, V.B.; Shiryaev, A.K. Rearrangement of thiazolo[3,2-a]pyrimidines into triazolo[4,3-a]pyrimidines induced by C=N bond reduction. Chem. Heterocycl. Compd. 2019, 55, 1217–1221. [Google Scholar] [CrossRef]
- Hwang, J.; Li, P.; Smith, M.D.; Warden, C.E.; Sirianni, D.A.; Vik, E.C.; Maier, J.M.; Yehl, C.J.; Sherrill, C.D.; Shimizu, K.D. Tipping the Balance between S-π and O-π Interactions. J. Am. Chem. Soc. 2018, 140, 13301–13307. [Google Scholar] [CrossRef] [PubMed]
- Caron, G.; Kihlberg, J.; Ermondi, G. Intramolecular hydrogen bonding: An opportunity for improved design in medicinal chemistry. Med. Res. Rev. 2019, 39, 1707–1729. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, B.; Mohr, P.; Stahl, M. Intramolecular hydrogen bonding in medicinal chemistry. J. Med. Chem. 2010, 53, 2601–2611. [Google Scholar] [CrossRef] [PubMed]
- Ayoup, M.S.; Wahby, Y.; Abdel-Hamid, H.; Ramadan, E.S.; Teleb, M.; Abu-Serie, M.M.; Noby, A. Design, synthesis and biological evaluation of novel α-acyloxy carboxamides via Passerini reaction as caspase 3/7 activators. Eur. J. Med. Chem. 2019, 168, 340–356. [Google Scholar] [CrossRef]
- Abdelwahid, E.; Rolland, S.; Teng, X.; Conradt, B.; Hardwick, J.M.; White, K. Mitochondrial involvement in cell death of non-mammalian eukaryotes. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2011, 1813, 597–607. [Google Scholar] [CrossRef]
- Orrenius, S.; Gogvadze, V.; Zhivotovsky, B. Mitochondrial oxidative stress: Implications for cell death. Annu. Rev. Pharmacol. Toxicol. 2007, 47, 143–183. [Google Scholar] [CrossRef]
- Carbone, R.; Pearson, M.; Minucci, S.; Pelicci, P.G. PML NBs associate with the hMre11 complex and p53 at sites of irradiation induced DNA damage. Oncogene 2002, 21, 1633–1640. [Google Scholar] [CrossRef]
- Lebedyeva, I.O.; Povstyanoy, M.V.; Ryabitskii, A.B.; Povstyanoy, V.M. Ternary condensation of Biginelli thiones, chloroacetic acid, and aldehydes as an effective approach towards thiazolo[3,2-a]pyrimidines and 5-arylidenethiazolidine-2,4-diones. J. Heterocycl. Chem. 2010, 47, 368–372. [Google Scholar] [CrossRef]
- Lazarenko, V.A.; Dorovatovskii, P.V.; Zubavichus, Y.V.; Burlov, A.S.; Koshchienko, Y.V.; Vlasenko, V.G.; Khrustalev, V.N. High-throughput small-molecule crystallography at the ‘Belok’ beamline of the Kurchatov synchrotron radiation source: Transition metal complexes with azomethine ligands as a case study. Crystals 2017, 7, 325. [Google Scholar] [CrossRef]
- Svetogorov, R.D.; Dorovatovskii, P.V.; Lazarenko, V.A. Belok/XSA diffraction beamline for studying crystalline samples at Kurchatov Synchrotron Radiation Source. Cryst. Res. Technol. 2020, 55, 1900184. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal-structure determination. Acta Crystallogr. Sect. A Found. Adv. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Farrugia, L.J. WinGX and ORTEP for Windows: An update. J. Appl. Crystallogr. 2012, 45, 849–854. [Google Scholar] [CrossRef]
- Spek, A.L. Structure validation in chemical crystallography. Acta Crystallogr. Sect. D Biol. Crystallogr. 2009, 65, 148–155. [Google Scholar] [CrossRef] [PubMed]
- Macrae, C.F.; Edgington, P.R.; McCabe, P.; Pidcock, E.; Shields, G.P.; Taylor, R.; Towler, M.; Streek, J.V.D. Mercury: Visualization and analysis of crystal structures. J. Appl. Crystallogr. 2006, 39, 453–457. [Google Scholar] [CrossRef]
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
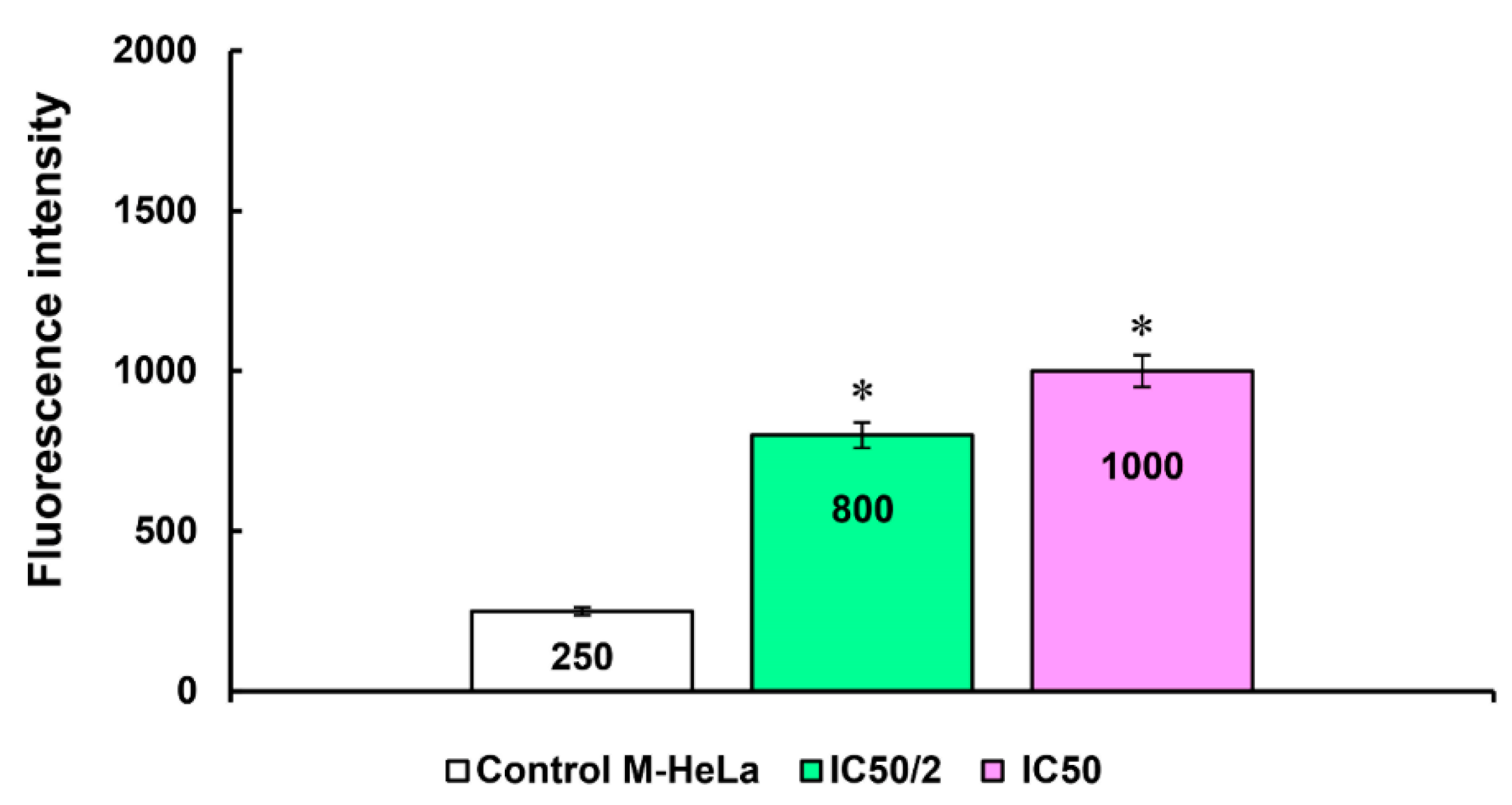{kind=link}
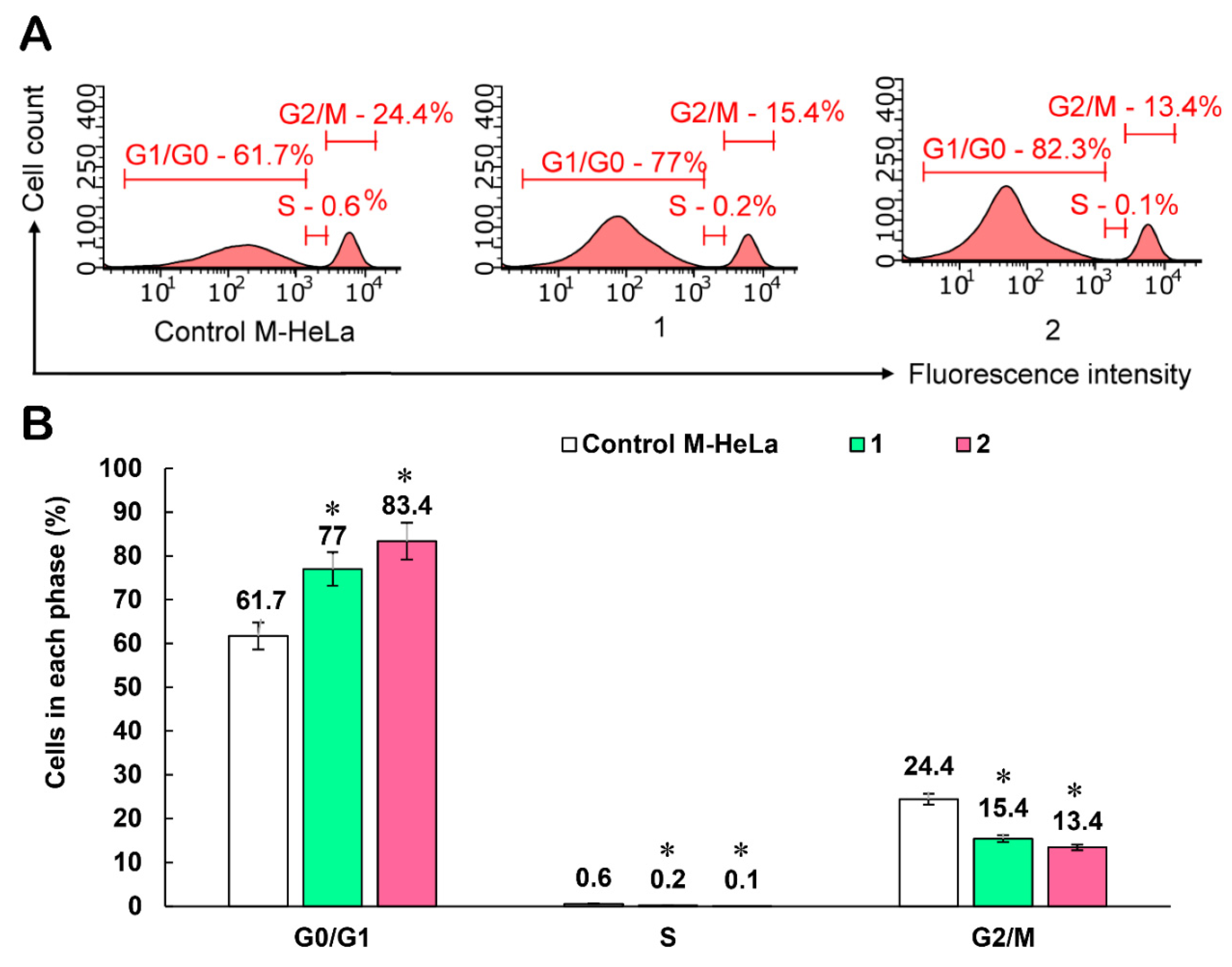{kind=link}
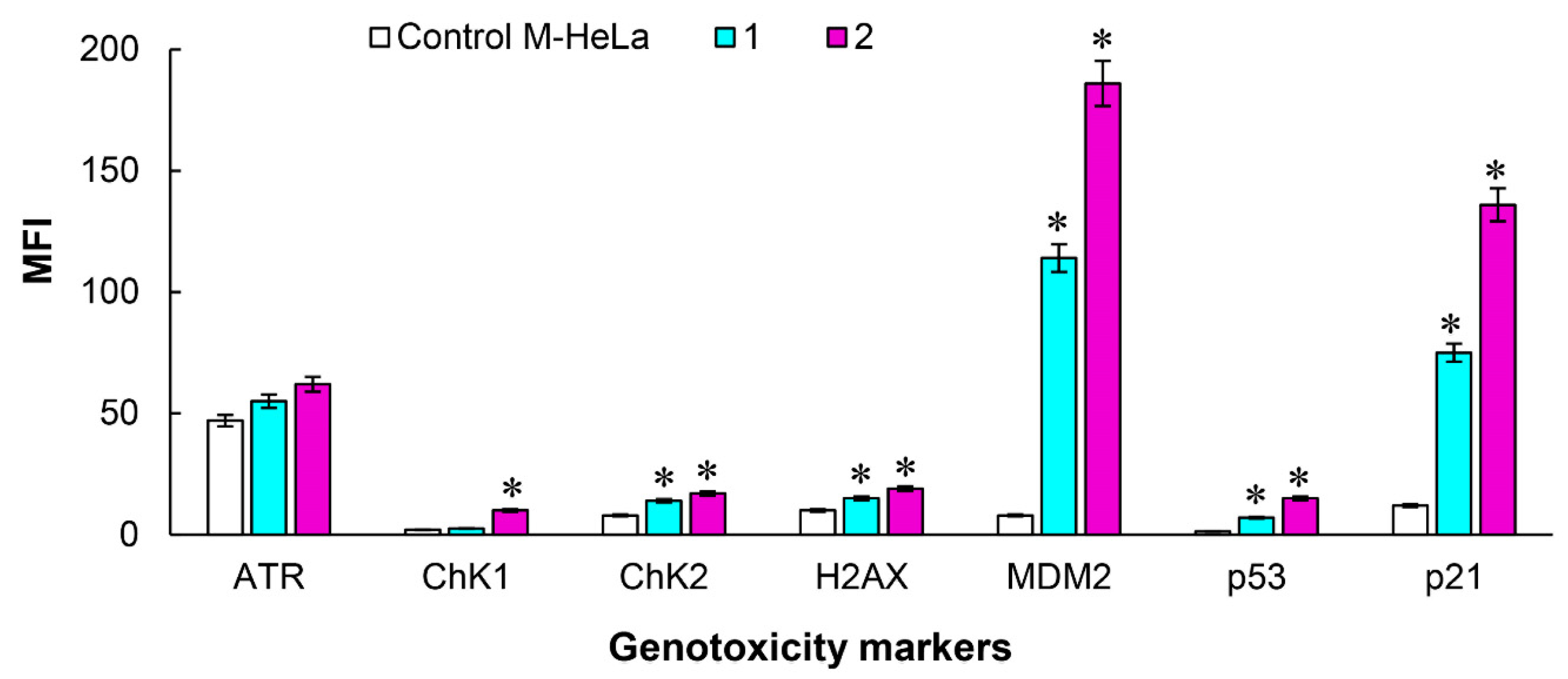{kind=link}
| Test Compounds | IC50 (µM) | ||||
|---|---|---|---|---|---|
| Cancer Cell Lines | Normal Cell Lines | ||||
| M-HeLa a | MCF-7 b | PC3 c | HuTu 80 d | Chang Liver (HeLa) e | |
| 7 | 11.9 ± 0.9 | 43.7 ± 3.5 | 77.6 ± 6.7 | 10.2 ± 0.8 | 75.0 ± 5.7 |
| 8 | 90 ± 8.3 | 93.3 ± 8.2 | 93.7 ± 8.5 | 85.9 ± 8.0 | 56.4 ± 4.5 |
| 9 | 20.6 ± 1.7 | 32.1 ± 2.5 | 23.3 ± 1.9 | 26.8 ± 2.1 | 22.7 ± 1.8 |
| 10 | 55.0 ± 4.4 | 56.0 ± 4.5 | 56.0 ± 4.4 | 60 ± 4.7 | 88.0 ± 7.0 |
| 11 | 56.4 ± 4.5 | 81.0 ± 7.4 | 54.0 ± 4.3 | 39.0 ± 3.1 | 64.0 ± 5.1 |
| 12 | 18.8 ± 1.5 | 22.3 ± 1.4 | 28.1 ± 2.2 | 23.7 ± 1.8 | 78.2 ± 6.2 |
| 13 | 70.0 ± 5.5 | >100 | 54.0 ± 4.3 | 43.3 ± 3.5 | 86.1 ± 6.8 |
| 14 | 31.0 ± 2.4 | 53.3 ± 4.2 | 49.3 ± 3.9 | 32.1 ± 2.5 | 53.2 ± 4.2 |
| 15 | >100 | >100 | 70.0 ± 5.5 | 74.5 ± 5.7 | 91.4 ± 7.2 |
Sorafenib  | 25.0 ± 1.8 | 27.5 ± 2.2 | 12.7 ± 1.1 | 13.0 ± 1.2 | 21.7 ± 1.7 |
| Test Compounds | Normal Cell Lines | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| M-HeLa a | MCF-7 b | PC3 c | HuTu 80 d | Chang Liver (HeLa) e | |||||
| IC50 | SI | IC50 | SI | IC50 | SI | IC50 | SI | ||
| 7 | 11.9 ± 0.9 | 6.3 | 43.7 ± 3.5 | 1.7 | 77.6 ± 6.7 | ns | 10.0 ± 0.8 | 7.5 | 75.0 ± 5.7 |
| 9 | 20.6 ± 1.7 | 1.1 | 32.1 ± 2.5 | ns | 23.3 ± 1.9 | ns | 26.8 ± 2.1 | ns | 22.7 ± 1.8 |
| 10 | 55.0 ± 4.4 | 1.6 | 56.0 ± 4.5 | 1.6 | 56.0 ± 4.4 | 1.6 | 14.6 ± 0.3 | 6.0 | 88.0 ± 7.0 |
| 12 | 18.8 ± 1.5 | 4.2 | 52.7 ± 4.2 | 1.5 | 28.1 ± 2.2 | 2.8 | 23.7 ± 1.8 | 3.3 | 78.2 ± 6.2 |
| Sorafenib | 25.0 ± 1.8 | ns | 27.5 ± 2.2 | ns | 27.5 ± 2.2 | ns | 13.0 ± 1.2 | 1.7 | 21.7 ± 1.7 |
| Compound | 10-MeOH | 11 | 12 | 12-DMSO |
|---|---|---|---|---|
| Molecular formula | C23H19N2O5S, CH4O | C28H22N2O4S | C23H19BrN2O4S | C23H19BrN2O4S, C2H6OS |
| Sum Formula | C24H23N2O5S | C28H22N2O4S | C23H19BrN2O4S | C26H28N2O6S2 |
| Formula Weight | 497.51 | 482.54 | 499.36 | 577.49 |
| Crystal System | triclinic | triclinic | triclinic | triclinic |
| Space group | P-1 (P1bar) | P-1 (P1bar) | P-1 (P1bar) | P-1 (P1bar) |
| Temp. of measurement, K | 100(2) | 105(2) | 100(2) | 105(2) |
| Cell parameters | a = 9.4229(5) Å, b = 9.4636(5) Å, c = 12.8810(7) Å; α = 82.651(2)° β = 89.551(2)° γ = 77.443(2)° | a = 9.7165(8) Å, b = 10.1644(9) Å, c = 13.4602(12) Å; α = 101.395(3)° β = 99.097(3)° γ = 113.423(3)° | a = 7.7700(16) Å, b = 11.950(2) Å, c = 12.540(3) Å; α = 64.59(3)° β = 82.44(3)° γ = 86.70(3)° | a = 8.5364(2) Å, b = 11.8152(3) Å, c = 13.4137(4) Å; α = 103.604(1)° β = 106.947(1)° γ = 92.508(1)° |
| V [Å3] | 1111.74(10) | 1153.65(18) | 1042.6(5) | 1248.63(6) |
| Z and Z′ | 2 and 1 | 2 and 1 | 2 and 1 | 2 and 1 |
| D(calc) [g/cm3] | 1.486 | 1.389 | 1.591 | 1.536 |
| λ (Å) | (MoKα) 0.71073 | (MoKα) 0.71073 | 0.7450 (synchrotron) | (MoKα) 0.71073 |
| μ [/mm] | 0.199 | 0.180 | 2.355 | 1.853 |
| F(000) | 520 | 504 | 508 | 592 |
| Theta Min-Max [Deg] | 2.2–28.0° | 2.3–30.0° | 1.9–31.0° | 1.8–32.0° |
| Reflections measured | 41,825 | 89,193 | 20,991 | 44,055 |
| Independent reflections | 5357 | 6713 | 5717 | 8644 |
| Observed reflections [I > 2σ(I)] | 4506 | 5883 | 5297 | 7568 |
| Goodness of fit | 1.138 | 1.030 | 1.076 | 1.048 |
| R [I > 2σ(I)] | R1 = 0.0391, wR2 = 0.1137 | R1 = 0.0340, wR2 = 0.0956 | R1 = 0.0326, wR2 = 0.0867 | R1 = 0.0356, wR2 = 0.1004 |
| R (all reflections) | R1 = 0.0491, wR2 = 0.1189 | R1 = 0.0397, wR2 = 0.0985 | R1 = 0.0361, wR2 = 0.0901 | R1 = 0.0418, wR2 = 0.1036 |
| Max. and Min. Resd. Dens. [e/Å−3] | 0.52 and −0.23 | 0.46 and −0.27 | 0.44 and −0.86 | 1.19 and −0.51 |
| Depositor numbers in CCDC | 2,213,096 | 2,213,093 | 2,213,097 | 2,213,094 |
| Compound | 13 | 14 | 14-DMSO | |
| Molecular formula | C23H20N2O4S | C23H19BrN2O4S | C23H19BrN2O4S, C2H6OS | |
| Sum Formula | C23H20N2O4S | C23H19BrN2O4S | C26H28N2O6S2 | |
| Formula Weight | 420.47 | 499.36 | 577.49 | |
| Crystal System | monoclinic | monoclinic | triclinic | |
| Space group | Pc | P21/n | P-1 (P1bar) | |
| Temp. of measurement, K | 110(2) | 110(2) | 105(2) | |
| Cell parameters | a = 11.0059(15) Å, b = 13.6530(15) Å, c = 14.8440(18) Å; β = 109.743(3)° | a = 11.3394(5) Å, b = 14.2710(7) Å, c = 13.6906(6) Å; β = 104.046(2)° | a = 8.8459(4) Å, b = 11.2404(4) Å, c = 12.8542(5) Å; α = 89.785(2)° β = 88.746(2)° γ = 83.140(2)° | |
| V [Å3] | 2099.4(4) | 2149.24(17) | 1268.66(9) | |
| Z and Z′ | 4 and 2 | 4 and 1 | 2 and 1 | |
| D(calc) [g/cm3] | 1.330 | 1.543 | 1.512 | |
| λ (Å) | (MoKα) 0.71073 | (MoKα) 0.71073 | (MoKα) 0.71073 | |
| μ [/mm] | 0.186 | 2.043 | 1.824 | |
| F(000) | 880 | 1016 | 592 | |
| Theta Min-Max [Deg] | 1.5–26.6° | 2.1–30.0°, | 1.8–30.0° | |
| Reflections measured | 69,218 | 104,197 | 89,171 | |
| Independent reflections | 8626 | 6257 | 7381 | |
| Observed reflections [I > 2σ(I)] | 6008 | 4741 | 6684 | |
| Goodness of fit | 1.062 | 1.009 | 1.027 | |
| R [I > 2σ(I)] | R1 0.0576, wR2 = 0.1249 | R1 = 0.0347, wR2 = 0.0713 | R1 = 0.0304, wR2 = 0.0761 | |
| R (all reflections) | R1 = 0.1017, wR2 = 0.1417 | R1 = 0.0578, wR2 = 0.0783 | R1 = 0.0355, wR2 = 0.0786 | |
| Max. and Min. Resd. Dens. [e/Å−3] | 0.35 and −0.55 | 0.55 and −0.41 | 0.68 and −0.61 | |
| Flack parameter | −0.35(8) (twin, BASF 0.52081) | - | - | |
| Depositor numbers in CCDC | 2,213,099 | 2,213,095 | 2,213,098 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Agarkov, A.S.; Nefedova, A.A.; Gabitova, E.R.; Ovsyannikov, A.S.; Amerhanova, S.K.; Lyubina, A.P.; Voloshina, A.D.; Dorovatovskii, P.V.; Litvinov, I.A.; Solovieva, S.E.; et al. Synthesis, Self-Assembly in Crystalline Phase and Anti-Tumor Activity of 2-(2-/4-Hydroxybenzylidene)thiazolo[3,2-a]pyrimidines. Molecules 2022, 27, 7747. https://doi.org/10.3390/molecules27227747
Agarkov AS, Nefedova AA, Gabitova ER, Ovsyannikov AS, Amerhanova SK, Lyubina AP, Voloshina AD, Dorovatovskii PV, Litvinov IA, Solovieva SE, et al. Synthesis, Self-Assembly in Crystalline Phase and Anti-Tumor Activity of 2-(2-/4-Hydroxybenzylidene)thiazolo[3,2-a]pyrimidines. Molecules. 2022; 27(22):7747. https://doi.org/10.3390/molecules27227747
Chicago/Turabian StyleAgarkov, Artem S., Anna A. Nefedova, Elina R. Gabitova, Alexander S. Ovsyannikov, Syumbelya K. Amerhanova, Anna P. Lyubina, Alexandra D. Voloshina, Pavel V. Dorovatovskii, Igor A. Litvinov, Svetlana E. Solovieva, and et al. 2022. "Synthesis, Self-Assembly in Crystalline Phase and Anti-Tumor Activity of 2-(2-/4-Hydroxybenzylidene)thiazolo[3,2-a]pyrimidines" Molecules 27, no. 22: 7747. https://doi.org/10.3390/molecules27227747
APA StyleAgarkov, A. S., Nefedova, A. A., Gabitova, E. R., Ovsyannikov, A. S., Amerhanova, S. K., Lyubina, A. P., Voloshina, A. D., Dorovatovskii, P. V., Litvinov, I. A., Solovieva, S. E., & Antipin, I. S. (2022). Synthesis, Self-Assembly in Crystalline Phase and Anti-Tumor Activity of 2-(2-/4-Hydroxybenzylidene)thiazolo[3,2-a]pyrimidines. Molecules, 27(22), 7747. https://doi.org/10.3390/molecules27227747