

Simultaneous Enantiodivergent Synthesis of Diverse Lactones and Lactams via Sequential One-Pot Enzymatic Kinetic Resolution–Ring-Closing Metathesis Reactions


Abstract
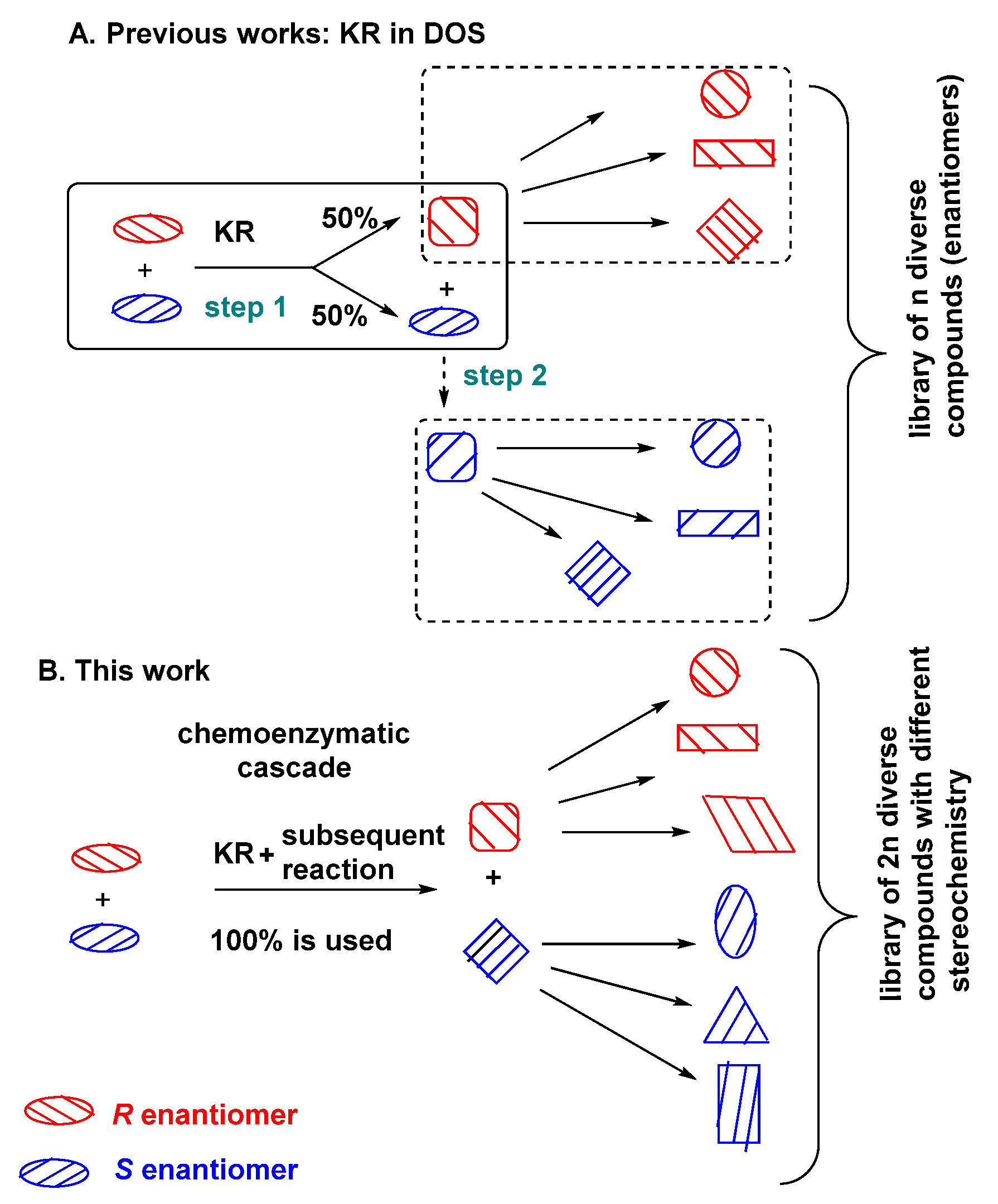
1. Introduction
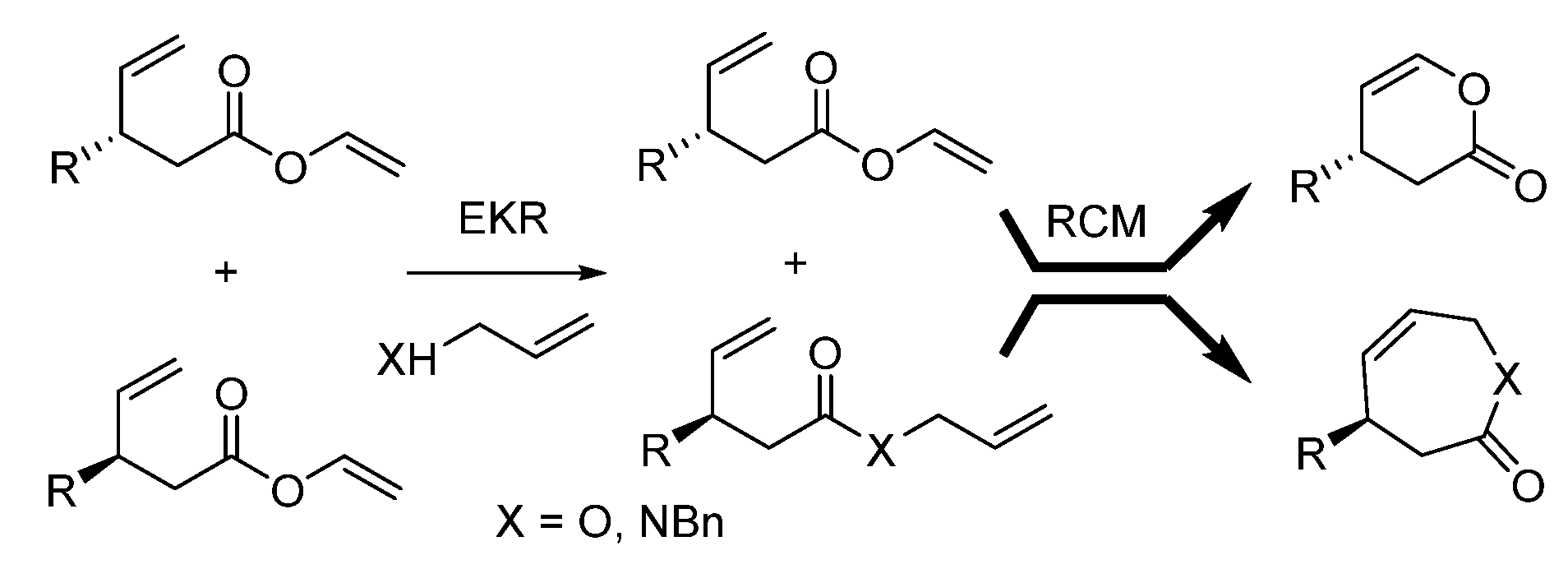
2. Results and Discussion
3. Materials and Methods
3.1. General
3.2. General Procedure of the Enzymatic Kinetic Resolution Reaction (EKR) of 1a–d with 2
3.3. General Procedure of the Enzymatic Kinetic Resolution Reaction (EKR) of 1a–d with 6
3.4. General Procedure of the Ring-Closing Metathesis Reaction (RCM)
3.4.1. 4-Phenyl-3,4-Dihydro-2H-Pyran-2-One (4a)
3.4.2. 4-(4-Bromophenyl)-3,4-Dihydro-2H-Pyran-2-One (4b)
3.4.3. 4-(4-Chlorophenyl)-3,4-Dihydro-2H-Pyran-2-One (4c)
3.4.4. 4-Propyl-3,4-Dihydro-2H-Pyran-2-One (4d)
3.4.5. 4-Phenyl-3,4-Dihydrooxepin-2(7H)-One (5a)
3.4.6. 4-(4-Bromophenyl)-3,4-Dihydrooxepin-2(7H)-One (5b)
3.4.7. 4-(4-Chlorophenyl)-3,4-Dihydrooxepin-2(7H)-One (5c)
3.4.8. 4-Propyl-3,4-Dihydrooxepin-2(7H)-One (5d)
3.4.9. 1-Benzyl-4-Phenyl-1,3,4,7-Tetrahydro-Azepin-2-One (8a)
3.4.10. 1-Benzyl-4-(4-Bromophenyl)-1,3,4,7-Tetrahydro-Azepin-2-One (8b)
3.4.11. 1-Benzyl-4-(4-Chloroophenyl)-1,3,4,7-Tetrahydro-Azepin-2-One (8c)
3.4.12. 1-Benzyl-4-Propyl-1,3,4,7-Tetrahydro-Azepin-2-One (8d)
3.5. General Procedure of the One-Pot EKR-RCM
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Schreiber, S.L. Target-Oriented and Diversity-Oriented Organic Synthesis in Drug Discovery. Science 2000, 287, 1964–1969. [Google Scholar] [CrossRef] [PubMed]
- Gray, B.L.; Schreiber, S.L. Skeletal Diversity in Small-Molecule Synthesis Using Ligand-Controlled Catalysis. J. Comb. Chem. 2007, 9, 1028–1035. [Google Scholar] [CrossRef] [PubMed]
- Burke, M.D.; Schreiber, S.L. A Planning Strategy for Diversity-Oriented Synthesis. Angew. Chem. Int. Ed. 2003, 43, 46–58. [Google Scholar] [CrossRef] [PubMed]
- Dandapani, S.; A Marcaurelle, L. Current strategies for diversity-oriented synthesis. Curr. Opin. Chem. Biol. 2010, 14, 362–370. [Google Scholar] [CrossRef] [PubMed]
- O’ Connor, C.J.; Beckmann, H.S.G.; Spring, D.R. Diversity-oriented synthesis: Producing chemical tools for dissecting biology. Chem. Soc. Rev. 2012, 41, 4444–4456. [Google Scholar] [CrossRef]
- Adriaenssens, L.V.; Austin, C.A.; Gibson, M.; Smith, D.; Hartley, R.C. Stereodivergent diversity oriented synthesis of pi-peridine alkaloids. Eur. J. Org. Chem. 2006, 2006, 4998–5001. [Google Scholar] [CrossRef]
- Guduru, S.K.R.; Chamakuri, S.; Raji, I.O.; MacKenzie, K.R.; Santini, C.; Young, D.W. Synthesis of Enantiomerically Pure 3-Substituted Piperazine-2-acetic Acid Esters as Intermediates for Library Production. J. Org. Chem. 2018, 83, 11777–11793. [Google Scholar] [CrossRef]
- Chamakuri, S.; Jain, P.; Guduru, S.K.R.; Arney, J.W.; MacKenzie, K.R.; Santini, C.; Young, D.W. Synthesis of Enantio-merically Pure 6-Substituted-Piperazine-2-Acetic Acid Esters as Intermediates for Library Production. J. Org. Chem. 2018, 83, 6541–6555. [Google Scholar] [CrossRef]
- Stavenger, R.; Schreiber, S.L. Asymmetric Catalysis in Diversity-Oriented Organic Synthesis: Enantioselective Synthesis of 4320 Encoded and Spatially Segregated Dihydropyrancarboxamides. Angew. Chem. Int. Ed. 2001, 40, 3417–3421. [Google Scholar] [CrossRef]
- Keith, J.M.; Larrow, J.F.; Jacobsen, E.N. Practical Considerations in Kinetic Resolution Reactions. Adv. Synth. Catal. 2001, 343, 5–26. [Google Scholar] [CrossRef]
- Ghanem, A.; Aboul-Enein, H.Y. Application of Lipases in Kinetic Resolution of Racemates. Chirality 2005, 17, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Seddigi, Z.S.; Malik, M.S.; Ahmed, S.A.; Babalghith, A.O.; Kamal, A. Lipases in asymmetric transformations: Recent ad-vances in classical kinetic resolution and lipase–metal combinations for dynamic processes. Coord. Chem. Rev. 2017, 348, 54–70. [Google Scholar] [CrossRef]
- Angoli, M.; Barilli, A.; Lesma, G.; Passarella, D.; Riva, S.; Silvani, A.; Danieli, B. Remote Stereocenter Discrimination in the Enzymatic Resolution of Piperidine-2-ethanol. Short Enantioselective Synthesis of Sedamine and Allosedamine. J. Org. Chem. 2003, 68, 9525–9527. [Google Scholar] [CrossRef] [PubMed]
- Perdicchia, D.; Christodoulou, M.S.; Fumagalli, G.; Calogero, F.; Marucci, C.; Passarella, D. Enzymatic Kinetic Resolution of 2-Piperidineethanol for the Enantioselective Targeted and Diversity Oriented Synthesis. Int. J. Mol. Sci. 2015, 17, 17. [Google Scholar] [CrossRef]
- Bhuniya, R.; Nanda, S. Enantiomeric scaffolding of α-tetralone and related scaffolds by EKR (Enzymatic Kinetic Resolution) and stereoselective ketoreduction with ketoreductases. Org. Biomol. Chem. 2011, 10, 536–547. [Google Scholar] [CrossRef]
- Reddy, U.C.; Manheri, M.K. 1-Hydroxymethyl-7-oxabicyclo [2.2.1]hept-2-ene skeleton in enantiopure form through enzy-matic kinetic resolution. Chirality 2019, 31, 336–347. [Google Scholar] [CrossRef]
- Pirkle, W.H.; Adams, P.E. Enantiomerically Pure Lactones. 3. Synthesis of and Stereospecific Conjugate Additions to α,β-Unsaturated Lactones. J. Org. Chem. 1980, 45, 4117–4121. [Google Scholar] [CrossRef]
- Hughes, G.; Kimura, M.; Buchwald, S.L. Catalytic Enantioselective Conjugate Reduction of Lactones and Lactams. J. Am. Chem. Soc. 2003, 125, 11253–11258. [Google Scholar] [CrossRef]
- Elliott, J.M.; Carlson, E.J.; Chicchi, G.G.; Dirat, O.; Dominguez, M.; Gerhard, U.; Jelley, R.; Jones, A.B.; Kurtz, M.M.; Tsao, K.L.; et al. NK1 antagonists based on seven membered lactam scaffolds. Bioorganic Med. Chem. Lett. 2006, 16, 2929–2932. [Google Scholar] [CrossRef]
- Clarke, A.K.; Unsworth, W.P. A happy medium: The synthesis of medicinally important medium-sized rings via ring ex-pansion. Chem. Sci. 2020, 11, 2876–2881. [Google Scholar] [CrossRef]
- Majumdar, K.C. Regioselective formation of medium-ring heterocycles of biological relevance by intramolecular cyclization. RSC Adv. 2011, 1, 1152–1170. [Google Scholar] [CrossRef]
- Kurouchi, H.; Ohwada, T. Synthesis of Medium-Ring-Sized Benzolactams by Using Strong Electrophiles and Quantitative Evaluation of Ring-Size Dependency of the Cyclization Reaction Rate. J. Org. Chem. 2019, 85, 876–901. [Google Scholar] [CrossRef]
- Tori, M.; Shiotani, Y.; Tanaka, M.; Nakashima, K.; Sono, M. Eremofarfugin A and eremopetasitenin B3, two new eremophi-lanolides from Farfugium japonicum. Tetrahedron Lett. 2000, 41, 1797–1799. [Google Scholar] [CrossRef]
- Seo, E.-K.; Wani, M.C.; E Wall, M.; Navarro, H.; Mukherjee, R.; Farnsworth, N.R.; Kinghorn, A.D. New bioactive aromatic compounds from Vismia guianensis. Phytochemistry 2000, 55, 35–42. [Google Scholar] [CrossRef]
- Schulz, A.G.; Pettus, L. Desymmetrization of Benzoic Acid in the Context of the Asymmetric Birch Reduction−Alkylation Protocol. Asymmetric Total Syntheses of (−)-Eburnamonine and (−)-Aspidospermidine. J. Org. Chem. 1997, 62, 6855–6861. [Google Scholar] [CrossRef]
- Jiménez-Tenorio, M.; Puerta, M.C.; Valerga, P.; Moreno-Dorado, F.J.; Guerra, F.M.; Massanet, G.M. Regioselective cyclization of α,ω-alkynoic acids catalysed by TpRu complexes: Synthesis of endocyclic enol lactones [Tp = hydrotris(pyrazolyl)borate]. Chem. Commun. 2001, 2324–2325. [Google Scholar] [CrossRef] [PubMed]
- Mao, B.; Fañanás-Mastral, M.; Feringa, B.L. Asymmetric Conjugate Addition of Grignard Reagents to Pyranones. Org. Lett. 2013, 15, 286–289. [Google Scholar] [CrossRef]
- Brodzka, A.; Borys, F.; Koszelewski, D.; Ostaszewski, R. Studies on the Synthesis of Endocyclic Enol Lactones via a RCM of Selected Vinyl Esters. J. Org. Chem. 2018, 83, 8655–8661. [Google Scholar] [CrossRef]
- Brodzka, A.; Koszelewski, D.; Ostaszewski, R. Dual Activity of Grubbs-Type Catalyst in the Transvinylation of Carboxylic Acids and Ring-Closing Metathesis Reactions. J. Org. Chem. 2020, 85, 15305–15313. [Google Scholar] [CrossRef]
- Koszelewski, D.; Paprocki, D.; Brodzka, A.; Ostaszewski, R. Enzyme mediated kinetic resolution of δ-hydroxy-α,β-unsaturated esters as a route to optically active lactones. Tetrahedron: Asymmetry 2017, 28, 809–818. [Google Scholar] [CrossRef]
- Koszelewski, D.; Borys, F.; Brodzka, A.; Ostaszewski, R. Synthesis of Enantiomerically Pure 5,6-Dihydropyran-2-ones via Chemoenzymatic Sequential DKR-RCM Reaction. Eur. J. Org. Chem. 2018, 2019, 1653–1658. [Google Scholar] [CrossRef]
- Riguet, E. Enantioselective Organocatalytic Friedel–Crafts Alkylation Reaction of Indoles with 5-Hydroxyfuran-2(5H)-one: Access to Chiral γ-Lactones and γ-Lactams via a Ugi 4-Center 3-Component Reaction. J. Org. Chem. 2011, 76, 8143–8150. [Google Scholar] [CrossRef] [PubMed]
- Tang, M.; Xing, D.; Huang, H.; Hu, W. Divergent synthesis of chiral heterocycles via sequencing of enantioselective three-component reactions and one-pot subsequent cyclization reactions. Chem. Commun. 2015, 51, 10612–10615. [Google Scholar] [CrossRef] [PubMed]
- Caputo, S.; Banfi, L.; Basso, A.; Galatini, A.; Moni, L.; Riva, R.; Lambruschini, C. Diversity-Oriented Synthesis of Various Enantiopure Heterocycles by Coupling Organocatalysis with Multicomponent Reactions. Eur. J. Org. Chem. 2017, 2017, 6619–6628. [Google Scholar] [CrossRef]
- Brodzka, A.; Koszelewski, D.; Ostaszewski, R. The studies on chemoenzymatic synthesis of Femoxetine. J. Mol. Catal. B: Enzym. 2012, 82, 96–101. [Google Scholar] [CrossRef]
- Koszelewski, D.; Cwiklak, M.; Ostaszewski, R. A new chemoenzymatic approach to the synthesis of chiral 4-aryl-1,4-dihydro-2H-isoquinolines via the enzymatic resolution of 2-acetyl-4-phenyl-1,4-dihydro-2H-isoquinolin-3-one. Tetrahedron: Asymmetry 2012, 23, 1256–1261. [Google Scholar] [CrossRef]
- Brodzka, A.; Koszelewski, D.; Zysk, M.; Ostaszewski, R. The mechanistic promiscuity of the enzymatic esterification of chiral carboxylic acids. Catal. Commun. 2018, 106, 82–86. [Google Scholar] [CrossRef]
- Żądło-Dobrowolska, A.; Koszelewski, D.; Paprocki, D.; Madej, A.; Wilk, M.; Ostaszewski, R. Enzyme-Promoted Asymmetric Tandem Passerini Reaction. ChemCatChem 2017, 9, 3047–3053. [Google Scholar] [CrossRef]
- Koszelewski, D.; Zysk, M.; Brodzka, A.; Żądło, A.; Paprocki, D.; Ostaszewski, R. Evaluation of a new protocol for enzymatic dynamic kinetic resolution of 3-hydroxy-3-(aryl)propanoic acids. Org. Biomol. Chem. 2015, 13, 11014–11020. [Google Scholar] [CrossRef]
- Koszelewski, D.; Brodzka, A.; Żądło, A.; Paprocki, D.; Trzepizur, D.; Zysk, M.; Ostaszewski, R. Dynamic Kinetic Resolution of 3-Aryl-4-pentenoic Acids. ACS Catal. 2016, 6, 3287–3292. [Google Scholar] [CrossRef]
- Sheldon, R.A. E factors, green chemistry and catalysis: An odyssey. Chem. Commun. 2008, 3352–3365. [Google Scholar] [CrossRef] [PubMed]
- Lapin, I. Phenibut (β-Phenyl-GABA): A Tranquilizer and Nootropic Drug. CNS Drug Rev. 2001, 7, 471–481. [Google Scholar] [CrossRef] [PubMed]
- Brodzka, A.; Koszelewski, D.; Cwiklak, M.; Ostaszewski, R. Studies on the chemoenzymatic synthesis of 3-phenyl-GABA and 4-phenyl-pyrrolid-2-one: The influence of donor of the alkoxy group on enantioselective esterification. Tetrahedron: Asymmetry 2013, 24, 427–433. [Google Scholar] [CrossRef]
- Saldívar-González, F.I.; Lenci, E.; Trabocchi, A.; Medina-Franco, J.L. Exploring the chemical space and the bioactivity profile of lactams: A chemoinformatic study. RSC Adv. 2019, 9, 27105–27116. [Google Scholar] [CrossRef]
- Persson, B.A.; Larsson, A.L.E.; Le Ray, M.; Ba1ckvall, J.-E. Ruthenium- and Enzyme-Catalyzed Dynamic Kinetic Resolution of Secondary Alcohols. J. Am. Chem. Soc. 1999, 121, 1645–1650. [Google Scholar] [CrossRef]
- Paravidino, M.; Hanefeld, U. Enzymatic acylation: Assessing the greenness of different acyl donors. Green Chem. 2011, 13, 2651–2657. [Google Scholar] [CrossRef]
- Izumi, T.; Tamura, F.; Akutsu, M. Enzymatic resolution of 4-methyl-, 4-phenyl- and 6-phenyltetrahydro-2H-pyran-2-one using esterases. J. Heterocycl. Chem. 1994, 31, 441–445. [Google Scholar] [CrossRef]
- Chen, L.-Y.; Zaks, A.; Chackalamannil, S.; Dugar, S. Asymmetric Synthesis of Substituted 2-Azaspiro [3.5]nonan-1-ones: An Enantioselective Synthesis of the Cholesterol Absorption Inhibitor (+)-SCH 54016. J. Org. Chem. 1996, 61, 8341–8343. [Google Scholar] [CrossRef]
- Clader, J.W. Ezetimibe and other azetidinone cholesterol absorption inhibitors. Curr. Top. Med. Chem. 2005, 5, 243–256. [Google Scholar] [CrossRef]
- Reynolds, N.T.; Rovis, T. The effect of pre-existing stereocenters in the intramolecular asymmetric Stetter reaction. Tetrahedron 2005, 61, 6368–6378. [Google Scholar] [CrossRef]
- Prandi, C.; Ferrali, A.; Guarna, A.; Venturello, P.; Occhiato, E.G. New Synthetic Approach to Cyclopenta-Fused Heterocycles Based upon a Mild Nazarov Reaction. 2. Further Studies on the Torquoselectivity. J. Org. Chem. 2004, 69, 7705–7709. [Google Scholar] [CrossRef] [PubMed]
- Reed, P.E.; Katzenellenbogen, J.A. beta-Substituted beta-phenylpropionyl chymotrypsins. Structural and stereochemical features in stable acyl enzymes. J. Med. Chem. 1991, 34, 1162–1176. [Google Scholar] [CrossRef] [PubMed]
- Ren, Q.; Sun, S.; Huang, J.; Li, W.; Wu, M.; Guo, H.; Wang, J. An enantioselective cascade reaction between α,β-unsaturated aldehydes and malonic half-thioesters: A rapid access to chiral δ-lactones. Chem. Commun. 2014, 50, 6137–6140. [Google Scholar] [CrossRef] [PubMed]
- Mellerup, E.T.; Plenge, P. High affinity binding of 3H-paroxetine and 3H-imipramine to rat neuronal membranes. Psychopharmacology 1986, 89, 436–439. [Google Scholar] [CrossRef] [PubMed]
- de Laszlo, S.E.; Bush, B.L.; Doyle, J.J.; Greenlee, W.J.; Hangauer, D.G.; Halgren, T.A.; Lynch, R.J.; Schom, T.W.; Siegl, P.K.S. Synthesis and use of 3-amino-4-phenyl-2-piperidones and 4-amino-2-benzazepin-3-ones as conformationally restricted phenylalanine isosteres in renin inhibitors. J. Med. Chem. 1992, 35, 833–846. [Google Scholar] [CrossRef]
- Moore, K.P.; Zhu, H.; Rajapakse, H.A.; McGaughey, G.B.; Colussi, D.; Price, E.A.; Sankaranarayanan, S.; Simon, A.J.; Pudvah, N.T.; Hochman, J.H.; et al. Strategies toward improving the brain penetration of macrocyclic tertiary carbinamine BACE-1 inhibitors. Bioorg. Med. Chem. Lett. 2007, 17, 5831–5835. [Google Scholar] [CrossRef]
- Gao, M.; Wang, D.-X.; Zheng, Q.-Y.; Wang, M.-X. An Unusual β-Vinyl Effect Leading to High Efficiency and Enantioselectivity of the Amidase, Nitrile Biotransformations for the Preparation of Enantiopure 3-Arylpent-4-enoic Acids and Amides and Their Applications in Synthesis. J. Org. Chem. 2006, 71, 9532–9535. [Google Scholar] [CrossRef] [PubMed]
- Dong, J.; Liu, Y.; Cui, Y. Chiral porous organic frameworks for asymmetric heterogeneous catalysis and gas chromatographic separation. Chem. Commun. 2014, 50, 14949–14952. [Google Scholar] [CrossRef]
- Basu, S.; Gupta, V.; Nickel, J.; Schneider, C. Organocatalytic Enantioselective Vinylogous Michael Reaction of Vinylketene Silyl-N,O-Acetals. Org. Lett. 2013, 16, 274–277. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
 | ||||||
|---|---|---|---|---|---|---|
| Entry | Enzyme | Yield of 4 [%] [b] | e.e.S [%] [c] | Yield of 5 [%] [b] | e.e.P [%] [c] | E [d] |
| 1 | - | 93 | 0 | 0 | nd | nd |
| 2 | Amano AK PfL | 58 | 35 | 36 | 52 | 4 |
| 3 | Amano PS PcL | 82 | 10 | 12 | 61 | 5 |
| 4 | RoL | 88 | 0 | 6 | 0 | nd |
| 5 | CALB | 81 | 16 | 12 | 95 | 46 |
| 6 | Novozym | 65 | 42 | 29 | 99 | >200 |
| 7 | Lipozyme | 55 | 23 | 40 | 33 | 2 |
| Entry | Solvent, Temp. | Yield of 4 [%] [b] | e.e.S [%] [c] | Yield of 5 [%] [b] | e.e.P [%] [c] | E [d] |
|---|---|---|---|---|---|---|
| 1 | Toluene 40 °C | 73 | 23 | 19 | 99 | >200 |
| 2 | Toluene 50 °C | 68 | 35 | 25 | 99 | >200 |
| 3 | Toluene 60 °C | 65 | 42 | 29 | 99 | >200 |
| 4 | Toluene 70 °C | 57 | 55 | 35 | 99 | >200 |
| 5 | TBME 40 °C | 42 | 30 | 47 | 21 | 2 |
| 6 | DCM 40 °C | 93 | nd | <5 | nd | nd |
| 7 | THF 40 °C | 91 | nd | <5 | nd | nd |
| 8 | CHCl3 40 °C | 93 | nd | <5 | nd | nd |
| 9 | MeCN 40 °C | 70 | 21 | 22 | 72 | 8 |
| 10 | Toluene 70 °C (48 h) | 47 | 99 | 43 | 99 | >200 |
 | ||||||
|---|---|---|---|---|---|---|
| Entry | Substrate | Yield of 4 [%] [b] | e.e.S [%] [c] | Yield of 5 [%] [b] | e.e.P [%] [c] | E [d] |
| 1 | 1a | 47 | >99 | 43 | >99 | >200 |
| 2 | 1b | 64 | 25 | 27 | 44 | 3 |
| 3 | 1c | 57 | Nd [e] | 22 | Nd [f] | nd |
| 4 | 1d | 52 | 72 | 16 | 78 | 17 |
 | |||||
|---|---|---|---|---|---|
| Entry | Enzyme | Yield of 7 [%] [b] | e.e.S [%] [c] | e.e.P [%] [c] | E [d] |
| 1 | - | 0 | 0 | nd | nd |
| 2 | Novozym | 50 | >99 | >99 | >200 |
| 3 | Lipozyme | 45 | 80 | 98 | >200 |
 | ||||||
|---|---|---|---|---|---|---|
| Entry | Substrate | Yield of 4 [%] [b] | e.e.S [%] [c] | Yield of 8 [%] [b] | e.e.P [%] [c] | E [d] |
| 1 | 1a | 48 | >99 | 45 | >99 | >200 |
| 2 | 1b | 62 | 23 | 32 | 37 | 3 |
| 3 | 1c | 55 | nd [e] | 25 | nd [f] | nd |
| 4 | 1d | 49 | 81 | 17 | 77 | 20 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brodzka, A.; Koszelewski, D.; Ostaszewski, R. Simultaneous Enantiodivergent Synthesis of Diverse Lactones and Lactams via Sequential One-Pot Enzymatic Kinetic Resolution–Ring-Closing Metathesis Reactions. Molecules 2022, 27, 7696. https://doi.org/10.3390/molecules27227696
Brodzka A, Koszelewski D, Ostaszewski R. Simultaneous Enantiodivergent Synthesis of Diverse Lactones and Lactams via Sequential One-Pot Enzymatic Kinetic Resolution–Ring-Closing Metathesis Reactions. Molecules. 2022; 27(22):7696. https://doi.org/10.3390/molecules27227696
Chicago/Turabian StyleBrodzka, Anna, Dominik Koszelewski, and Ryszard Ostaszewski. 2022. "Simultaneous Enantiodivergent Synthesis of Diverse Lactones and Lactams via Sequential One-Pot Enzymatic Kinetic Resolution–Ring-Closing Metathesis Reactions" Molecules 27, no. 22: 7696. https://doi.org/10.3390/molecules27227696
APA StyleBrodzka, A., Koszelewski, D., & Ostaszewski, R. (2022). Simultaneous Enantiodivergent Synthesis of Diverse Lactones and Lactams via Sequential One-Pot Enzymatic Kinetic Resolution–Ring-Closing Metathesis Reactions. Molecules, 27(22), 7696. https://doi.org/10.3390/molecules27227696