


Electrochemically Induced Synthesis of Imidazoles from Vinyl Azides and Benzyl Amines



Abstract
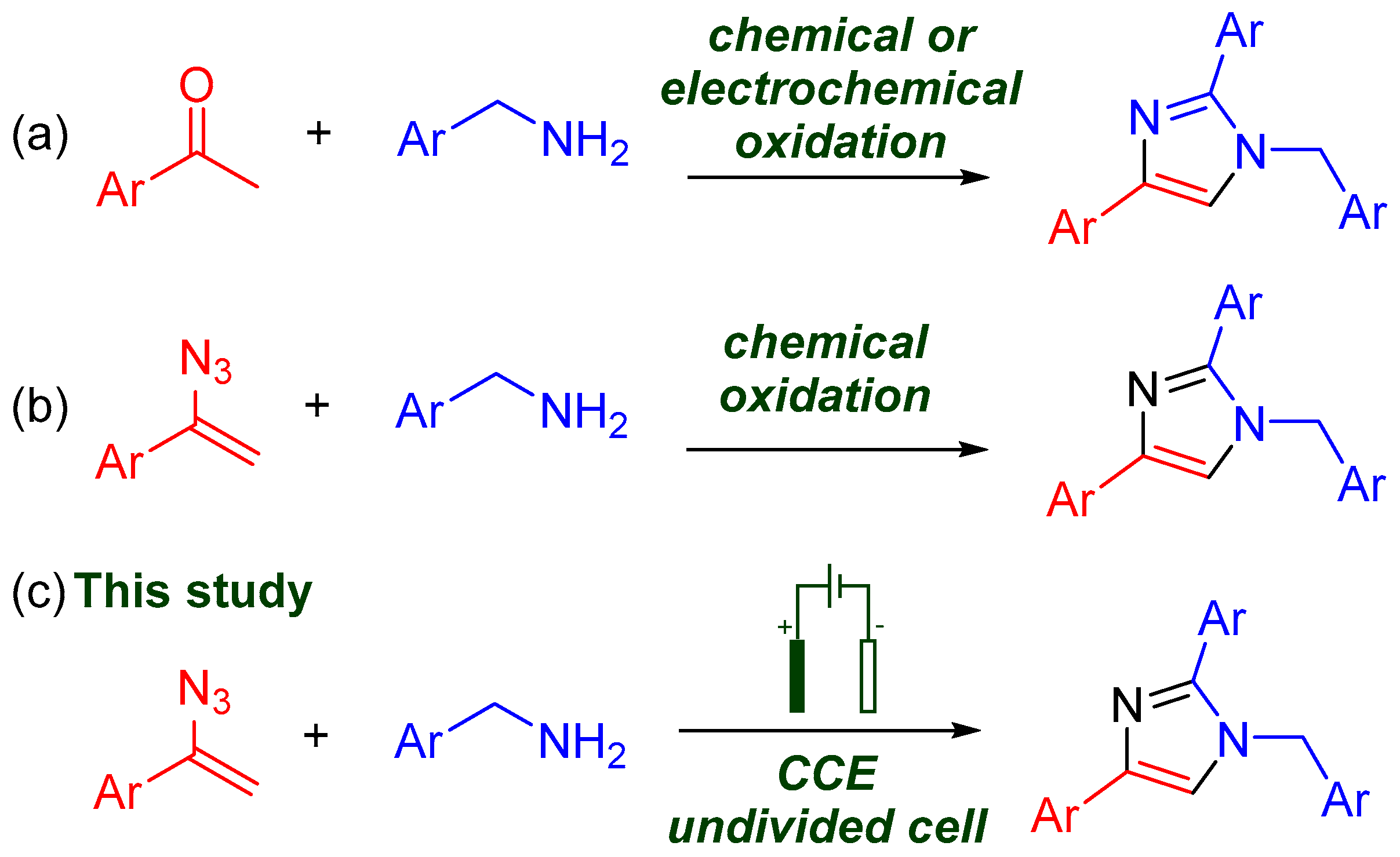
1. Introduction
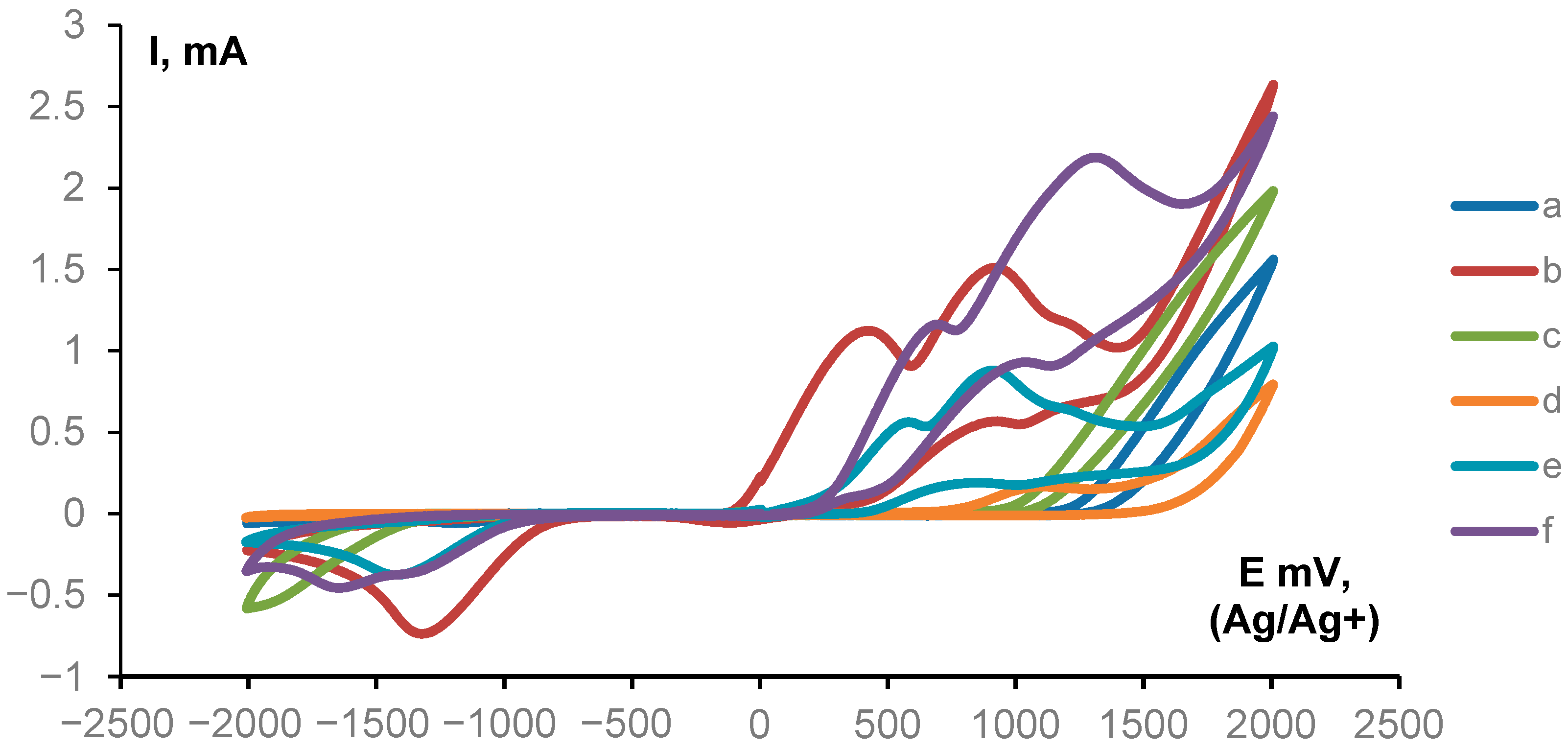
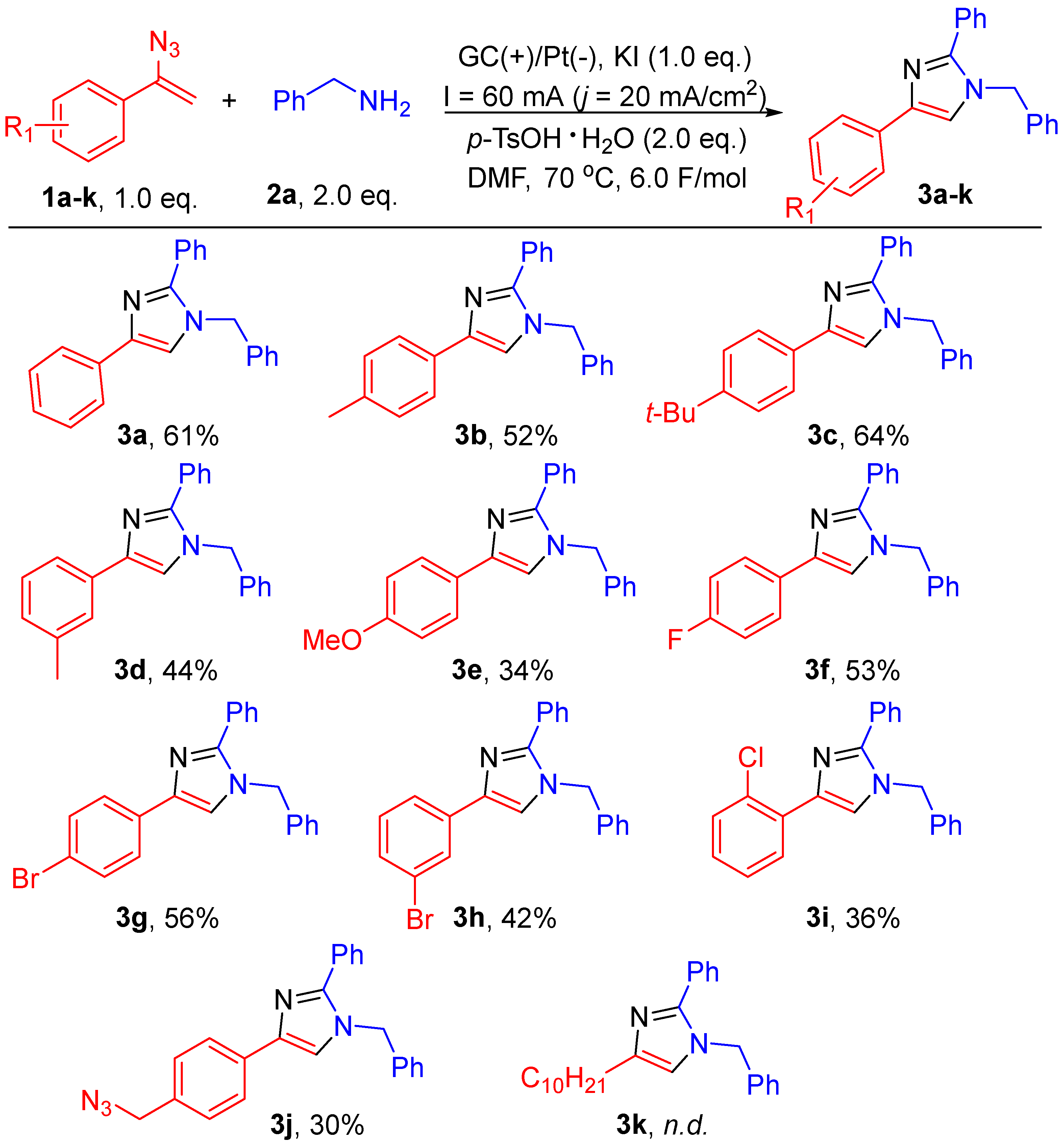
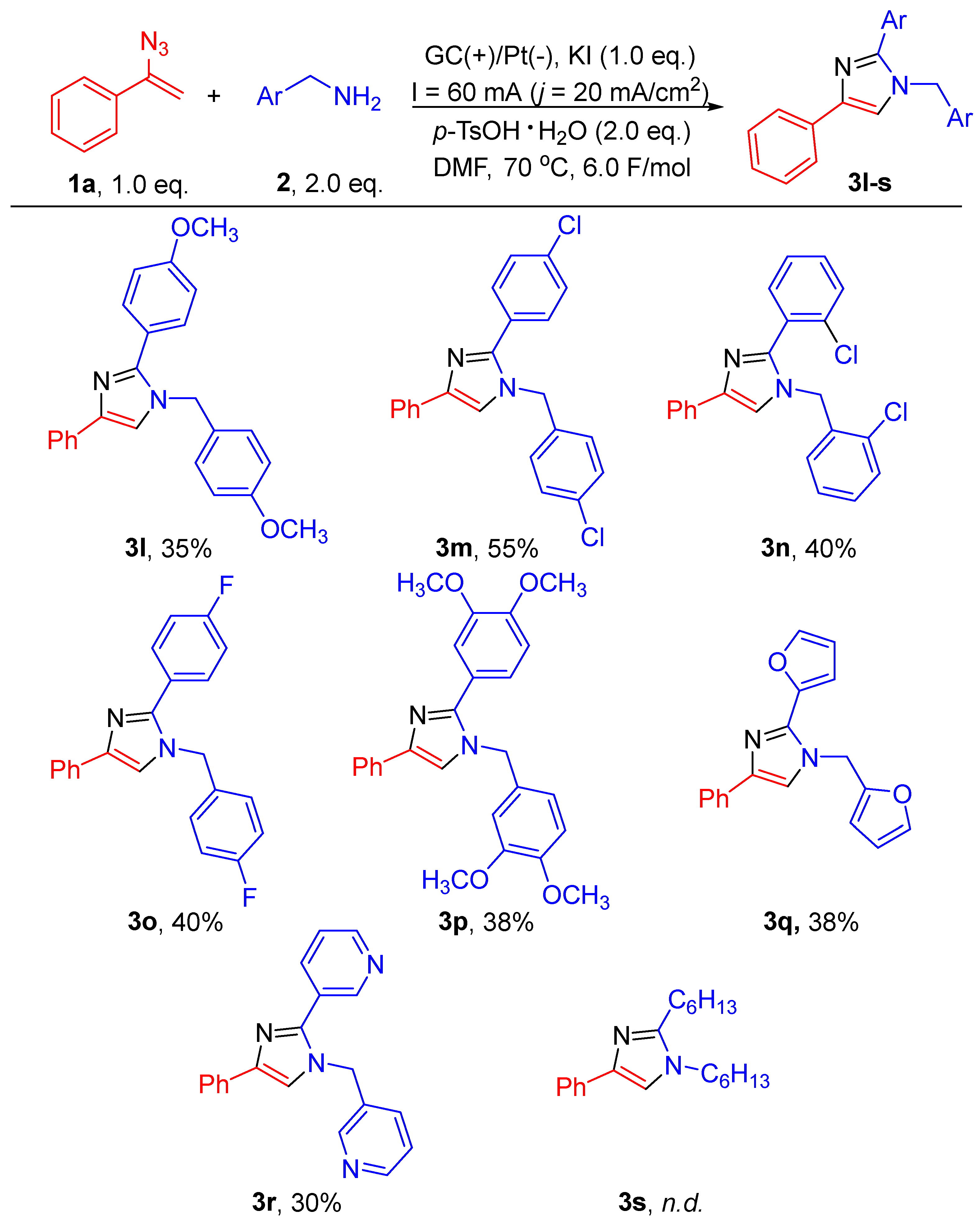
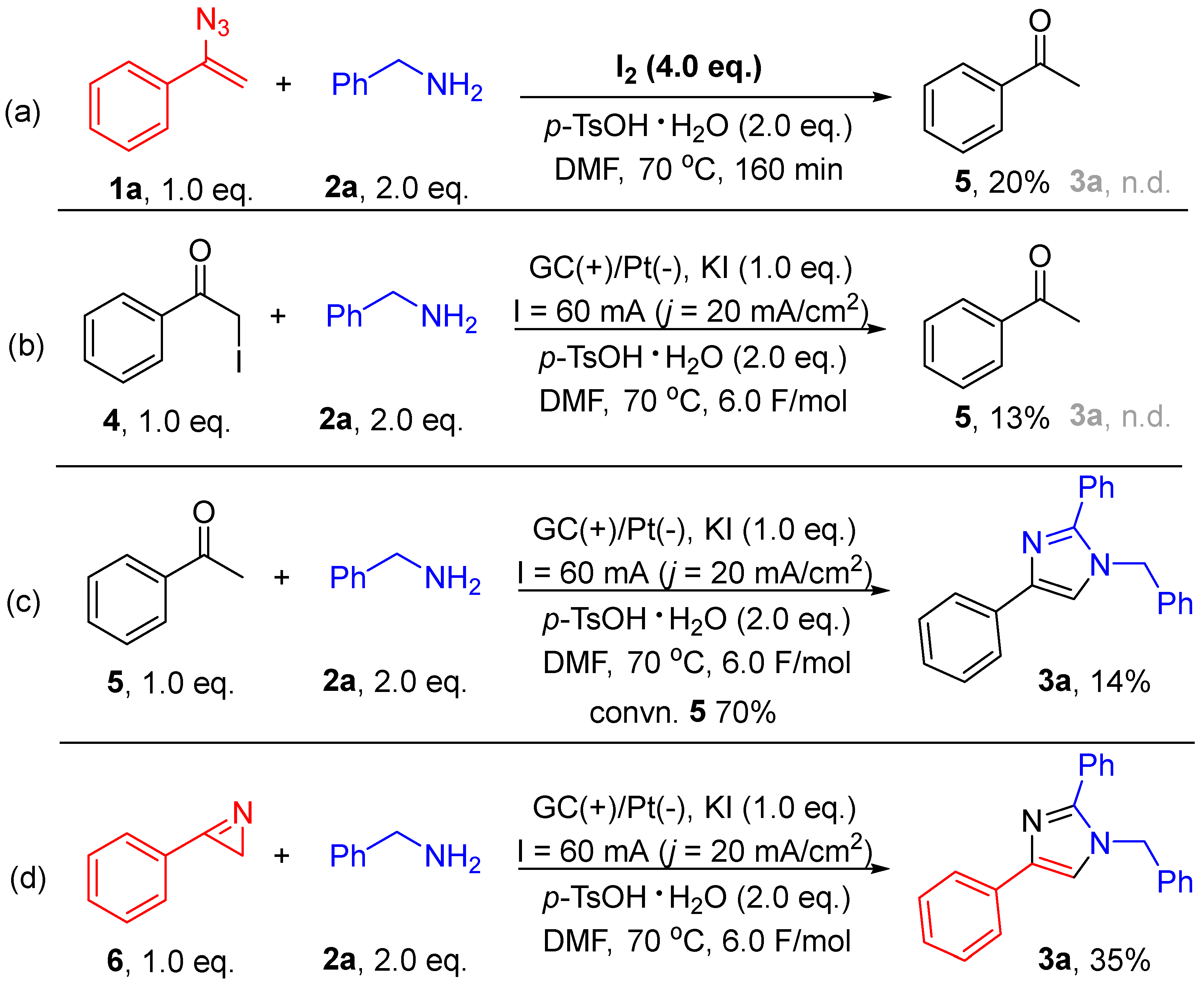
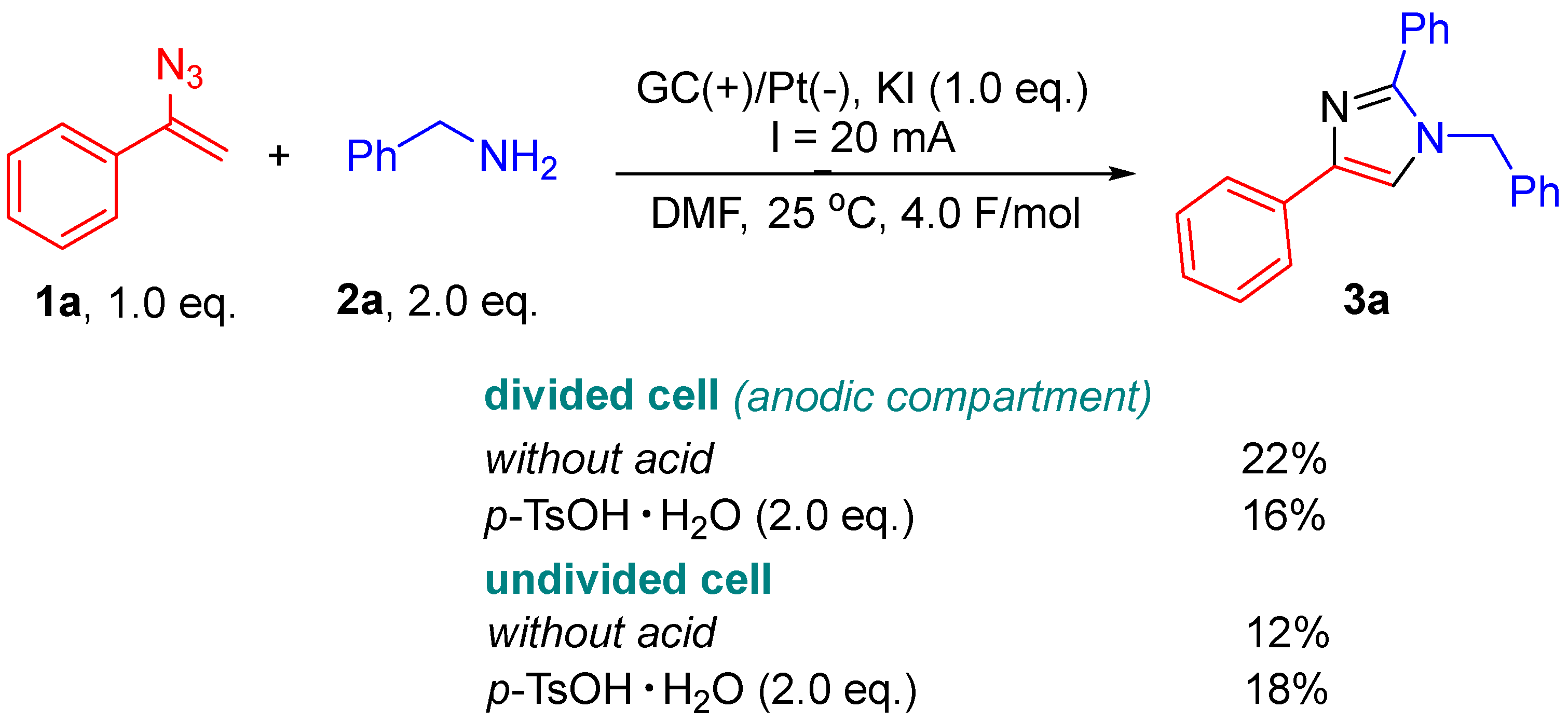
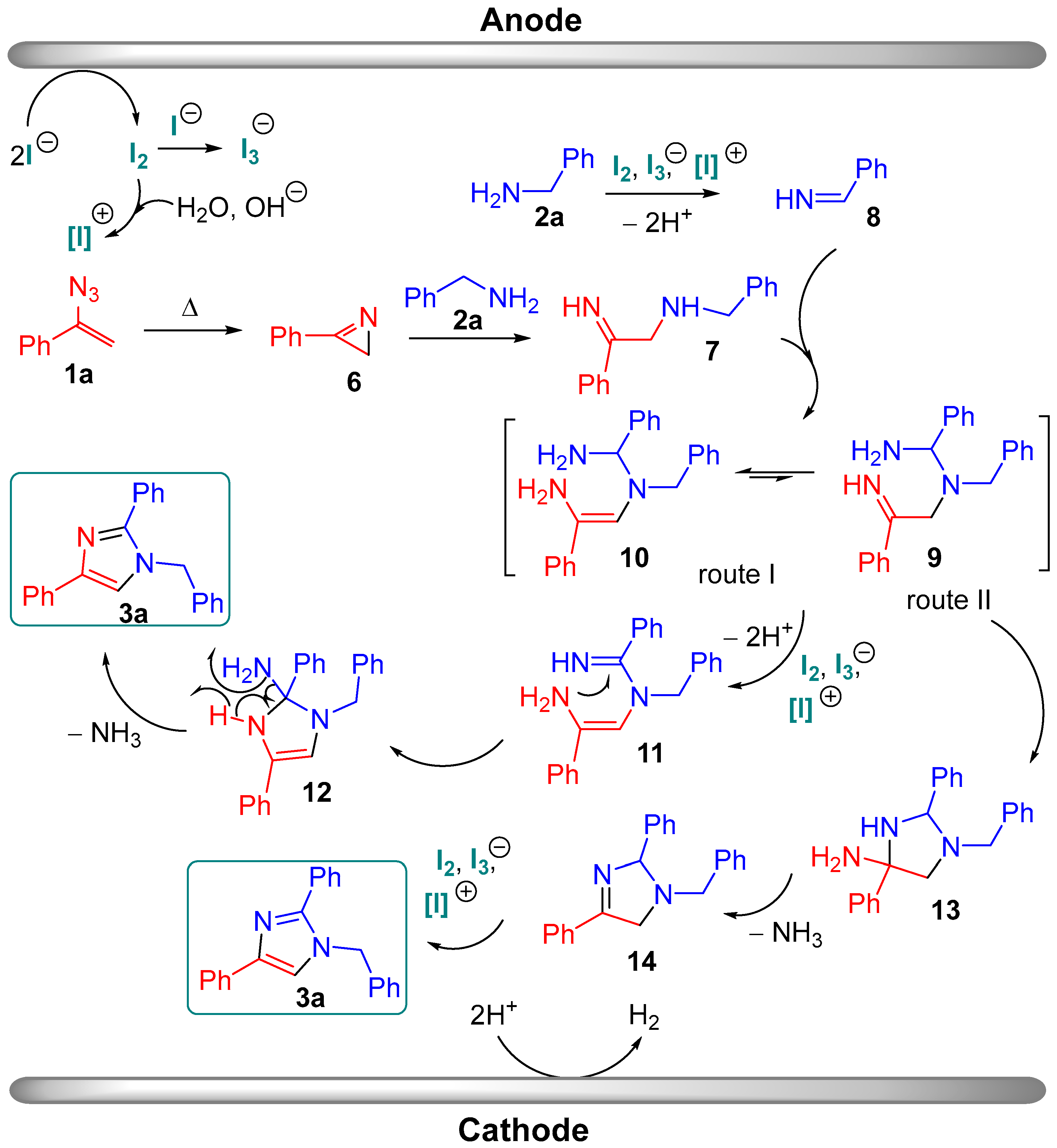
2. Results and Discussion
3. Materials and Methods
3.1. General Materials and Methods
3.2. Synthesis of Starting Compounds
3.3. Electrochemical Cell
3.4. General Experimental Procedure for Scheme 2 and Scheme 3
3.4.1. 1-Benzyl-2,4-diphenyl-1H-imidazole (3a)
3.4.2. 1-Benzyl-2-phenyl-4-(p-tolyl)-1H-imidazole (3b)
3.4.3. 1-Benzyl-4-(4-(tert-butyl)phenyl)-2-phenyl-1H-imidazole (3c)
3.4.4. 1-Benzyl-2-phenyl-4-(m-tolyl)-1H-imidazole (3d)
3.4.5. 1-Benzyl-4-(4-methoxyphenyl)-2-phenyl-1H-imidazole (3e)
3.4.6. 1-Benzyl-4-(4-fluorophenyl)-2-phenyl-1H-imidazole (3f)
3.4.7. 1-Benzyl-4-(4-bromophenyl)-2-phenyl-1H-imidazole (3g)
3.4.8. 1-Benzyl-4-(3-bromophenyl)-2-phenyl-1H-imidazole (3h)
3.4.9. 1-Benzyl-4-(2-chlorophenyl)-2-phenyl-1H-imidazole (3i)
3.4.10. 4-(4-(Azidomethyl)phenyl)-1-benzyl-2-phenyl-1H-imidazole (3j)
3.4.11. 1-(4-Methoxybenzyl)-2-(4-methoxyphenyl)-4-phenyl-1H-imidazole (3l)
3.4.12. 1-(4-Chlorobenzyl)-2-(4-chlorophenyl)-4-phenyl-1H-imidazole (3m)
3.4.13. 1-(2-Chlorobenzyl)-2-(2-chlorophenyl)-4-phenyl-1H-imidazole (3n)
3.4.14. 1-(4-Fluorobenzyl)-2-(4-fluorophenyl)-4-phenyl-1H-imidazole (3o)
3.4.15. 1-(3,4-Dimethoxybenzyl)-2-(3,4-dimethoxyphenyl)-4-phenyl-1H-imidazole (3p)
3.4.16. 2-(Furan-2-yl)-1-(furan-2-ylmethyl)-4-phenyl-1H-imidazole (3q)
3.4.17. 3-(4-Phenyl-1-(Pyridin-3-Ylmethyl)-1H-Imidazol-2-yl)Pyridine (3r)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Sharma, P.; LaRosa, C.; Antwi, J.; Govindarajan, R.; Werbovetz, K.A. Imidazoles as Potential Anticancer Agents: An Update on Recent Studies. Molecules 2021, 26, 4213. [Google Scholar] [CrossRef]
- Fan, Y.-L.; Jin, X.-H.; Huang, Z.-P.; Yu, H.-F.; Zeng, Z.-G.; Gao, T.; Feng, L.-S. Recent advances of imidazole-containing derivatives as anti-tubercular agents. Eur. J. Med. Chem. 2018, 150, 347–365. [Google Scholar] [CrossRef] [PubMed]
- Siwach, A.; Verma, P.K. Synthesis and therapeutic potential of imidazole containing compounds. BMC Chem. 2021, 15, 12. [Google Scholar] [CrossRef]
- Beltran-Hortelano, I.; Alcolea, V.; Font, M.; Pérez-Silanes, S. The role of imidazole and benzimidazole heterocycles in Chagas disease: A review. Eur. J. Med. Chem. 2020, 206, 112692. [Google Scholar] [CrossRef] [PubMed]
- Jin, Z. Muscarine, imidazole, oxazole and thiazole alkaloids. Nat. Prod. Rep. 2009, 26, 382–445. [Google Scholar] [CrossRef] [PubMed]
- Ouakki, M.; Galai, M.; Cherkaoui, M. Imidazole derivatives as efficient and potential class of corrosion inhibitors for metals and alloys in aqueous electrolytes: A review. J. Mol. Liq. 2022, 345, 117815. [Google Scholar] [CrossRef]
- Chen, W.-C.; Zhu, Z.-L.; Lee, C.-S. Organic Light-Emitting Diodes Based on Imidazole Semiconductors. Adv. Opt. Mater. 2018, 6, 1800258. [Google Scholar] [CrossRef]
- Rossi, R.; Angelici, G.; Casotti, G.; Manzini, C.; Lessi, M. Catalytic Synthesis of 1,2,4,5-Tetrasubstituted 1H-Imidazole Derivatives: State of the Art. Adv. Synth. Catal. 2019, 361, 2737–2803. [Google Scholar] [CrossRef]
- Daraji, D.G.; Prajapati, N.P.; Patel, H.D. Synthesis and Applications of 2-Substituted Imidazole and Its Derivatives: A Review. J. Heterocycl. Chem. 2019, 56, 2299–2317. [Google Scholar] [CrossRef]
- Shabalin, D.A.; Camp, J.E. Recent advances in the synthesis of imidazoles. Org. Biomol. Chem. 2020, 18, 3950–3964. [Google Scholar] [CrossRef]
- Alanthadka, A.; Elango, S.D.; Thangavel, P.; Subbiah, N.; Vellaisamy, S.; Chockalingam, U.M. Construction of substituted imidazoles from aryl methyl ketones and benzylamines via N-heterocyclic carbene-catalysis. Catal. Commun. 2019, 125, 26–31. [Google Scholar] [CrossRef]
- Cai, Z.-J.; Wang, S.-Y.; Ji, S.-J. CuI/BF3·Et2O Cocatalyzed Aerobic Dehydrogenative Reactions of Ketones with Benzylamines: Facile Synthesis of Substituted Imidazoles. Org. Lett. 2012, 14, 6068–6071. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Ji, X.; Wu, W.; Jiang, H. Practical Synthesis of Polysubstituted Imidazoles via Iodine- Catalyzed Aerobic Oxidative Cyclization of Aryl Ketones and Benzylamines. Adv. Synth. Catal. 2013, 355, 170–180. [Google Scholar] [CrossRef]
- Geng, F.; Wu, S.; Gan, X.; Hou, W.; Dong, J.; Zhou, Y. TEMPO mediated oxidative annulation of aryl methyl ketones with amines/ammonium acetate for imidazole synthesis. Org. Biomol. Chem. 2022, 20, 5416–5422. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Zhang, J.; Hu, L.; Li, A.; Li, L.; Liu, K.; Yang, T.; Zhou, C. Electrochemical HI-mediated Intermolecular C–N Bond Formation to Synthesize Imidazoles from Aryl Ketones and Benzylamines. J. Org. Chem. 2020, 85, 5952–5958. [Google Scholar] [CrossRef] [PubMed]
- Zeng, L.; Li, J.; Gao, J.; Huang, X.; Wang, W.; Zheng, X.; Gu, L.; Li, G.; Zhang, S.; He, Y. An electrochemical oxidative multicomponent cascade annulation of ketones and amines used to produce imidazoles. Green Chem. 2020, 22, 3416–3420. [Google Scholar] [CrossRef]
- Cao, J.; Zhou, X.; Ma, H.; Shi, C.; Huang, G. A facile and efficient method for the synthesis of 1,2,4-trisubstituted imidazoles with enamides and benzylamines. RSC Adv. 2016, 6, 57232–57235. [Google Scholar] [CrossRef]
- Wang, W.; Zhang, S.; Shi, G.; Chen, Z. Electrochemical synthesis of 1,2,4,5-tetrasubstituted imidazoles from enamines and benzylamines. Org. Biomol. Chem. 2021, 19, 6682–6686. [Google Scholar] [CrossRef]
- Fu, J.; Zanoni, G.; Anderson, E.A.; Bi, X. α-Substituted vinyl azides: An emerging functionalized alkene. Chem. Soc. Rev. 2017, 46, 7208–7228. [Google Scholar] [CrossRef]
- Nguyen, T.K.; Titov, G.D.; Khoroshilova, O.V.; Kinzhalov, M.A.; Rostovskii, N.V. Light-induced one-pot synthesis of pyrimidine derivatives from vinyl azides. Org. Biomol. Chem. 2020, 18, 4971–4982. [Google Scholar] [CrossRef]
- Yin, W.; Wang, X. Recent advances in iminyl radical-mediated catalytic cyclizations and ring-opening reactions. New J. Chem. 2019, 43, 3254–3264. [Google Scholar] [CrossRef]
- Mulina, O.M.; Zhironkina, N.V.; Paveliev, S.A.; Demchuk, D.V.; Terent’ev, A.O. Electrochemically Induced Synthesis of Sulfonylated N-Unsubstituted Enamines from Vinyl Azides and Sulfonyl Hydrazides. Org. Lett. 2020, 22, 1818–1824. [Google Scholar] [CrossRef] [PubMed]
- Paveliev, S.A.; Churakov, A.I.; Alimkhanova, L.S.; Segida, O.O.; Nikishin, G.I.; Terent’ev, A.O. Electrochemical Synthesis of O-Phthalimide Oximes from α-Azido Styrenes via Radical Sequence: Generation, Addition and Recombination of Imide-N-Oxyl and Iminyl Radicals with C−O/N−O Bonds Formation. Adv. Synth. Catal. 2020, 362, 3864–3871. [Google Scholar] [CrossRef]
- Nayl, A.A.; Aly, A.A.; Arafa, W.A.A.; Ahmed, I.M.; Abd-Elhamid, A.I.; El-Fakharany, E.M.; Abdelgawad, M.A.; Tawfeek, H.N.; Bräse, S. Azides in the Synthesis of Various Heterocycles. Molecules 2022, 27, 3716. [Google Scholar] [CrossRef] [PubMed]
- Xiang, L.; Niu, Y.; Pang, X.; Yang, X.; Yan, R. I2-catalyzed synthesis of substituted imidazoles from vinyl azides and benzylamines. Chem. Commun. 2015, 51, 6598–6600. [Google Scholar] [CrossRef]
- Yoshida, J.-I.; Kataoka, K.; Horcajada, R.; Nagaki, A. Modern Strategies in Electroorganic Synthesis. Chem. Rev. 2008, 108, 2265–2299. [Google Scholar] [CrossRef]
- Frontana-Uribe, B.A.; Little, R.D.; Ibanez, J.G.; Palma, A.; Vasquez-Medrano, R. Organic electrosynthesis: A promising green methodology in organic chemistry. Green Chem. 2010, 12, 2099–2119. [Google Scholar] [CrossRef]
- Horn, E.J.; Rosen, B.R.; Baran, P.S. Synthetic Organic Electrochemistry: An Enabling and Innately Sustainable Method. ACS Cent. Sci. 2016, 2, 302–308. [Google Scholar] [CrossRef]
- Sperry, J.B.; Wright, D.L. The application of cathodic reductions and anodic oxidations in the synthesis of complex molecules. Chem. Soc. Rev. 2006, 35, 605–621. [Google Scholar] [CrossRef]
- Wiebe, A.; Gieshoff, T.; Möhle, S.; Rodrigo, E.; Zirbes, M.; Waldvogel, S.R. Electrifying Organic Synthesis. Angew. Chem. Int. Ed. 2018, 57, 5594–5619. [Google Scholar] [CrossRef]
- Yan, M.; Kawamata, Y.; Baran, P.S. Synthetic Organic Electrochemical Methods Since 2000: On the Verge of a Renaissance. Chem. Rev. 2017, 117, 13230–13319. [Google Scholar] [CrossRef] [PubMed]
- Waldvogel, S.R.; Janza, B. Renaissance of Electrosynthetic Methods for the Construction of Complex Molecules. Angew. Chem. Int. Ed. 2014, 53, 7122–7123. [Google Scholar] [CrossRef] [PubMed]
- Vil’, V.A.; Merkulova, V.M.; Ilovaisky, A.I.; Paveliev, S.A.; Nikishin, G.I.; Terent’ev, A.O. Electrochemical Synthesis of Fluorinated Ketones from Enol Acetates and Sodium Perfluoroalkyl Sulfinates. Org. Lett. 2021, 23, 5107–5112. [Google Scholar] [CrossRef] [PubMed]
- Francke, R. Recent advances in the electrochemical construction of heterocycles. Beilstein J. Org. Chem. 2014, 10, 2858–2873. [Google Scholar] [CrossRef]
- Jiang, Y.; Xu, K.; Zeng, C. Use of Electrochemistry in the Synthesis of Heterocyclic Structures. Chem. Rev. 2018, 118, 4485–4540. [Google Scholar] [CrossRef]
- Sbei, N.; Listratova, A.V.; Titov, A.A.; Voskressensky, L.G. Recent Advances in Electrochemistry for the Synthesis of N-Heterocycles. Synthesis 2019, 51, 2455–2473. [Google Scholar] [CrossRef]
- Liang, S.; Zeng, C.-C. Organic electrochemistry: Anodic construction of heterocyclic structures. Curr. Opin. Electrochem. 2020, 24, 31–43. [Google Scholar] [CrossRef]
- Ye, Z.; Wu, Y.; Chen, N.; Zhang, H.; Zhu, K.; Ding, M.; Liu, M.; Li, Y.; Zhang, F. Enantiospecific electrochemical rearrangement for the synthesis of hindered triazolopyridinone derivatives. Nat. Commun. 2020, 11, 3628. [Google Scholar] [CrossRef]
- Li, Y.; Ye, Z.; Chen, N.; Chen, Z.; Zhang, F. Intramolecular electrochemical dehydrogenative N–N bond formation for the synthesis of 1,2,4-triazolo[1,5-a]pyridines. Green Chem. 2019, 21, 4035–4039. [Google Scholar] [CrossRef]
- Ye, Z.; Zhang, F. Recent Advances in Constructing Nitrogen-Containing Heterocycles via Electrochemical Dehydrogenation. Chin. J. Chem. 2019, 37, 513–528. [Google Scholar] [CrossRef]
- Feng, M.-L.; Li, S.-Q.; He, H.-Z.; Xi, L.-Y.; Chen, S.-Y.; Yu, X.-Q. Electrochemically initiated intermolecular C–N formation/cyclization of ketones with 2-aminopyridines: An efficient method for the synthesis of imidazo[1,2-a]pyridines. Green Chem. 2019, 21, 1619–1624. [Google Scholar] [CrossRef]
- Qian, P.; Yan, Z.; Zhou, Z.; Hu, K.; Wang, J.; Li, Z.; Zha, Z.; Wang, Z. Electrocatalytic Intermolecular C(sp3)–H/N–H Coupling of Methyl N-Heteroaromatics with Amines and Amino Acids: Access to Imidazo-Fused N-Heterocycles. Org. Lett. 2018, 20, 6359–6363. [Google Scholar] [CrossRef] [PubMed]
- Qian, P.; Yan, Z.; Zhou, Z.; Hu, K.; Wang, J.; Li, Z.; Zha, Z.; Wang, Z. Electrocatalytic Tandem Synthesis of 1,3-Disubstituted Imidazo[1,5-a]quinolines via Sequential Dual Oxidative C(sp3)–H Amination in Aqueous Medium. J. Org. Chem. 2019, 84, 3148–3157. [Google Scholar] [CrossRef] [PubMed]
- Vil’, V.A.; Grishin, S.S.; Baberkina, E.P.; Alekseenko, A.L.; Glinushkin, A.P.; Kovalenko, A.E.; Terent’ev, A.O. Electrochemical Synthesis of Tetrahydroquinolines from Imines and Cyclic Ethers via Oxidation/Aza-Diels-Alder Cycloaddition. Adv. Synth. Catal. 2022, 364, 1098–1108. [Google Scholar] [CrossRef]
- El-Hallag, I.S. Electrochemical oxidation of iodide at a glassy carbon electrode in methylene chloride at various temperatures. J. Chilean Chem. Soc. 2010, 55, 67–73. [Google Scholar] [CrossRef]
- Sun, J.; Nie, Q.; Fang, X.; He, Z.; Zhang, G.; Li, Y.; Li, Y. Vinyl azide as a synthon for DNA-compatible divergent transformations into N-heterocycles. Org. Biomol. Chem. 2022, 20, 5045–5049. [Google Scholar] [CrossRef]
- Wen, J.; Shi, W.; Zhang, F.; Liu, D.; Tang, S.; Wang, H.; Lin, X.-M.; Lei, A. Electrooxidative Tandem Cyclization of Activated Alkynes with Sulfinic Acids To Access Sulfonated Indenones. Org. Lett. 2017, 19, 3131–3134. [Google Scholar] [CrossRef]
- Liu, K.; Song, C.; Lei, A. Recent advances in iodine mediated electrochemical oxidative cross-coupling. Org. Biomol. Chem. 2018, 16, 2375–2387. [Google Scholar] [CrossRef]
- Dey, R.; Banerjee, P. Lewis Acid Catalyzed Diastereoselective Cycloaddition Reactions of Donor–Acceptor Cyclopropanes and Vinyl Azides: Synthesis of Functionalized Azidocyclopentane and Tetrahydropyridine Derivatives. Org. Lett. 2017, 19, 304–307. [Google Scholar] [CrossRef]
- Andresini, M.; Degannaro, L.; Luisi, R. A sustainable strategy for the straightforward preparation of 2H-azirines and highly functionalized NH-aziridines from vinyl azides using a single solvent flow-batch approach. Beilstein J. Org. Chem. 2021, 17, 203–209. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Entry | Cathode/Anode | Electrolyte (eq) | Additive (eq.) | Solvent | Current Density, mA/cm2 | Electricity Passed per 1a, F/mol | Yield 3a, % |
|---|---|---|---|---|---|---|---|
| 1 | Pt/GC | TBAI (1.0) | - | DMF | 10.0 | 4.0 | 24 |
| 2 | Pt/GC | KI (1.0) | - | DMF | 10.0 | 4.0 | 37 |
| 3 | Pt/GC | LiClO4 (1.0) | - | DMF | 10.0 | 4.0 | 22 |
| 4 2 | Pt/GC | KI (1.0) | - | DMF | 10.0 | 4.0 | 33 |
| 5 | Pt/GC | KI (1.0) | p-TsOH·H2O (2.0) | DMF | 10.0 | 4.0 | 48 |
| 6 | Pt/GC | KI (1.0) | p-TsOH·H2O (2.0) | DMF | 20.0 | 4.0 | 39 |
| 7 | Pt/GC | KI (1.0) | p-TsOH·H2O (2.0) | DMF | 20.0 | 6.0 | 55 |
| 8 | Pt/GC | KI (1.0) | p-TsOH·H2O (2.0) | DMF | - | - | 7 |
| 9 | Pt/GC | KI (1.0) | H2SO4 (2.0) | DMF | 20.0 | 6.0 | - |
| 10 | Pt/GC | KI (1.0) | CH3SO3H (2.0) | DMF | 20.0 | 6.0 | - |
| 11 | Pt/GC | KI (1.0) | Amberlyst-15 (2.0) | DMF | 20.0 | 6.0 | 46 |
| 12 | GC/GC | KI (1.0) | p-TsOH·H2O (2.0) | DMF | 20.0 | 6.0 | 15 |
| 13 | SS/GC | KI (1.0) | p-TsOH·H2O (2.0) | DMF | 20.0 | 6.0 | 34 |
| 14 | Ni/GC | KI (1.0) | p-TsOH·H2O (2.0) | DMF | 20.0 | 6.0 | 48 |
| 15 | Pt/C | KI (1.0) | p-TsOH·H2O (2.0) | DMF | 20.0 | 6.0 | 36 |
| 16 | Pt/Pt | KI (1.0) | p-TsOH·H2O (2.0) | DMF | 20.0 | 6.0 | 22 |
| 17 | Pt/GC | KI (1.0) | p-TsOH·H2O (2.0) | DMSO | 20.0 | 6.0 | 34 |
| 18 | Pt/GC | KI (1.0) | p-TsOH·H2O (2.0) n-Bu4NClO4 (1.0) | PhCl | 20.0 | 6.0 | 18 |
| 19 3 | Pt/GC | KI (1.0) | p-TsOH·H2O (2.0) | DMF | 20.0 | 6.0 | 61 |
| 20 4 | Pt/GC | KI (1.0) | p-TsOH·H2O (2.0) | DMF | 20.0 | 6.0 | 31 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vil’, V.A.; Grishin, S.S.; Terent’ev, A.O. Electrochemically Induced Synthesis of Imidazoles from Vinyl Azides and Benzyl Amines. Molecules 2022, 27, 7721. https://doi.org/10.3390/molecules27227721
Vil’ VA, Grishin SS, Terent’ev AO. Electrochemically Induced Synthesis of Imidazoles from Vinyl Azides and Benzyl Amines. Molecules. 2022; 27(22):7721. https://doi.org/10.3390/molecules27227721
Chicago/Turabian StyleVil’, Vera A., Sergei S. Grishin, and Alexander O. Terent’ev. 2022. "Electrochemically Induced Synthesis of Imidazoles from Vinyl Azides and Benzyl Amines" Molecules 27, no. 22: 7721. https://doi.org/10.3390/molecules27227721
APA StyleVil’, V. A., Grishin, S. S., & Terent’ev, A. O. (2022). Electrochemically Induced Synthesis of Imidazoles from Vinyl Azides and Benzyl Amines. Molecules, 27(22), 7721. https://doi.org/10.3390/molecules27227721