
Synthesis of 2,5-Dialkyl-1,3,4-oxadiazoles Bearing Carboxymethylamino Groups



Abstract
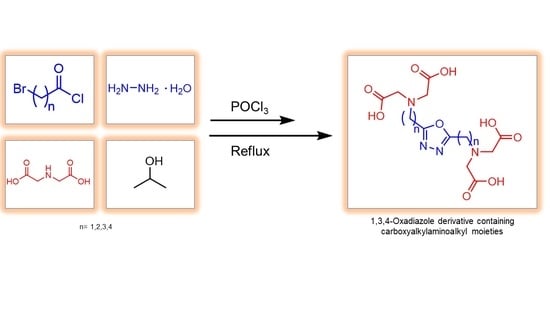
1. Introduction
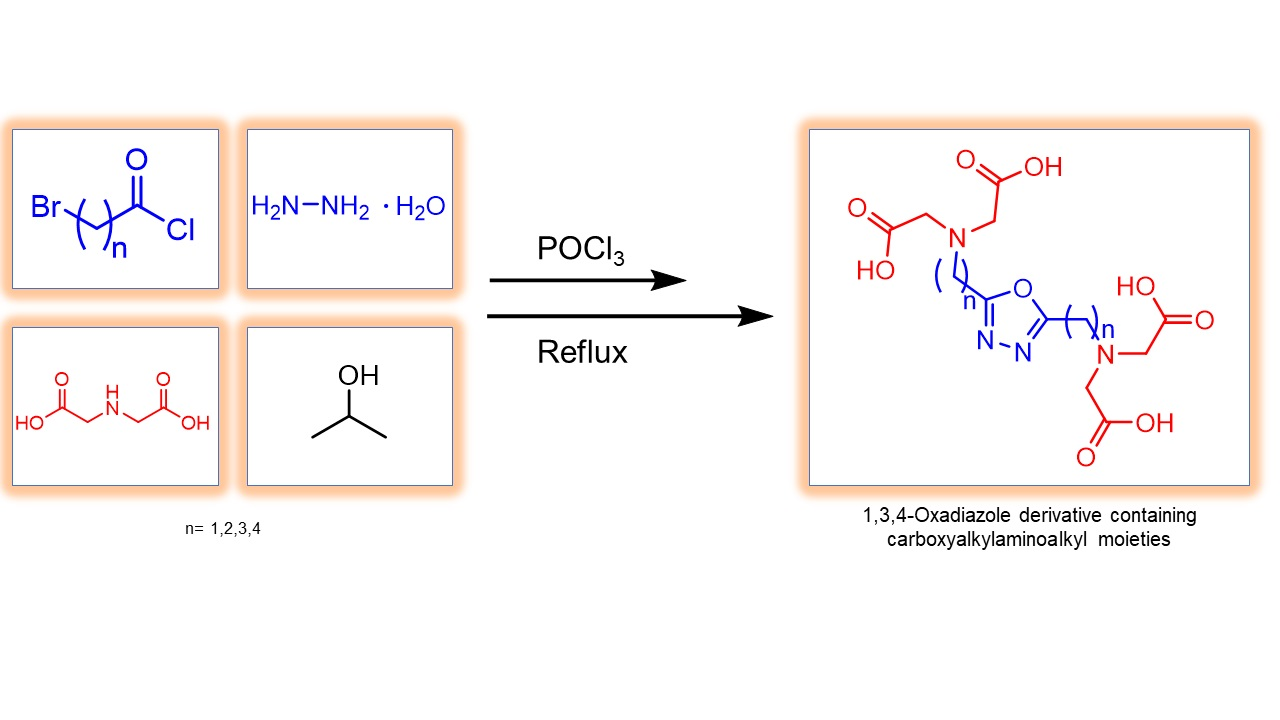
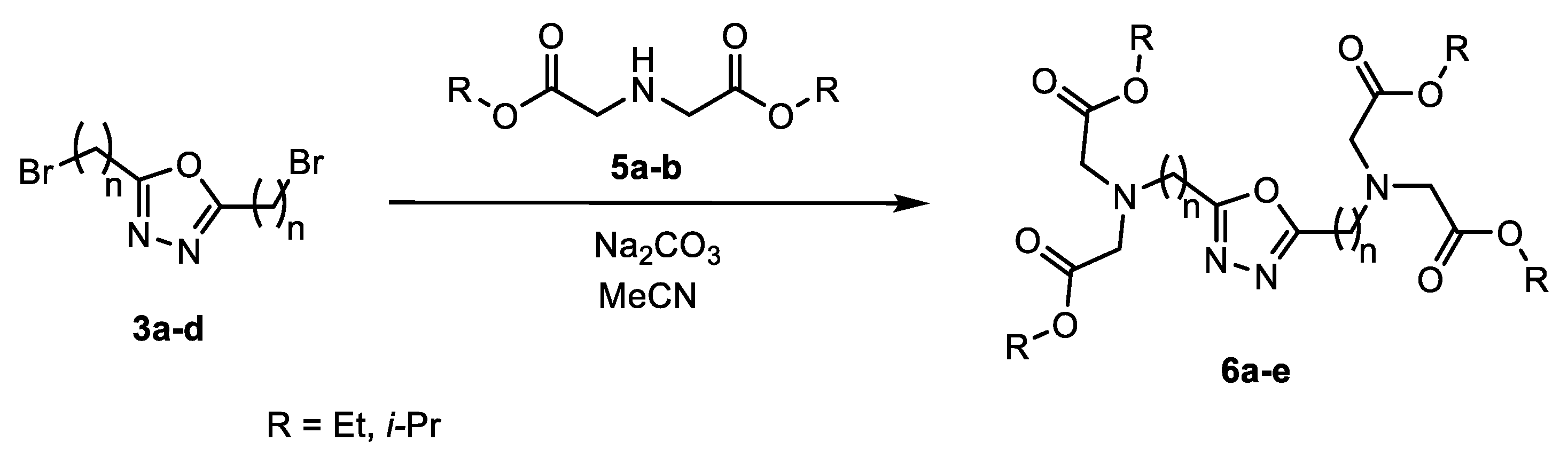
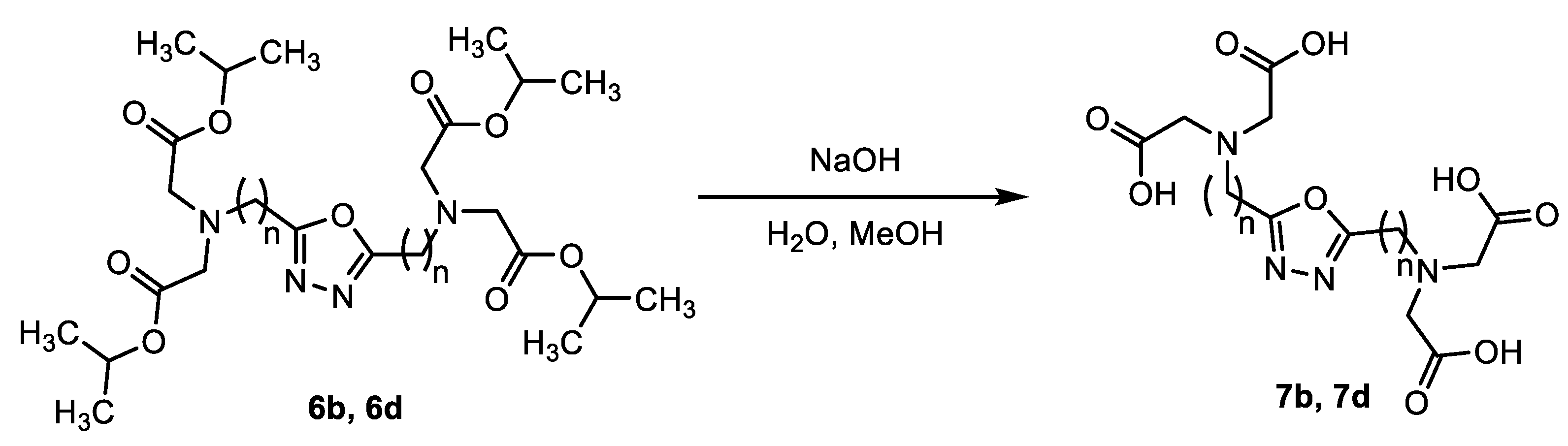
2. Results
3. Experimental Section
3.1. General Information
3.2. Synthesis and Characterization
3.2.1. Synthesis of N,N′-Diacylhydrazine Derivatives (2a–d)
- 2-Bromo-N′-(2-bromoacetyl)acetohydrazide (2a)
- 3-Bromo-N′-(3-bromopropanoyl)propanehydrazide (2b)
- 4-Bromo-N′-(4-bromobutanoyl)butanehydrazide (2c)
- 5-Bromo-N′-(5-bromopentanoyl)pentanehydrazide (2d)
3.2.2. Synthesis of 1,3,4-Oxadiazole Derivatives (3a–d)
- 2,5-Bis(bromomethyl)-1,3,4-oxadiazole (3a)
- 2,5-Bis(2-bromoethyl)-1,3,4-oxadiazole (3b)
- 2,5-Bis(3-bromopropyl)-1,3,4-oxadiazole (3c)
- 2,5-Bis(4-bromobutyl)-1,3,4-oxadiazole (3d)
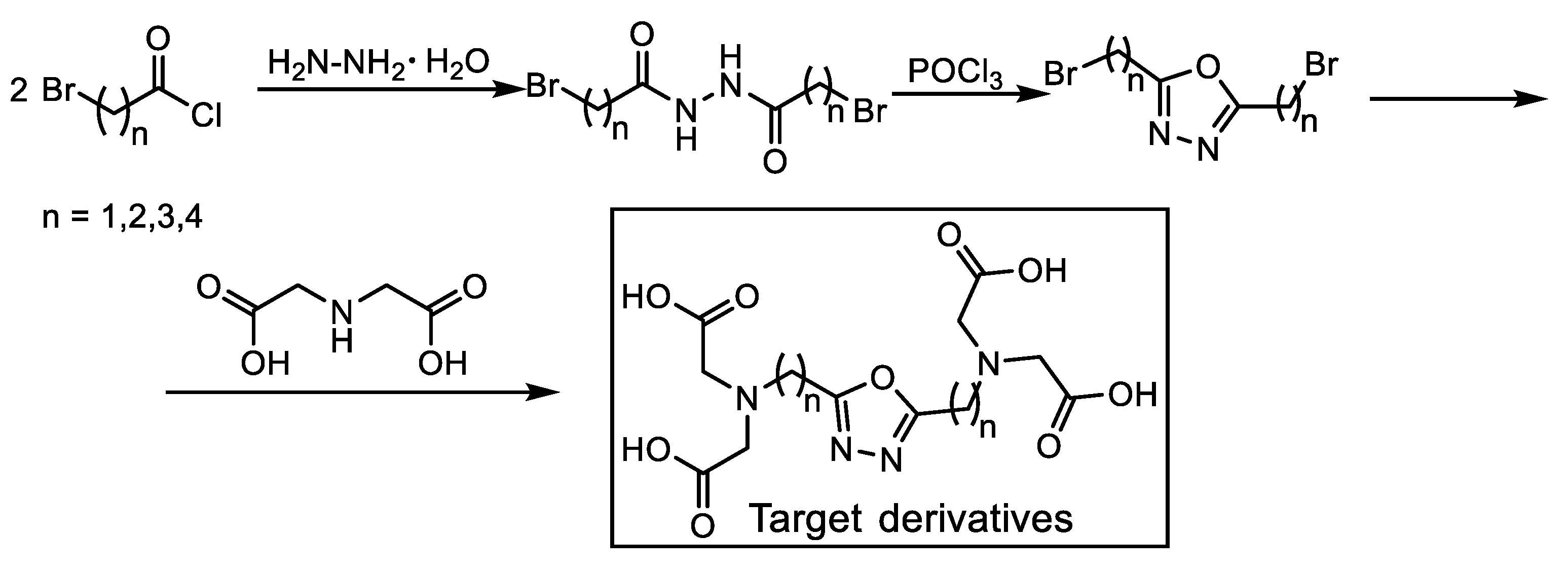
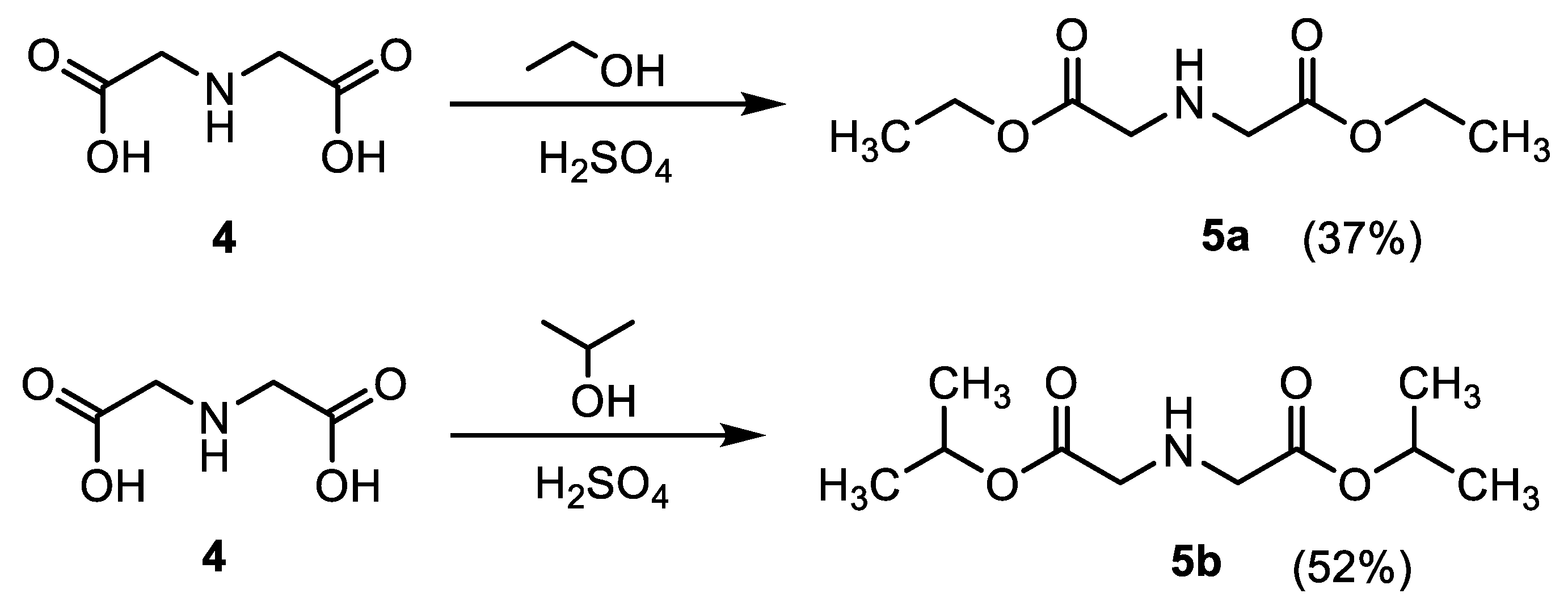
3.2.3. Synthesis of Iminodiacetic Acid Ester Derivatives (5a,b)
- Diethyl iminodiacetate (5a)
- Diisopropyl iminodiacetate (5b)
3.2.4. Synthesis of Ester Derivatives of 2,5-Dialkyl-1,3,4-oxadiazole (6a–e)
- Tetraethyl 2,2′,2″,2‴-(((1,3,4-oxadiazole-2,5-diyl)bis(methylene))bis(azanetriyl))tetraacetate (6a)
- Tetraisopropyl 2,2′,2″,2‴-(((1,3,4-oxadiazole=2,5-diyl)bis(methylene))bis(azanetriyl))tetraacetate (6b)
- Tetraisopropyl 2,2′,2″,2‴-(((1,3,4-oxadiazole-2,5-diyl)bis(ethane-2,1-diyl))bis(azanetriyl))tetraacete (6c)
- Tetraisopropyl 2,2′,2″,2‴-(((1,3,4-oxadiazole-2,5-diyl)bis(propane-3,1-diyl))bis(azanetriyl))tetraacetate (6d)
- Tetraisopropyl 2,2′,2″,2‴-(((1,3,4-oxadiazole-2,5-diyl)bis(butane-4,1-diyl))bis(azanetriyl))tetraacetate (6e)
3.2.5. Synthesis of 2,5-Dialkyl-1,3,4-oxadiazole Derivatives Containing Carboxymethylamino Groups (7b, 7d)
- 2,2′,2″,2‴-(((1,3,4-oxadiazole-2,5-diyl)bis(methylene))bis(azanetriyl))tetraacetic acid (7b)
- 2,2′,2″,2‴-(((1,3,4-oxadiazole-2,5-diyl)bis(propane-3,1-diyl))bis(azanetriyl))tetraacetic acid (7d)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Shukla, C.; Sanchit, S. Biologically active oxadiazole. J. Drug Deliv. Ther. 2015, 5, 8–13. [Google Scholar] [CrossRef]
- Kumar, K.A.; Jayaroopa, P.; Kumar, V. Comprehensive review on the chemistry of 1,3,4-oxadiazoles and their applications. Int. J. ChemTech Res. 2012, 4, 1782–1791. [Google Scholar]
- Akhter, M.; Husain, A.; Azad, B.; Ajmal, M. Aroylpropionic acid based 2,5-disubstituted-1,3,4-oxadiazoles: Synthesis and their anti-inflammatory and analgesic activities. Eur. J. Med. Chem. 2009, 44, 2372–2378. [Google Scholar] [CrossRef] [PubMed]
- Albratty, M.; El-Sharkawy, K.A.; Alhazmi, H.A. Synthesis and evaluation of some new 1,3,4-oxadiazoles bearing thiophene, thiazole, coumarin, pyridine and pyridazine derivatives as antiviral agents. Acta Pharm. 2019, 69, 261–276. [Google Scholar] [CrossRef]
- Ahsan, M.J.; Samy, J.G.; Khalilullah, H.; Nomani, S.; Saraswat, P.; Gaur, R.; Singh, A. Molecular properties prediction and synthesis of novel 1,3,4-oxadiazole analogues as potent antimicrobial and antitubercular agents. Bioorganic Med. Chem. Lett. 2011, 21, 7246–7250. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Li, Z.; Han, Y. Synthesis and fungicidal activity against Rhizoctonia solani of 2-alkyl(Alkylthio)-5-pyrazolyl-1,3,4-oxadiazoles (Thiadiazoles). J. Agric. Food Chem. 2000, 48, 5312–5315. [Google Scholar] [CrossRef]
- Ahsan, M.J.; Choupra, A.; Sharma, R.K.; Jadav, S.S.; Padmaja, P.; Hassan, Z.; Al-Tamimi, A.B.S.; Geesi, M.H.; Bakht, M.A. Rationale Design, Synthesis, Cytotoxicity Evaluation, and Molecular Docking Studies of 1,3,4-oxadiazole Analogues. Anticancer Agents Med. Chem. 2017, 18, 121–138. [Google Scholar] [CrossRef] [PubMed]
- Lelyukh, M.; Martynets, M.; Kalytovska, M.; Drapak, I.; Harkov, S.; Chaban, T.; Matiychuk, V. Approaches for synthesis and chemical modification of non-condensed heterocyclic systems based on 1,3,4-oxadiazole ring and their biological activity: A review. J. Appl. Pharm. Sci. 2020, 10, 151–165. [Google Scholar] [CrossRef]
- Zheng, X.; Li, Z.; Wang, Y.; Chen, W.; Huang, Q.; Liu, C.; Song, G. Syntheses and insecticidal activities of novel 2,5-disubstituted 1,3,4-oxadiazoles. J. Fluor. Chem. 2003, 123, 163–169. [Google Scholar] [CrossRef]
- Zou, X.J.; Lai, L.H.; Jin, G.Y.; Zhang, Z.X. Synthesis, Fungicidal Activity, and 3D-QSAR of Pyridazinone-Substituted 1,3,4-Oxadiazoles and 1,3,4-Thiadiazoles. Agric. Food Chem. 2002, 50, 3757–3760. [Google Scholar] [CrossRef] [PubMed]
- Luczynski, M.; Kudelko, A. Synthesis and Biological Activity of 1,3,4-Oxadiazoles Used in Medicine and Agriculture. Appl. Sci. 2022, 12, 3756. [Google Scholar] [CrossRef]
- Tajik, H.; Dadras, A. Synthesis and herbicidal activity of novel 5-chloro-3-fluoro-2-phenoxypyridines with a 1,3,4-oxadiazole ring. J. Pestic. Sci. 2011, 36, 27–32. [Google Scholar] [CrossRef]
- Wróblowska, M.; Kudelko, A.; Kuźnik, N.; Łaba, K.; Łapkowski, M. Synthesis of Extended 1,3,4-Oxadiazole and 1,3,4-Thiadiazole Derivatives in the Suzuki Cross-coupling Reactions. J. Heterocycl. Chem. 2017, 54, 1550–1557. [Google Scholar] [CrossRef]
- Bhujabal, Y.B.; Vadagaonkar, K.S.; Kapdi, A.R. Pd/PTABS: Catalyst for Efficient C-H (Hetero_Arylation of 1,3,4-Oxadiazoles Using Bromo(Hetero)Arenes. Asian J. Org. Chem. 2019, 8, 289–295. [Google Scholar] [CrossRef]
- Zhang, M.; Hu, Z.; He, T. Conducting probe atomic force microscopy investigation of anisotropic charge transport in solution cast PBD single crystals Induced by an external field. J. Phys. Chem. B. 2004, 108, 19198–19204. [Google Scholar] [CrossRef]
- Homocianu, M.; Airinei, A. 1,3,4-Oxadiazole Derivatives. Optical Properties in Pure and Mixed Solvents. J. Fluoresc. 2016, 26, 1617–1635. [Google Scholar] [CrossRef] [PubMed]
- Schulz, B.; Orgzall, I.; Freydank, A.; Xu, C. Self-organization of substituted 1,3,4-oxadiazoles in the solid state and at surfaces. Adv. Colloid Interface Sci. 2005, 116, 143–164. [Google Scholar] [CrossRef]
- Tamoto, N.; Adachi, C.; Nagai, K. Electroluminescence of 1,3,4-Oxadiazole and Triphenylamine-Containing Molecules as an Emitter in Organic Multilayer Light Emitting Diodes. Chem. Mater. 1997, 9, 1077–1085. [Google Scholar] [CrossRef]
- Chen, Z.K.; Meng, H.; Lai, Y.H.; Huang, W. Photoluminescent poly(p-phenylenevinylene)s with an aromatic oxadiazole moiety as the side chain: Synthesis, electrochemistry, and spectroscopy study. Macromolecules 1999, 32, 4351–4358. [Google Scholar] [CrossRef]
- Kedzia, A.; Jasiak, K.; Kudelko, A. An Efficient Synthesis of New 2-Aryl-5-phenylazenyl-1,3,4-oxadiazole Derivatives from N,N′-Diarylcarbonohydrazides. Syn. Lett. 2018, 29, 1745–1748. [Google Scholar] [CrossRef]
- Tully, W.R.; Gardner, C.R.; Gillespie, R.J.; Westwood, R. 2-(Oxadiazolyl)- and 2-(Thiazolyl)imidazo[1,2-a]pyrimidines as Agonists and Inverse Agonists at Benzodiazepine Receptors. J. Med. Chem. 1991, 34, 2060–2067. [Google Scholar] [CrossRef] [PubMed]
- Short, F.W.; Long, L.M. Synthesis of 5-aryl-2-oxazolepropionic acids and analogs. Antiinflammatory agents. J. Heterocycl. Chem. 1969, 6, 707–712. [Google Scholar] [CrossRef]
- Theocharis, A.B.; Alexandrou, N.E. Synthesis and spectral data of 4,5-bis[5-aryl-1,3,4-oxadiazol-2-yl]-1-benzyl-1,2,3-triazoles. J. Heterocycl. Chem. 1990, 27, 1685–1688. [Google Scholar] [CrossRef]
- Saeed, A. An expeditious, solvent-free synthesis of some 5-aryl-2-(2-hydroxyphenyl)-1,3,4-oxadiazoles. Chem. Heterocycl. Compd. 2007, 43, 1072–1075. [Google Scholar] [CrossRef]
- Liras, S.; Allen, M.P.; Segelstein, B.E. A mild method for the preparation of 1,3,4-oxadiazoles: Traffic anhydride promoted cyclization of diacylhydrazines. Synth. Commun. 2000, 30, 437–443. [Google Scholar] [CrossRef]
- Carlsen, P.H.J.; Jorgensen, K.B. Synthesis of unsymmetrically substituted 4H-1,2,4-triazoles. J. Heterocycl. Chem. 1994, 31, 805–807. [Google Scholar] [CrossRef]
- Tandon, V.K.; Chhor, R.B. An efficient one pot synthesis of 1,3,4-oxadiazoles. Synth. Commun. 2001, 31, 1727–1732. [Google Scholar] [CrossRef]
- Brain, C.T.; Paul, J.M.; Loong, Y.; Oakley, P.J. Novel procedure for the synthesis of 1,3,4-oxadiazoles from 1,2-diacylhydrazines using polymer-supported Burgess reagent under microwave conditions. Tetrahedron Lett. 1999, 40, 3275–3278. [Google Scholar] [CrossRef]
- Dabiri, M.; Salehi, P.; Baghbanzadeh, M.; Bahramnejad, M. A facile procedure for the one-pot synthesis of unsymmetrical 2,5-disubstituted 1,3,4-oxadiazoles. Tetrahedron Lett. 2006, 47, 6983–6986. [Google Scholar] [CrossRef]
- Majji, G.; Rout, S.K.; Guin, S.; Gogoi, A.; Patel, B.K. Iodine-catalysed oxidative cyclisation of acylhydrazones to 2,5-substituted 1,3,4-oxadiazoles. RSC Adv. 2014, 4, 5357–5362. [Google Scholar] [CrossRef]
- Rostamizadeh, S.; Housaini, A.G. Microwave assisted synthesis of 2,5-disubstituted 1,3,4-oxadiazoles. Tetrahedron Lett. 2004, 45, 8753–8756. [Google Scholar] [CrossRef]
- Milcent, R.; Barbier, G. Oxidation of hydrazones with lead dioxide: New synthesis of 1,3,4-oxadiazoles and 4-amino-1,2,4-triazol-5-one derivatives. Chem. Informationsd. 1983, 14, 80–81. [Google Scholar] [CrossRef]
- Jedlovska, E.; Lesko, J. A simple one-pot procedure for the synthesis of 1,3,4-oxadiazoles. Synth. Commun. 1994, 24, 1879–1885. [Google Scholar] [CrossRef]
- Jasiak, K.; Kudelko, A. Oxidative cyclization of N-aroylhydrazones to 2-(2-arylthenyl)-1,3,4-oxadiazoles using DDQ as an efficient oxidant. Tetrahedron Lett. 2015, 56, 5878–5881. [Google Scholar] [CrossRef]
- Shang, Z.; Reiner, J.; Chang, J.; Zhao, K. Oxidative cyclization of aldazines with bis(trifuloroacetoxy)iodobenzene. Tetrahedron Lett. 2005, 46, 2701–2704. [Google Scholar] [CrossRef]
- Oviedo, C.; Rodriguez, J. EDTA: The chelating agent under environmental scrutiny. Qumica Nova 2003, 26, 901–905. [Google Scholar] [CrossRef]
- Shi, Y.; Campbell, J.A. Study of cyclization of chelating compounds using electrospray ionization mass spectrometry. J. Radioanal. Nucl. Chem. 2000, 245, 293–300. [Google Scholar] [CrossRef]
- Yunta, F.; Garcia-Marco, S.; Lucena, J.J.; Gomez-Gallego, M.; Alcazar, R.; Sierra, M.A. Chelating agents related to ethylenediamine bis(2-hydroxyphenyl)acetic acid (EDDHA): Synthesis, characterization, and equilibrium studies of the free ligands and their Mg2+, Ca2+, Cu2+, Fe3+ chelates. Inorg. Chem. 2003, 42, 5412–5421. [Google Scholar] [CrossRef]
- Surgutskaia, N.S.; Di Martino, A.; Zednik, J.; Ozaltin, K.; Lovecka, L.; Domincova-Bergerova, E.; Kimmer, D.; Svoboda, J.; Sedlarik, V. Efficient Cu2+, Pb2+ and Ni2+ ion removal from wastewater using electrospun DTPA-moified chitosan/polyethylene oxide nanofibers. Sep. Purif. Technol. 2020, 247, 116914. [Google Scholar] [CrossRef]
- Knecht, S.; Ricklin, D.; Eberle, A.N.; Ernst, B. Oligohis-tags: Mechanisms of binding to Ni2+-NTA surfaces. J. Mol. Recognit. 2009, 22, 270–279. [Google Scholar] [CrossRef]
- Shaddox, T.W.; Unruh, J.B.; Kruse, J.K.; Restuccia, N.G. Solubility of Iron, Manganese, and Magnesium Sulfates and Glucoheptonates in Two Alkaline Soils. Soil Sci. Soc. Am. J. 2016, 80, 765–770. [Google Scholar] [CrossRef]
- Pozdnyakov, I.P.; Tyutereva, Y.E.; Mikheilis, A.V.; Grivin, V.P.; Plyusnin, V.F. Primary photoprocesses for Fe(III) complexes with citric and hlycolic acids in aqueous solutions. J. Photochem. Photobiol. A Chem. 2022, 434, 114274. [Google Scholar] [CrossRef]
- Sekhon, B.S. Chelates for Micronutrient Nutrition among Crops. Resonance 2003, 8, 46–53. [Google Scholar] [CrossRef]
- Mrozek-Niecko, A.; Pernak, J. Biodegradacja soli tetrasodowej kwasu N-(1,2-dikarboksyetyleno)-D,L-asparaginowego i jego chelatów. Przem. Chem. 2006, 85, 635–637. [Google Scholar]
- Santos, M.A.; Marques, S.M.; Tuccinardi, T.; Carelli, P.; Panelli, L.; Rossello, A. Design, synthesis and molecular modelling study of iminodiacetyl monohydroxamic acid derivatives as MMP inhibitors. Bioorganic Med. Chem. 2006, 14, 7539–7550. [Google Scholar] [CrossRef]
- Elsinger, F.; Schreiber, J.; Eschenmoser, A. Notiz uber die Selektivitat der Spaltung von Carbonsauremethylestern mit Lithiumjodid. Chim. Acta 1960, 43, 113–118. [Google Scholar] [CrossRef]
- Tanemura, K.; Rohand, T. Activated charcoal as an effective additive for alkaline and acidic hydrolysis of esters in water. Tetrahedron Lett. 2020, 61, 152467. [Google Scholar] [CrossRef]
- Cież, D.; Svetlik, J. A One-Pot Preparation of 5-Oxo 2,4-Disubstituted 2,5-Dihydro-1H-imidazol-2-carboxylates from α-Bromo Esters. Synlett 2011, 3, 315–318. [Google Scholar] [CrossRef]
- Gallagher, J.A.; Levine, L.A.; Williams, M.E. Anion Effects in Cu-Crosslinked Palindromic Artificial Tripeptides with Pendant Bpy Ligands. Eur. J. Inorg. Chem. 2011, 27, 4168–4174. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Product | n | Base | Solvent | Yield [%] |
|---|---|---|---|---|---|
| 1 | 2a | 1 | TEA | Chloroform | 26 |
| 2 | Diethyl ether | 11 | |||
| 3 | Na2CO3 | Chloroform | 47 | ||
| 4 | Diethyl ether | 65 | |||
| 5 | 2b | 2 | TEA | Chloroform | 33 |
| 6 | Diethyl ether | 44 | |||
| 7 | Na2CO3 | Chloroform | 59 | ||
| 8 | Diethyl ether | 73 | |||
| 9 | 2c | 3 | TEA | Chloroform | 37 |
| 10 | Diethyl ether | 35 | |||
| 11 | Na2CO3 | Chloroform | 66 | ||
| 12 | Diethyl ether | 76 | |||
| 13 | 2d | 4 | TEA | Chloroform | 28 |
| 14 | Diethyl ether | 20 | |||
| 15 | Na2CO3 | Chloroform | 74 | ||
| 16 | Diethyl ether | 79 |
| Entry | Product | n | Solvent | Yield (%) |
|---|---|---|---|---|
| 1 | 3a | 1 | Toluene | 36 |
| 2 | - | 51 | ||
| 3 | 3b | 2 | Toluene | 15 |
| 4 | - | 40 | ||
| 5 | 3c | 3 | Toluene | 39 |
| 6 | - | 44 | ||
| 7 | 3d | 4 | Toluene | 59 |
| 8 | - | 76 |
| Entry | Solvent | Temp. (°C) | Base | Yield (%) |
|---|---|---|---|---|
| 1 | DMF | 25 | TEA | 18 |
| 2 | 60 | 22 | ||
| 3 | 25 | Na2CO3 | 14 | |
| 4 | 60 | 69 | ||
| 5 | DMSO | 25 | TEA | 9 |
| 6 | 60 | 24 | ||
| 7 | 25 | Na2CO3 | 18 | |
| 8 | 60 | 55 | ||
| 9 | Acetonitrile | 25 | TEA | 21 |
| 10 | 60 | 39 | ||
| 11 | 25 | Na2CO3 | 23 | |
| 12 | 60 | 91 |
| Entry | n | R | Product | Yield (%) |
|---|---|---|---|---|
| 1 | 1 | Ethyl | 6a | 91 |
| 2 | 1 | i-Propyl | 6b | 84 |
| 3 | 2 | i-Propyl | 6c | 71 |
| 4 | 3 | i-Propyl | 6d | 68 |
| 5 | 4 | i-Propyl | 6e | 73 |
| 6 | 1 | i-Propyl | 7b | 54 |
| 7 | 3 | i-Propyl | 7d | 68 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Łuczyński, M.; Kubiesa, K.; Kudelko, A. Synthesis of 2,5-Dialkyl-1,3,4-oxadiazoles Bearing Carboxymethylamino Groups. Molecules 2022, 27, 7687. https://doi.org/10.3390/molecules27227687
Łuczyński M, Kubiesa K, Kudelko A. Synthesis of 2,5-Dialkyl-1,3,4-oxadiazoles Bearing Carboxymethylamino Groups. Molecules. 2022; 27(22):7687. https://doi.org/10.3390/molecules27227687
Chicago/Turabian StyleŁuczyński, Marcin, Kornelia Kubiesa, and Agnieszka Kudelko. 2022. "Synthesis of 2,5-Dialkyl-1,3,4-oxadiazoles Bearing Carboxymethylamino Groups" Molecules 27, no. 22: 7687. https://doi.org/10.3390/molecules27227687
APA StyleŁuczyński, M., Kubiesa, K., & Kudelko, A. (2022). Synthesis of 2,5-Dialkyl-1,3,4-oxadiazoles Bearing Carboxymethylamino Groups. Molecules, 27(22), 7687. https://doi.org/10.3390/molecules27227687