
Synthesis, Biosynthesis, and Biological Activity of Diels–Alder Adducts from Morus Genus: An Update


 ,
,  ,
,  , ,
, ,  and
and 
Abstract
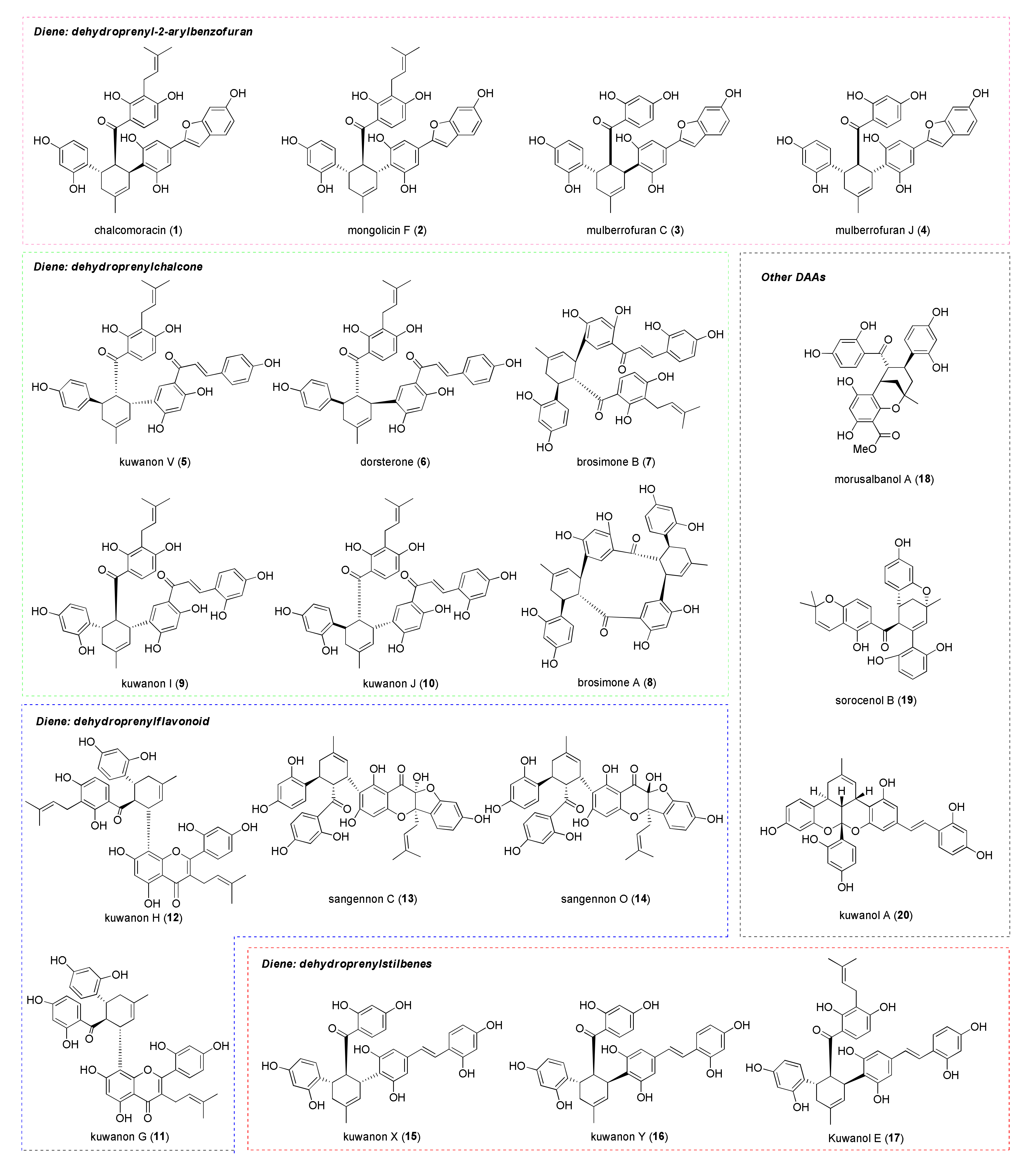
1. Introduction
2. The [4+2] Cycloaddition Reaction as a Powerful Tool for Biomimetic Synthesis of DAAs
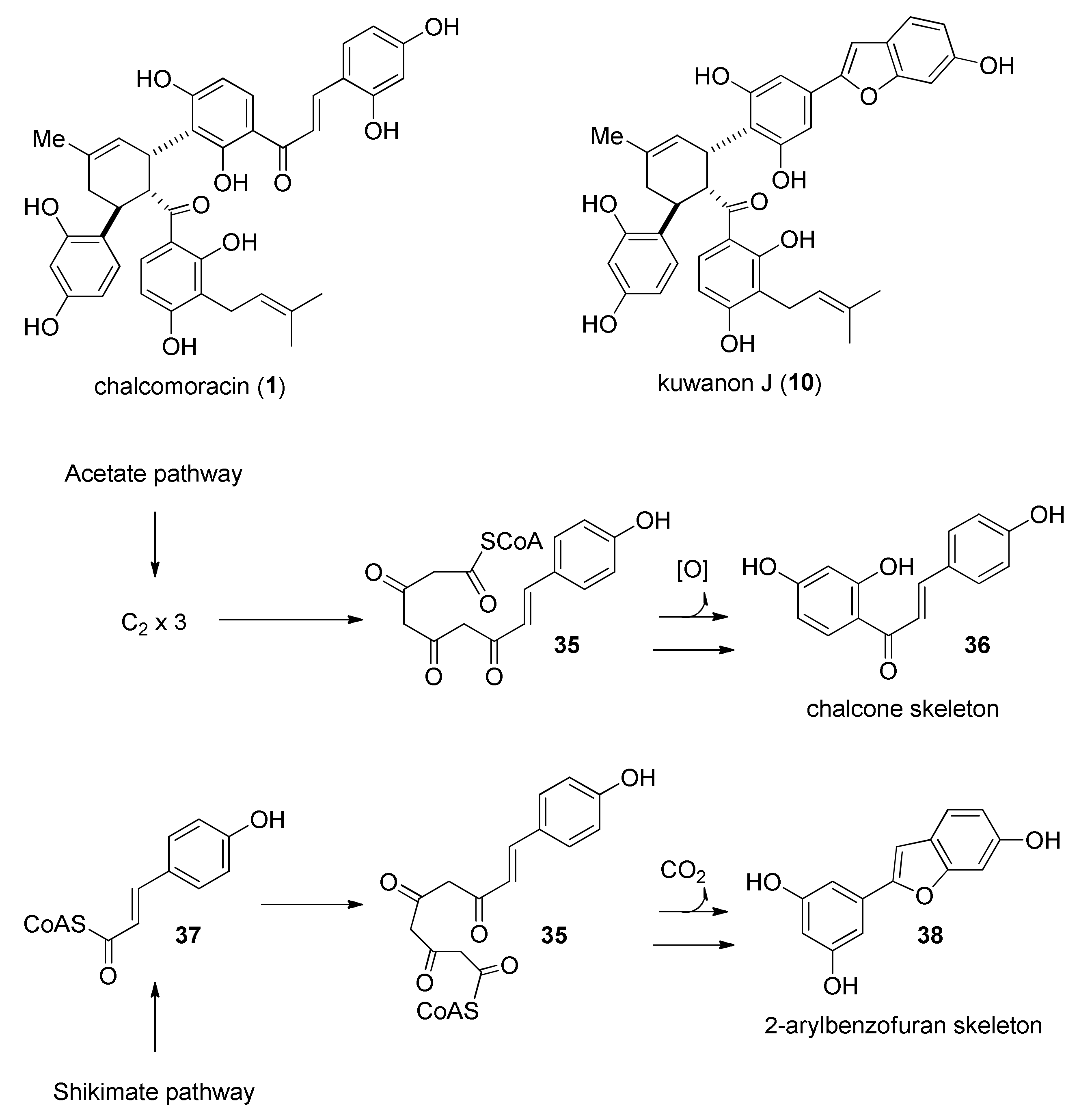
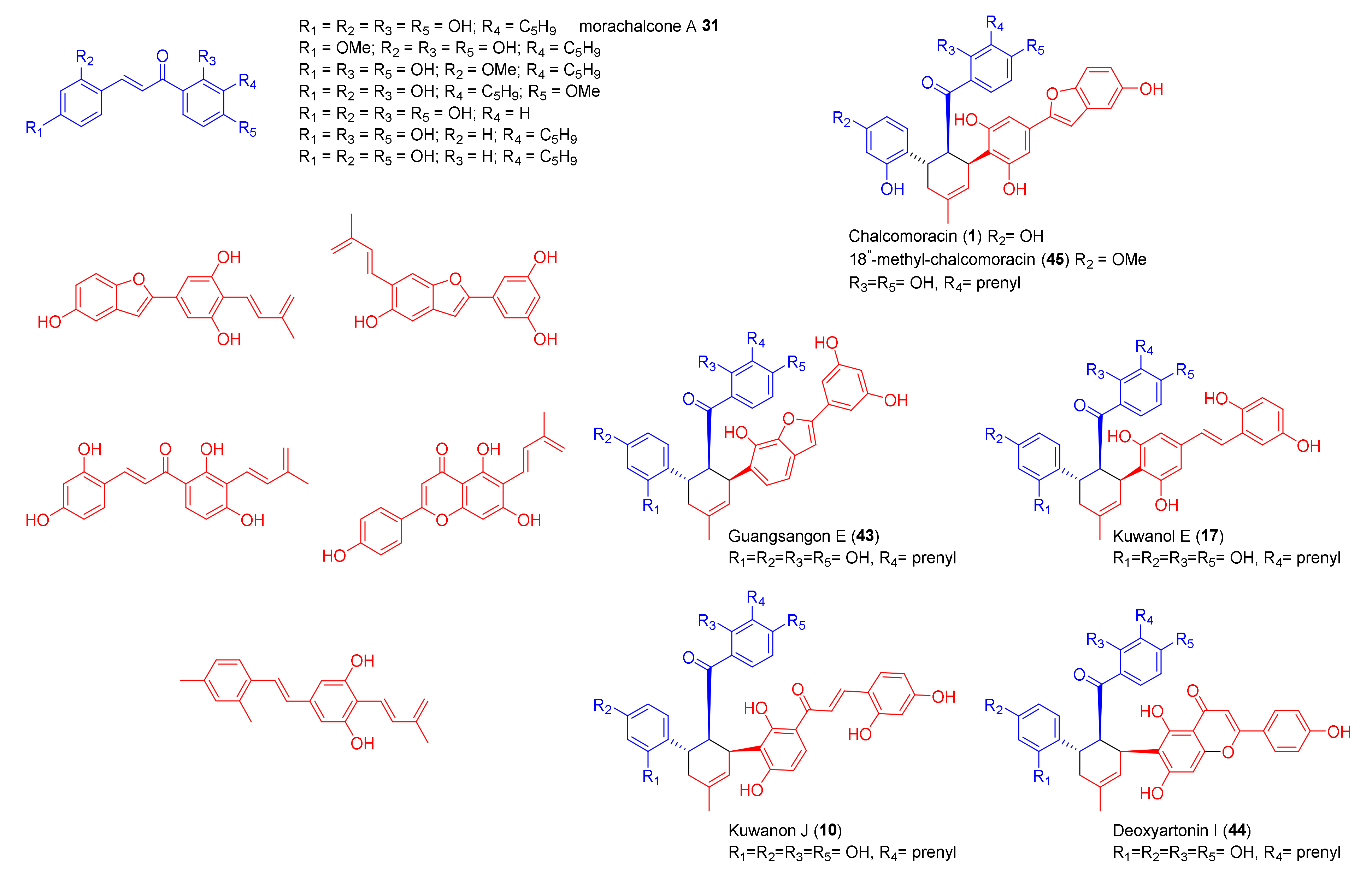
3. Biosynthesis of Mulberry DAAs
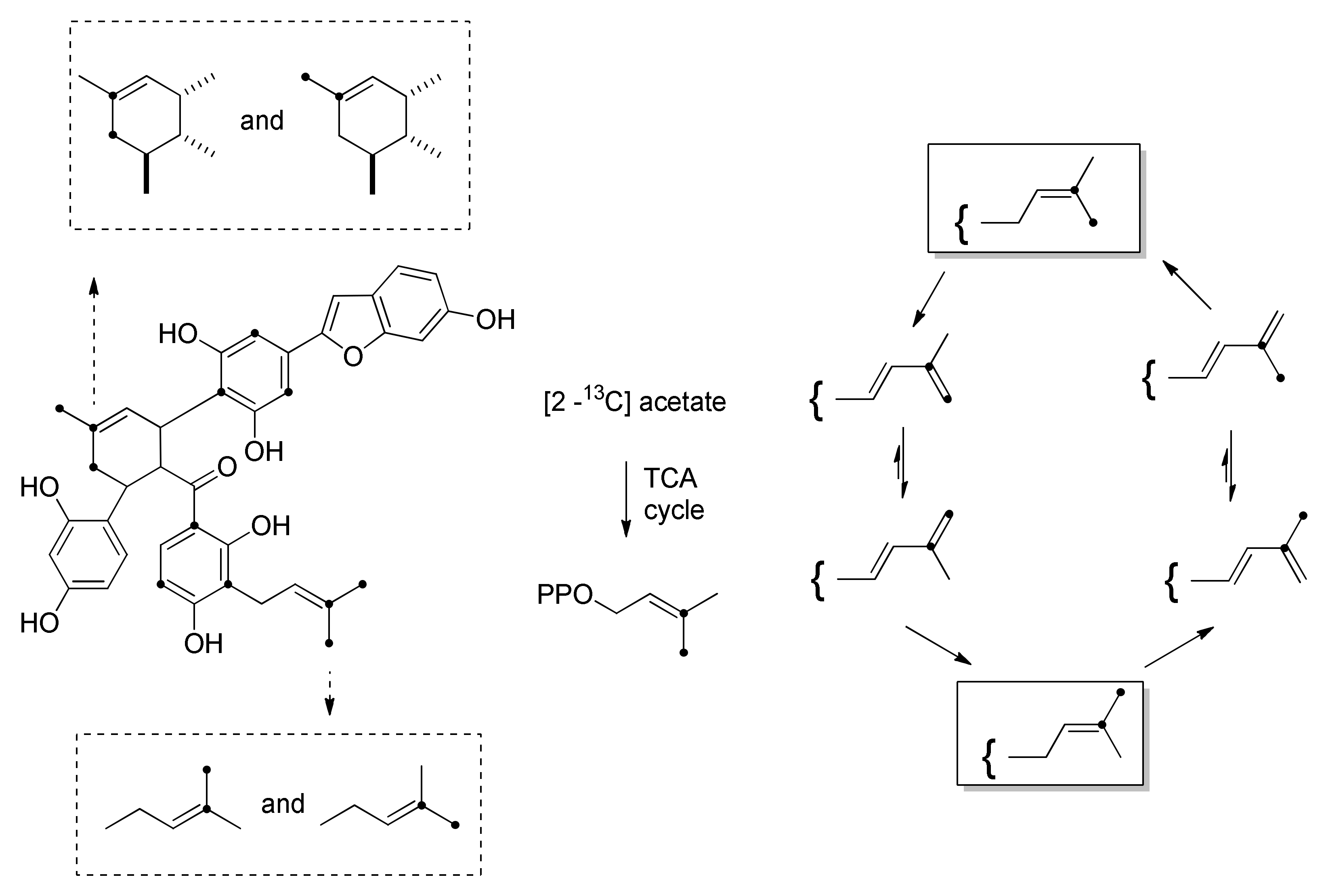
3.1. Incorporation Studies with 13C-Labelled Acetate
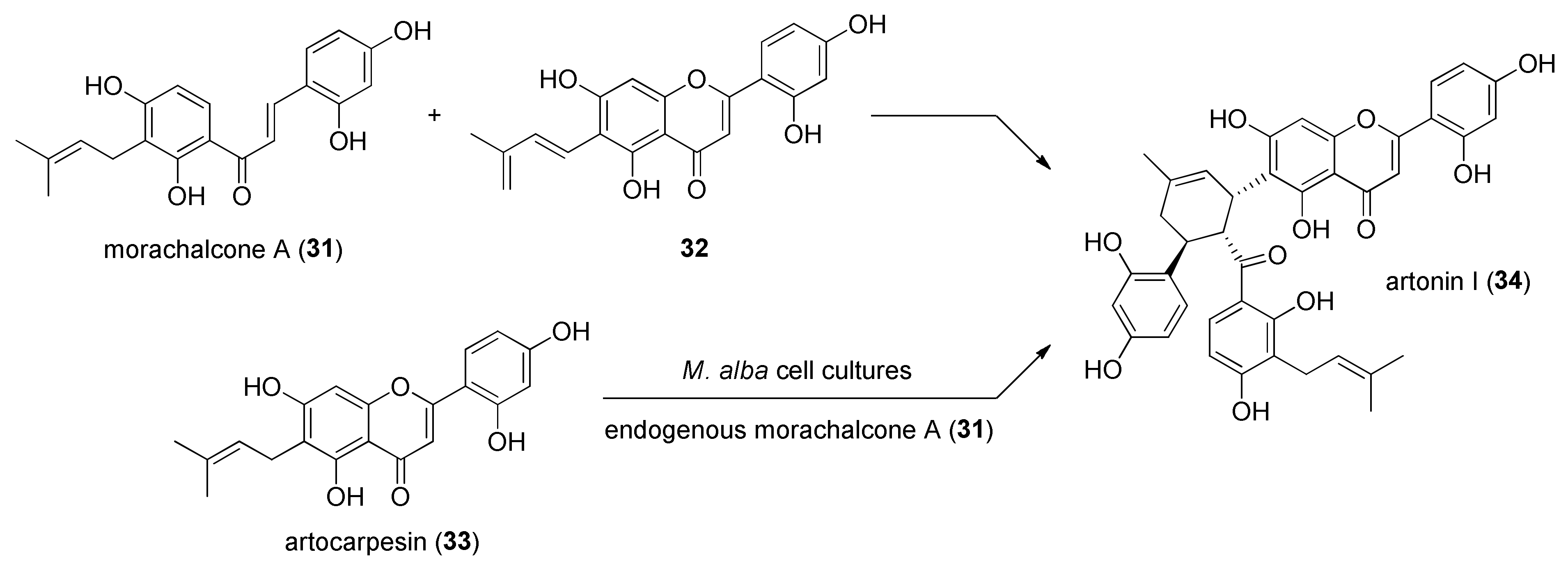
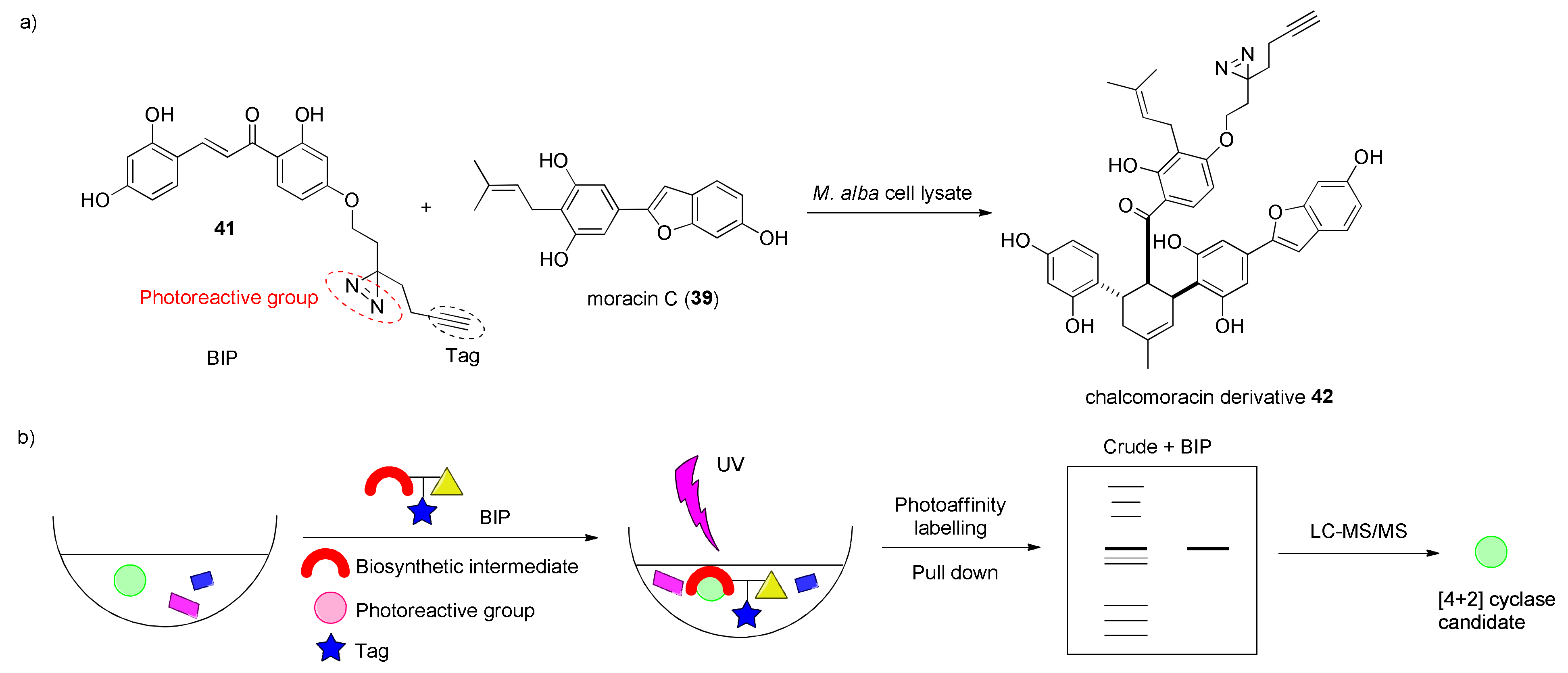
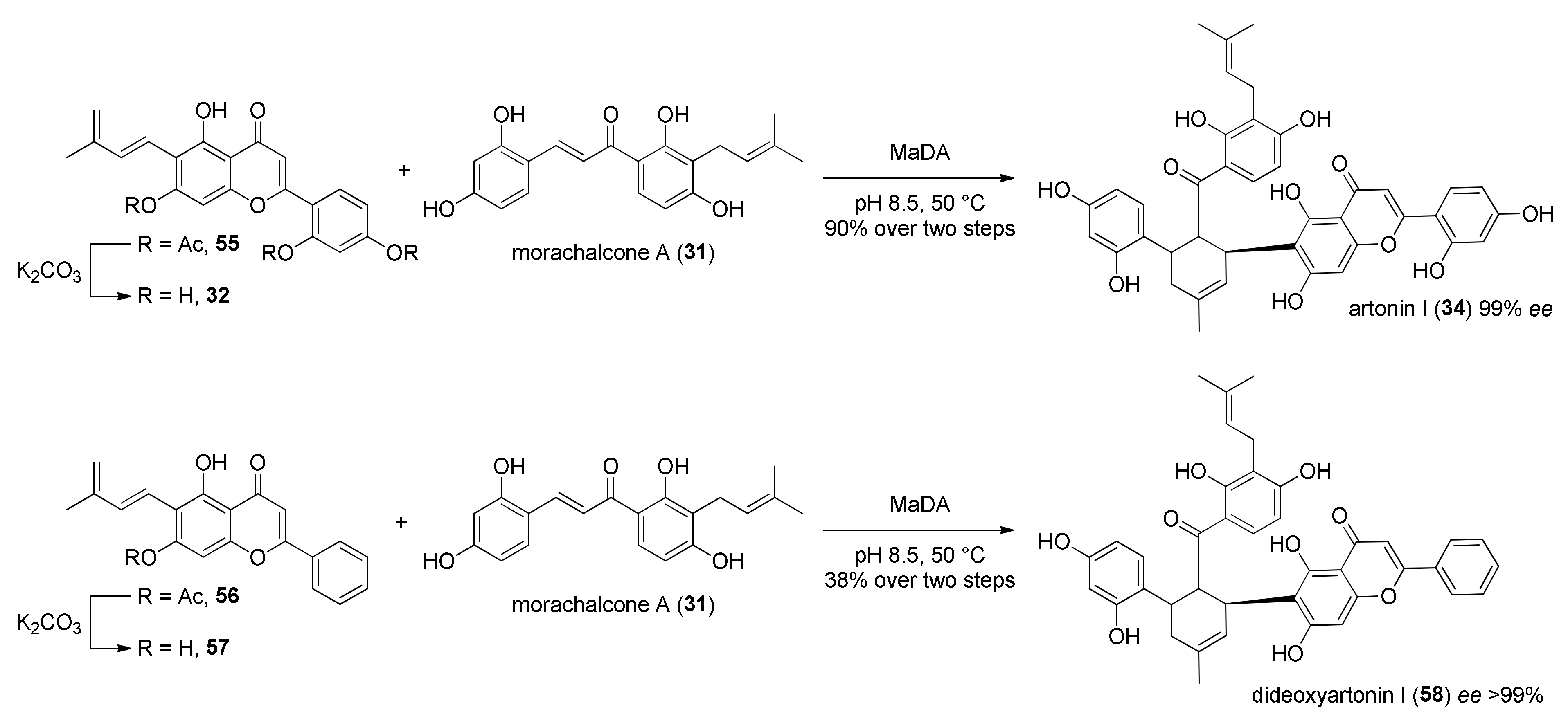
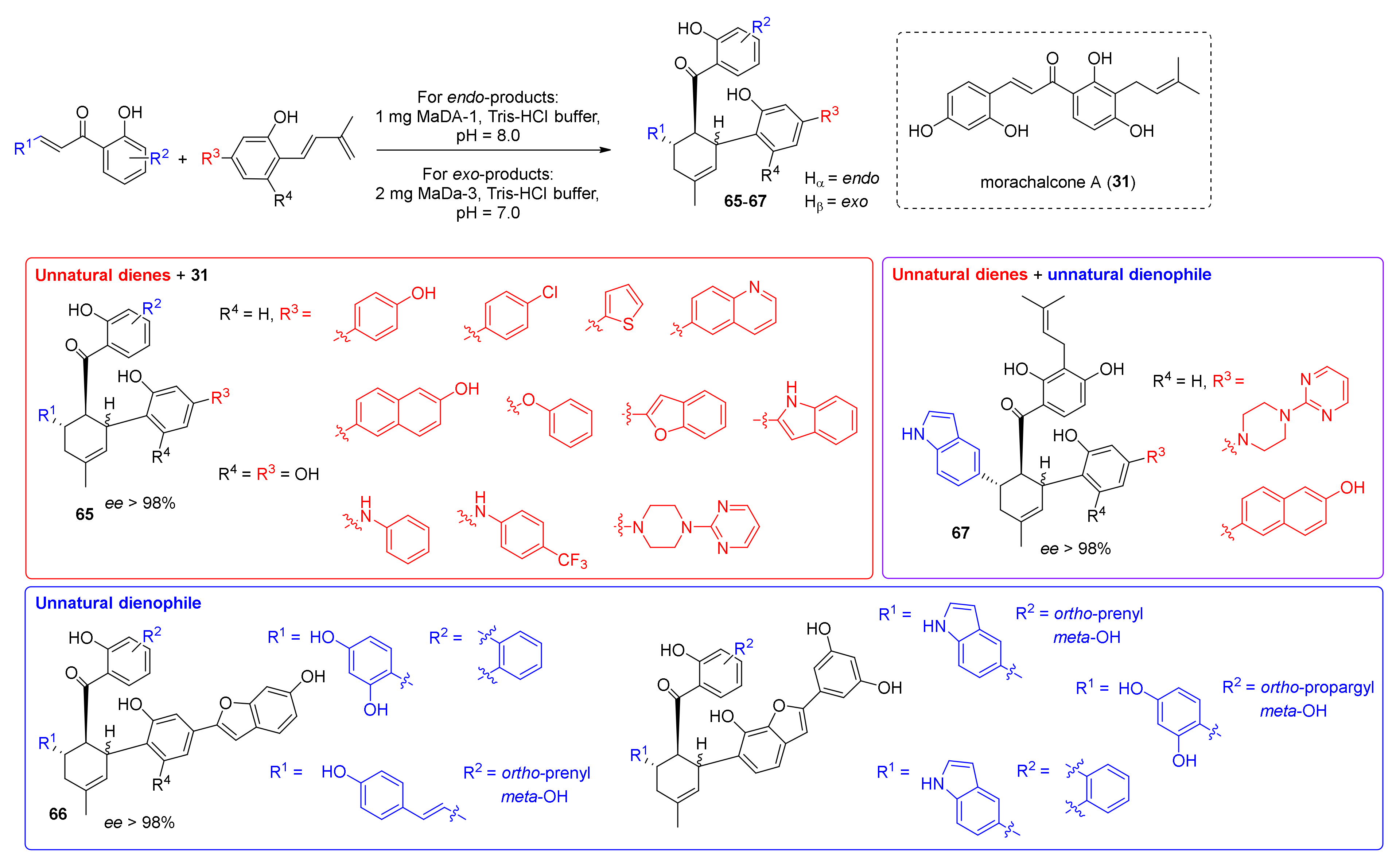
3.2. Discovery of the First Intermolecular Diels–Alderase from M. alba
4. Chemoenzymatic Total Syntheses of Natural and Unnatural Mulberry DAAs
5. Total Synthesis of Mulberry DAAs
5.1. Racemic Total Synthesis of Mulberry DAAs
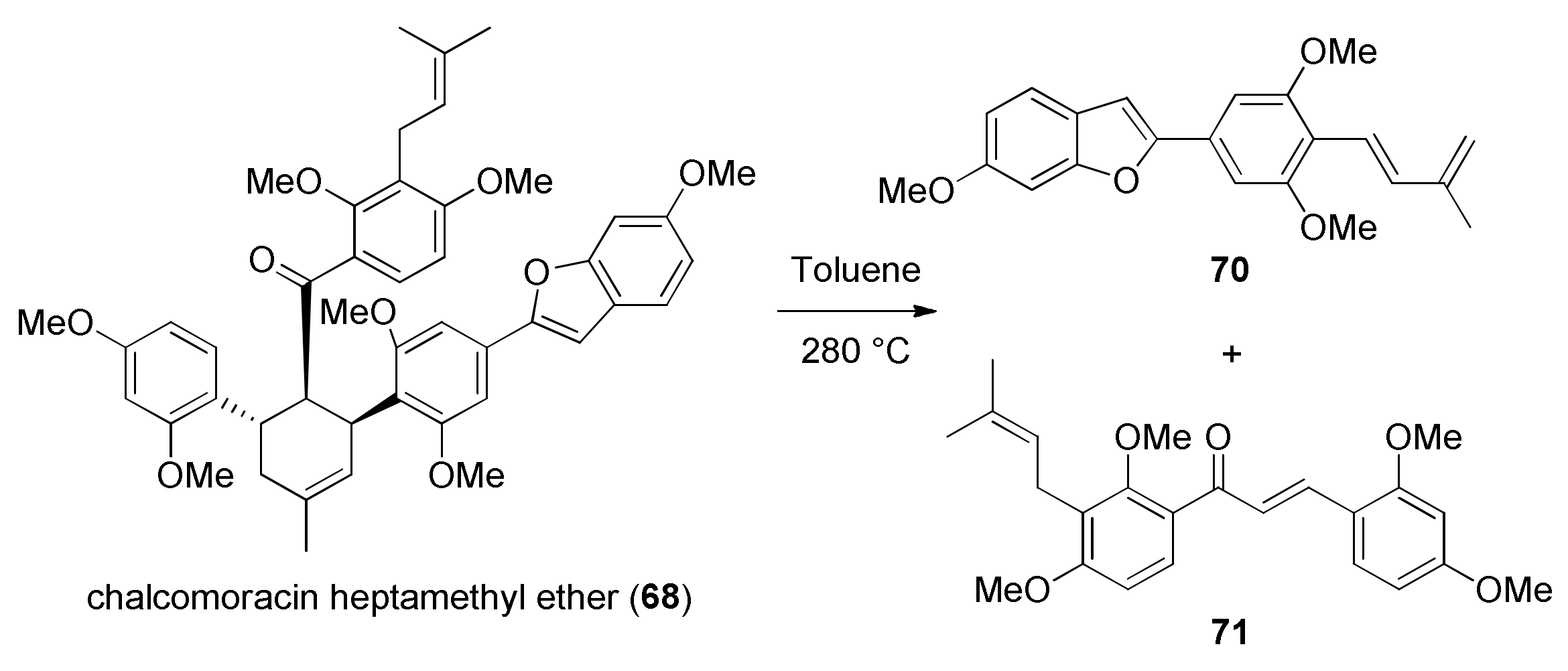
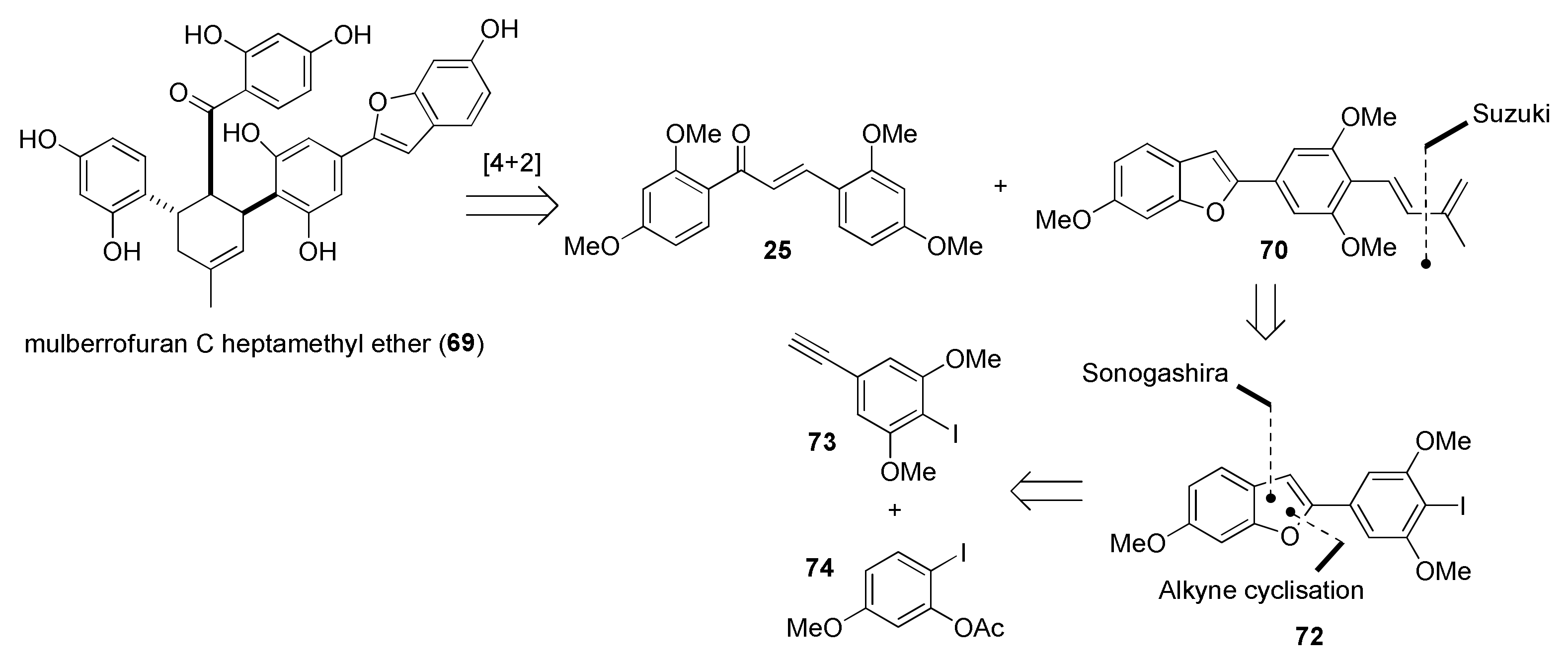
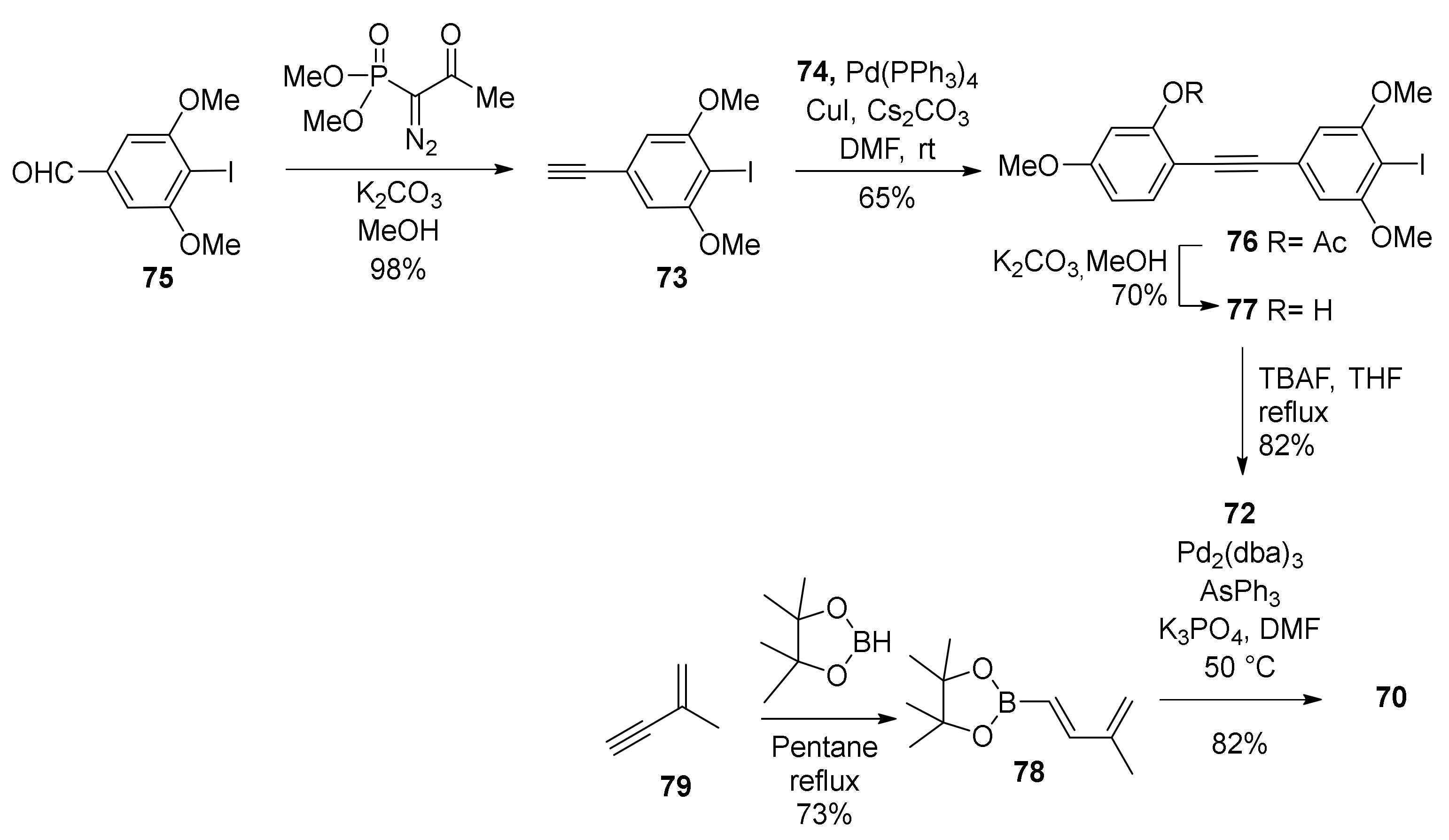
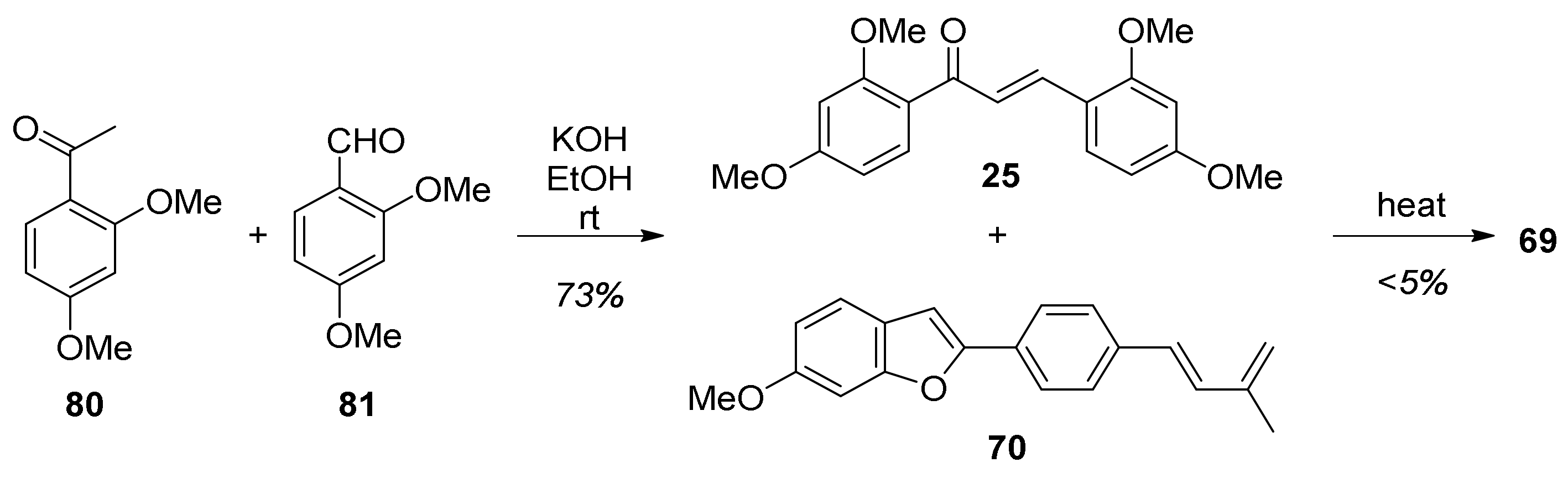
5.1.1. Chalcomoracin, Mongolicin F, Mulberrofurans C, and J Methyl Ethers
5.1.2. Kuwanon V, Dorsterone Methyl Ethers
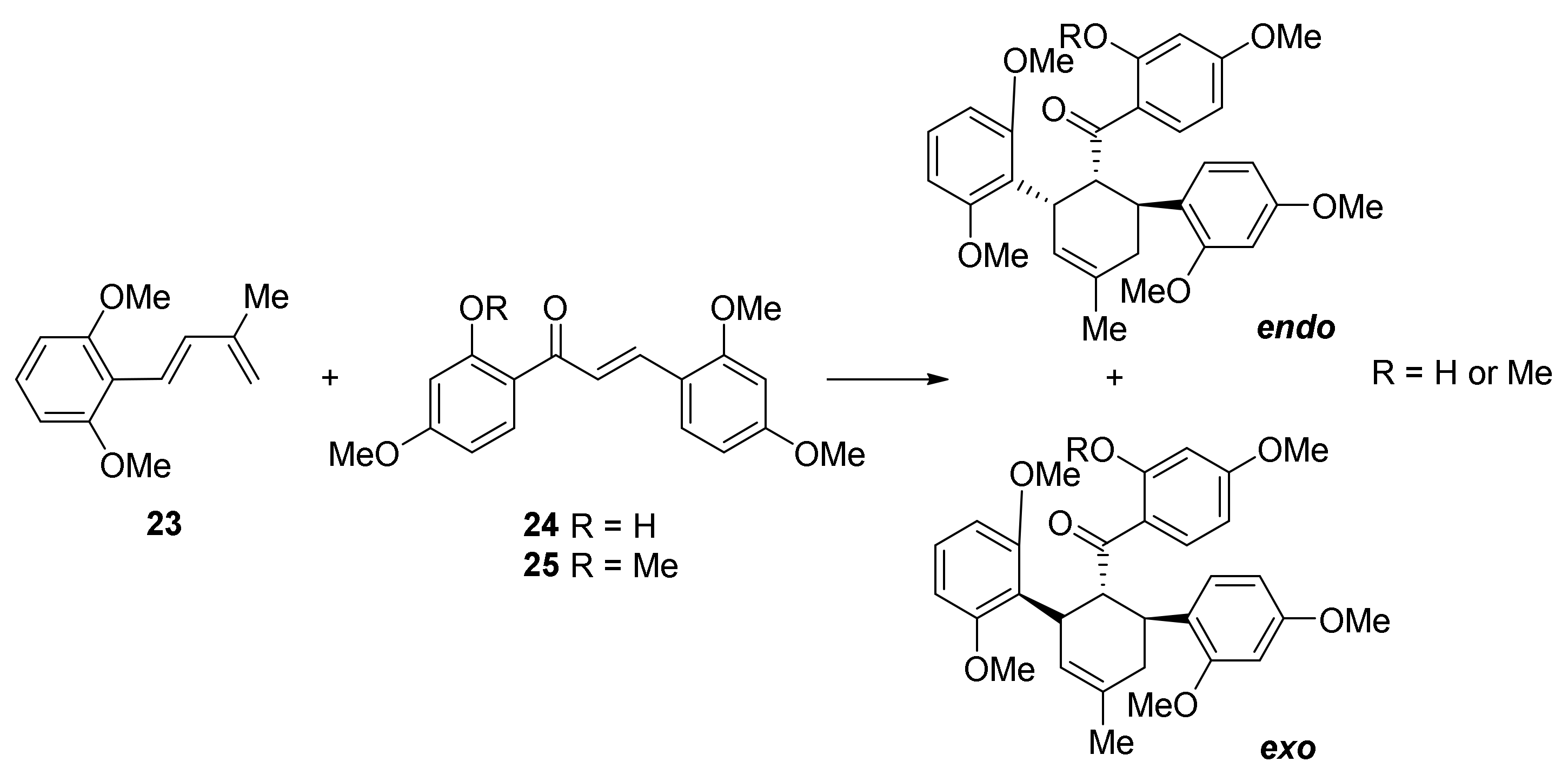
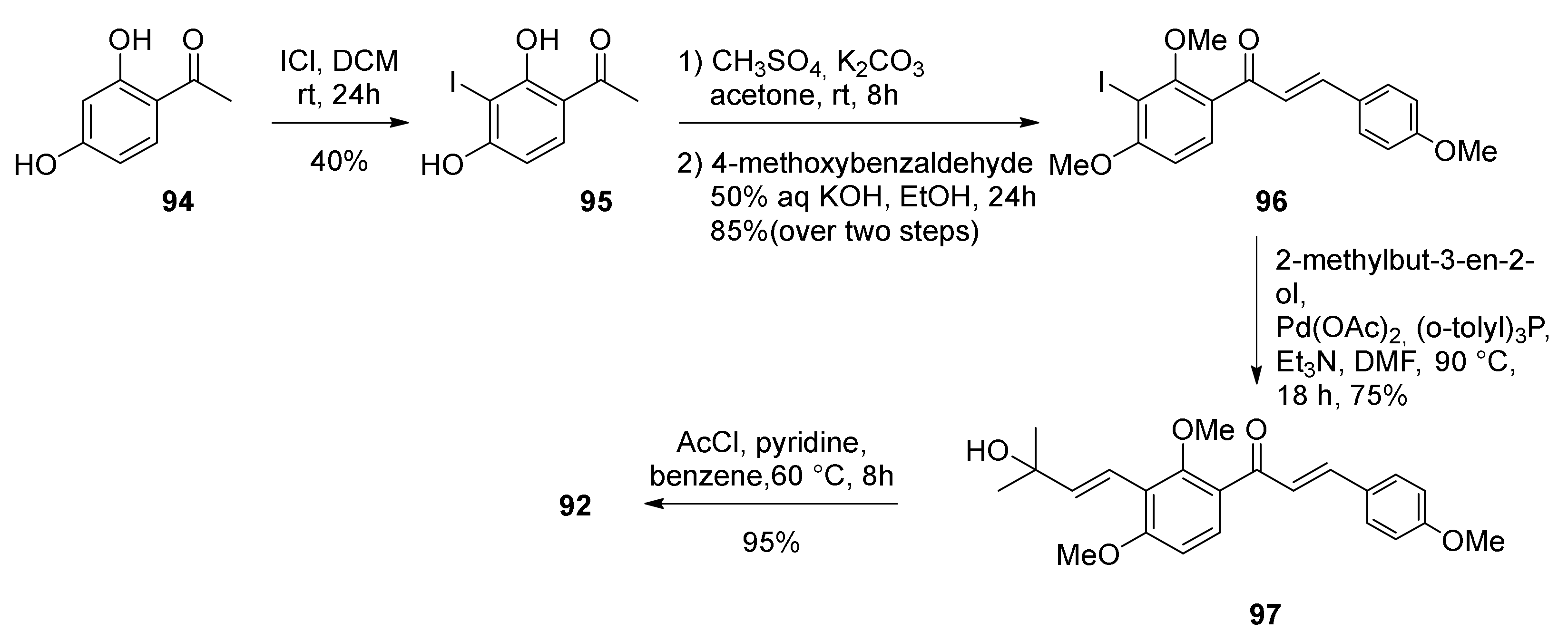
5.1.3. Sorocenol B
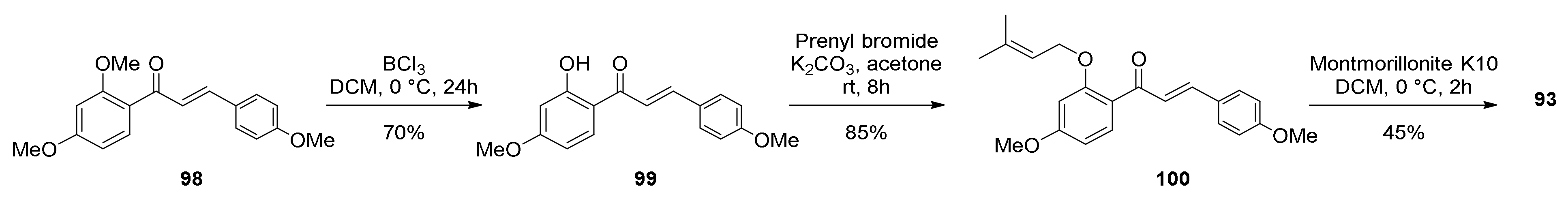
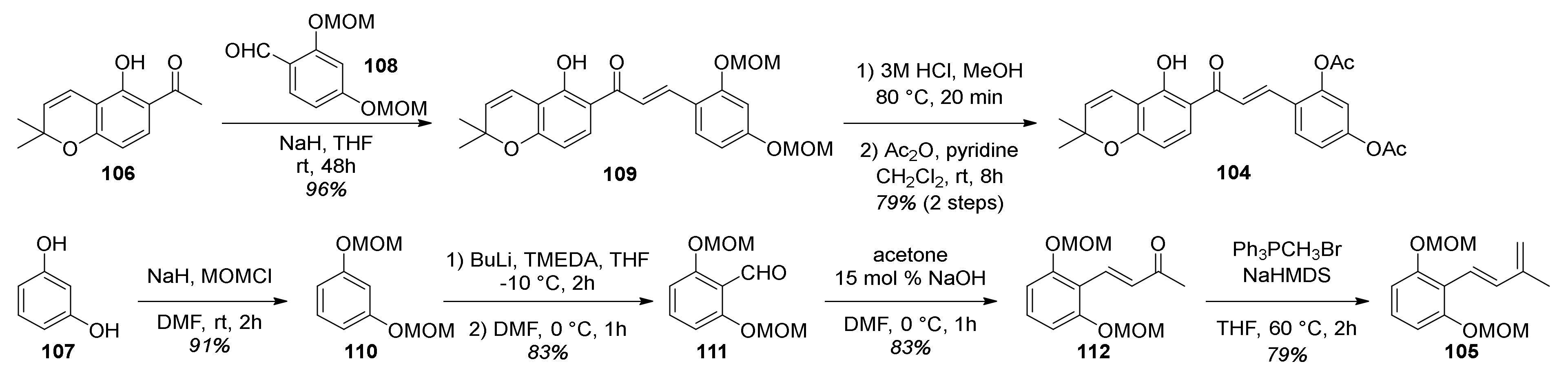
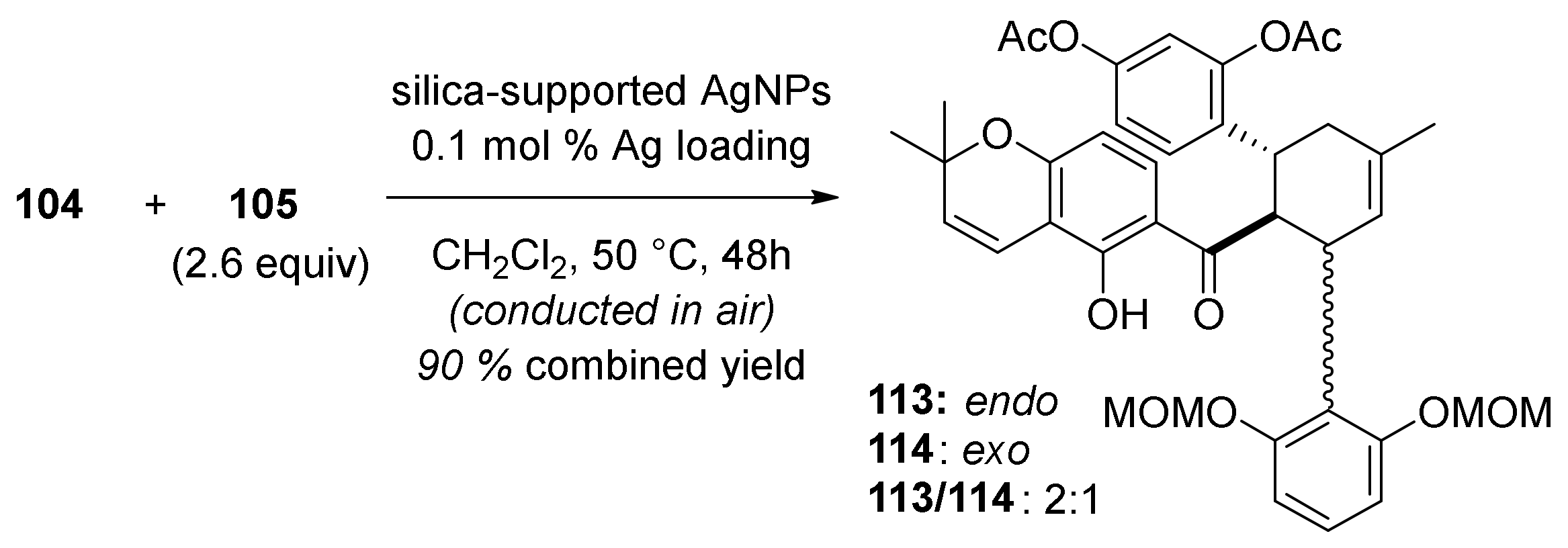
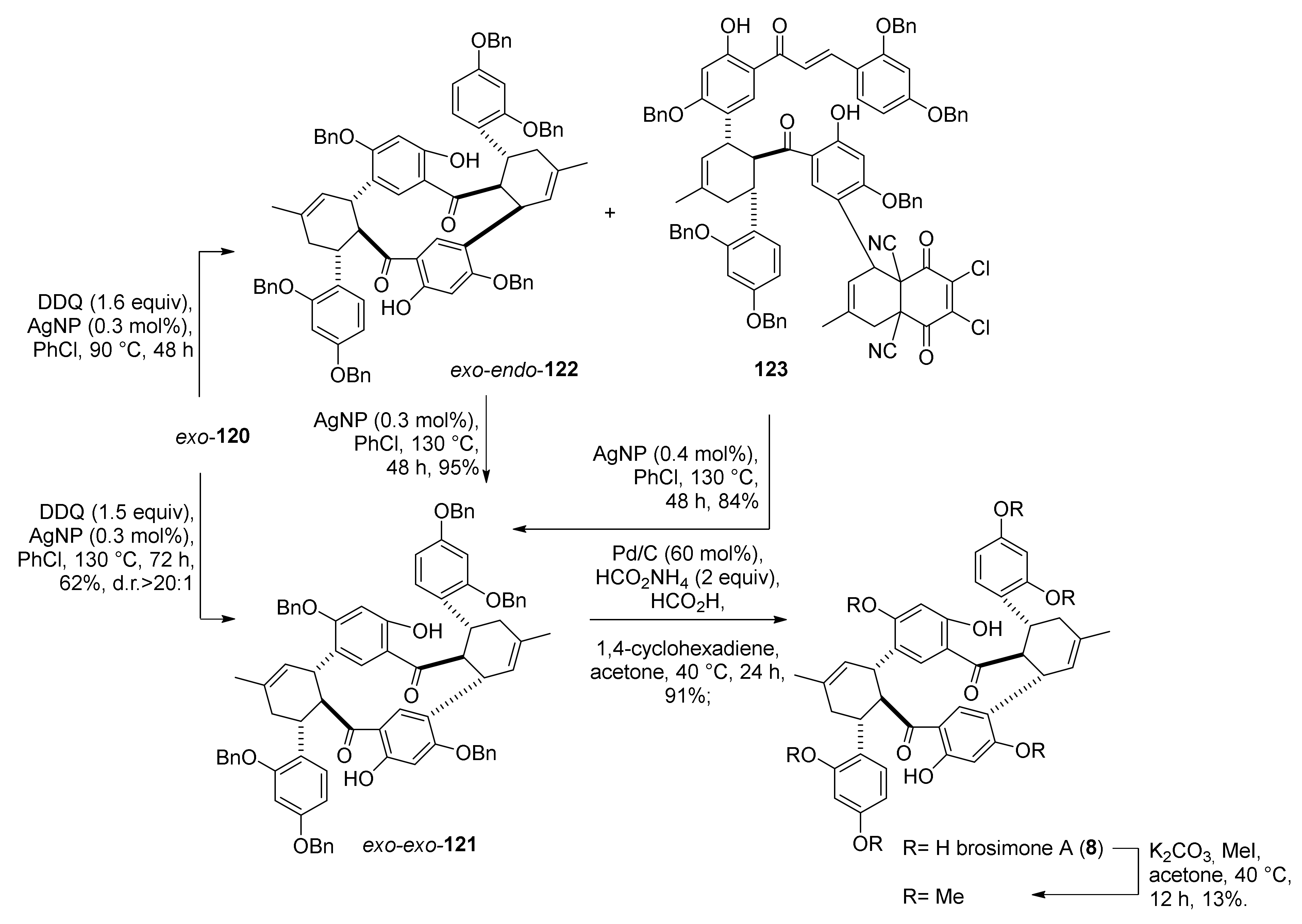
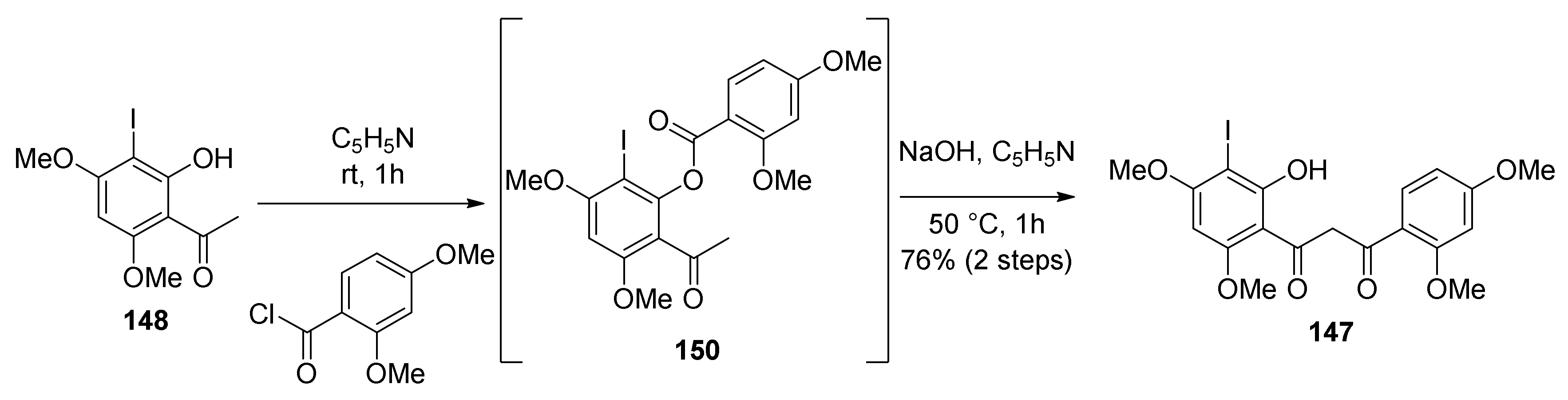
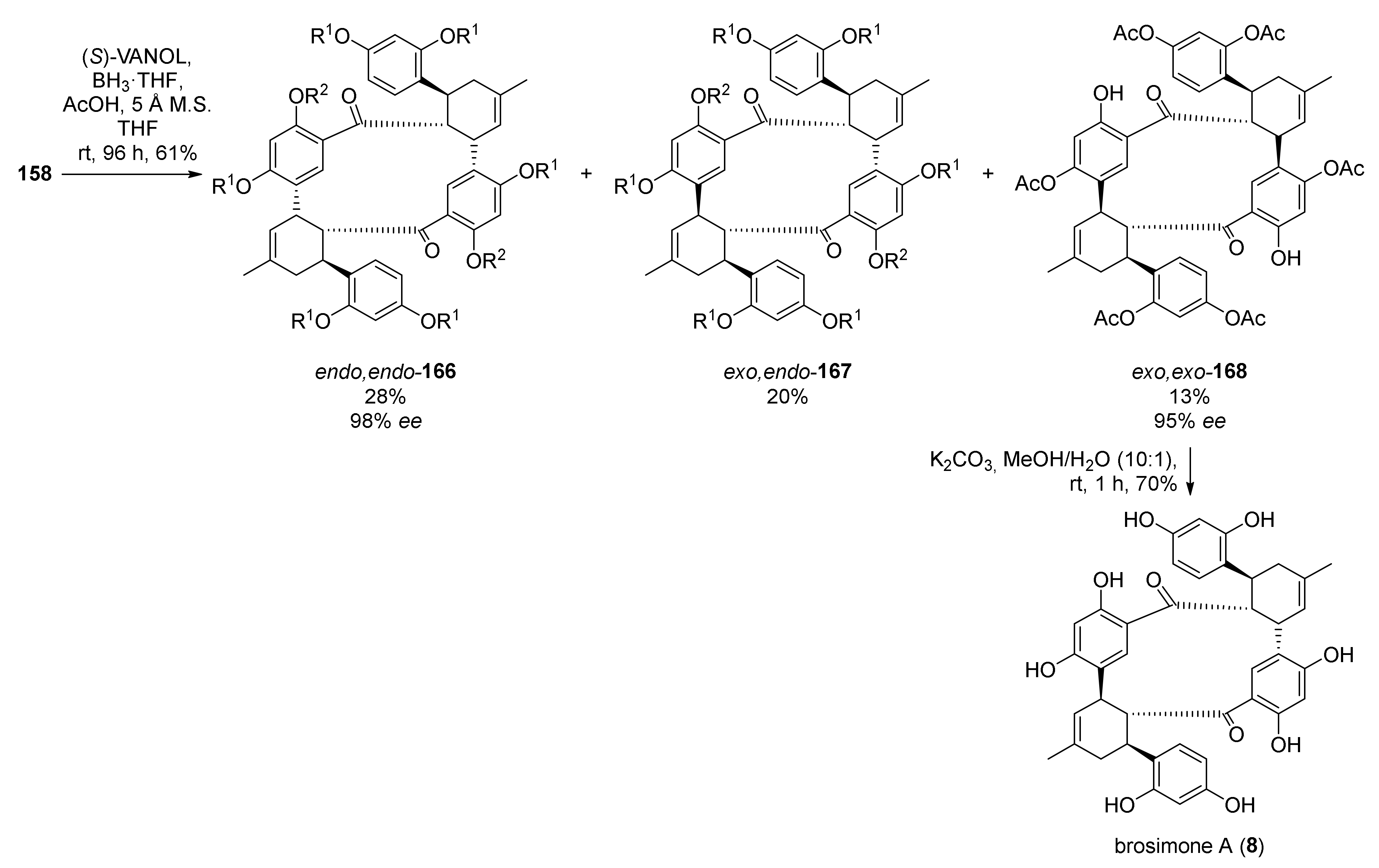
5.1.4. Brosimones A and B
5.1.5. Morusalbanol A Pentamethyl Ether
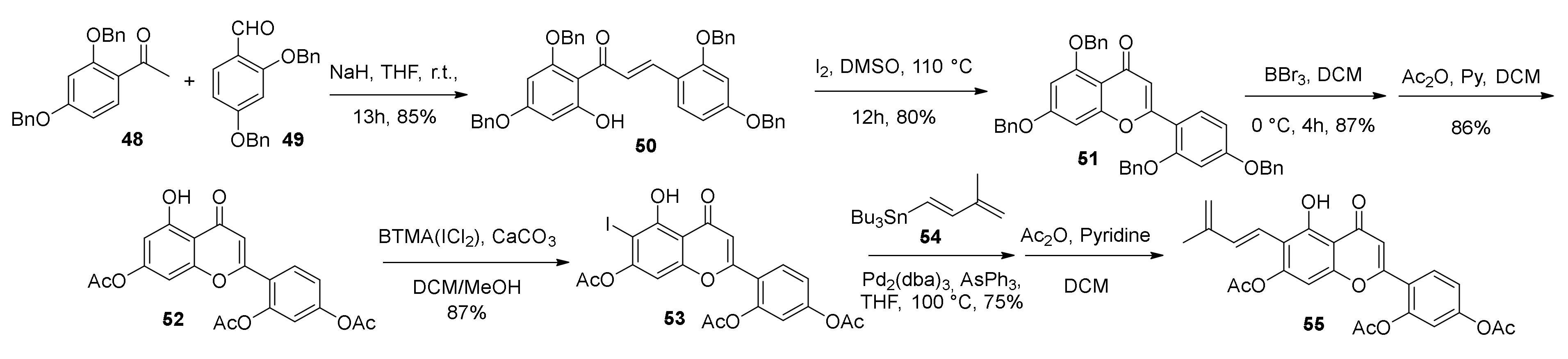
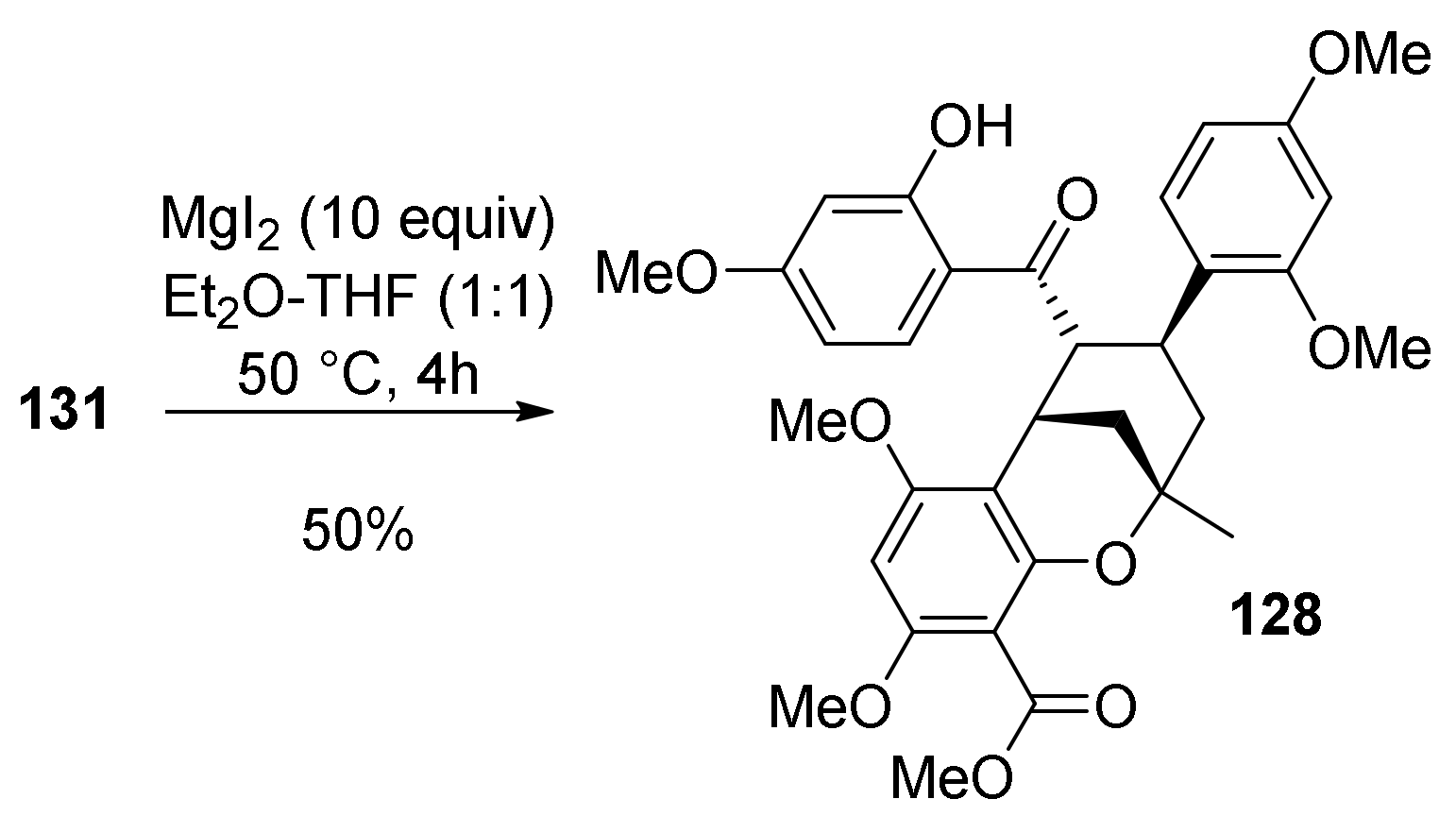
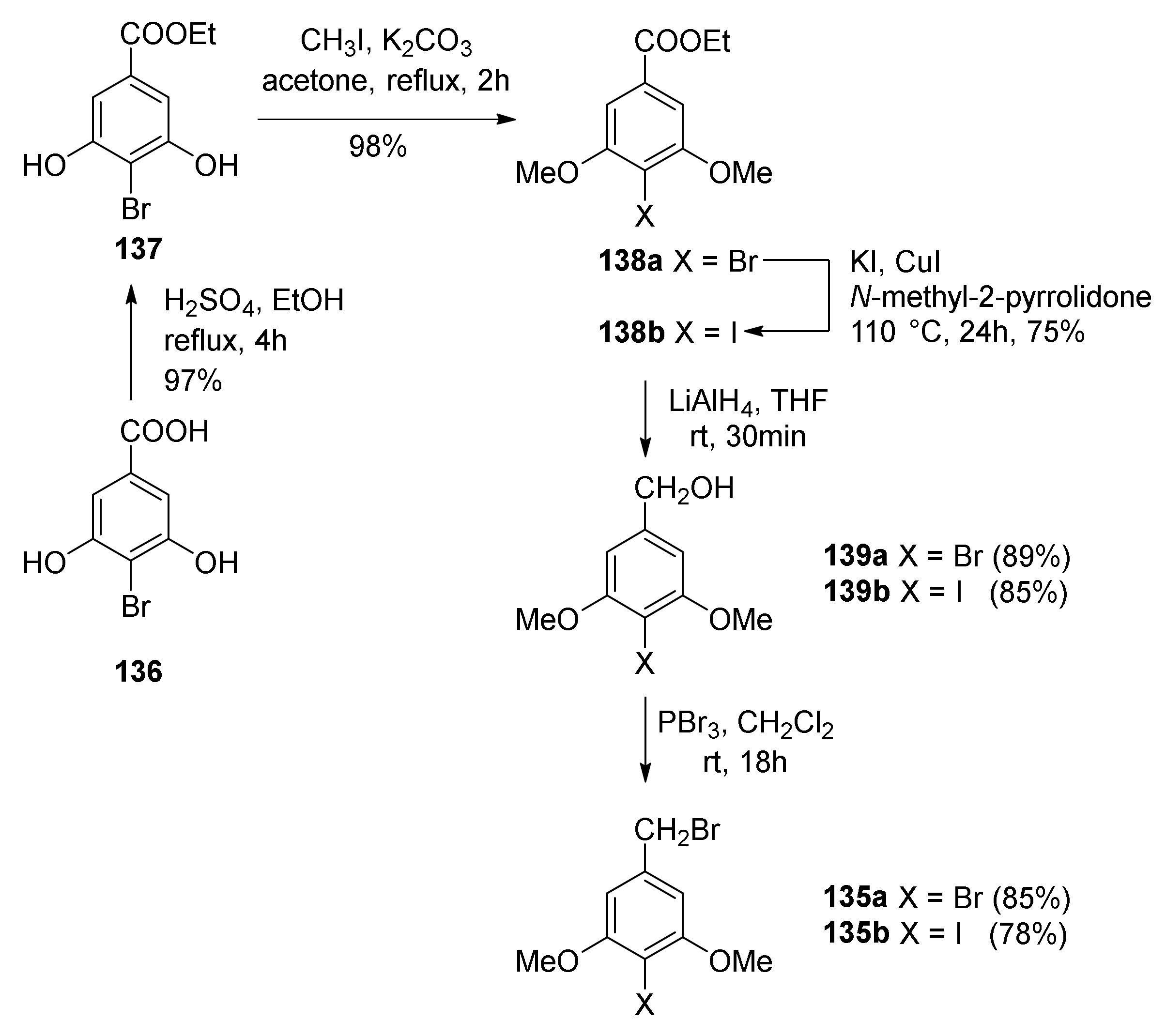
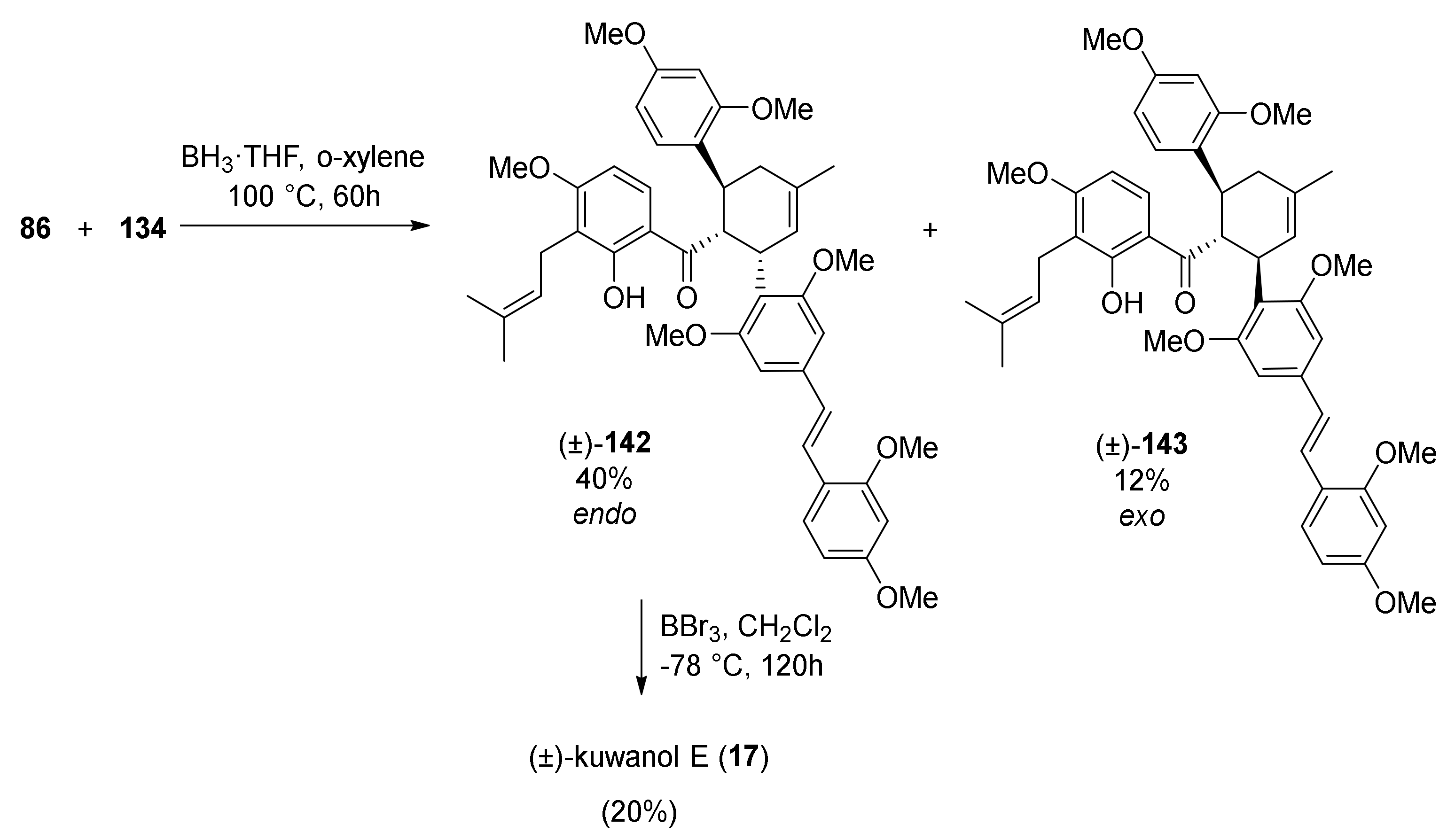
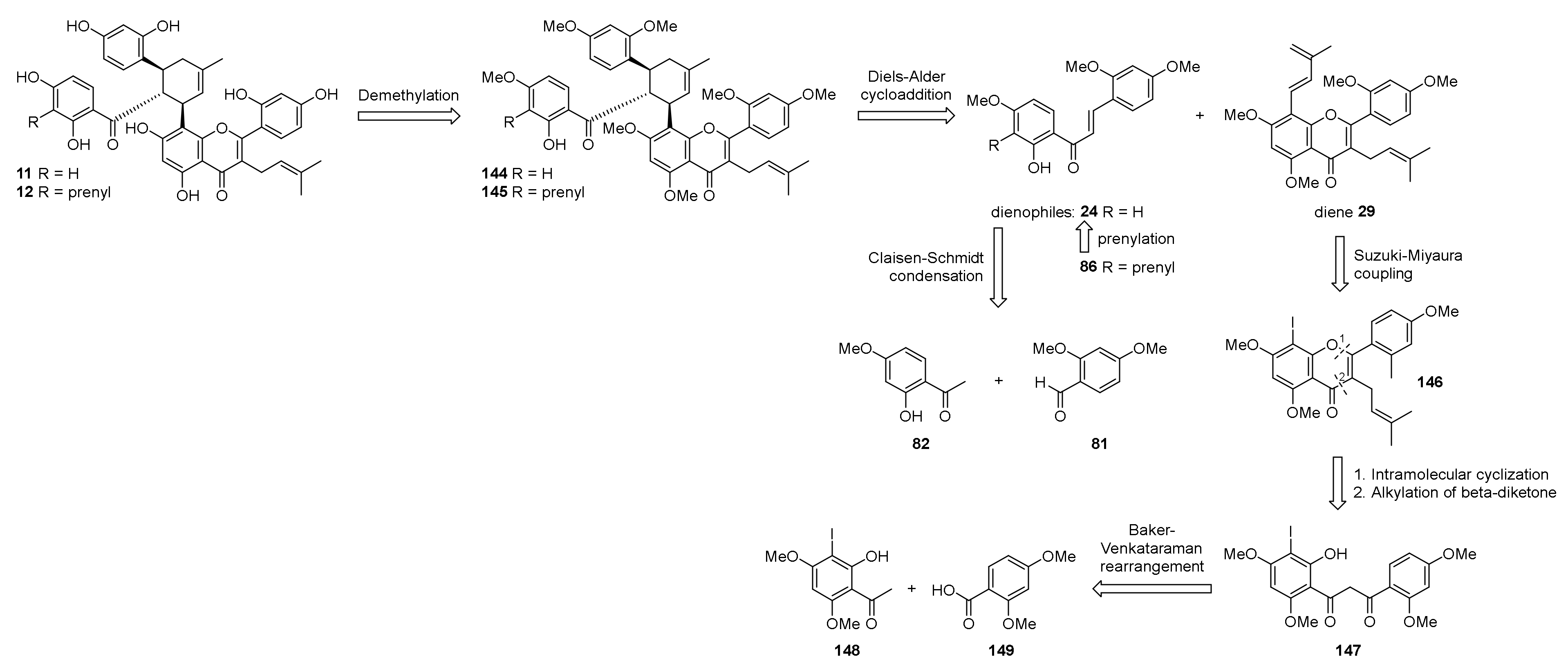
5.1.6. Kuwanol E
5.1.7. Kuwanon G and H
5.2. Enantioselective Total Synthesis of Mulberry DAAs
5.2.1. Brosimones A and B and Kuwanons I and J
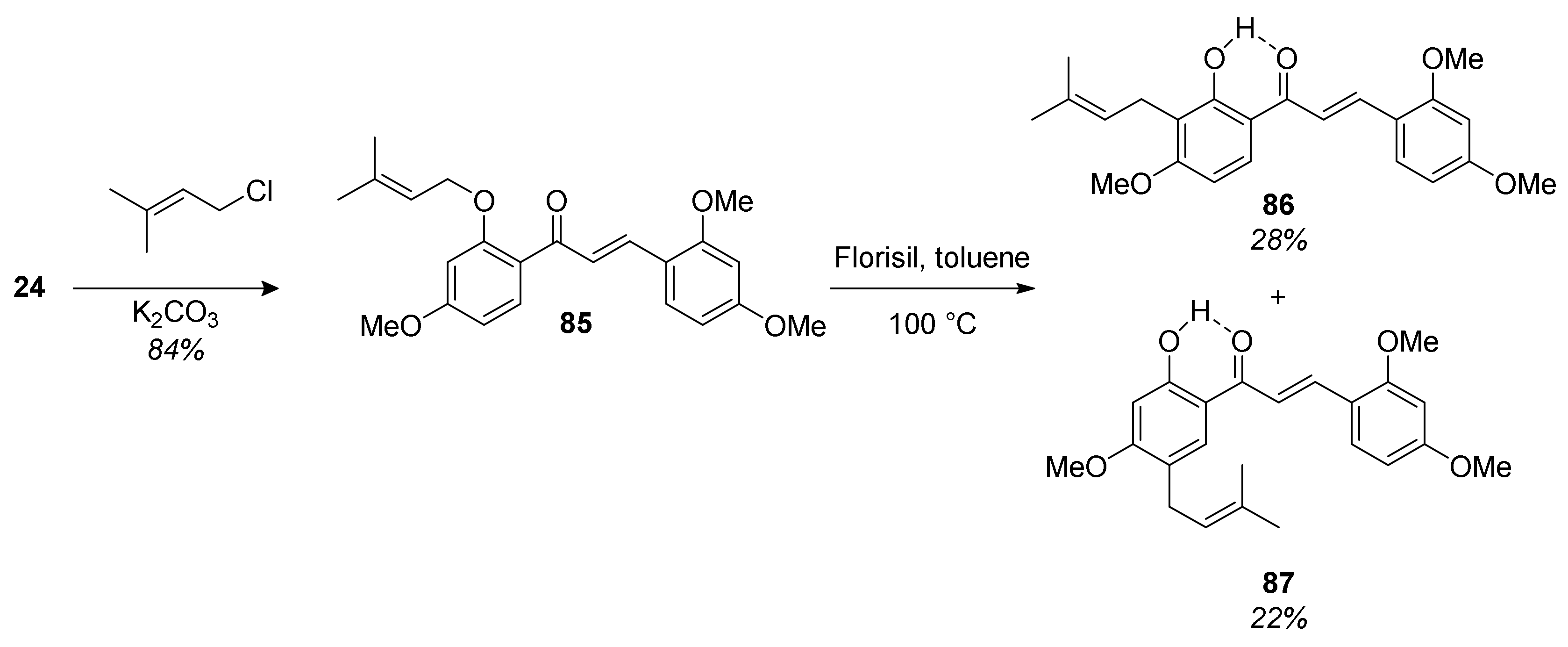
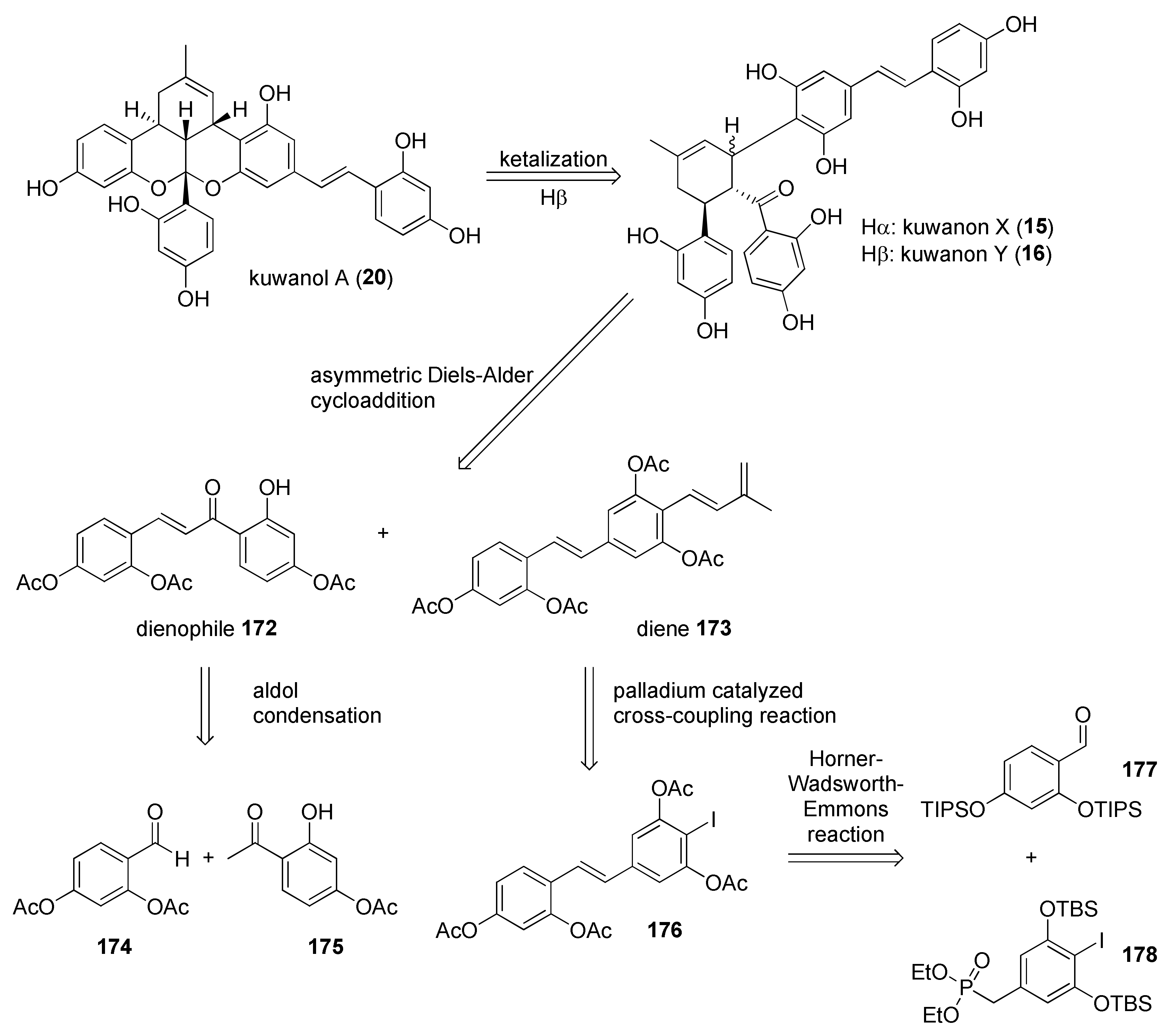
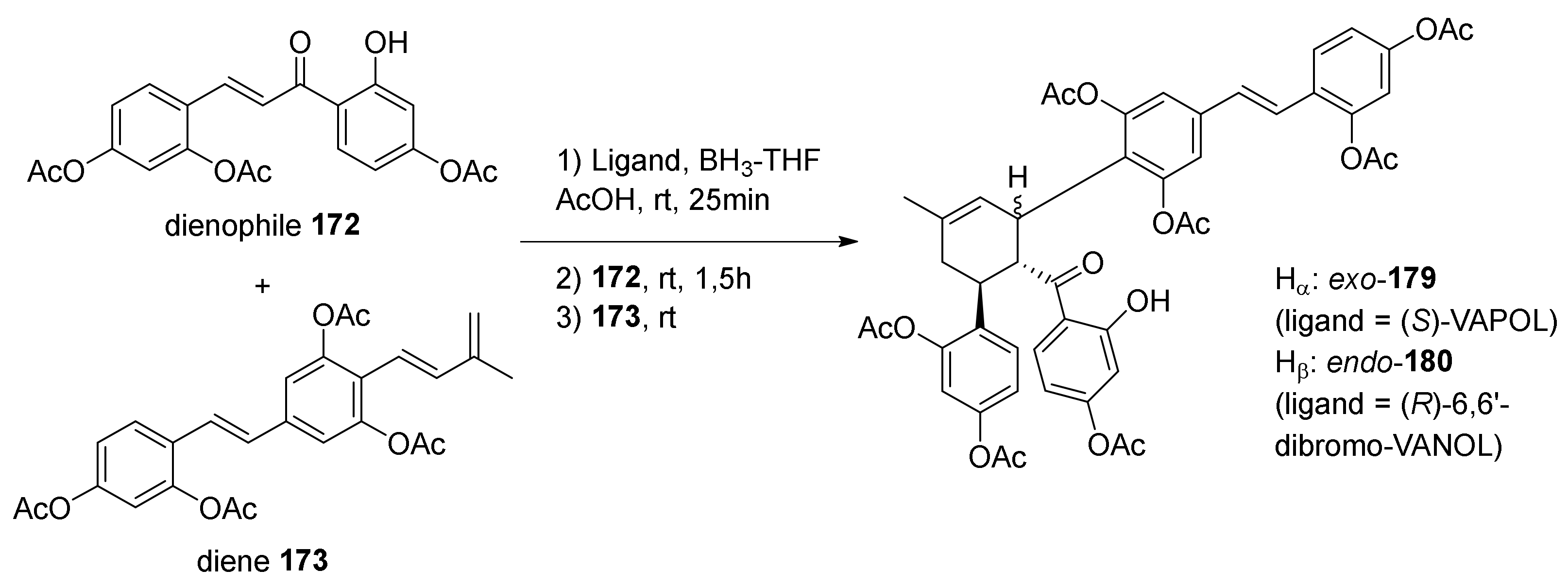
5.2.2. Kuwanon X, Kuwanon Y, and Kuwanol A
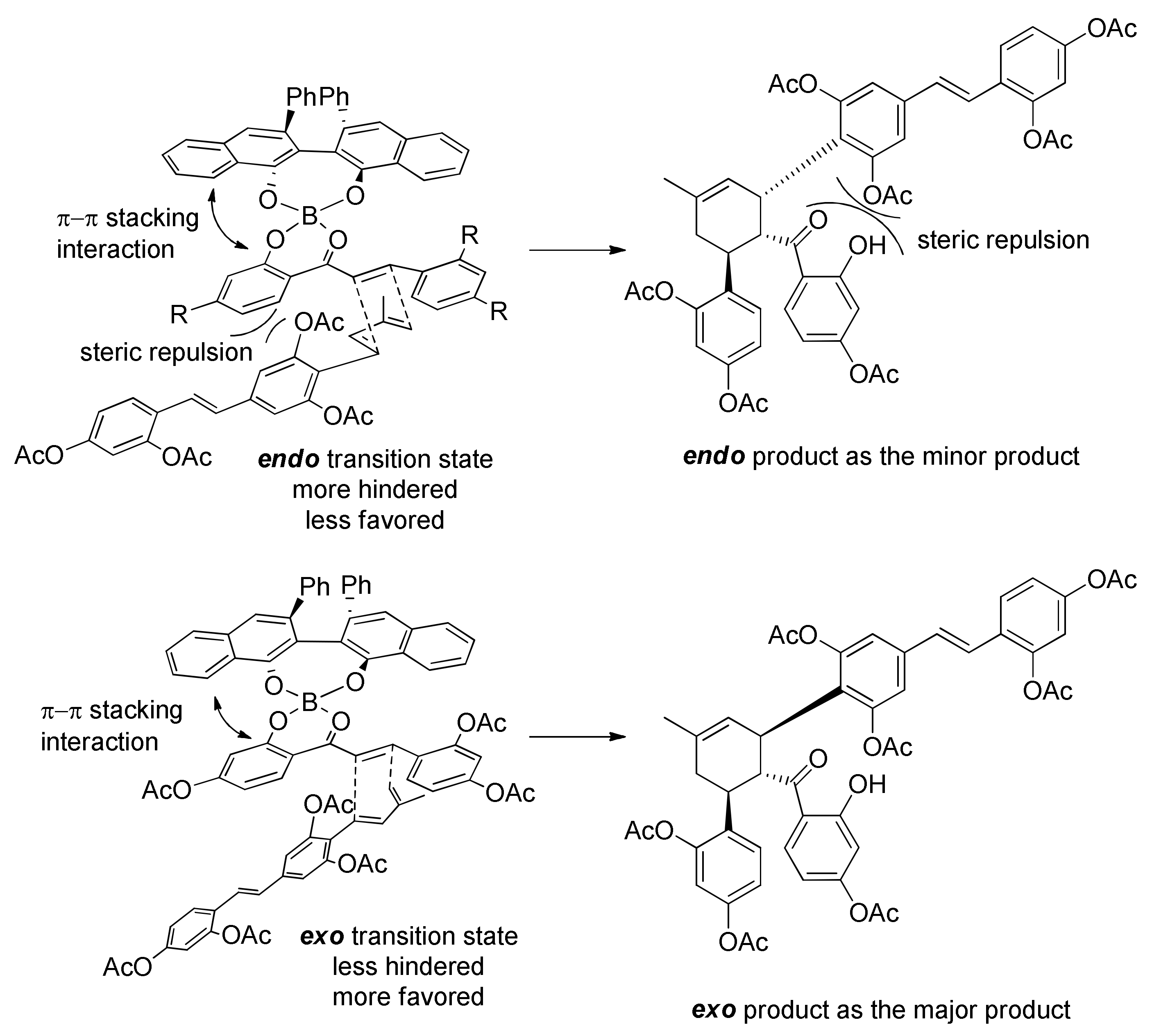
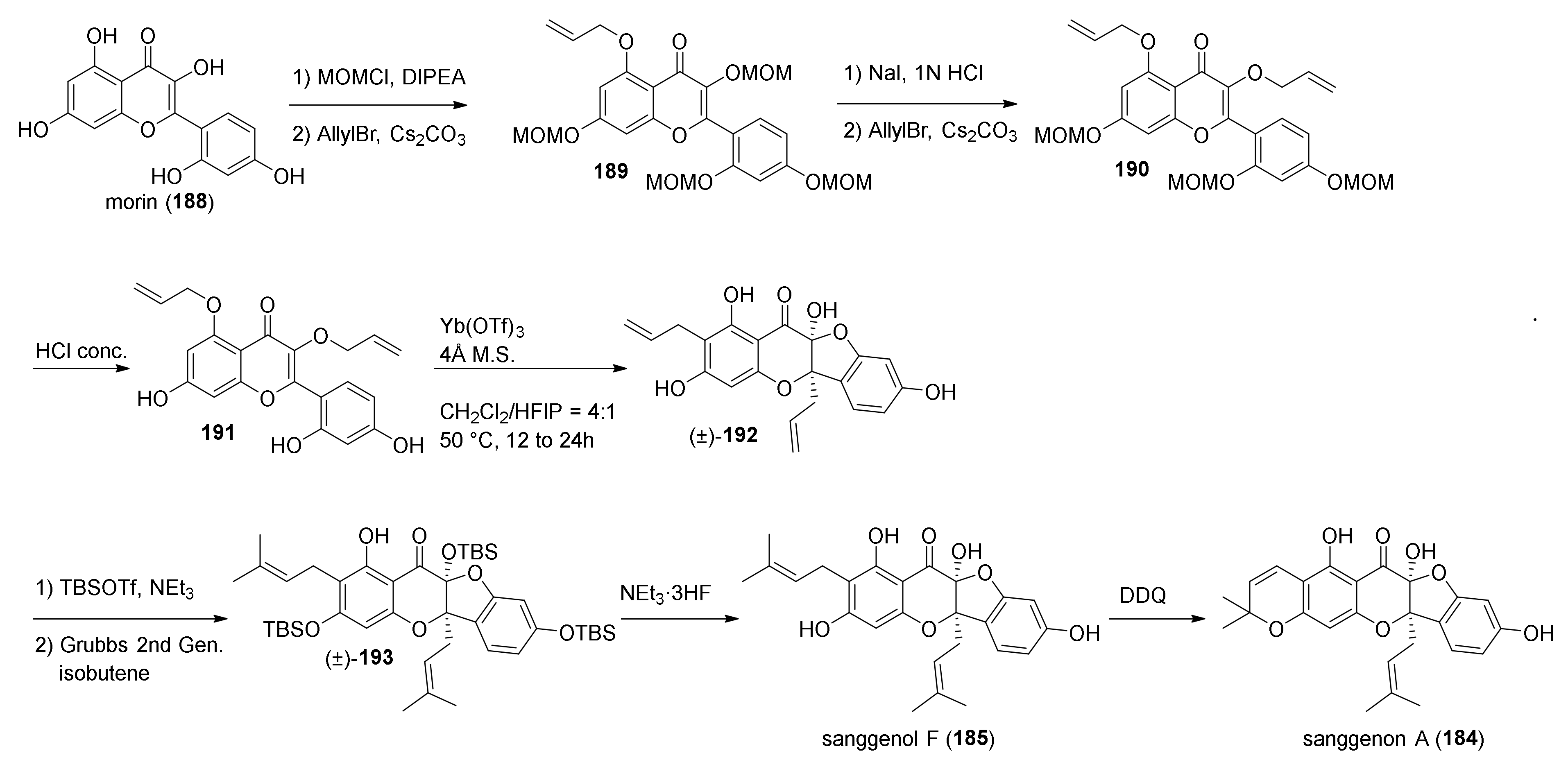
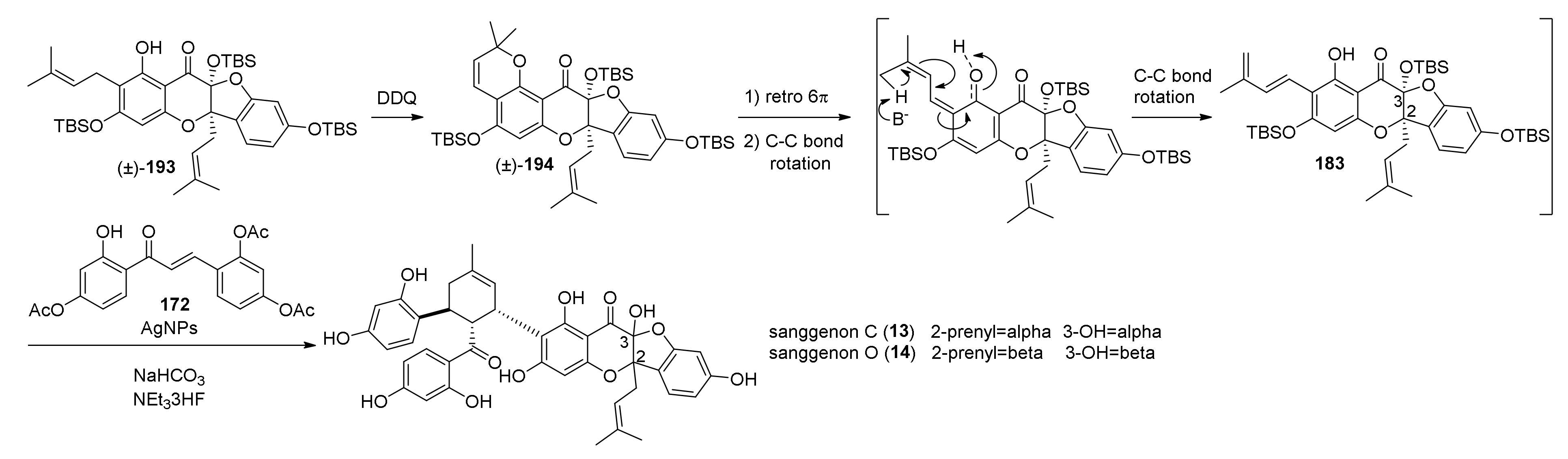
5.2.3. Sanggenons C and O
6. Biological Activity of Mulberry DAAs
6.1. Antioxidant Activity
6.2. Anti-Inflammatory Activity
6.3. Cytotoxic Activity
6.4. Antimicrobial Activity
6.5. Miscellaneous Biological Activities
7. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Nomura, T.; Hano, Y. Chemistry, Biosynthesis, and Biological Activity of Natural Diels-Alder Type Adducts from Moraceous Plants. In Plant Polyphenols 2: Chemistry, Biology, Pharmacology, Ecology; Gross, G.G., Hemingway, R.W., Yoshida, T., Branham, S.J., Eds.; Springer US: Boston, MA, USA, 1999; pp. 279–297. [Google Scholar]
- Yang, Y.; Tan, Y.-X.; Chen, R.-Y.; Kang, J. The latest review on the polyphenols and their bioactivities of Chinese Morus plants. J. Asian Nat. Prod. Res. 2014, 16, 690–702. [Google Scholar] [CrossRef] [PubMed]
- Nomura, T.; Hano, Y.; Yamanaka, J.; Momose, Y. Sorocenols A and B, Two New Isoprenylated Phenols from the Root Bark of Sorocea bonplandii Baillon. Heterocycles 1995, 41, 1035–1043. [Google Scholar] [CrossRef]
- Nomura, T.; Hano, Y.; Suzuko, S.; Kohno, H. Absolute Configuration of Kuwanon L, a Natural Diels-Alder Type Adduct from the Morus Root Bark. Heterocycles 1988, 27, 75–81. [Google Scholar] [CrossRef]
- Hano, Y.; Nomura, T.; Ueda, S. Biosynthesis of chalcomoracin and kuwanon J, the Diels-Alder type adducts, in Morus Alba L. cell coltures. Chem. Pharm. Bull. 1989, 37, 554–556. [Google Scholar] [CrossRef][Green Version]
- Gao, L.; Zou, Y.; Liu, X.; Yang, J.; Du, X.; Wang, J.; Yu, X.; Fan, J.; Jiang, M.; Li, Y.; et al. Enzymatic control of endo- and exo-stereoselective Diels–Alder reactions with broad substrate scope. Nat. Catal. 2021, 4, 1059–1069. [Google Scholar] [CrossRef]
- Gunawan, C.; Rizzacasa, M.A. Mulberry Diels−Alder Adducts: Synthesis of Chalcomoracin and Mulberrofuran C Methyl Ethers. Org. Lett. 2010, 12, 1388–1391. [Google Scholar] [CrossRef]
- Kelly, T.R.; Whiting, A.; Chandrakumar, N.S. Rationally designed, chiral Lewis acid for the asymmetric induction of some Diels-Alder reactions. J. Am. Chem. Soc. 1986, 108, 3510–3512. [Google Scholar] [CrossRef]
- Snyder, S.A.; Tang, Z.-Y.; Gupta, R. Enantioselective Total Synthesis of (−)-Napyradiomycin A1 via Asymmetric Chlorination of an Isolated Olefin. J. Am. Chem. Soc. 2009, 131, 5744–5745. [Google Scholar] [CrossRef]
- Han, J.; Li, X.; Guan, Y.; Zhao, W.; Wulff, W.D.; Lei, X. Enantioselective Biomimetic Total Syntheses of Kuwanons I and J and Brosimones A and B. Angew. Chem. Int. Ed. 2014, 53, 9257–9261. [Google Scholar] [CrossRef]
- Li, X.; Han, J.; Jones, A.X.; Lei, X. Chiral Boron Complex-Promoted Asymmetric Diels–Alder Cycloaddition and Its Application in Natural Product Synthesis. J. Org. Chem. 2016, 81, 458–468. [Google Scholar] [CrossRef]
- Gao, L.; Han, J.; Lei, X. Enantioselective Total Syntheses of Kuwanon X, Kuwanon Y, and Kuwanol A. Org. Lett. 2016, 18, 360–363. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Jones, A.X.; Lei, X. Recent Advances in the Total Synthesis of Prenylflavonoid and Related Diels–Alder Natural Products. Synthesis 2015, 47, 1519–1533. [Google Scholar] [CrossRef]
- Luo, S.-Y.; Zhu, J.-Y.; Zou, M.-F.; Yin, S.; Tang, G.-H. Mulberry Diels–Alder-type adducts: Isolation, structure, bioactivity, and synthesis. Nat. Prod. Bioprospect. 2022, 12, 31. [Google Scholar] [CrossRef]
- Wasserman, A. Diels Alder Reactions; Elsevier: Amsterdam, The Netherlands, 1965. [Google Scholar]
- Nicolaou, K.C.; Snyder, S.A.; Montagnon, T.; Vassilikogiannakis, G. The Diels–Alder Reaction in Total Synthesis. Angew. Chem. Int. Ed. 2002, 41, 1668–1698. [Google Scholar] [CrossRef]
- Butz, L.W.; Rytina, A.W. The Diels-Alder reaction. Quinones and other cyclenones. In Organic Reactions; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 1949; Volume 5, pp. 136–192. [Google Scholar]
- Kloetzel, M.C. The Diels-Alder reactions with maleic anhydride. In Organic Reactions; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 1948; Volume 4, pp. 1–59. [Google Scholar]
- Kraka, E.; Wu, A.; Cremer, D. Mechanism of the Diels−Alder Reaction Studied with the United Reaction Valley Approach: Mechanistic Differences between Symmetry-Allowed and Symmetry-Forbidden Reactions. J. Phys. Chem. A 2003, 107, 9008–9021. [Google Scholar] [CrossRef]
- Woodward, R.B.; Katz, T.J. The mechanism of the Diels-Alder reaction. Tetrahedron 1959, 5, 70–89. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Lim, D.; Blake, J.F. Ab initio study of Diels-Alder reactions of cyclopentadiene with ethylene, isoprene, cyclopentadiene, acrylonitrile, and methyl vinyl ketone. J. Am. Chem. Soc. 1993, 115, 2936–2942. [Google Scholar] [CrossRef]
- Kobuke, Y.; Sugimoto, T.; Furukawa, J.; Fueno, T. Role of attractive interactions in endo-exo stereoselectivities of Diels-Alder reactions. J. Am. Chem. Soc. 1972, 94, 3633–3635. [Google Scholar] [CrossRef]
- Williamson, K.L.; Hsu, Y.-F.L. Stereochemistry of the Diels-Alder reaction. II. Lewis acid catalysis of syn-anti isomerism. J. Am. Chem. Soc. 1970, 92, 7385–7389. [Google Scholar] [CrossRef]
- Berson, J.A.; Hamlet, Z.; Mueller, W.A. The Correlation of Solvent Effects on the Stereoselectivities of Diels-Alder Reactions by Means of Linear Free Energy Relationships. A New Empirical Measure of Solvent Polarity. J. Am. Chem. Soc. 1962, 84, 297–304. [Google Scholar] [CrossRef]
- Houk, K.N.; Luskus, L.J. Influence of steric interactions on endo stereoselectivity. J. Am. Chem. Soc. 1971, 93, 4606–4607. [Google Scholar] [CrossRef]
- Nomura, T.; Hano, Y. Isoprenoid-substituted phenolic compounds of moraceous plants. Nat. Prod. Rep. 1994, 11, 205–218. [Google Scholar] [CrossRef] [PubMed]
- Boonsri, S.; Gunawan, C.; Krenske, E.H.; Rizzacasa, M.A. Synthetic studies towards the mulberry Diels–Alder adducts: H-bond accelerated cycloadditions of chalcones. Org. Biomol. Chem. 2012, 10, 6010–6021. [Google Scholar] [CrossRef]
- Vermeeren, P.; Hamlin, T.A.; Fernández, I.; Bickelhaupt, F.M. How Lewis Acids Catalyze Diels–Alder Reactions. Angew. Chem. Int. Ed. 2020, 59, 6201–6206. [Google Scholar] [CrossRef] [PubMed]
- Vermeeren, P.; Tiezza, M.D.; van Dongen, M.; Fernández, I.; Bickelhaupt, F.M.; Hamlin, T.A. Lewis Acid-Catalyzed Diels-Alder Reactions: Reactivity Trends across the Periodic Table. Chem.-A Eur. J. 2021, 27, 10610–10620. [Google Scholar] [CrossRef] [PubMed]
- Bañuelos, P.; García, J.M.; Gómez-Bengoa, E.; Herrero, A.; Odriozola, J.M.; Oiarbide, M.; Palomo, C.; Razkin, J. (1R)-(+)-Camphor and Acetone Derived α′-Hydroxy Enones in Asymmetric Diels−Alder Reaction: Catalytic Activation by Lewis and Brønsted Acids, Substrate Scope, Applications in Syntheses, and Mechanistic Studies. J. Org. Chem. 2010, 75, 1458–1473. [Google Scholar] [CrossRef]
- Ikuta, J.; Fukai, T.; Nomura, T.; Ueda, S. Constituents of Morus alba L. Cell Cultures. (1).: Structures of Four New Natural Diels-Alder Type Adducts, Kuwanons J, Q, R, and V. Chem. Pharm. Bull. 1986, 34, 2471–2478. [Google Scholar]
- Mitsuo, T.; Shigemitsu, N.; Shoji, U.; Tadashi, M.; Akira, S.; Kokichi, T. Moracin C and D, new phytoalexins from diseased mulberry. Chem. Lett. 1978, 7, 1239–1240. [Google Scholar]
- Mitsuo, T.; Shigemitsu, N.; Tadashi, M.; Akira, S.; Kokichi, T. Chalcomoracin a natural Diels-Alder adduct from diseased Mulberry. Chem. Lett. 1980, 9, 1573–1576. [Google Scholar]
- Ueda, S.-i.; Matsumoto, J.; Nomura, T. Four new natural Diels―Alder type adducts, mulberrofuran E, kuwanon Q, R, and V from callus culture of Morus alba L. Chem. Pharm. Bull. 1984, 32, 350–353. [Google Scholar] [CrossRef]
- Nomura, T. Phenolic compounds of the mulberry tree and related plants. Fortschr. Chem. Org. Nat. 1988, 53, 87–201. [Google Scholar]
- Nomura, T.; Hano, Y.; Suzuki, S.; Iitaka, Y. Absolute Configuration of Natural Diels-Alder Type Adducts from the Morus Root Bark. Heterocycles 1988, 27, 2315–2325. [Google Scholar] [CrossRef]
- Nomura, T.; Hano, Y.; Ueda, S. Chemistry and Biosynthesis of Natural Diels-Alder Type Adducts from Moraceous Plants. In Studies in Natural Products Chemistry; Atta Ur, R., Ed.; Elsevier: Amsterdam, The Netherlands, 1995; Volume 17, pp. 451–478. [Google Scholar]
- Hano, Y.; Nomura, T.; Ueda, S. Biosynthesis of optically active Diels–Alder type adducts revealed by an aberrant metabolism of O-methylated precursors in Morus alba cell cultures. J. Chem. Soc. Chem. Commun. 1990, 8, 610–613. [Google Scholar] [CrossRef]
- Hano, Y.; Aida, M.; Nomura, T.; Ueda, S. A novel way of determining the structure of artonin I, an optically active Diels–Alder type adduct, with the aid of an enzyme system of Morus alba cell cultures. J. Chem. Soc. Chem. Commun. 1992, 17, 1177–1178. [Google Scholar] [CrossRef]
- Vitali, A.; Giardina, B.; Delle Monache, G.; Rocca, F.; Silvestrini, A.; Tafi, A.; Botta, B. Chalcone dimethylallyltransferase from Morus nigra cell cultures. Substrate specificity studies. FEBS Lett. 2004, 557, 33–38. [Google Scholar] [CrossRef]
- Wang, R.; Chen, R.; Li, J.; Liu, X.; Xie, K.; Chen, D.; Yin, Y.; Tao, X.; Xie, D.; Zou, J.; et al. Molecular characterization and phylogenetic analysis of two novel regio-specific flavonoid prenyltransferases from Morus alba and Cudrania tricuspidata. J. Biol. Chem. 2014, 289, 35815–35825. [Google Scholar] [CrossRef]
- De Bruijn, W.J.C.; Levisson, M.; Beekwilder, J.; van Berkel, W.J.H.; Vincken, J.-P. Plant Aromatic Prenyltransferases: Tools for Microbial Cell Factories. Trends Biotechnol. 2020, 38, 917–934. [Google Scholar] [CrossRef]
- Gao, L.; Su, C.; Du, X.; Wang, R.; Chen, S.; Zhou, Y.; Liu, C.; Liu, X.; Tian, R.; Zhang, L.; et al. FAD-dependent enzyme-catalysed intermolecular [4+2] cycloaddition in natural product biosynthesis. Nat. Chem. 2020, 12, 620–628. [Google Scholar] [CrossRef]
- Chakrabarty, S.; Romero, E.O.; Pyser, J.B.; Yazarians, J.A.; Narayan, A.R.H. Chemoenzymatic Total Synthesis of Natural Products. Acc. Chem. Res. 2021, 54, 1374–1384. [Google Scholar] [CrossRef]
- Gao, L.; Yang, J.; Lei, X. Enzymatic intermolecular Diels-Alder reactions in synthesis: From nature to design. Tetrahedron Chem 2022, 2, 100013. [Google Scholar] [CrossRef]
- Li, J.; Amatuni, A.; Renata, H. Recent advances in the chemoenzymatic synthesis of bioactive natural products. Curr. Opin. Chem. Biol. 2020, 55, 111–118. [Google Scholar] [CrossRef] [PubMed]
- Moore, B.S.; Gulder, T.A.M. Enzymes in natural product total synthesis. Nat. Prod. Rep. 2020, 37, 1292–1293. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Yang, J.; Gao, L.; Zhang, L.; Lei, X. Chemoenzymatic Total Syntheses of Artonin I with an Intermolecular Diels–Alderase. Biotechnol. J. 2020, 15, 2000119. [Google Scholar] [CrossRef] [PubMed]
- Dai, S.-J.; Wu, Y.; Wang, Y.-H.; He, W.-Y.; Chen, R.-Y.; Yu, D.-Q. New Diels-Alder type adducts from Morus macroura and their anti-oxidant activities. Chem. Pharm. Bull. 2004, 52, 1190–1193. [Google Scholar] [CrossRef]
- Esposito, F.; Tintori, C.; Martini, R.; Christ, F.; Debyser, Z.; Ferrarese, R.; Cabiddu, G.; Corona, A.; Ceresola, E.R.; Calcaterra, A.; et al. Kuwanon-L as a New Allosteric HIV-1 Integrase Inhibitor: Molecular Modeling and Biological Evaluation. ChemBioChem 2015, 16, 2507–2512. [Google Scholar] [CrossRef]
- Fukai, T.; Kaitou, K.; Terada, S. Antimicrobial activity of 2-arylbenzofurans from Morus species against methicillin-resistant Staphylococcus aureus. Fitoterapia 2005, 76, 708–711. [Google Scholar] [CrossRef]
- Martini, R.; Esposito, F.; Corona, A.; Ferrarese, R.; Ceresola, E.R.; Visconti, L.; Tintori, C.; Barbieri, A.; Calcaterra, A.; Iovine, V.; et al. Natural Product Kuwanon-L Inhibits HIV-1 Replication through Multiple Target Binding. ChemBioChem 2017, 18, 374–377. [Google Scholar] [CrossRef]
- Mascarello, A.; Mori, M.; Chiaradia-Delatorre, L.D.; Menegatti, A.C.O.; Monache, F.D.; Ferrari, F.; Yunes, R.A.; Nunes, R.J.; Terenzi, H.; Botta, B.; et al. Discovery of Mycobacterium tuberculosis Protein Tyrosine Phosphatase B (PtpB) Inhibitors from Natural Products. PLoS ONE 2013, 8, e77081. [Google Scholar] [CrossRef]
- Mascarello, A.; Orbem Menegatti, A.C.; Calcaterra, A.; Martins, P.G.A.; Chiaradia-Delatorre, L.D.; D’Acquarica, I.; Ferrari, F.; Pau, V.; Sanna, A.; De Logu, A.; et al. Naturally occurring Diels-Alder-type adducts from Morus nigra as potent inhibitors of Mycobacterium tuberculosis protein tyrosine phosphatase B. Eur. J. Med. Chem. 2018, 144, 277–288. [Google Scholar] [CrossRef]
- Phung, T.X.B.; Tran, T.H.H.; Dan, T.T.H.; Chau, V.M.; Hoang, T.H.; Nguyen, T.D. Chalcone-derived Diels–Alder adducts as NF-κB inhibitors from Morus alba. J. Asian Nat. Prod. Res. 2012, 14, 596–600. [Google Scholar] [CrossRef]
- Zhang, Q.-J.; Tang, Y.-B.; Chen, R.-Y.; Yu, D.-Q. Three New Cytotoxic Diels–Alder-Type Adducts from Morus australis. Chem. Biodivers. 2007, 4, 1533–1540. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Z.-P.; Cheng, K.-W.; Zhu, Q.; Wang, X.-C.; Lin, Z.-X.; Wang, M. Tyrosinase Inhibitory Constituents from the Roots of Morus nigra: A Structure−Activity Relationship Study. J. Agric. Food Chem. 2010, 58, 5368–5373. [Google Scholar] [CrossRef] [PubMed]
- Hideaki, O. Involvement of the Diels–Alderases in the Biosynthesis of Natural Products. Bull. Chem. Soc. Jpn. 2005, 78, 537–554. [Google Scholar]
- Stocking, E.M.; Williams, R.M. Chemistry and Biology of Biosynthetic Diels–Alder Reactions. Angew. Chem. Int. Ed. 2003, 42, 3078–3115. [Google Scholar] [CrossRef]
- Miyaura, N.; Suzuki, A. Palladium-Catalyzed Cross-Coupling Reactions of Organoboron Compounds. Chem. Rev. 1995, 95, 2457–2483. [Google Scholar] [CrossRef]
- Sonogashira, K.; Tohda, Y.; Hagihara, N. A convenient synthesis of acetylenes: Catalytic substitutions of acetylenic hydrogen with bromoalkenes, iodoarenes and bromopyridines. Tetrahedron Lett. 1975, 16, 4467–4470. [Google Scholar] [CrossRef]
- Müller, S.; Liepold, B.; Roth, G.J.; Bestmann, H.J. An Improved One-pot Procedure for the Synthesis of Alkynes from Aldehydes. Synlett 1996, 1996, 521–522. [Google Scholar] [CrossRef]
- Rathwell, D.C.K.; Yang, S.-H.; Tsang, K.Y.; Brimble, M.A. An Efficient Formal Synthesis of the Human Telomerase Inhibitor (±)-γ-Rubromycin. Angew. Chem. Int. Ed. 2009, 48, 7996–8000. [Google Scholar] [CrossRef]
- Hiroya, K.; Suzuki, N.; Yasuhara, A.; Egawa, Y.; Kasano, A.; Sakamoto, T. Total syntheses of three natural products, vignafuran, 2-(4-hydroxy-2-methoxyphenyl)-6-methoxybenzofuran-3-carboxylic acid methyl ester, and coumestrol from a common starting material. J. Chem. Soc. Perkin Trans. 2000, 1, 4339–4346. [Google Scholar] [CrossRef]
- Vaultier, M.; Truchet, F.; Carboni, B.; Hoffmann, R.W.; Denne, I. Diels-Alder reactions of 1,3-dienylboronates as a new route to functionalized carbocycles. Tetrahedron Lett. 1987, 28, 4169–4172. [Google Scholar] [CrossRef]
- Tucker, C.E.; Davidson, J.; Knochel, P. Mild and stereoselective hydroborations of functionalized alkynes and alkenes using pinacolborane. J. Org. Chem. 1992, 57, 3482–3485. [Google Scholar] [CrossRef]
- Talamás, F.X.; Smith, D.B.; Cervantes, A.; Franco, F.; Cutler, S.T.; Loughhead, D.G.; Morgans, D.J.; Weikert, R.J. The Florisil® catalyzed [1,3]-sigmatropic shift of allyl phenyl ethers—An entryway into novel mycophenolic acid analogues. Tetrahedron Lett. 1997, 38, 4725–4728. [Google Scholar] [CrossRef]
- Kang, J.; Chen, R.-Y.; Yu, D.-Q. Five New Diels-Alder Type Adducts from the Stem and Root Bark of Morus mongolica. Planta Med. 2006, 72, 52–59. [Google Scholar] [CrossRef] [PubMed]
- Chee, C.F.; Lee, Y.K.; Buckle, M.J.C.; Rahman, N.A. Synthesis of (±)-kuwanon V and (±)-dorsterone methyl ethers via Diels–Alder reaction. Tetrahedron Lett. 2011, 52, 1797–1799. [Google Scholar] [CrossRef]
- Hano, Y.; Fukai, T.; Nomura, T.; Uzawa, J.; Fukushima, K. Structure of mulberrofuran I, a novel 2-arylbenzofuran derivative from the cultivated mulberry tree (Morus bombycis Koidz.). Chem. Pharm. Bull. 1984, 32, 1260–1263. [Google Scholar] [CrossRef][Green Version]
- Cong, H.; Porco, J.A. Total Synthesis of (±)-Sorocenol B Employing Nanoparticle Catalysis. Org. Lett. 2012, 14, 2516–2519. [Google Scholar] [CrossRef]
- Cong, H.; Becker, C.F.; Elliott, S.J.; Grinstaff, M.W.; Porco, J.A. Silver Nanoparticle-Catalyzed Diels−Alder Cycloadditions of 2′-Hydroxychalcones. J. Am. Chem. Soc. 2010, 132, 7514–7518. [Google Scholar] [CrossRef]
- Cong, H.; Ledbetter, D.; Rowe, G.T.; Caradonna, J.P.; Porco, J.A. Electron Transfer-Initiated Diels−Alder Cycloadditions of 2′-Hydroxychalcones. J. Am. Chem. Soc. 2008, 130, 9214–9215. [Google Scholar] [CrossRef]
- Ballerini, E.; Minuti, L.; Piermatti, O.; Pizzo, F. High Pressure Diels−Alder Approach to Hydroxy-Substituted 6a-Cyano-tetrahydro-6H-benzo[c]chromen-6-ones: A Route to Δ6-Cis-Cannabidiol. J. Org. Chem. 2009, 74, 4311–4317. [Google Scholar] [CrossRef]
- Floreancig, P.E. Development and Applications of Electron-Transfer-Initiated Cyclization Reactions. Synlett 2007, 2007, 191–203. [Google Scholar] [CrossRef]
- Li, C.-J. Cross-Dehydrogenative Coupling (CDC): Exploring C−C Bond Formations beyond Functional Group Transformations. Acc. Chem. Res. 2009, 42, 335–344. [Google Scholar] [CrossRef] [PubMed]
- Porco, J.A.; Schoenen, F.J.; Stout, T.J.; Clardy, J.; Schreiber, S.L. Transannular Diels-Alder route to systems related to dynemicin A. J. Am. Chem. Soc. 1990, 112, 7410–7411. [Google Scholar] [CrossRef]
- Wang, D.H.; Engle, K.M.; Shi, B.F.; Yu, J.Q. Ligand-enabled reactivity and selectivity in a synthetically versatile aryl C-H olefination. Science 2010, 327, 315–319. [Google Scholar] [CrossRef] [PubMed]
- Stoltz, B.M. Palladium Catalyzed Aerobic Dehydrogenation: From Alcohols to Indoles and Asymmetric Catalysis. Chem. Lett. 2004, 33, 362–367. [Google Scholar] [CrossRef]
- Trend, R.M.; Ramtohul, Y.K.; Ferreira, E.M.; Stoltz, B.M. Palladium-Catalyzed Oxidative Wacker Cyclizations in Nonpolar Organic Solvents with Molecular Oxygen: A Stepping Stone to Asymmetric Aerobic Cyclizations. Angew. Chem. Int. Ed. 2003, 42, 2892–2895. [Google Scholar] [CrossRef]
- Trend, R.M.; Ramtohul, Y.K.; Stoltz, B.M. Oxidative Cyclizations in a Nonpolar Solvent Using Molecular Oxygen and Studies on the Stereochemistry of Oxypalladation. J. Am. Chem. Soc. 2005, 127, 17778–17788. [Google Scholar] [CrossRef]
- Qi, C.; Cong, H.; Cahill, K.J.; Müller, P.; Johnson, R.P.; Porco, J.A., Jr. Biomimetic Dehydrogenative Diels–Alder Cycloadditions: Total Syntheses of Brosimones A and B. Angew. Chem. Int. Ed. 2013, 52, 8345–8348. [Google Scholar] [CrossRef]
- Cong, H.; Porco, J.A. Chemical Synthesis of Complex Molecules Using Nanoparticle Catalysis. ACS Catal. 2012, 2, 65–70. [Google Scholar] [CrossRef]
- Chen, H.-D.; Ding, Y.-Q.; Yang, S.-P.; Li, X.-C.; Wang, X.-J.; Zhang, H.-Y.; Ferreira, D.; Yue, J.-M. Morusalbanol A, a neuro-protective Diels–Alder adduct with an unprecedented architecture from Morus alba. Tetrahedron 2012, 68, 6054–6058. [Google Scholar] [CrossRef]
- Tee, J.T.; Chee, C.F.; Buckle, M.J.C.; Lee, V.S.; Chong, W.L.; Khaledi, H.; Rahman, N.A. Model studies on construction of the oxabicyclic [3.3.1] core of the mulberry Diels–Alder adducts morusalbanol A and 441772-64-1. Tetrahedron Lett. 2015, 56, 5082–5085. [Google Scholar] [CrossRef]
- Wang, H.; Yan, Z.; Lei, Y.; Sheng, K.; Yao, Q.; Lu, K.; Yu, P. Concise synthesis of prenylated and geranylated chalcone natural products by regiospecific iodination and Suzuki coupling reactions. Tetrahedron Lett. 2014, 55, 897–899. [Google Scholar] [CrossRef]
- Arkoudis, E.; Lykakis, I.N.; Gryparis, C.; Stratakis, M. Biomimetic Synthesis of Dimeric Metabolite Acremine G via a Highly Regioselective and Stereoselective Diels−Alder Reaction. Org. Lett. 2009, 11, 2988–2991. [Google Scholar] [CrossRef] [PubMed]
- Iovine, V.; Benni, I.; Sabia, R.; D’Acquarica, I.; Fabrizi, G.; Botta, B.; Calcaterra, A. Total Synthesis of (±)-Kuwanol E. J. Nat. Prod. 2016, 79, 2495–2503. [Google Scholar] [CrossRef] [PubMed]
- Sugamoto, K.; Kurogi, C.; Matsushita, Y.-i.; Matsui, T. Synthesis of 4-hydroxyderricin and related derivatives. Tetrahedron Lett. 2008, 49, 6639–6641. [Google Scholar] [CrossRef]
- Yamashita, K.-I.; Tsuboi, M.; Asano, M.S.; Sugiura, K.-I. Facile Aromatic Finkelstein Iodination (AFI) Reaction in 1,3-Dimethyl-2-imidazolidinone (DMI). Synth. Commun. 2012, 42, 170–175. [Google Scholar] [CrossRef]
- Kim, S.; Ko, H.; Park, J.E.; Jung, S.; Lee, S.K.; Chun, Y.-J. Design, Synthesis, and Discovery of Novel trans-Stilbene Analogues as Potent and Selective Human Cytochrome P450 1B1 Inhibitors. J. Med. Chem. 2002, 45, 160–164. [Google Scholar] [CrossRef]
- Nicolaou, K.C.; Kang, Q.; Wu, T.R.; Lim, C.S.; Chen, D.Y.K. Total Synthesis and Biological Evaluation of the Resveratrol-Derived Polyphenol Natural Products Hopeanol and Hopeahainol A. J. Am. Chem. Soc. 2010, 132, 7540–7548. [Google Scholar] [CrossRef]
- Pereira, S.; Srebnik, M. Hydroboration of Alkynes with Pinacolborane Catalyzed by HZrCp2Cl. Organometallics 1995, 14, 3127–3128. [Google Scholar] [CrossRef]
- Hano, Y.; Nomura, T.; Ueda, S. Two New Diels-Alder Type Adducts, Mulberrofuran T and Kuwanol E, from Callus Tissues of Morus alba L. Heterocycles 1989, 29, 2035–2041. [Google Scholar]
- Luo, S.-Y.; Tang, Z.-Y.; Li, Q.; Weng, J.; Yin, S.; Tang, G.-H. Total Synthesis of Mulberry Diels–Alder-Type Adducts Kuwanons G and H. J. Org. Chem. 2021, 86, 4786–4793. [Google Scholar] [CrossRef]
- Jun, N.; Hong, G.; Jun, K. Synthesis and evaluation of 2′,4′,6′-trihydroxychalcones as a new class of tyrosinase inhibitors. Bioorg. Med. Chem. 2007, 15, 2396–2402. [Google Scholar] [CrossRef] [PubMed]
- Lei, X.; Porco, J.A. Synthesis of a Polymer-Supported Anthracene and Its Application as a Dienophile Scavenger. Org. Lett. 2004, 6, 795–798. [Google Scholar] [CrossRef] [PubMed]
- Qi, C.; Xiong, Y.; Eschenbrenner-Lux, V.; Cong, H.; Porco, J.A. Asymmetric Syntheses of the Flavonoid Diels–Alder Natural Products Sanggenons C and O. J. Am. Chem. Soc. 2016, 138, 798–801. [Google Scholar] [CrossRef] [PubMed]
- Dai, S.-J.; Mi, Z.-M.; Ma, Z.-B.; Li, S.; Chen, R.-Y.; Yu, D.-Q. Bioactive Diels-Alder Type Adducts from the Stem Bark of Morus macroura. Planta Med. 2004, 70, 758–763. [Google Scholar] [CrossRef] [PubMed]
- Sharma, R.; Sharma, A.; Shono, T.; Takasugi, M.; Shirata, A.; Fujimura, T.; Machii, H. Mulberry Moracins: Scavengers of UV Stress-generated Free Radicals. Biosci. Biotechnol. Biochem. 2001, 65, 1402–1405. [Google Scholar] [CrossRef]
- Kimura, Y.; Okuda, H.; Nomura, T.; Fukai, T.; Arichi, S. Effects of Phenolic Constituents from the Mulberry Tree on Arachidonate Metabolism in Rat Platelets. J. Nat. Prod. 1986, 49, 639–644. [Google Scholar] [CrossRef]
- Rollinger, J.M.; Bodensieck, A.; Seger, C.; Ellmerer, E.P.; Bauer, R.; Langer, T.; Stuppner, H. Discovering COX-Inhibiting Constituents of Morus Root Bark: Activity-Guided versus Computer-Aided Methods. Planta Med. 2005, 71, 399–405. [Google Scholar] [CrossRef]
- Li, L.-C.; Shen, F.; Hou, Q.; Cheng, G.-F. Inhibitory effect and mechanism of action of sanggenon C on human polymorphonuclear leukocyte adhesion to human synovial cells. Acta Pharmacol. Sin. 2002, 23, 138–142. [Google Scholar]
- Lim, H.J.; Jin, H.-G.; Woo, E.-R.; Lee, S.K.; Kim, H.P. The root barks of Morus alba and the flavonoid constituents inhibit airway inflammation. J. Ethnopharmacol. 2013, 149, 169–175. [Google Scholar] [CrossRef]
- Shi, Y.-Q.; Fukai, T.; Sakagami, H.; Chang, W.-J.; Yang, P.-Q.; Wang, F.-P.; Nomura, T. Cytotoxic Flavonoids with Isoprenoid Groups from Morus mongolica. J. Nat. Prod. 2001, 64, 181–188. [Google Scholar] [CrossRef]
- Wei, H.; Zhu, J.-J.; Liu, X.-Q.; Feng, W.-H.; Wang, Z.-M.; Yan, L.-H. Review of bioactive compounds from root barks of Morus plants (Sang-Bai-Pi) and their pharmacological effects. Cogent Chem. 2016, 2, 1212320. [Google Scholar] [CrossRef]
- Huang, H.; Liu, N.; Zhao, K.; Zhu, C.; Lu, X.; Li, S.; Lian, W.; Zhou, P.; Dong, X.; Zhao, C.; et al. Sanggenon C decreases tumor cell viability associated with proteasome inhibition. Front. Biosci. 2011, 3, 1315–1325. [Google Scholar] [CrossRef]
- Dat, N.T.; Jin, X.; Lee, K.; Hong, Y.-S.; Kim, Y.H.; Lee, J.J. Hypoxia-Inducible Factor-1 Inhibitory Benzofurans and Chalcone-Derived Diels−Alder Adducts from Morus Species. J. Nat. Prod. 2009, 72, 39–43. [Google Scholar] [CrossRef]
- Zhou, P.; Dong, X.-X.; Tang, P. Sanggenon C induces apoptosis of prostate cancer PC3 cells by activating caspase 3 and caspase 9 pathways. J. South. Med. Univ. 2017, 37, 1206–1210. [Google Scholar]
- Park, K.M.; You, J.S.; Lee, H.Y.; Baek, N.I.; Hwang, J.K. Kuwanon G: An antibacterial agent from the root bark of Morus alba against oral pathogens. J. Ethnopharmacol. 2003, 84, 181–185. [Google Scholar] [CrossRef]
- Liang, J.-H.; Fu, Y.-W.; Zhang, Q.-Z.; Xu, D.-H.; Wang, B.; Lin, D.-J. Identification and Effect of Two Flavonoids from Root Bark of Morus alba against Ichthyophthirius multifiliis in Grass Carp. J. Agric. Food Chem. 2015, 63, 1452–1459. [Google Scholar] [CrossRef] [PubMed]
- Rollinger, J.M.; Spitaler, R.; Menz, M.; Marschall, K.; Zelger, R.; Ellmerer, E.P.; Schneider, P.; Stuppner, H. Venturia inaequalis-Inhibiting Diels−Alder Adducts from Morus Root Bark. J. Agric. Food Chem. 2006, 54, 8432–8436. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Feng, T.; Guo, D.; Zhang, M.; Chen, L.; Zhou, Y. Sanggenon C Stimulates Osteoblastic Proliferation and Differentiation, Inhibits Osteoclastic Resorption, and Ameliorates Prednisone-Induced Osteoporosis in Zebrafish Model. Molecules 2018, 23, 2343. [Google Scholar] [CrossRef]
- Gu, Y.; Gao, L.; Chen, Y.; Xu, Z.; Yu, K.; Zhang, D.; Zhang, G.; Zhang, X. Sanggenon C protects against cardiomyocyte hypoxia injury by increasing autophagy. Mol. Med. Rep. 2017, 16, 8130–8136. [Google Scholar] [CrossRef]
- Xiao, L.; Gu, Y.; Gao, L.; Shangguan, J.; Chen, Y.; Zhang, Y.; Li, L. Sanggenon C protects against pressure overload-induced cardiac hypertrophy via the calcineurin/NFAT2 pathway. Mol. Med. Rep. 2017, 16, 5338–5346. [Google Scholar] [CrossRef]
- Nomura, T.; Fukai, T.; Matsumoto, J.; Fukushima, K.; Momose, Y. Structure of Mulberrofuran C, a Natural Hypotensive Diels-Alder Adduct from Root Barks of the Cultivated Mulberry Tree (Morus bombycis Koidzumi). Heterocycles 1981, 16, 759–765. [Google Scholar] [CrossRef]
- Nomura, T.; Fukai, T. Kuwanon G, a New Flavone Derivative from the Root Barks of the Cultivated Mulberry Tree (Morus alba L.). Chem. Pharm. Bull. 1980, 28, 2548–2552. [Google Scholar] [CrossRef]
- Xia, C.-L.; Tang, G.-H.; Guo, Y.-Q.; Xu, Y.-K.; Huang, Z.-S.; Yin, S. Mulberry Diels-Alder-type adducts from Morus alba as multi-targeted agents for Alzheimer’s disease. Phytochemistry 2019, 157, 82–91. [Google Scholar] [CrossRef] [PubMed]
- Shi-De, L.; Nemec, J.; Bing-Mei, N. Anti-HIV flavonoids from Morus alba. Plant Divers. 1995, 17, 89–95. [Google Scholar]



































































{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| DDAs Isolated from M. alba Cell Cultures | Prenylchalcone Precursor | Dehydroprenylphenol Precursor |
|---|---|---|
| kuwanon J | morachalcone A | dehydroprenylmorachalcone A |
| kuwanon Q | isobavachalcone | dehydroprenylmorachalcone A |
| kuwanon R | morachalcone A | dehydroprenylisobavachalcone |
| kuwanon V | isobavachalcone | dehydroprenylisobavachalcone |
| chalcomoracin | morachalcone A | dehydroprenylmoracin C |
| mulberrofuran E | isobavachalcone | dehydroprenylmoracin C |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tortora, C.; Pisano, L.; Vergine, V.; Ghirga, F.; Iazzetti, A.; Calcaterra, A.; Marković, V.; Botta, B.; Quaglio, D. Synthesis, Biosynthesis, and Biological Activity of Diels–Alder Adducts from Morus Genus: An Update. Molecules 2022, 27, 7580. https://doi.org/10.3390/molecules27217580
Tortora C, Pisano L, Vergine V, Ghirga F, Iazzetti A, Calcaterra A, Marković V, Botta B, Quaglio D. Synthesis, Biosynthesis, and Biological Activity of Diels–Alder Adducts from Morus Genus: An Update. Molecules. 2022; 27(21):7580. https://doi.org/10.3390/molecules27217580
Chicago/Turabian StyleTortora, Carola, Luca Pisano, Valeria Vergine, Francesca Ghirga, Antonia Iazzetti, Andrea Calcaterra, Violeta Marković, Bruno Botta, and Deborah Quaglio. 2022. "Synthesis, Biosynthesis, and Biological Activity of Diels–Alder Adducts from Morus Genus: An Update" Molecules 27, no. 21: 7580. https://doi.org/10.3390/molecules27217580
APA StyleTortora, C., Pisano, L., Vergine, V., Ghirga, F., Iazzetti, A., Calcaterra, A., Marković, V., Botta, B., & Quaglio, D. (2022). Synthesis, Biosynthesis, and Biological Activity of Diels–Alder Adducts from Morus Genus: An Update. Molecules, 27(21), 7580. https://doi.org/10.3390/molecules27217580