1. Introduction
Isoginkgetin (Iso) is a natural bioflavonoid isolated from the leaves of
Ginkgo biloba [
1], which has manifested numerous healing properties, among which the anti-inflammatory, antioxidant, and anti-tumor properties certainly stand out. For thousands of years,
Ginkgo biloba L., a famous Chinese herb, has been used to treat respiratory ailments such as asthma and bronchitis [
2]; it was also famous for neuroprotective activity [
3]. Furthermore,
Ginkgo biloba L. determines an improvement of peripheral cardiovascular and vascular disorders, as well as influencing the platelet aggregation acting as a platelet antiplatelet [
4]. A large number of documented studies have shown that G. biloba (EGB) leaf extracts show inhibitory effects on various types of cancer cells [
5]. There is also experimental evidence that Iso, Gingko’s flavonoid extract, damages the extracellular matrix in cells by altering the production of metalloproteinase 9 (MMP-9), a protein known for modulating tumor metastasis and invasion. In fact, it is known that in human fibrosarcoma [
6], the metalloproteases (MMP-9) is activated by the tissue metalloproteinase 1 (TIMP-1) and the inhibiting factor of the nuclear enhancer of the light chain kappa of activated B lymphocyte’s “NFκB” signaling [
6,
7].
Gingko biloba extract also has an inhibitory growth effect on MCF-7 and MDA-MB-231 human breast cancer [
8], this property in particular motivated us to study its effect on glioblastoma cells. Glioblastoma multiforme (GBM) is the most aggressive and most frequent neoplasm originating from the cerebral nervous system. Its average incidence (i.e., the number of new cases per year) in Italy is eight cases per 100,000 inhabitants and represents 54% of all diagnosed gliomas with a median survival of about 18 months [
9]. GBM is characterized by high neo-angiogenesis, pronounced mitotic activity, cellular heterogeneity, high rates of proliferation, and necrosis. The presence of cancer stem cells, capable of proliferating and generating glial neoplastic cells, contributes to the poor prognosis of patients with GBM [
10], whose average survival is approximately 12 months from diagnosis. The therapeutic standard for all patients under 70 years of age and with good performance status after surgery or surgical biopsy is the combination of temozolomide 75 mg/m
2/day for the duration of the entire radiotherapy (60 Gy/30 fractions) for a total of 7 weeks. This is followed by 6 cycles (which in some selected cases can be extended to 12 cycles) of adjuvant temozolomide at a dosage of 150–200 mg/m
2/day for 5 days repeated every 28 days [
11]. To date, clinical studies on the use of brain implant devices in the area of cancer, the so-called WAFERS of BCNU (carmustine) [
12], an alkylating chemotherapeutic agent, should not be considered as a first-choice therapeutic option, given their little impact on survival.
The recurrence of GBM is unfortunately an almost inevitable event; therapeutic options range from the possible re-surgery to the resumption of treatment with temozolomide if there has been a sufficiently long interval of at least a few months or the use of chemotherapy regimens such as PCV (procarbazine, carmustine, and vincristine) or nitrosureas such as fotemustina. In conclusion, GBM patients have few therapeutic options, especially after failure of the initial standard treatment; however, there are many ongoing studies that are evaluating the use of new drugs. Furthermore, the intratumoral heterogeneity of GBM complicates the clinical outcome of the therapy [
13]. The reconfiguration of the diagnostic criteria with the introduction of the WHO classification of CNS tumors in 2021 represented a first decisive change, which stratifies the tumors in molecularly different subsets towards a personalized therapy [
14]. However, despite the efforts to classify GBM, patients currently have few therapeutic options, especially after the failure of the initial standard treatment. The ineffectiveness of chemotherapy agents can be attributed to the challenges of overcoming the blood–brain barrier. A growing body of evidence indicates that natural substances have shown high anticancer activity, which can be used successfully in the treatment of GBM as adjuvant therapy [
15,
16]. Relevant previous studies have shown valid inhibitory effects in vitro and in vivo of natural substances such as lactoferrin and aloe emodin [
17,
18]. The limit of these adjuvant substances is that after the scientific demonstration of the efficacy of the substance in vitro and in vivo, it is difficult to access to clinical trials that are fast, excessively complex for patient recruitment and too expensive. Here, we report the effect of isoginkgetin treatment on the continuous U87MG glioblastoma cell line. We found that Iso treatment at very low concentration (15 and 25 µM) determines an inhibition of growth and a reduction in migration in U87MG glioblastoma cells, two very interesting, combined effects.
3. Discussion
Although progress has been made in the classification of brain tumors and new molecular targets have been identified, glioblastoma remains a disease with a poor prognosis and the therapeutic resources remain limited to the Stupp protocol: temozolomide and radiotherapy. This pushes more and more to study natural substances that can act in conjunction with chemotherapy drugs, trying to act on growth of neoplastic cells in a cooperative way, in order to stop their proliferation through multiple mechanisms; this will be able to overcome the variegated response of glioblastoma to canonical treatments. In this study, we wanted to evaluate the effect of the Iso on U87MG glioblastoma cells. As described above, isoginkgetin is a natural substance from the leaves of Ginkgo biloba. Numerous properties are attributed to the leaves’ extract, among which, certainly, the anti-inflammatory and antioxidant ones stand out. More specifically, the anti-inflammatory action attributed to this plant is traceable to the ginkgolides contained in it. In particular, some studies have shown that ginkgolide B is able to inhibit the activity of PAF (platelet-activating factor) through the antagonization of its receptor. The platelet activation factor, in fact, plays an important role in inflammatory processes and variations in vascular permeability.
Among these stands a relatively recent study (2001), which highlighted how ginkgo extract is able to increase the efficacy and tolerability of 5-fluorouracil (5-FU) in patients with colon cancer straight from refractory to treatment with only 5-FU [
20]. This clinical study drove us to investigate the effect of
Ginkgo biloba isoginkentine leaf derivatives on U87MG glioblastoma cells.
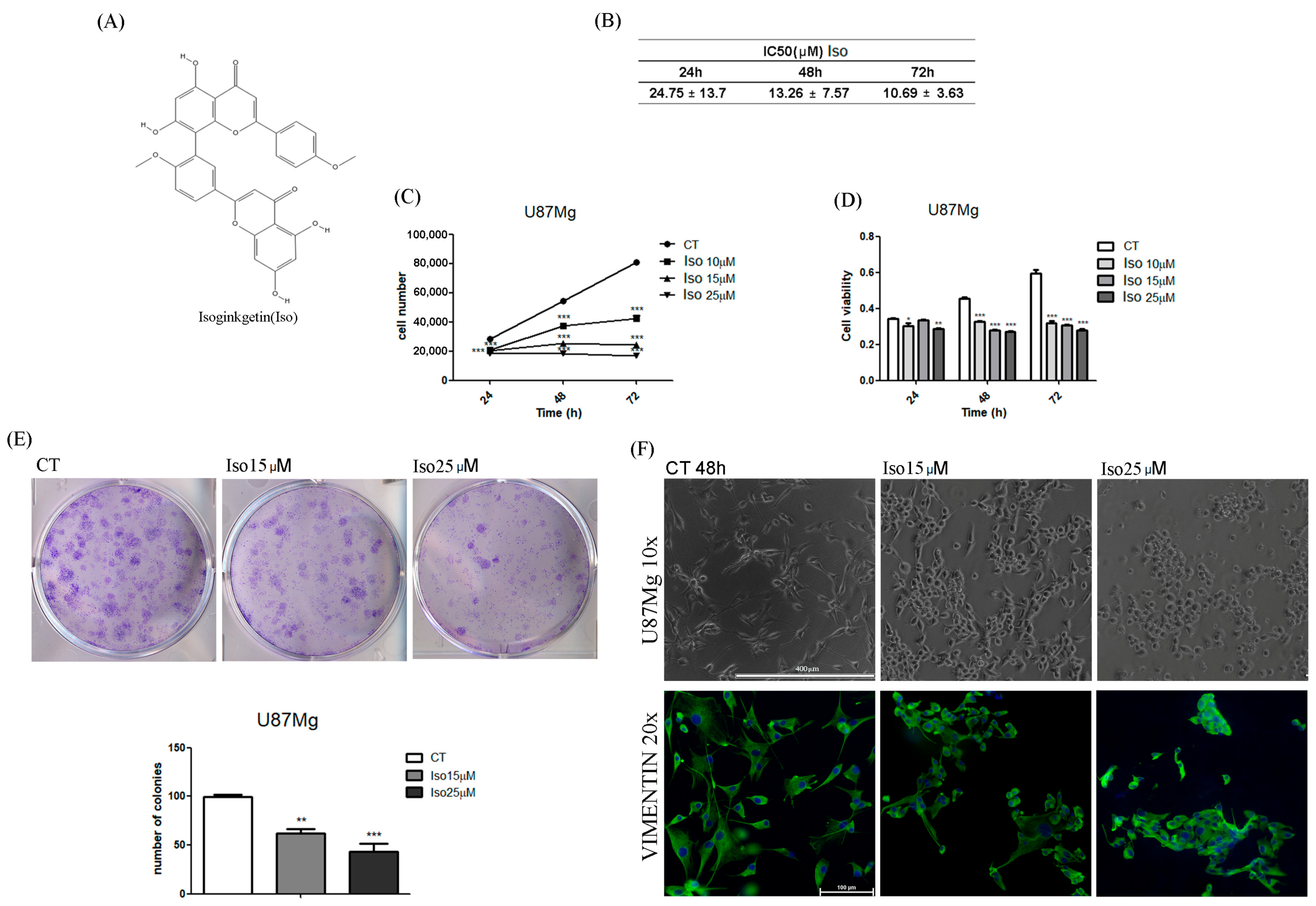
In the first experiments, the effect of Iso on cell growth was analyzed; in particular, U87MG growth curves and MTT toxicity assays after treatments with Iso cells showed a time (24, 48, 72 h)- and dose (15, 25 µM)-dependent inhibition of growth. Iso treatment modified glioblastoma morphology, the cell widespread with extensions become rounded and devoid of extensions after 48 h of treatment with 15 or 25 µM Iso. This morphological change is further highlighted by immunofluorescence staining with anti-vimentin antibody, the treatment with Iso 15 and 25 µM determines a redistribution of the vimentin, which no longer appears distributed in the extensions but thickened in a roundish cell similar to a rounded apoptotic-like cell. These results indicate that the link between the vimentin protein and DNA may be functional to the protein itself to grow and move cancer cells [
21]. Growth inhibition of U87MG glioblastoma cells was further investigated by colony formation assay after 24 h of treatment with Iso 15 µM. The number of colonies was reduced by approximately 35% after 15 days in GBM cells treated with 15 µM Iso, and about 60% with 25 µM Iso compared to control cells not subjected to treatments (
Figure 1E). FACS analysis of cell samples treated with Iso 15 μM, at different times (24, 48, and 72 h), shows a decrease in the percentage of cells in S-phase as early as 24 h after treatment, becoming significant at 48 and 72 h of treatment. The number of cells in S phase decreases with time exposition.
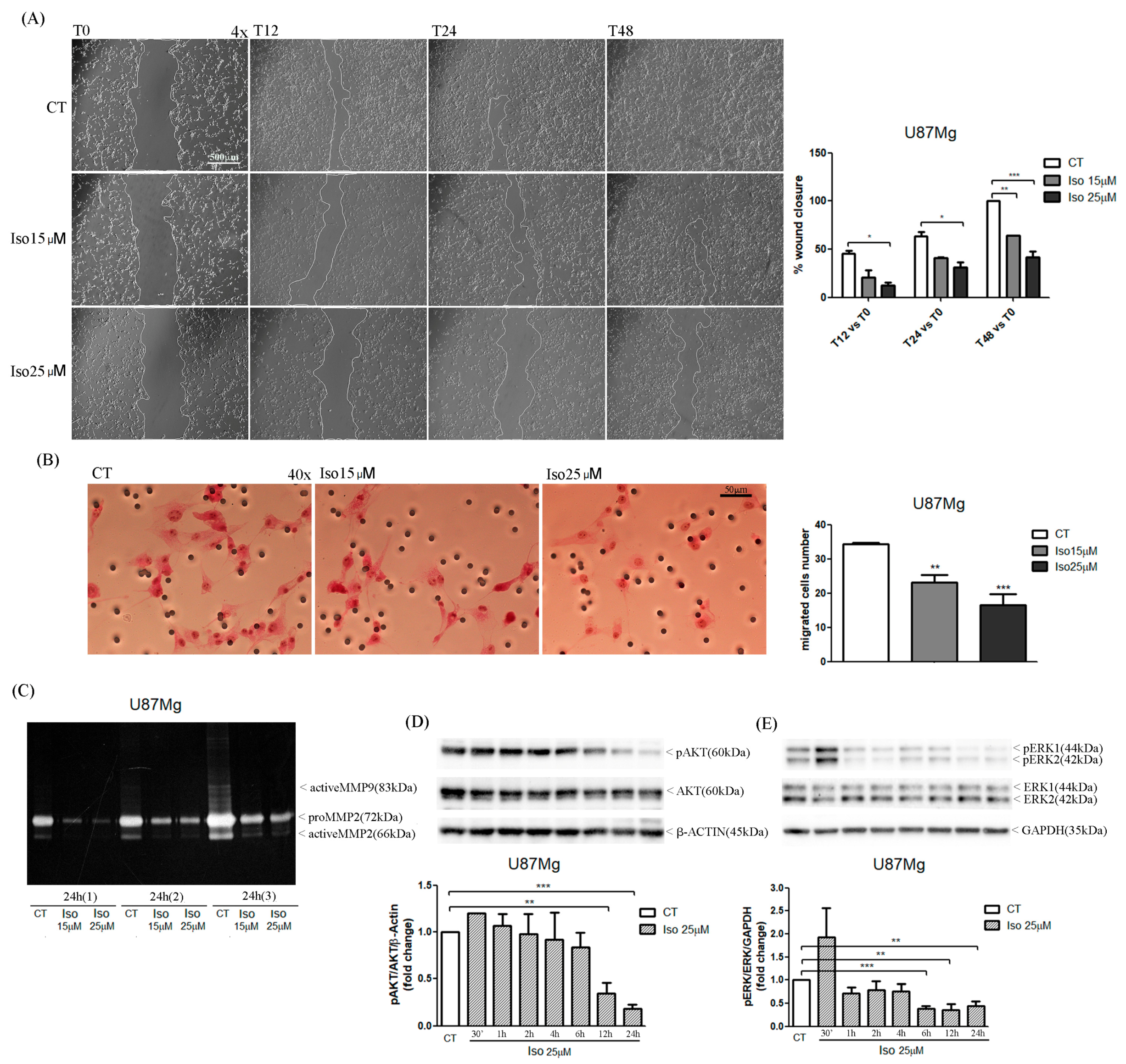
The migration of glioma cells within the brain parenchymal represents one of the mechanisms that makes glioblastoma one of the most aggressive and lethal tumors. In this paper we reported a very interesting effect of the treatment of glioblastoma cells with Iso represented by a reduction in cell migration as evidenced by a scratch test. Iso-treated U87MG glioblastoma cells repaired the scratch slower than untreated cells, on average percentage of closure of the scratch area of about 25% compared to 90% of the control cells. The migration assay in-chamber slides also strengthened and confirmed that after 24 h of treatment with Iso 15 and 25 µM, U87MG cell migration decreased by 30% and 45%, respectively. The proteolytic activity of metalloproteinase (MMPs) influenced tumor progression: primary tumor growth, angiogenesis, extravasation and invasion of neoplastic cells, migration, and invasion of metastatic cells in the secondary organ. In particular, it has been previously demonstrated in breast cancer that MMPs can also suppress tumor progression through the decreasing of proteolytic activity [
22,
23]. The U87MG zymography assay showed that treatment with 25 µM Iso for 24 h determined a decrease in the proteolytic activity of MMP2. Moreover, in U87MG glioblastoma cells, treatment with Iso determined an intensification of the proteolytic activity of the metalloproteases after 24 h of treatment; the levels of MMP-2 were reduced with an increase of MMP-9 form. This proteolytic activity induced by Iso-reduced cell migration could make Iso an excellent remedy for blocking the dissemination of neoplastic glial cells in the brain as a control strategy in the development of relapse, which complicates the patient’s prognosis, leading to a quick death. Finally, we investigated the mechanisms involved in the growth block by studying the main phenomena that control cell growth homeostasis: apoptosis and autophagy. Autophagy and apoptosis represent distinct physiological processes within the cell. Apoptosis is the best-known form of programmed cell death (Type I cell death). This occurs through the activation of catabolic enzymes by signal cascades, which leads to the rapid breakdown of cells structures and organelles [
24]. In some circumstances, therefore, apoptosis and autophagy can exert synergistic effects, while in other situations autophagy can be induced only when apoptosis is suppressed [
25]. In general terms, it seems that similar stimuli can induce both autophagy and apoptosis, so it is possible to find a mixed phenotype. In fact, in some settings, autophagy and apoptosis seem to be interconnected and the idea that “molecular switches” exist between the two processes has been hypothesized [
7,
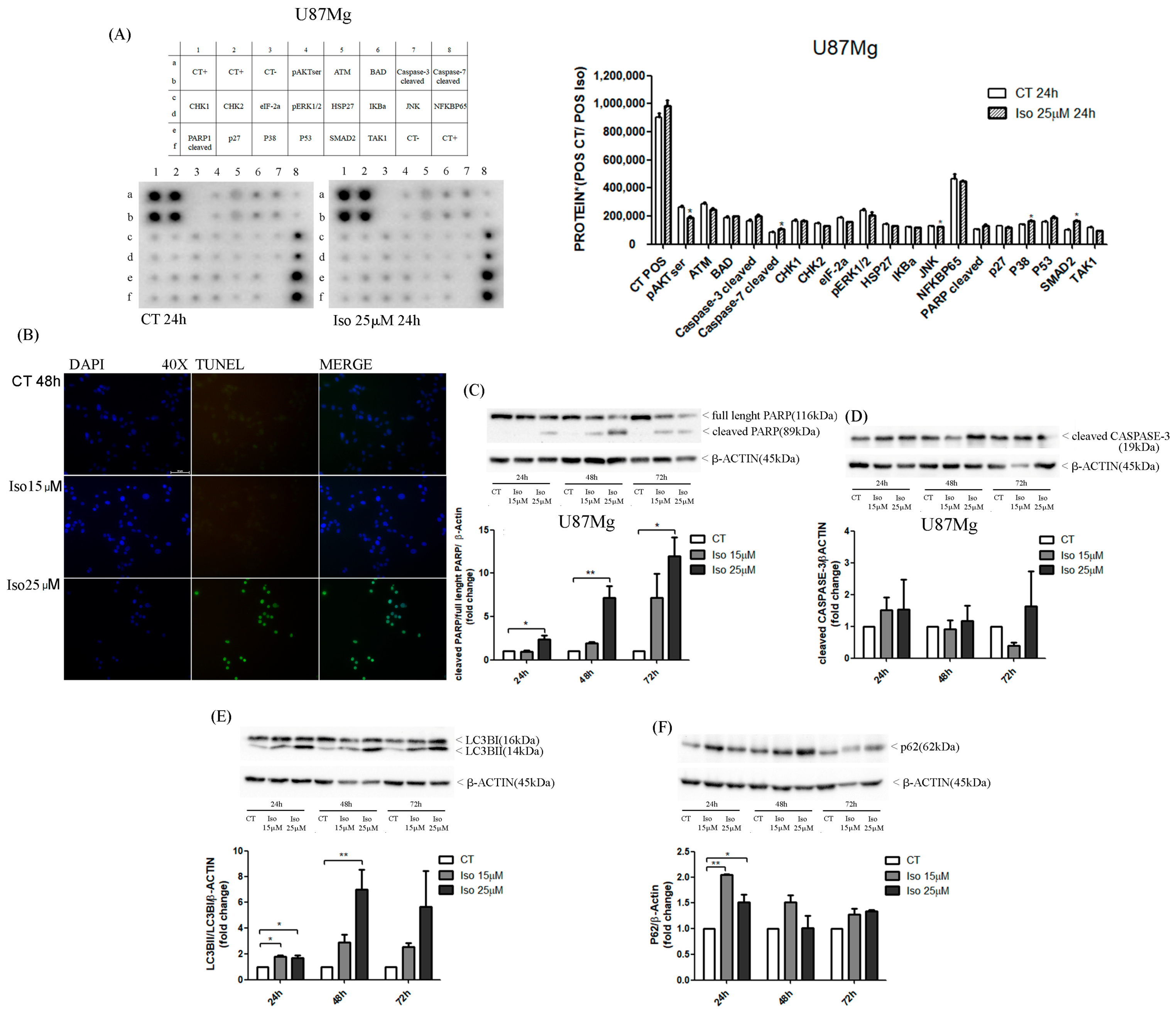
26]. Similarly, in the present study, Western blot results pointed out an increase in cleaved band 89KD9 PARP (PARP activated) and a strong increase in caspase 3-cleaved bands after Iso treatment. The activation of apoptosis was also endorsed by DNA fragmentation evidenced in the U87MG Iso-treated by tunnel assay. The contemporary role of the presence of the autophagy process in Iso-treated U87MG glioblastoma cells was manifested by an increased in active band LCB3-II (16 kDa) and altered the levels of p62. This confirms that Iso may also activate a protagonist of the autophagy process. The adapter protein p62, also called sequestosome 1 (SQSTM1), is a ubiquitin-binding scaffold protein that colocalizes with ubiquitinate; generally, p62 accumulates when autophagy is inhibited, and decreases in level when autophagy is induced. Monitoring of the autophagic degradation of p62/SQSTM1 represents the indicator of the autophagic process. In our contest the decrease in p62 levels, after Iso 15 and 25 μM treatment, which contrasts with the intensification of the autophagic process, can be explained as the impairment of cell protein degradation machinery of cancer cells [
27]. Iso could represent an excellent adjuvant for glioblastoma care, together with traditional temozolomide treatments and radiotherapy.
5. Materials and Methods
5.1. Cell Culture
The continuous human glioblastoma cell line U87MG was purchased from Sigma Aldrich Collection (LGCPromochem, Teddington, UK); cells were growth in Dulbecco’s Modified Eagle’s Medium supplemented with 10% fetal bovine serum, 2 mmol/L-glutamine, 100 IU/mL penicillin, 100 µg streptomycin, at 37 °C, 5% CO2 and 95% of humidity. In vitro treatments were performed with pure molecule Isoginkgetin (Iso) from MedChemExpress, South Brunswick, NJ, USA, and Temozolomide (TMZ) from Sigma Aldrich (St. Louis, MI, USA).
5.2. Determination of Half-Maximal Inhibitory Concentration (IC50) of Isoginkgetin in U87MG Cells
To estimate the IC50-values of Isoginkgetin in U87MG at 24, 48 and 72 h, cells were plated in 96-well plates (5 × 103 cells/well) and treated with Iso at different concentration (0.001, 0.01, 0.1, 1, 10, 25 and 50 µM), by only one induction. The IC50-values were calculated using GraphPad Prism (GraphPad Software Inc., San Diego, CA, USA).
5.3. Proliferation Assay
To evaluate the effect of isoginkgetin on U87MG proliferation, cells were plated in 48-well plates (1 × 104 cells/well) in DMEM 10% FBS and incubated at 37 °C, 5% CO2. The day after, cells were treated with Iso at three different concentrations (10, 15, and 25 µM) by only one induction; DMSO 0.1% was used as vehicle control. Cell count was performed at 24, 48, and 72 h of treatment using a Burker chamber.
5.4. Cell Viability Assay
U87MG cells were seeded in 96-well plates (5 × 103 cells/well) and treated with Iso at three different concentrations (10, 15 and 25 µM) by only one induction; DMSO 0.1% was added in the control. At 24, 48, and 72 h of treatment, cell viability was evaluated by MTT assay. Briefly, 10 µL of 5 mg/mL MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide; Sigma-Aldrich) was added in each well with 100 µL of medium. After 1 h of incubation at 37 °C, formazan crystals were dissolved with 100 µL isopropanol/HCl 0.4% and the absorbance was measured at 570 nm with a plate reading spectrophotometer.
5.5. Immunofluorescence
To analyze the morphological change of U87MG induced by isoginkgetin, cells were plated in 8-well chamber slides (10 × 104 cells/well) in DMEM 0.5% FBS. After 48 h the medium was replaced with DMEM 10% FBS and cells were treated with Iso 15 and 25 µM for 48 h. At the end of treatment, cells were fixed in 4% formalin pH 7.4 (Diapath, Martinengo, Italy) for 20 min and permeabilized with 0.1% Triton (Invitrogen, Carlsbad, CA, USA) for 30 min. After blocking with 10% horse serum (Vector) for 1 h, cells were incubated with a mouse monoclonal antibody anti-vimentin (prediluted; Roche Diagnostic, Mannheim, Germany) overnight at 4 °C. After washing with 0.025% PBS-Tween-20 (Sigma-Aldrich, St. Louis, Missouri, USA) cells were incubated with secondary antibody anti-mouse fluorescein (1:100; Vector) in 2% serum for 1 h at room temperature. The slide was counterstained with DAPI mounting medium (Vectashield) and analyzed with a fluorescence microscope (Axiophot 2 Zeiss, Gottigen, Germany) at 20× magnification.
5.6. Wound Healing Assay
To evaluate the effects of Iso on U87MG motility, cells were seeded into 6-well culture plates (5 × 103 cells/well); when the cells reached confluence, a scratch was made by sterile 100 µL pipette tips and detached cells were washed with PBS 1×. After adding of DMEM 10% FBS, an induction with Iso 15 and 25 µM was performed. Cell migration was followed by observation at a phase contrast microscope (Evos, Life Technologies, Carlsbad, CA, USA) at 4× magnification. The scratch area was quantized by image analysis with ImageJ software.
5.7. Migration Assay
U87MG cells were seeded (15 × 104 cells/well) in a serum-free medium in Boyden chambers (BD Falcon, Schaffhausen, Switzerland) immersed in 2 mL of FBS. After adhesion to the bottom of the chambers, cells were treated with Iso 15 and 25 µM and incubated at 37 °C for 48 h. The cells migrated through the filter were fixed with methanol for 5 min and stained with hematoxylin and eosin. The count of migrated cells was carried out by an optical microscope at 40× magnification in 5 different fields.
5.8. Clonogenic Assay
Colony formation assay was performed seeding U87MG cells in 6-well plates (103 cells/well) in DMEM 10% FBS for 48 h. Cells were treated with Iso 15 and 25 µM for 24 h and medium was replaced every 3 days for 14 days. Cells were fixed with 4% paraformaldehyde solution for 5 min, washed with PBS 1× and stained with crystal violet 0.05% for 30 min. The colony count was carried out by an optical microscope at 40× magnification.
5.9. Zymography Assay
The effect of Iso on metalloprotease enzymatic activity was investigated with a Zymography assay. U87MG cells were plated in DMEM 0.5% FBS (4 × 104 cells/well) for 48 h; at 70–80% confluency, FBS medium was replaced with FBS-free medium and the cells were induced with Iso15 and 25 µM for 24 h. After treatment, culture medium was collected and concentrated in a SpeedVac centrifuge (Thermo Scientific, Waltham, Massachusetts). Different concentrations of protein from culture medium (150 ηg, 300 ηg, and 600 ηg), in 10 µL of non-reducing sample buffer (4% SDS, 20% glycerol, 0.01% bromophenol blue, 125 mM Tris-HCl pH 6.8), were separated on a 10% polyacrylamide gel containing porcine gelatin (4 mg/mL; Sigma-Aldrich, Missouri, St. Louis, MO, USA). The gel was immersed for 30 min in washing buffer (2.5% Triton X-100, 50 mM Tris-HCl pH 7.5, 5 mM CaCl2, 1 µM ZnCl2) and stirred in incubation buffer (1% Triton X-100, 50 mM Tris-HCl pH 7.5, 5 mM CaCl2, 1 µM ZnCl2) for 24 h at 37 °C. Finally, the gel was stained with staining solution (40 mL methanol, 10 mL acetic acid, 50 mL H2O, 0.5 g Coomassie blue) for 30 min, rinsed with H2O and incubated in destaining solution (40 mL methanol, 10 mL acetic acid, 50 mL H2O) until bands were clearly seen. Areas of enzyme activity appeared as white bands against a dark blue background.
5.10. Cell Cycle Analysis by Flow Cytometry
U87MG cells were plated in DMEM 0.5% FBS (5 × 105 cells/well) for 48h and treated with Iso 15 and 25 µM. At 24, 48, and 72 h of treatment, cells were washed in sample buffer (0.1% Glucose in HBSS without Ca++ and Mg++), resuspended in sample buffer and fixed in ice-cold 70% ethanol overnight at 4 °C. After centrifugation, cells were incubated in propidium iodide (PI) staining solution (50 µg/mL of PI) for 30 min at room temperature and analyzed within 24 h in a flow cytometer (Gallios Instrument, Beckman Coulter, Brea, CA, USA). Cell cycle analysis was performed by using Kaluza software for analysis v. 2.1 (Beckman Coulter).
5.11. DNA Fragmentation’s Detection by TUNEL Assay
U87MG cells were plated in 8-well chamber slides (10 × 104 cells/well), in DMEM 0.5% FBS, for 48 h and treated with Iso 15 and 25 µM for 48 h. After treatment, cells were fixed in 4% methanol-free formaldehyde solution pH 7.4 for 25 min at 4 °C and washed twice with PBS 1×. DNA fragmentation was detected by TUNEL assay with commercial kit DeadEnd Fluorometric TUNEL System (Promega, Madison, WI, USA), following the manufacturer’s instructions. Cells were counterstained with DAPI mounting medium (Vectashield) and analyzed with a fluorescence microscope (Axiophot 2 Zeiss) at 40× magnification.
5.12. Human Apoptosis Signaling Pathway Array
The analysis of several proteins involved in the apoptosis pathway was carried out in U87MG control cells and treated with Iso 25 µM for 24 h using the human apoptosis signaling pathway array (RayBiotech, Norcross, GA, Peachtree Corners, GA, USA). Two antibody arrays were incubated in blocking buffer for 30 min at room temperature and then with protein samples (200 µg proteins in 1ml of blocking buffer) overnight at 4 °C. After washing in a specific buffer, each membrane was incubated with a detection antibody cocktail and then with HRP-anti-rabbit IgG for 2 h at room temperature, respectively. Two membranes were washed twice with specific buffer, incubated with chemiluminescence detection buffer for 2 min and analyzed with a ChemiDoc XRS imaging system (Bio-Rad Laboratories, CA, USA). Each membrane had a positive control to normalize the signal of all antibodies. The membrane with proteins from U87MG treated was considered as the “Reference Array” to which the other membranes were normalized too.
5.13. Western Blot Analysis of Proteins Involved in Apoptosis and Autophagy
U87MG cells were seeded in DMEM 0.5% FBS (5 × 105 cells/well) for 48 h and treated with Iso 15 and 25 µM. A short-term treatment (15 min, 30 min, 1 h, 2 h, 4 h, 6 h, and 12 h) was performed for analysis of phosphorylated proteins (pAKT and pERK1/2) and a long-term treatment (24, 48, 72 h) for analysis of all other proteins (PARP, caspase 3, LC3 and P62). Proteins were extracted from U87MG cells with Triton X-100 lysis buffer (10 mM Tris-HCl, 1 mM EDTA, 150 mM NaCl, 1% Triton X-100, 1 mM NaF, 1 mM Na4P2O7, 1 mM Na3VO4, and 1x protease inhibitors). Protein lysates were resolved on 10–12% SDS-PAGE and transferred to PVDF membranes (Bio-Rad, California, USA) by electroblotting. Each blot was incubated for 1h at room temperature in 5% non-fat dry milk or bovine serum albumin (BSA) diluted in 1× Tris-buffered saline–0.1% Tween-20 and then was incubated overnight at 4 °C with primary antibody, diluted in 2% milk or BSA. After washing in 1 × TBS-0.1% Tween the membrane was incubated for 1 h at room temperature with a specific HRP-conjugated secondary antibody (anti-mouse or anti-rabbit 1:7000; Calbiochem, St. Louis, MO, USA) and then for 1 min with ECL (Amersham Pharmacia Biotech Italia Spa, Cologno Monzese, Italy). Blot was analyzed with a ChemiDoc XRS Imaging System (Bio-Rad) and the digital signals were quantified by Image Lab Software (Bio-Rad Laboratories). For analysis of phosphorylated proteins, we used two primary antibodies, anti-pERK1/2, and anti-pAKT, (Cell Signaling; 1:1000, Danvers, MA, USA) in 2.5% BSA, while for the proteins involved in apoptosis and autophagy, we used different primary antibodies (anti-PARP, anti-caspase 3, anti-LC3, and anti-P62), all from Cell Signaling, 1:1000 in 2.5% milk. For protein normalization, each membrane was incubated with a mouse monoclonal antibody, anti-beta-actin (Cell Signaling;1:1000) or a mouse monoclonal antibody anti-GAPDH (Cell Signaling; 1:1000).
5.14. Statistical Analysis
Data were expressed as means ± SEM of three individual experiments; statistical significance was determined by one-way ANOVA test and Dunnett’s multiple comparison test, considering p-values < 0.05 statistically significant, according to GraphPad Prism.



{kind=link}
{kind=link}
{kind=link}
{kind=link}



