Abstract
Two unique structures were isolated from the phosphorylation reaction of 10H-phenothiazine. The 5,5-dimethyl-2-(10H-phenothiazin-10-yl)-1,3,2-dioxaphosphinane 2-oxide (2a) illustrates the product of N-phosphorylation of phenothiazine. Moreover, a potential product of 2a instability, a thiophosphoric acid 2b, was successfully isolated and structurally characterized. Molecule 2a, similarly to sulfoxide derivative 3, possesses interesting phosphorescence properties due to the presence of d-pπ bonds. The X-ray, NMR, and DFT computational studies indicate that compound 2a exhibits an anomeric effect. Additionally, the syntheses of selected symmetrical and unsymmetrical pyridine-embedded phenazines were elaborated. To compare the influence of phosphorus and sulfur atoms on the structural characteristics of 10H-phenothiazine derivatives, the high-quality crystals of (4a,12a-dihydro-12H-benzo[5,6][1,4]thiazino[2,3-b]quinoxalin-12-yl)(phenyl)methanone (1) and selected phenazines 5,12-diisopropyl-3,10-dimethyldipyrido[3,2-a:3′,2′-h]phenazine (5) and 5-isopropyl-N,N,3-trimethylpyrido[3,2-a]phenazin-10-amine (6a) were obtained. The structures of molecules 1, 2a, 2-mercapto-5,5-dimethyl-1,3,2-dioxaphosphinane 2-oxide (2b), 3,7-dinitro-10H-phenothiazine 5-oxide (3), 5 and 6a were determined by single-crystal X-ray diffraction measurements.
1. Introduction
The phenothiazines and the phenazines are exceptional and versatile classes of electron-rich nitrogen heterocycles with broad pharmaceutical profiles and rich materials sciences applications [1,2,3]. A seemingly small difference between these types of molecules, i.e., the presence of a six-membered ring with nitrogen and sulfur atoms for phenothiazines and two nitrogen atoms for phenazines, manifests itself profoundly through their physicochemical and structural properties. The unique characteristics of both systems are present in (4a,12a-dihydro-12H-benzo[5,6][1,4]thiazino[2,3-b]quinoxalin-12-yl)(phenyl)methanone (1) and its analogs.
The phenazine class of N-heterocycles, numbering over 6000 compounds, is an important structural motif in functional materials, pharmaceuticals, agricultural, and natural products. Therefore, developing new efficient methods for preparing molecules with this privileged moiety is an essential endeavor in synthetic chemistry [4]. Their broad range of biological activity is thought to be due to their ability to undergo redox cycling in the presence of various reducing agents and molecular oxygen [5,6]. Phenazine natural products constitute biologically active metabolites produced by soil habitats and marine microorganisms [4]. The blue pigment pyocyanine, the fluorescent siderophore pyoverdine, phenazine-1-carboxylic acid, and the siderophore pyochelin are the most famous representatives. The biological properties of this class of natural products include antibiotic, antitumor, antimalarial, and antiparasitic activities [4]. They play an important role in microbial competitiveness, the suppression of soilborne plant pathogens, and virulence in human and animal hosts [5]. The described class of compounds is an intriguing topic of research.
Concerning the phenothiazines, many representatives of these electron-rich tricyclic nitrogen–sulfur heterocycles are the active components in various sedatives, tranquilizers, antiepileptics, antituberculosis, antipyretics, antitumor agents, bactericides, and parasiticides [7,8,9,10,11]. In addition to their physiological activities, the phenothiazines’ reversible oxidative properties give rise to deeply colored radical cations, making them attractive spectroscopic probes in molecular arrangements for photoinduced electron transfer studies and as scientific motif materials [12,13]. Phenothiazine derivatives can participate in reduction and oxidation processes manifested in their various oxidation states, such as neutral, cation radical, and oxygenated sulfoxide forms, providing synthetic perspectives, especially in pharmaceuticals [13,14,15]. Phenothiazines can act as antioxidants for many easily oxidizable substrates, including lubricants, rubber, polymers, and biological materials [16,17,18]. In particular, 10H-phenothiazine has been found to inhibit the autoxidation of methyl linoleate and phenoxazine to retard lipid peroxidation in rat brains [19]. It has also been suggested that the pharmacological activity of phenothiazines might be somehow related to their antioxidant or radical-trapping ability [20]. Another important application of phenothiazine and related compounds concerns their use as polymerization inhibitors to stop the reactions of radical polymerization, which may occur during the preparation and workup of acrylic monomers or during their storage [21,22]. N-Alkylation of phenothiazine has led to pharmaceutically essential substances, such as promethazine (antihistaminic), ethopropazine (anti-Parkinson), and dimethoxanate (antitussive). The discovery of the antipsychotic agent chlorpromazine in the early 1950s and the appearance of even more potent phenothiazine psychopharmacological agents constitute a landmark event in the history of the medical and psychiatric sciences. As a result of their numerous medicinal and industrial applications, extensive derivatization and/or functionalization of these parent structures continue unabated.
The current study presents the syntheses and spectroscopic characterization of selected phenothiazines and phenazines rationalized based on density functional theory (DFT) calculations.
2. Results and Discussion
2.1. Synthesis and Structural Characterization of Studied Compounds
The N-benzoylated, N-alkylated, or N-arylated phenothiazines and related compounds are stable molecules [23,24,25,26,27]. Some of their representatives are highly potent inhibitors of cancer cell growth and tubulin polymerization [26]. The summary of molecular structures of compounds discussed in this paper is presented in Scheme 1.
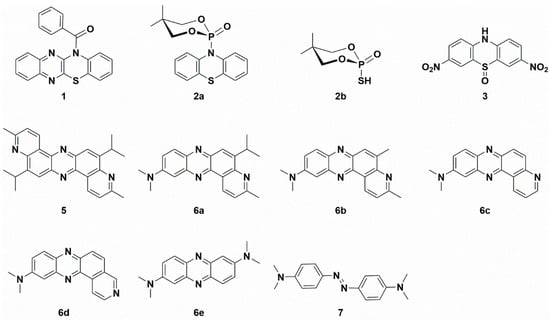
Scheme 1.
Summary of the molecular structures of studied compounds.
Keglevich and co-workers obtained the N-phosphorylated phenothiazine, i.e., O,O-diethyl (10H-phenothiazine-10-yl)phosphonate. The authors claimed that the phosphorylated phenothiazine was not stable and decomposed. They reported difficulties during its separation from the other components, especially the N-ethylated phenothiazine, i.e., 10-ethyl-10H-phenothiazine. The authors realized that the distribution profile and product yields strongly depend on the used base, solvents, and heating mode (conventional or microwave irradiation). They determined the product’s composition by LC-MS techniques. The highest O,O-diethyl (10H-phenothiazine-10-yl)phosphonate yield was observed in the case of using NaH as a base in refluxing DMF (52%), and lower values were observed for refluxing toluene (15%). Under other reaction conditions, the authors did not identify the N-phosphorylated product. The results obtained from phosphorylation reactions were opposite to N-ethylation, where the highest yields were observed for applying K2CO3 under microwave irradiation conditions (80%), and the lowest yields were observed for NaH and toluene with conventional heating (5%) [25]. Inspired by the work of Prof. Keglevich’s team, we performed a similar type of reaction; however, as a phosphorylating agent, we chose 2-chloro-5,5-dimethyl-1,3,2-dioxaphosphinane 2-oxide. This electrophile is known for its high tendency to crystallize (Scheme 2) [28,29]. In order to synthesize the 5,5-dimethyl-2-(10H-phenothiazin-10-yl)-1,3,2-dioxaphosphinane 2-oxide (2a), we performed a reaction between the in situ generated sodium salt of phenothiazine and 2-chloro-5,5-dimethyl-1,3,2-dioxaphosphinane 2-oxide in THF. The synthetic route with the structures of reagents is presented in Scheme 2. Product 2a was obtained with a good 58% yield. The unexpected side-product 2-mercapto-5,5-dimethyl-1,3,2-dioxaphosphinane 2-oxide (2b) was only isolated to prove its presence (Scheme 2). The radical rearrangement of product 2a may explain the formation of molecule 2b through the disintegration of the P-N bond and the establishment of a new P-S bond. To elucidate the possible origin of side-product and the relationship between the molecular structures of stable acyl or aryl phenothiazine derivatives, high-quality single crystals of (4a,12a-dihydro-12H-benzo[5,6][1,4]thiazino[2,3-b]quinoxalin-12-yl)(phenyl)methanone (1) and compound 2a were cultivated by slow evaporation from a hot DMSO or a hot mixture of THF and hexane, respectively.
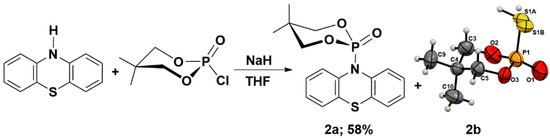
Scheme 2.
Synthesis of the 5,5-dimethyl-2-(10H-phenothiazin-10-yl)-1,3,2-dioxaphosphinane 2-oxide (2a).
The phenothiazine molecule possesses a unique non-planar butterfly structure. Tian and co-workers drew attention to the derivatives having an S=O (or SO2) and P=O group, which contains the d-pπ bond that could hinder a possible intense intermolecular π-π stacking, which is beneficial for inhibiting excited triplet-triplet annihilation (TTA) [27]. Tian et al. presented some examples of molecules with a S=O bond (in the SO2 group), which shows a sharp increase in TTA by 19 times compared to that without this group.
Compound 1 crystallizes in the monoclinic system with the P21/n space group (Figure 1) and demonstrates layered packing similar to the phenothiazine model system described by Tian and co-workers [27]. Intermolecular π-π stacking distances vary in the range of 3.670-3.736 Å (Figure 1c). Additionally, a rigid 2D network structure of 1 is stabilized via intermolecular C-H⋯N interactions with distances of 2.690 Å. The bending angles in the phenothiazine core of molecule 1 were defined as the ∠N12−S11−C10 and ∠N5−N6−C7, as shown in Figure 1a,b, and were found to be 130.88(5)° and 131.92(8)°. The distance between the centroids of the phenyl and azine rings is equal to 3.712(1)Å.
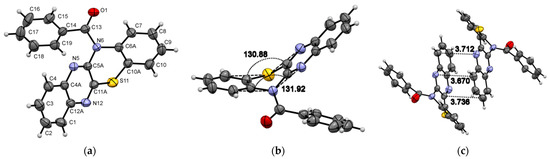
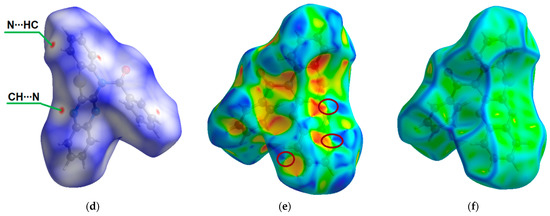
Figure 1.
ORTEP (Oak Ridge thermal ellipsoid plot) drawings of (4a,12a-dihydro-12H-benzo[5,6][1,4]thiazino[2,3-b]quinoxalin-12-yl)(phenyl)methanone (1) with 50% probability. Top view (a), angle view (b) and packing (c) [23]. Hirshfeld surfaces for molecule 1 are mapped with dnorm (d), shape index (e), and curvedness (f).
The X-ray bond lengths and bond angles for molecule 1 are provided in detail in Supplementary Materials. The computational DFT results confirm a bent-core structure of compound 1 and are consistent with a crystal structure in bending angles and bond length presented in Supplementary Materials. The calculated bending angles (135.4° and 135.8°, respectively) are close to X-ray data, indicating that the bending structure is not primarily caused by lattice packing but is due to the steric hindrance inherent in phenothiazines.
The intermolecular interactions between atoms in the crystal of molecule 1 were also studied using Hirshfeld surfaces analysis [30,31] via the dnorm, shape index, and curvedness (Figure 1d,e). The surfaces are shown as transparent to visualize the molecular skeleton. The dnorm surface exhibits two intensive red spots, which correspond to short intermolecular CH…N interactions with distances of 2.690 Å (Figure 1d). The presence of π-π stacking interactions is indicated by red and blue triangles on the shape-index surface (brown ellipses in Figure 1e) and by a flat region on the curvedness (Figure 1f). A network of intermolecular CH…N and π-π stacking interactions hinder molecular movement in crystal 1, which helps to reduce possible energy losses through non-radiative relaxation channels.
Molecules 2a crystallize to a monoclinic system, P21/n, with unit cell dimensions of a = 8.8248(3) Å, b = 16.5758(4) Å, c = 11.8216(4) Å, V = 1678.23(9) Å3, and Z = 4; the monoclinic angle (β) is 103.951(3)°, and the unit cell contains 4 molecules (Figure 2a–d). The ∠N10−P1−O1 and ∠O2−P1−O3 in compound 2a were found to be 103.67° and 112.26°, respectively (Figure 2b). The bending angle ∠C1−S9−C8 in the phenothiazine core of N-phosphorylated phenothiazine 2a is smaller than in molecule 1a and was found to be 126.87°. The DFT calculations of the phenothiazine derivative 2a structure by the B3LYP/6-31G(d,p) method indicate a minimal lattice packing effect in the dioxaphosphinane fragment in terms of ∠N10−P1−O1 (102.96°) and ∠O2−P1−O3 (113.70°) and bond lengths presented in Supplementary Materials. The calculated bending angle in the phenothiazine core (∠C1−S9−C8) of molecule 2a is 131.17°. The bending angles in the middle phenothiazine ring are denoted as ΘS and ΘN and were calculated to be 98.30° (exp.: 97.90°) and 117.13° (exp.: 115.60°) (Figure 2e). The Hirshfeld dnorm surfaces of 2a indicate the stabilization of crystal packing with numerous CH…O and CH…S intermolecular interactions (Figure 2f,g). The shape index visualizes the complementarity of molecules in the crystal and shows that there are no π-π stacking interactions in the volume (Figure 2h,i).
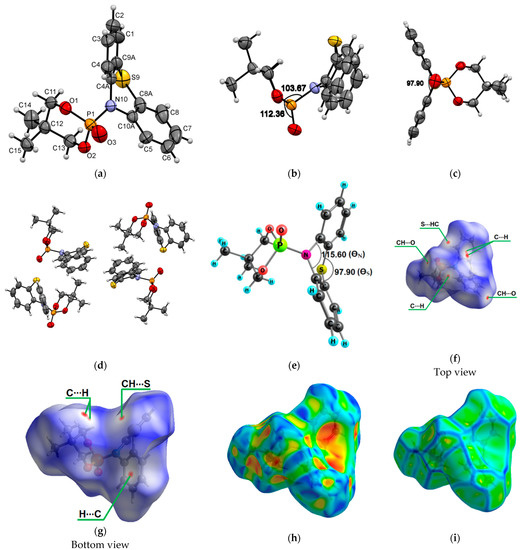
Figure 2.
ORTEP drawings of 5,5-dimethyl-2-(10H-phenothiazin-10-yl)-1,3,2-dioxaphosphinane 2-oxide (2a) with 50% probability. Top view (a), angle view (b,c) and packing (d) [23]. Optimized structure of 2a with the bending angles in the middle phenothiazine ring marked (e). Hirshfeld surfaces for molecule 2a are mapped with dnorm (f,g), shape index (h) and curvedness (i).
The thiophosphoric acid of > P(=S)OH type could exist in one of two tautomeric structures, either with P=S and P-OH bonds or with P=O and P-SH. The second tautomeric form of molecule 2b, which we suggest, is much less proposed in the chemical literature (Scheme 2). The P=O (P1–O1 1.446(4) Å) and P-S (P1–S1B 2.0006(10) Å, P1–S1A 2.0006(10) Å) bond lengths in molecule 2b (Supplementary Materials) are in good agreement with those reported for a similar type of compounds [32,33,34]. Molecules 2b crystallize into an orthorhombic system, Pbca, with unit cell dimensions of a = 11.1587(17) Å, b = 11.8846(13) Å, c = 13.1114(16) Å, V = 1738.8(4) Å3, and Z = 8; the orthorhombic angle (β) is 90°, and the unit cell contains 8 molecules. The -SH group is disordered over two positions with a refined occupancy ratio of 0.49(5):0.51(5).
Compound 3 crystallizes to a monoclinic system, P21/n, with unit cell dimensions of a = 8.2753(3) Å, b = 19.9331(7) Å, c = 8.5875(3) Å, V= 1398.32(9) Å3, and Z = 4; the monoclinic angle (β) is 99.197(3)°, and the unit cell contains 4 molecules (Figure 3c). The oxygen atom from the -SO group is disordered over two positions with a refined occupancy ratio of 0.949(5):0.051(5). Therefore, Hirshfeld surfaces analysis is not applicable for 3. Both molecules 2a and 3 crystallize in the monoclinic system and have no π-π interactions (for 2a) or are weak (for 3, Figure 3b). This can be explained by a reduction in the electron density on the benzene rings on both sides of phenothiazine upon the N-phosphorylation for compound 2a or the oxidation of sulfur (electron-donating group) to sulfoxides (electron-withdrawing group) for molecule 3. Experimental bending angles in the middle phenothiazine ring are 98.31°for ΘS and 124.47° for ΘN (Figure 3d). We can find some similarities in physicochemical properties among our molecule 3 and sulfonyl derivatives published by Tian et al. [27]. They also crystallize into the monoclinic system and have abundant intermolecular C-H⋯π and C-H⋯O interactions in the crystal. The multiple intermolecular interactions help to suppress molecular motions and thereby reduce the possible energy loss via non-radiative relaxation channels.
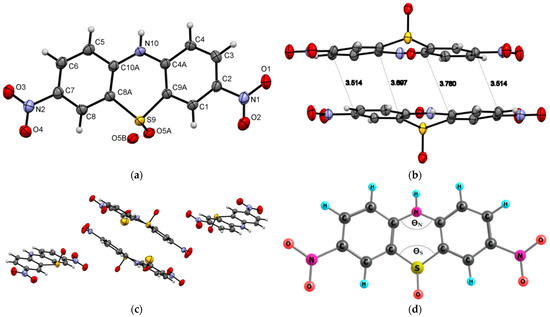
Figure 3.
ORTEP diagram of 3,7-dinitro-10H-phenothiazine 5-oxide (3) with 50% probability. Top view (a), packing (b,c) and structure with denoted bending angles in the middle phenothiazine ring ΘS and ΘN (d).
In the final step, we decided to replace heteroatoms of the phenothiazine core to establish the influence of such modification on the structural properties of studied compounds. According to the literature, changing the nitrogen atom into a sulfur atom, which leads to thianthrene derivatives, would preserve the bent spatial arrangement of the atoms, hindering π-π stacking interactions. Such a configuration is abundant in molecules already described in previous paragraphs. Aiming to increase the number of π-stacks in the crystal structure, we chose more planar phenazines as synthetic targets, where nitrogen is placed instead of a sulfur atom. There are already published examples of synthesizing this class of compounds in the literature. Therefore, further experiments were conducted to explore some synthetic routes that gave the aforementioned compounds. Additionally, we tried to explain the mechanism of producing symmetrical and unsymmetrical phenazines.
We have recently reported the direct oxidative condensation to introduce the carbon-nitrogen bond by a one-step method [35]. To synthesize the symmetrical phenazine 5 and the unsymmetrical phenazines 6, we applied two other types of chemical transformations. One led to symmetrical molecules and was based on the oxidative condensation of two identical molecules, i.e., 8-isopropyl-2-methylquinolin-5-amine (4a) (Scheme 3). In the second route, we chose the oxidative condensation between two electron-rich amines (ERA) N1,N1-dimethylbenzene-1,4-diamine and selected 8-(alkyl)-2-methylquinolin-5-amines 4 (Scheme 4). We chose silver carbonate adsorbed on celite as an oxidizing agent in both methods. The transformation occurred with a good yield for symmetrical phenazine 5 (Scheme 3) and a poor yield for the unsymmetrical phenazines 6 (Scheme 4). The deposition of metallic silver during the reaction progress suggests the involvement of electron transfer processes from ERA to silver(I) salt. This assumption is grounded on the preparation of the 1,2,4,5-tetrakis(dimethylamino)benzene dication iodide-triiodide through iodination of 1,2,4,5-tetrakis(dimethylamino)benzene patented by Staab et al. [36].
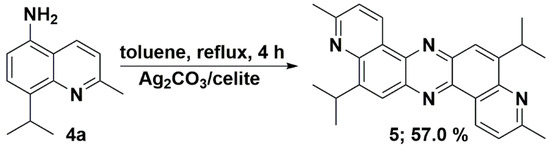
Scheme 3.
Synthetic route of symmetrical 5,12-diisopropyl-3,10-dimethyldipyrido[3,2-a:3′,2′-h]phenazine (5).
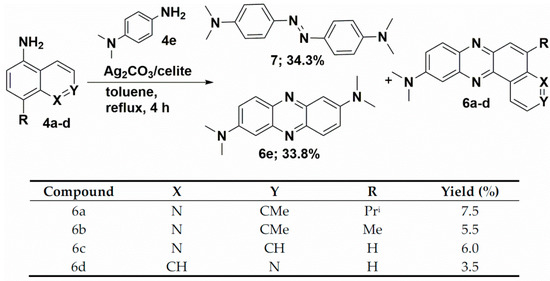
Scheme 4.
Synthetic route of phenazines 6.
Seth and co-workers reported the synthesis of symmetrical phenazines on non-radical C-H activation mode of C-N coupling during the oxidative self-coupling of anilines catalyzed by Pd-Ag and Cu-Ag nanoclusters [37,38]. They show the superiority of Pd-Ag over Cu-Ag nanoclusters during the synthesis of phenazines, with yields up to 87%. As another product, they isolated azoaryls with good yields (up to 89%) by applying the same nanoclusters, with superiority of Cu-Ag. In order to discern the character of the reaction mechanism, ionic or radical, they used radical scavenger (2,2,6,6-tetramethylpiperidin-1-yl)oxyl (TEMPO). Due to the lack of evidence of the impact of TEMPO, they excluded the radical mechanism of phenazine formation. Inspired by the work of Seth and co-workers [37,38], we employed only silver carbonate as a mildly basic oxidizing agent. We observed a similar double C-N bond formation involving two different amine molecules. However, the synthesis of unsymmetrical phenazines 6 is more complicated. Both used amine molecules 4 and N1,N1-dimethylbenzene-1,4-diamine could participate in electron transfer to silver carbonate, however, at a different rate, due to their redox potentials (Scheme 5). EPR techniques do not readily detect free radicals 4a’ derived from ERA [39,40]. The evidence of their presence may be an azoarene of the 7 types (Scheme 5), the formation of which accompanied the synthesis of phenazines, similar to Seth and co-workers’ studies [37,38]. For any synthesis of unsymmetrical phenazine 6, we identified compounds 6e and 7, which were confirmed by the original sample addition. This observation partially explains the much lower yields of unsymmetrical phenazines 6 (Scheme 4). On the basis of the literature and our present results, we propose a plausible mechanism for the formation of phenazines (symmetrical 5 and unsymmetrical 6) under applied reaction conditions. For simplicity, the postulated pathways are based on amine 4e (Scheme 5). The presented mechanism illustrates the influence of the silver(I) cation as an oxidizing agent and oxygen from the air. During transformation, molecules of water are created.
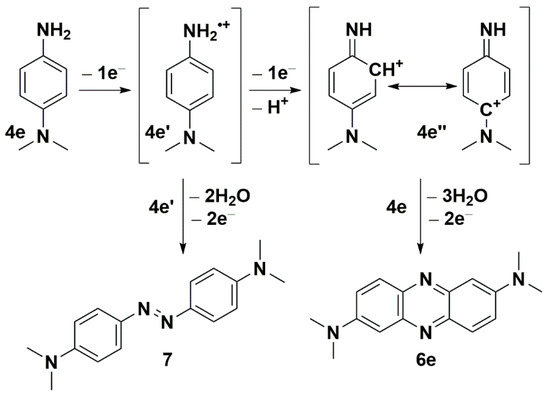
Scheme 5.
The postulated pathways for the formation of phenazine 6e and (E)-4,4′-(diazene-1,2-diyl)bis(N,N-dimethylaniline) (7) from amine 4e.
Compound 5 crystallizes in the tetragonal system (P42/n space group) with a = 21.1688(8) Å, b = 21.1688(8) Å, c = 4.6776(3) Å, and α = β = γ = 90°. Figure 4 presents the molecular (a) and crystal (b) structure of phenazine 5. Figure 4a–c shows that the planar molecular skeleton of molecule 5 promotes the formation of π-stacks with intermolecular distances of 3.652–3.690 Å. Hirshfeld’s dnorm surface indicates these regions as white or light blue colors in Figure 4d. More clearly, these π-stacking arrangements are identified in the shape index map with red and blue triangles (brown ellipses in Figure 4e) and the curvedness map as large green flat areas along the aromatic structural fragments of compound 5 (Figure 4f).
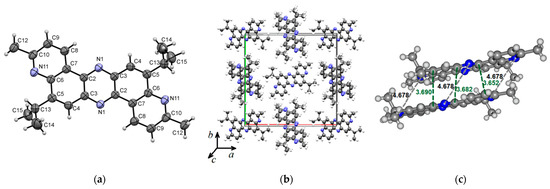
Figure 4.
ORTEP diagram of 5,12-diisopropyl-3,10-dimethyldipyrido[3,2-a:3′,2′-h]phenazine (5) with 50% probability. Top view (a), packing (b) and distances (c). Hirshfeld surfaces are mapped with dnorm (d), shape index (e), and curvedness (f).
Compound 6a crystallizes in the triclinic system (P-1 space group) with a = 9.0731(6) Å, b = 9.3169(7) Å, and c = 11.5962(8) Å. Figure 5 presents the molecular (a) and packing (b) structure of phenazine 6a. The distance between the centroids of the azine rings of neighboring molecules is equal to 3.628(1) Å.
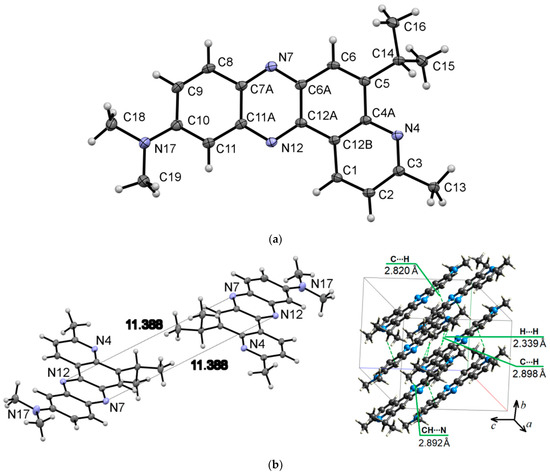
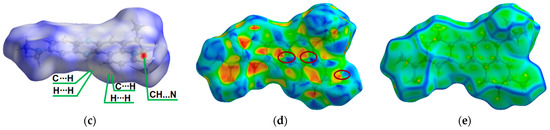
Figure 5.
ORTEP diagram of 5-isopropyl-N,N,3-trimethylpyrido[3,2-a]phenazin-10-amine (6a) with 50% probability. Top view (a), and packing (b). Hirshfeld surfaces for molecule 6a are mapped with dnorm (c), shape index (d), and curvedness (e).
The Hirshfeld dnorm surface for molecule 6a illustrates the prominent red spot, which corresponds to the intermolecular CH⋯N interaction with a distance of 2.892 Å (Figure 5b,c). The intermolecular H…H and C⋯H contacts (Figure 5b,c) appear as pale red areas in the dnorm. The latter belongs to the C-H⋯π interactions. The shape index (Figure 5d) confirms the presence of π-π stacking interactions as closely spaced red and blue triangles encircled by brown ellipses. Figure 5e shows π-π stacking with a flat zone in a curvedness map.
Mortzfeld et al. [41] reported the alkylation methodology of phenazine. As alkylation reagents, they chose methyl iodide, trimethyloxonium tetrafluoroborate, and dimethyl sulfate. They received stable mono N-alkylated products with good to poor yield: 86% for dimethyl sulfate, 25% for trimethyloxonium tetrafluoroborate, and only 19% for methyl iodine
2.2. Anomeric Effect of Compound 2a
The fundamental impact of stereochemical properties on a molecule’s biological activity makes the conformational aspect of research essential in many fields of organic chemistry. One of the crucial phenomena altering conformational behavior and, consequently, the reactivity of saturated heterocyclic systems is the anomeric effect, also known as the Edward–Lemieux effect [42,43]. It characterizes the propensity of heteroatomic substituents neighboring a heteroatom embedded in the cyclohexane ring to favor the axial position instead of the less sterically hindered equatorial orientation. In the IUPAC Compendium of Chemical Terminology, this is “a special case of a general preference for synclinal (gauche) conformations about the bond C-Y in the system X-C-Y-C where X and Y are heteroatoms having non-bonding electron pairs, commonly at least one of which is nitrogen, oxygen or fluorine” [44].
In the 1970s, Bentrude et al. initiated conformation studies concerning saturated 1,3,2-dioxaphosphinane rings [45,46], which structures mirror features of the carbohydrate molecules. As inferred, the preferential axial position of substituents is the one adjacent to oxygen. Hence, the anomeric effect was determined to be the most dominant conformation-controlling factor for compounds with 1,3,2-dioxaphosphinane rings.
Our results show that compound 2a also undergoes the anomeric effect. Both forms of chair conformers were conveniently identified by 31P{1H}-NMR spectroscopy and X-ray diffraction measurements (Figure 2, Figure 6 and Figure 7). For molecule 2a, the non-bonding p oxygen orbitals interact with phosphorus, and the charge is transferred through the nitrogen atom to the thiazine-type ring, which possesses empty π-accepting orbitals. Moreover, the energies of the two possible chair conformers differ by about 1.45 kcal/mol or 6.07 kJ/mol (gas-phase B3LYP/6-31g(d,p) calculation, Figure 6), which makes the existence of an equilibrium between these two forms at room temperature possible. As shown in the 31P{1H}-NMR spectrum (Figure 7), compound 2a has two separated conformers, δ1 = −6.91 and δ2 = −7.02 ppm. The relative signal intensity of both molecules 2a is in a ratio (ca. 72:1).
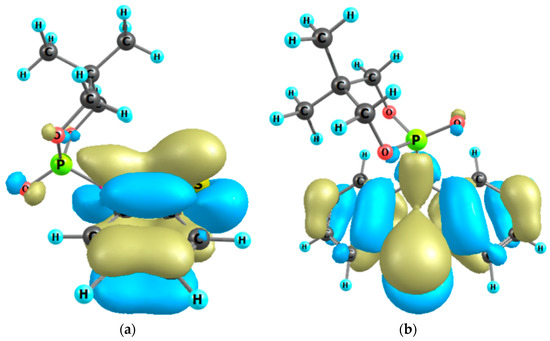
Figure 6.
The highest occupied MO of molecule 2a: chair conformer 1, side view (a); chair conformer 2, front view (b). The stabilizing hyperconjugation effect of non-bonding 2p oxygen orbitals via phosphorus with antibonding π*-accepting orbitals of the thiazine-type ring.
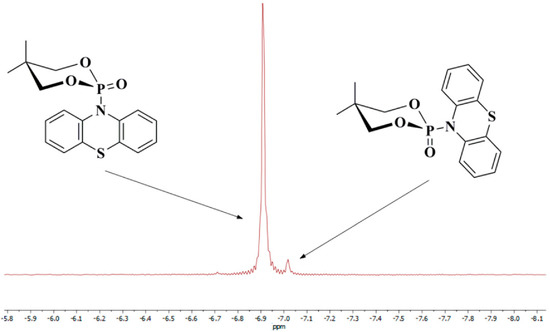
Figure 7.
31P{1H}−NMR spectrum for molecule 2a (two chair conformers) in CDCl3.
The nature of the anomeric effect is still a matter of controversy, and it is interesting to note that this challenging situation has attracted the attention of chemists and physicists for over half a century. It has been shown that steric, electrostatic, and stereoelectronic factors are all important forces that influence the anomeric effect. In the literature, there are several proposed explanations for the validity of the anomeric effect. One could be a stabilizing hyperconjugation contribution between the unshared electron pair on the oxygen atom in a 1,3,2-dioxaphosphinane ring and the σ* orbital for the P-C bond in the axial position (Figure 6). For conformer 2, the stabilizing hyperconjugation effect is weaker. Therefore, this form is less stable and was not isolated but only identified in the 31P{1H}-NMR spectrum (Figure 7).
The occurrence of the anomeric phenomenon entails certain geometric features that are consistent with the above-noted hyperconjugative delocalization: some bonds are lengthened, others are shortened, and the angles are widened or reduced [47]. The assignments of conformation were based on NMR, X-ray, literature analysis, and our previous results [28,32,45]. According to the Karplus equation and the couplings constant, the order of chemical shifts for CH2 protons, δHaxial > δHequatorial, was determined. The methylene protons of compound 2a showed two distinct double doublets in the 1H-NMR spectrum at δ = 3.60 and 3.74 ppm. The expected long-range couplings (3JP,Haxial = 3.7 Hz and 3JP,Hequatorial = 22.9 Hz) between phosphorus and one of the bridged methylene protons are observed.
2.3. Photophysical Properties of the Phenothiazine Derivatives 2a and 3
Figure 8a depicts the absorption spectra of molecule 2a in MeCN in the 600–190 nm region. Phenothiazine 2a has two absorption bands located at 280 and 234 nm, which can be ascribed to the n-π* and π-π* transitions, respectively, typical of many phenothiazines [48]. In addition, we can observe a band below 200 nm, similar to other phenothiazines [49]. In comparison with 10H-phenothiazine (λmax = 252 nm and 316 nm [50]), the presence of N-phosphoryl substituent in compound 2a caused a hypsochromic effect. Non-planar structure of 2a makes the n-π assignment questionable.
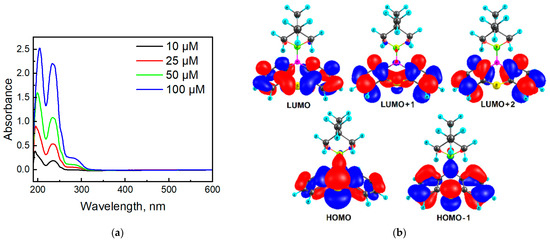
Figure 8.
Absorption spectra of molecule 2a in MeCN (a) and orbital diagrams for compound 2a calculated at the B3LYP/6-31G(d,p) level of theory (b).
According to quantum chemical calculations by the TDDFT/B3LYP/6-31G(d,p) method, the weak absorption in the range 300–275 nm (absorption band 280 nm in Figure 8) in the experimental spectrum of molecule 2a is manifested as a broadening on the right side of the spectrum and corresponds to a series of low-intensity transitions S0 → S1, S0 → S2 and S0 → S3 at 286 (f = 0.0148), 273 (f = 0.0141), and 265 nm (f = 0.0401), respectively. The electronic transitions and frontier molecular orbitals (HOMO—highest occupied molecular orbital; LUMO—lowest unoccupied molecular orbital) are presented in Table S1 and Figure 8b, Figures S9 and S10 in Supplementary Materials. All these three transitions into S1, S2, and S3 states of compound 2a possess a similar orbital nature, S0 → S1 (HOMO → LUMO), S0 → S2 (HOMO → LUMO + 1), and S0 → S3 (HOMO → LUMO + 2, HOMO-1→ LUMO), and are due to the charge transfer from the phenothiazine core to benzene fragments (Figure 8b). The same orbital nature of the first absorption band is also characteristic of N-methyl phenothiazine [48]. The next intense absorption band (234 nm) was calculated in the range of 250–220 nm with a maximum at 238 nm (Figure S7), and it can be assigned to the electronic transitions into the S4, S5, and S6 excited states (Table S1 in Supplementary Materials).
The fluorescent band of compound 2a (Figure 9) is located at 374 nm, and it is also blue-shifted in comparison with the emission band of 10H-phenothiazine (λem = 454 nm [50]) and N-methyl-phenothiazine (λem = 450 nm [51]). The TDDFT calculations for molecule 2a clearly indicate the observed emission band at 374 nm (Figure 9), which corresponds to the vertical S1→S0 transition calculated at 386 nm (Table S2, Supplementary Materials).
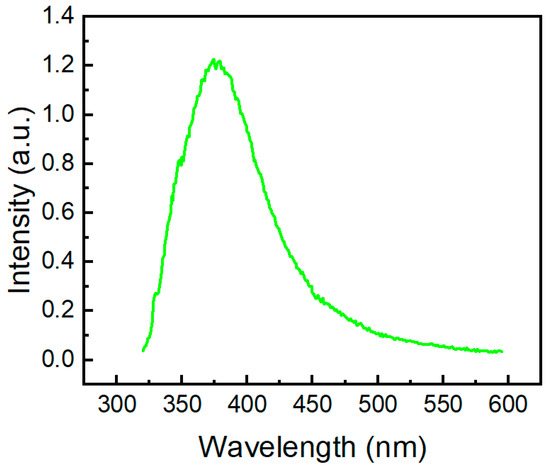
Figure 9.
Fluorescence spectrum of molecule 2a in DMSO excited at 300 nm.
We also investigated the emission behavior of compound 2a at 77 K (Figure 10). When excited at 291 nm, molecule 2a shows an emission band with two peaks at 434 nm and 457 nm. Its profile of phosphorescence is similar to the phosphorescence spectra of pure 10H-phenothiazine (λ 500 nm [50], λ 535 nm [52]). Similar to absorption and fluorescence, it is clear that the phosphoryl moiety in compound 2a causes a large hypsochromic effect (c.a. 70 nm) in the phosphorescence spectrum. Phosphorescence bands of molecule 2a are located in the same region as the phosphorescence of two N-acylphenothiazine derivatives, namely 10H-phenothiazin-10-yl)(phenyl)methanone and 1-(10H-phenothiazin-10-yl)ethan-1-one) [27].
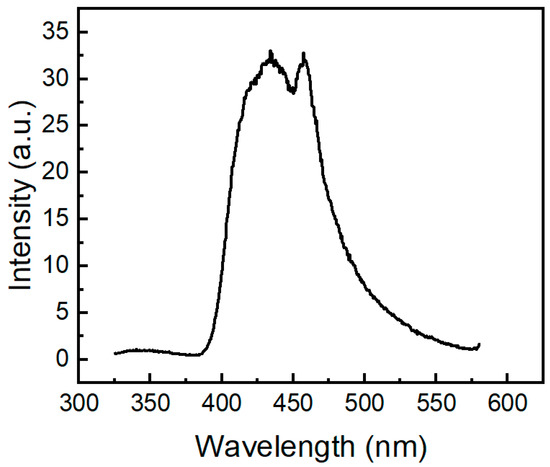
Figure 10.
Phosphorescence spectrum of molecule 2a (200 µM) in 2-methyltetrahydrofuran glass at 77 K excited at 291 nm.
We also recorded the phosphorescence spectrum of compound 2a in crystals at 298 K (Figure 11). Molecule 2a exhibits a relatively low-intensity fluorescence peak at 373 nm and an intense phosphorescence peak centered at 540 nm. Phosphorescence characteristics of N-phosphorylated phenothiazine 2a are similar to phosphorescence spectra of 10H-phenothiazin-10-yl)(phenyl)methanone and 1-(10H-phenothiazin-10-yl)ethan-1-one) that have phosphorescence emission at 460–600 nm, with the maximum located at 530 nm and 525 nm, respectively [8]. The TDDFT results for compound 2a show that the lowest triplet (T1) state lies at 2.46 eV and involves mixed electronic configurations HOMO → LUMO, HOMO-1 → LUMO + 2, HOMO-2→ LUMO + 3, HOMO-3 → LUMO + 1. Similar to other phenothiazines [52,53], for molecule 2a, S1, S2, S3, and T1 possess charge transfer characters with a certain degree of locally excited nature. Thus, a thermally activated reverse singlet-to-triplet intersystem crossing (RISC) can probably occur from the upper triplet to singlet levels providing phosphorescence in compound 2a since the singlet-triplet (S1-T1) energy gap is large (0.76 eV).
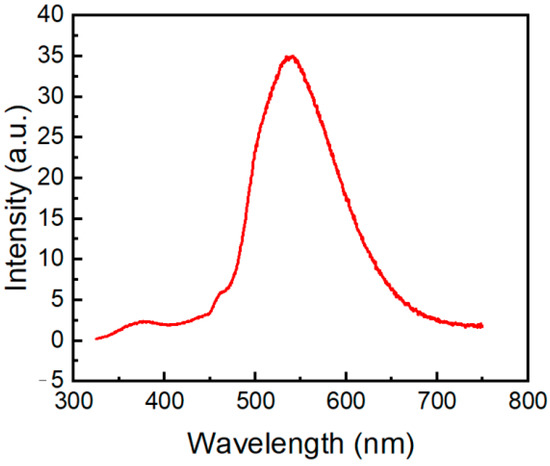
Figure 11.
Phosphorescence spectrum of compound 2a in solid state excited at 300 nm.
The absence of π-π stacking contacts in molecule 2a and strong charge-transfer character of the T1 state (Figure 8b) provide efficient σ-π orbital mixing, which is responsible for the increase in spin-orbit coupling and intersystem crossing in this compound affecting high phosphorescence [54].
Figure 12 shows the steady-state emission spectra of the phenothiazine derivative 2a under different conditions.
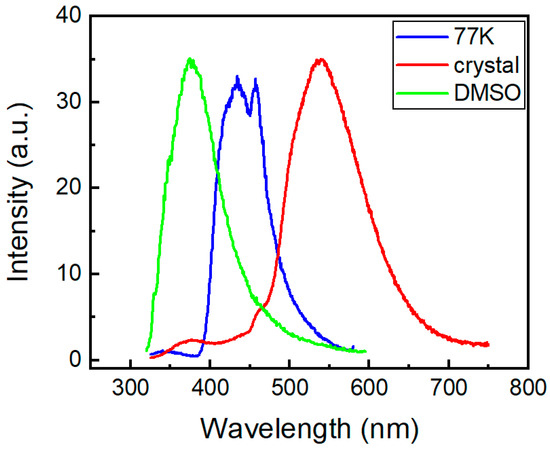
Figure 12.
Steady-state emission spectra of compound 2a under different conditions.
Dinitrophenothiazine S-oxide 3 has four absorption bands located at 271 nm, 359 nm, 398 nm, and 528 nm (Figure 13a). In comparison with phenothiazine S-oxide (λmax 229 nm, 271 nm, 303 nm, 336 nm [55]), the two additional nitro groups presented in the phenothiazine S-oxide skeleton caused a large bathochromic effect. TDDFT calculations predict that the first high-intensity absorption maximum in molecule 3 (Figure 13) belongs to the S0→S1 transition calculated at 376 nm (f = 0.6989) in DMSO solution (Table S1). The S0→S1 transition is provided by the HOMO→LUMO configuration and possesses a charge transfer (CT) nature; this is mainly CT from sulfur to nitro groups (Figure 13b). For comparison, the first S0→S1 transition for phenothiazine S-oxide was calculated at 305 nm, which confirms the presence of a bathochromic effect due to nitro groups. The difference in the calculated and experimental values of the wavelengths can be explained by an incomplete allowance for solvation effects within the framework of the TDDFT/B3LYP/6-31G(d,p) level of theory. Figure 13b indicates that the HOMO is an antibonding orbital with respect to C-N and C-S bonds in the central ring, showing that there is a strong C-C bonding character, whereas the LUMO is mostly a non-bonding orbital in the center. Thus, the HOMO-LUMO excitation leads to much shorter and stronger C-N and C-S bonds in the S1 excited state, but C-C bonds are becoming weaker (Figure S4).
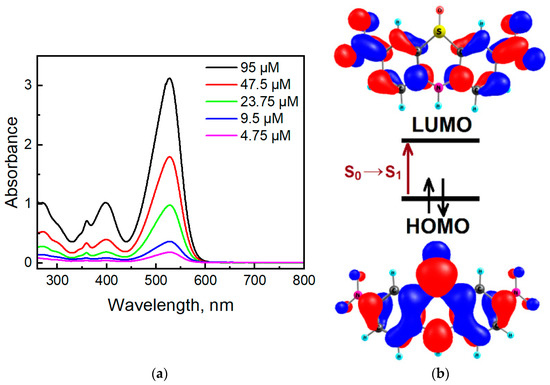
Figure 13.
Absorption spectra of compound 3 in DMSO (a) and orbital diagrams for compound 3 calculated at the B3LYP/6-31G(d,p) level of theory (b).
Compound 3 shows a fluorescent band located at 647 nm (Figure 14). The phenothiazine S-oxide or its N-alkyl analogs exhibit fluorescence in the 350–450 nm region [56,57]. For molecule 3, TDDFT calculations indicate the observed emission band at 647 nm as the S1 → S0 transition calculated at 568 nm (Table S2). Two electron-withdrawing nitro groups remove the electron density from the sulfur atom and decrease the LUMO energy, and the emission band of dinitrophenothiazine S-oxide is red-shifted by 200 nm. Moreover, molecule 3 shows a large Stokes shift (122 nm), which indicates that the geometry of the singlet excited state differs from the geometry of the ground state. In the S0 state of molecule 3, the bending angles in the middle phenothiazine ring denoted as ΘS and ΘN were calculated to be 95.08° and 122.78°, respectively, while in S1, these angles are 104.50 and 127.27 (Figure S4, SI). Much larger distortions are calculated in the C-N and C-S bond lengths upon the S1 → S0 transition (Figure S4) according to the strong differences in the bonding-nonbonding characters of the frontier orbitals (Figure 13b). Typical distortions also occur in the nitro groups since LUMO is bonding for the C-N links and anti-bonding for the N-O chemical bonds.
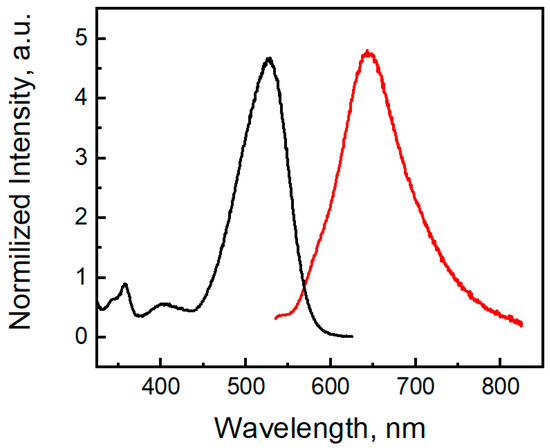
Figure 14.
Excitation (black) and emission (red) spectra of compound 3 (4.75 µM) in DMSO.
In contrast to molecule 2a and 10H-phenothiazine, the 5,5-dioxide-based push-pull chromophore [27] compound 3 shows no room-temperature phosphorescence, which is typical for nitroaromatic compounds. Asymmetric NO2 stretching and hindered rotation of nitro-groups represent accepting and promoting modes, respectively, for efficient phosphorescence quenching of compound 3.
3. Materials and Methods
3.1. Materials
All experiments were carried out in an atmosphere of dry argon, and flasks were flame dried. Solvents were dried by usual methods (diphenyl ether, diethyl ether, and THF over benzophenoneketyl, CHCl3, and CH2Cl2 over P4O10, hexane, and pyridine over sodium-potassium alloy) and distilled. Chromatographic purification was carried out on silica gel 60 (0.15–0.3 mm, Macherey-Nagel GmbH & Co. KG, Dueren, Germany). Crotonaldehyde, 4a,12a-dihydro-12H-benzo[5,6][1,4]thiazino[2,3-b]quinoxaline, N1,N1-dimethylbenzene-1,4-diamine, 2,2-dimethylpropane-1,3-diol, 10H-phenothiazine, phosphoryl chloride, 2-isopropylaniline, isoquinolin-5-amine, quinolin-5-amine, sodium hydride (dry, 95%), stannous chloride, silver carbonate on Celite and o-toluidine were purchased from Sigma–Aldrich (Poznań, Poland), and were used without further purification.
3.2. Instrumentation
NMR spectra were obtained with Avance 400 and 500 spectrometers (Bruker, Billerica, MA, USA) operating at 500.2 or 400.2 MHz (1H) and 125.8 or 100.6 MHz (13C) and 202.47 (31P) at 21 °C. Chemical shifts referenced to ext. TMS (tetramethylsilane) (1H, 13C) or 85% H3PO4 (31P), or using the residual CHCl3 signal (δH 7.26 ppm) and CDCl3 (δC 77.1 ppm) as internal references for 1H and 13C-NMR, respectively. Coupling constants are given in Hz. The LCMS-IT-TOF analysis was performed on an Agilent 1200 Series binary LC system coupled to a micrOTOF-Q system mass spectrometer (BrukerDaltonics, Bremen, Germany). High-resolution mass spectrometry (HRMS) measurements were performed using a Synapt G2-Si mass spectrometer (Waters, New Castle, DE, USA) equipped with an ESI source and quadrupole-time-of-flight mass analyzer. To ensure accurate mass measurements, data were collected in centroid mode, and mass was corrected during acquisition using leucine enkephalin solution as an external reference (Lock-Spray) (Waters, New Castle, DE, USA). The measurement results were processed using the MassLynx 4.1 software (Waters, Milford, MA, USA) incorporated within the instrument. A Nicolet iS50 FTIR spectrometer was used for recording spectra in the IR range of 4000–400 cm−1. FTIR spectra were recorded on a Perkin Elmer (Schwerzenbach, Switzerland) spectrophotometer in the spectral range of 4000–450 cm−1 with the samples in the form of KBr pellets. Elementary analysis was performed using a Vario EL III apparatus (Elementar, Langenselbold, Germany). Differential scanning calorimetry (DSC) measurements were performed using a Q2000 calorimeter (TA Instruments, New Castle, DE, USA) in a nitrogen stream at a scanning rate of 10 °C/min. Samples were analyzed in aluminum pans in the temperature range of 50 to 350 °C. Melting points were determined on an MPA100 OptiMelt melting point apparatus (Stanford Research Systems, Sunnyvale, CA, USA) and are uncorrected. UV/Vis absorption spectra were measured on a SHIMADZU UV-VIS-NIR spectrophotometer using quartz (Suprasil) cuvettes (10 mm path length). Fluorescence spectra were recorded on an FLS-900 spectrofluorimeter (Edinburgh Instruments, Edinburgh, UK) and were measured in quartz (Suprasil) cuvettes (10 mm path length) at 25 °C. The phosphorescence spectrum was recorded on an FLS-900 spectrofluorimeter (Edinburgh Instruments, Edinburgh, UK) at 77 K using a 3 mm (inner diameter) quartz tube inside a quartz liquid nitrogen Dewar flask.
3.3. Synthesis of (4a,12a-Dihydro-12H-benzo[5,6][1,4]thiazino[2,3-b]quinoxalin-12-yl)(phenyl)methanone Followed Our Procedure Described in the Literature [23]
(4a,12a-dihydro-12H-benzo[5,6][1,4]thiazino[2,3-b]quinoxalin-12-yl)(phenyl)methanone (1) [23], m.p. = 200.0–200.5 °C; 1H-NMR (DMSO-d6; 400.2 MHz) δ = 7.25 (t, 3JH,H = 7.4 Hz, 2H, aromatic), 7.31 (t, 3JH,H = 7.0 Hz, 1H, aromatic), 7.41 (d, 3JH,H = 7.7 Hz, 3H, aromatic), 7.50 (t, 3JH,H = 8.8 Hz, 2H, aromatic), 7.64 (t, 3JH,H = 7.7 Hz, 1H, aromatic), 7.69 (d, 3JH,H = 7.8 Hz, 1H, aromatic), 7.73 (t, 3JH,H = 7.7 Hz, 1H, aromatic), 7.95 (d, 3JH,H = 8.3 Hz, 1H, aromatic), 8.11 (d, 3JH,H = 8.1 Hz, 1H, aromatic); 13C{1H}-NMR (DMSO-d6; 100.6 MHz) δ = 124.8, 125.1, 127.2, 127.5, 127.7, 127.9, 128.0, 128.1, 128.4, 130.2, 130.3, 130.9, 134.8, 135.9, 138.6, 139.7, 145.3, 149.8, 169.5; CCDC (The Cambridge Crystallographic Data Centre) 2177507.
3.4. Phosphorylation of Phenothiazine
The synthesis of 2-chloro-5,5-dimethyl-1,3,2-dioxaphosphinane 2-oxide followed our procedure described in the literature [28]. The synthesis of 5,5-dimethyl-2-(10H-phenothiazin-10-yl)-1,3,2-dioxaphosphinane 2-oxide (2a) was based on procedures described in the literature [58].
To a suspension of NaH (0.96 g, 40.0 mmol) in THF (100 mL), 10H-phenothiazine (4.00 g, 20.1 mmol) was added and stirred until the evolution of H2 ceased. Reagents were stirred under reflux for 30 min. under argon. 2-Chloro-5,5-dimethyl-1,3,2-dioxaphosphinane 2-oxide (3.70 g, 20.1 mmol) was then added to the reaction mixture, which was refluxed overnight. After evaporation of the solvent to give a solid, water (20 mL) and chloroform (100 mL) were added. The organic layer was separated, and the aqueous layer was extracted four times with chloroform. The combined organic layers were dried over MgSO4. After the solvent evaporated, the crude product was purified by column chromatography on silica gel using methanol/dichloromethane as eluent to afford a crude solid, and finally, crystallization from a mixture of CH2Cl2 and hexane occurred to yield solids as follows:
5,5-Dimethyl-2-(10H-phenothiazin-10-yl)-1,3,2-dioxaphosphinane 2-oxide (2a) 0.66 g (2.78 mmol, 58 %), white powder, m.p. = 183–185 °C; 1H-NMR (CDCl3; 500.2 MHz) δ = 0.74 (s, 3H, CH3), 1.25 (s, 3H, CH3), 3.60 (ddd, 3JP,H = 22.9 Hz, 3JH,H = 11.2 Hz, 4JH,H = 1.5 Hz, 2H, CH), 3.74 (dd, 3JH,H = 10.5 Hz, 3JP,H = 3.7 Hz, 2H, CH), 7.17 (td, 3JH,H = 7.6 Hz, 4JH,H = 1.2 Hz, 2H, aromatic), 7.26 (dd, 3JH,H = 7.7 Hz, 4JH,H = 1.5 Hz, 2H, aromatic), 7.34 (dd, 3JH,H = 7.7 Hz, 4JH,H = 1.4 Hz, 2H, aromatic), 7.60 (dd, 3JH,H = 8.0 Hz, 4JH,H = 0.9 Hz, 2H, aromatic); 13C{1H}-NMR (CDCl3; 125.8 MHz) δ = 20.5, 22.0, 32.1 (d, 3JP,C = 32.1 Hz), 77.5 (d, 3JP,C = 6.9 Hz), 123.6, 126.0, 126.8, 127.7, 127.6, 130.0 (d, 3JP,C = 5.9 Hz), 139.5 (d, 3JP,C = 6.9 Hz); 31P-NMR (CDCl3; 202.5 MHz) δ1 = −6.91 (t, 3JP,H = 22.7 Hz), 31P{1H}-NMR (CDCl3; 202.47 MHz) δ1 = −6.91 and δ2 = −7.02; GC-MS: tr = 9.6 min, (EI) M+ = 347.1 (68%), (M-C5H10O3P)+ = 198.1 (100); UV-Vis (metanol; λ [nm] (logε)): 273 (3.12), 252 (3.58), 230 (4.01), 204(3.99); IR (KBr): ν = 3059.24, 2984.65, 2969.43, 2886.37, 1463.03, 1298.53, 1269.99, 1225.79, 1055.73, 1008.36, 995.08, 981.81, 841.68, 795.30, 773.30, 759.03, 509.49, 552.50 cm−1; CCDC (The Cambridge Crystallographic Data Centre) 2177508. 2-Mercapto-5,5-dimethyl-1,3,2-dioxaphosphinane 2-oxide (2b) CCDC (The Cambridge Crystallographic Data Centre) 2178722.
3.5. Synthesis of 3,7-Dinitro-10H-phenothiazine 5-oxide (3)
10H-Phenothiazine (10.0 g, 50.2 mmol) was added in one portion to 100 mL of 65% nitric acid stirred in a wide beaker at room temperature. The immediate evolution of brown vapor was observed, and the reaction mixture’s temperature increased to 45 °C. After stirring for 1h, the deep red solution was filtered through a sintered glass funnel under reduced pressure, and the obtained solid was flushed with 200 mL of water and 100 mL of methanol. The crude product was boiled for 5 min in 300 mL of THF, filtered, and dried under reduced pressure. To obtain the crystals, the saturated solution of the purified product in CHCl3 was kept at −20 °C for a week.
3,7-Dinitro-10H-phenothiazine 5-oxide (3) [24] 10.5 g (34.4 mmol, 68.5 %) as a yellow powder, mpdec. = 310.0-316.5 °C; 1H-NMR (DMSO-d6; 500.2 MHz) δ = 7.61 (d, 3JH,H = 9.1 Hz, 2H, aromatic), 8.48 (dd, 3JH,H = 9.1 Hz, 4JH,H = 2.6 Hz, 2H, aromatic), 8.96 (d, 4JH,H = 2.6 Hz, 1H, aromatic), 12.32 (s, 1H, NH); 1H-NMR (DMSO-d6/KOD; 500.2 MHz) δ = 7.56 (d, 3JH,H = 9.1 Hz, 2H, aromatic), 8.42 (dd, 3JH,H = 9.1 Hz, 4JH,H = 2.7 Hz, 2H, aromatic), 8.89 (d, 4JH,H = 2.6 Hz, 1H, aromatic); 13C{1H}-NMR (DMSO-d6; 100.6 MHz) δ = 118.8, 121.5, 128.1, 128.2, 139.9, 141.7; 13C{1H}-NMR(DMSO-d6/KOD; 100.6 MHz) δ = 120.2, 121.2, 127.7, 128.4, 140.9, 141.9; CCDC (The Cambridge Crystallographic Data Centre) 2177509.
3.6. General Procedure for the Synthesis of 8-(Alkyl)-2-methylquinolin-5-amines
The synthesis of 2,8-dimethylquinoline, 8-(isopropyl)-2-methylquinolin-5-amine (4a) and 2,8-dimethylquinolin-5-amine (4b) followed our procedure described in the literature [35].
Synthesis of 2,8-dimethylquinoline.
Toluene (50 mL) and crotonaldehyde (2.6 mL, 2.2 g, 31.4 mmol) were added to the solution of o-toluidine (1.7 g, 15.7 mmol) in aqueous 6 M HCl (200 mL) and were heated under reflux for 16 h. The mixture was allowed to cool down to room temperature. The aqueous layer was separated and neutralized with an aqueous solution of K2CO3. After extraction with CH2Cl2 (3 × 50 mL), the organic layer was separated and dried over MgSO4, then filtered and distilled bp 100–110 °C/3 mmHg. The liquid mixture was dissolved in concentrated 36% HCl (100 mL) at 5 °C, and ZnCl2 (2.7 g, 20.0 mmol) was added with vigorous stirring for one hour. The precipitate was filtered and washed with cold 3 M aq. HCl and dried in air. The solid was washed with iPrOH and dried. The received white solid was added to a 10% ammonia solution and extraction with Et2O (3 × 50 mL). The organic layer was separated and dried over MgSO4 to afford:
2,8-Dimethylquinoline as a colorless oil, 1.8 g (11.6 mmol, 74%), bp 98–100 °C/3 mmHg; 1H-NMR (CDCl3; 400.2 MHz) δ = 2.74 (s, 3H, CH3), 2.80 (s, 3H, CH3), 7.24 (d, 3JH,H = 8.3 Hz, 1H, aromatic), 7.34 (dd, 3JH,H = 8.1 Hz, 3JH,H = 7.0 Hz, 1H, aromatic), 7.50 (d, 3JH,H = 7.0 Hz, 1H, aromatic), 7.58 (d, 3JH,H = 8.0 Hz, 1H, aromatic), 7.97 (d, 3JH,H = 8.3 Hz, 1H, aromatic); 13C{1H}-NMR (CDCl3; 125.8 MHz) δ = 18.0, 25.6, 26.9, 121.6, 125.2, 125.5, 126.3, 129.4, 136.2, 136.5, 147.0, 157.8.
Synthesis of 2,8-dimethyl-5-nitroquinoline.
2,8-Dimethylquinoline (1.2 g, 7.5 mmol) was dissolved in a mixture of concentrated H2SO4 and HNO3 (4.5 and 10.5 mL, respectively) at 5 °C. After stirring for 1 h at room temperature, no evolution of gas was observed, so the reaction mixture was heated up to 70 °C and stirred overnight. After this time, the reaction mixture was poured down into a beaker containing 25 g of ice and 25 mL of water, and the precipitated solid was filtered off, washed with 10 mL of cold water, and dried on air, giving:
2,8-Dimethyl-5-nitroquinoline as a beige solid, 1.1 g (5.6 mmol, 74%); DSC = 78.80–78.86 °C; 1H-NMR (CDCl3; 500.2 MHz) δ = 2.78 (s, 3H, CH3), 2.87 (d, 4JH,H = 1.0 Hz, 3H, CH3), 7.49 (d, 3JH,H = 8.9 Hz, 1H, aromatic), 7.59 (dd, 3JH,H = 7.9 Hz, 4JH,H = 1.0 Hz, 1H, aromatic), 8.22 (d, 3JH,H = 7.9 Hz, 1H, aromatic), 8.91 (d, 3JH,H = 8.9 Hz, 1H, aromatic); 13C{1H}-NMR (CDCl3; 125.8 MHz) δ = 19.0, 25.4, 119.3, 123.6, 124.6, 127.3, 132.0, 143.8, 145.6, 146.6, 159.2; HRMS (TOF-ES+): m/z Calcd for C11H11N2O2 (M + H)+ = 203.0821, Found 203.0818.
Synthesis of 2,8-dimethylquinolin-5-amine (4b).
Stannous chloride (47.4 g, 250.0 mmol) was added to a stirred solution of 2,8-dimethyl-5-nitroquinoline (5.0 g, 25.0 mmol) in 6M hydrochloric acid (100 mL) and methanol (300 mL). After stirring for 30 min at room temperature, the reaction mixture was brought to reflux and stirred for 3 h. After cooling to room temperature, the mixture was basified with aqueous ammonia and extracted with chloroform (3 × 50 mL). The combined extract was dried over MgSO4 and evaporated to afford:
2,8-Dimethylquinolin-5-amine (4b) as a beige solid, 3.5 g (20.2 mmol, 81%); DSC = 62.89–63.54 °C; 1H-NMR (CDCl3; 500.2 MHz) δ = 2.67 (d, 4JH,H = 1.0 Hz, 3H, CH3), 2.73 (s, 3H, CH3), 3.97 (bs, 2H, NH2), 6.66 (d, 3JH,H = 7.5 Hz, 1H, aromatic), 7.21 (d, 3JH,H = 8.6 Hz, 1H, aromatic), 7.30 (dd, 3JH,H = 7.6 Hz, 4JH,H = 1.0 Hz, 1H, aromatic), 8.03 (d, 3JH,H = 8.6 Hz, 1H, aromatic); 13C{1H}-NMR (CDCl3; 125.8 MHz) δ = 17.5, 25.5, 109.3, 117.2, 120.1, 127.0, 129.66, 129.67, 140.1, 147.4, 157.6; HRMS (TOF-ES+): m/z Calcd for C11H11N2O2 (M + H)+ = 173.1079, Found 173.1074.
3.7. Synthesis of 5,12-Diisopropyl-3,10-dimethyldipyrido[3,2-a:3′,2′-h]phenazine (5)
The solution of 8-isopropyl-2-methylquinolin-5-amine (4a) (400 mg, 2.00 mmol) in toluene (50 mL) was brought to gentle reflux, and silver carbonate on celite (2.5 g, 50% w/w, 4.50 mmol Ag2CO3) was added in small portions over 30 min. The reaction mixture was stirred under reflux for a further 4 h and filtered under reduced pressure without cooling. The solid residue was flushed with ca. 50 mL of ethyl acetate until a clean, colorless filtrate was observed. The obtained solution was left to stand in the refrigerator overnight. The next day, the formed light yellow precipitate was collected by filtration, flushed with warm methanol, and recrystallized from chloroform to afford yellow crystals:
5,12-Diisopropyl-3,10-dimethyldipyrido[3,2-a:3′,2′-h]phenazine (5) 224 mg (0.57 mmol, 57.0%); DSC = 289.70 °C; 1H-NMR (CDCl3; 500.2 MHz) δ = 1.54 (2d, 3JH,H = 6.9 Hz, 12H, CH(CH3)2), 2.85 (s, 6H, CH3), 4.44 (2 septet, 3JH,H = 6.9 Hz, 2H, CH(CH3)2), 7.58 (2d, 3JH,H = 8.2 Hz, 2H, aromatic), 8.21 (2s, 2H, aromatic), 9.57 (2d, 3JH,H = 8.3 Hz, 2H, aromatic); 13C{1H}-NMR (CDCl3; 125.8 MHz) δ = 23.3, 25.7, 28.0, 122.4, 124.2, 125.5, 133.2, 140.6, 141.3, 148.2, 151.7, 160.1; IR (KBr):ν = 2956, 1586, 1472, 1420, 1373, 1233, 1136, 1050, 885, 847, 807cm−1; HRMS (TOF-ES+): m/z Calcd for C26H27N4 (M + H)+ = 395.2236, Found 395.2230; CCDC (The Cambridge Crystallographic Data Centre) 2177510.
3.8. Synthesis of Unsymmetrical Phenazines (6a–d)
The solution of appropriate quinoline (4a–c) or isoquinoline 4d (2.00 mmol) and N1,N1-dimethylbenzene-1,4-diamine (286 mg, 2.10 mmol) in toluene (50 mL) was brought to a gentle reflux, and silver carbonate on celite (2.5 g, 50% w/w, 4.50 mmol Ag2CO3) was added in small portions over 30 min. The reaction mixture was stirred under reflux for a further 4 h and filtered under reduced pressure without cooling. The solid residue was flushed with ca. 50 mL of ethyl acetate until a clean, colorless filtrate was observed. The obtained solution was concentrated and purified by column chromatography on silica gel using chloroform and/or ethyl acetate/hexane eluent system to afford:
5-Isopropyl-N,N,3-trimethylpyrido[3,2-a]phenazin-10-amine (6a) as orange crystals, 50 mg (0.15 mmol, 7.5%); DSC = 209.35 °C; 1H-NMR (CDCl3; 500.2 MHz) δ = 1.47 (d, 3JH,H = 6.9 Hz, 6H, 2CH3), 2.82 (s, 3H, CH3), 3.22 (s, 6H, NCH3), 4.33 (septet, 3JH,H = 6.9 Hz, 1H, CH(CH3)2), 7.23 (d, 3JH,H = 2.8 Hz, 1H, aromatic), 7.51 (d, 3JH,H = 8.3 Hz, 1H, aromatic), 7.57 (dd, 3JH,H = 9.5 Hz, 4JH,H = 2.8 Hz, 1H, aromatic), 8.00 (d, 4JH,H = 0.8 Hz, 1H, aromatic), 8.08 (d,3JH,H = 9.5 Hz, 1H, aromatic), 9.48 (d, 3JH,H = 8.2 Hz, 1H, aromatic); 13C{1H}-NMR (CDCl3; 125.8 MHz) δ = 23.3, 25.6, 27.7, 40.7, 104.3, 121.5, 122.0, 124.1, 125.6, 129.8, 133.3, 138,2, 140.1, 141.7, 144.1, 148.7, 149.4, 151.0, 159.9; IR (KBr): ν =3872, 3650, 2954, 2922, 2864, 2801, 2366, 1734, 1628, 1601, 1497, 1461, 1407, 1356, 1313, 1225, 1186, 1145, 1081, 1011, 966, 916, 884, 851, 810 cm−1; HRMS (TOF-ES+): m/z Calcd for C17H15N4 (M + H)+ = 331.1923, Found 331.1919; CCDC (The Cambridge Crystallographic Data Centre) 2177511.
N,N,3,5-Tetramethylpyrido[3,2-a]phenazin-10-amine (6b) as a brown powder, 33 mg (0.11 mmol, 5.5%); DSC = 217.68 °C; 1H-NMR (CDCl3; 500.2 MHz) δ = 2.83 (s, 3H, CH3), 2.87 (d, 4JH,H = 1.2 Hz, 3H, CH3), 3.23 (s, 6H, NCH3), 7.23 (d, 3JH,H = 2.8 Hz, 1H, aromatic), 7.52 (d, 3JH,H = 8.2 Hz, 1H, aromatic), 7.57 (dd, 3JH,H = 9.5 Hz, 3JH,H = 2.8 Hz, 1H, aromatic), 7.98 (dd, 4JH,H = 1.3 Hz,1H, aromatic), 8.08 (d, 3JH,H = 9.5 Hz, 1H, aromatic), 9.48 (d, 3JH,H = 8.2 Hz, 1H, aromatic); 13C{1H}-NMR (CDCl3; 125.8 MHz) δ = 19.0, 25.6, 40.7, 104.3, 121.5, 122.1, 124.0, 129.4, 129.9, 133.2, 138.2, 139.3, 140.0, 142.0, 144.1, 149.8, 151.1, 160.2; IR (KBr): ν = 3788, 3435, 2918, 1632, 1608, 1505, 1468, 1437, 1405, 1358, 1313, 1262, 1188, 1151, 1087, 1151, 1087, 1014, 914, 869, 803 cm−1; HRMS (TOF-ES+): m/z calcd for C19H19N4 (M + H)+ = 303.1610, Found 303.1606.
N,N-Dimethylpyrido[3,2-a]phenazin-10-amine (6c) as a red powder, 32 mg (0.12 mmol, 6.0%); DSC = 189.46 °C; 1H-NMR (CDCl3; 500.2 MHz) δ = 3.25 (s, 6H, NCH3), 7.24 (d, 3JH,H = 2.5 Hz, 1H, aromatic), 7.62 (ddd, 3JH,H = 9.5 Hz, 4JH,H = 2.8 Hz, 4JH,H = 1.1 Hz, 1H, aromatic), 8.11 (d,3JH,H = 4.3 Hz, 1H, aromatic), 8.13 (d, 3JH,H = 4.4 Hz, 1H, aromatic), 8.18 (d, 3JH,H = 9.5 Hz,1H, aromatic), 9.06 (dd, 3JH,H = 4.5 Hz, 3JH,H = 1.8 Hz, 1H, aromatic), 9.63 (dd, 3JH,H = 8.2 Hz, 4JH,H = 1.1 Hz, 1H, aromatic); 13C{1H}-NMR (CDCl3; 125.8 MHz) δ = 40.5, 103.7, 121.9, 122.0, 126.7, 130.0, 130.6, 131.4, 133.3, 138.4, 139.6, 141.9, 144.4, 149.9, 151.29, 151.34; IR (KBr): ν = 3340, 2923, 1630, 1601, 1509, 1472, 1413, 1358, 1261, 1149, 1096, 1024, 918, 808 cm−1; HRMS (TOF-ES+): m/z Calcd for C17H15N4 (M + H)+ = 275.1297, found 275.1290.
N,N-Dimethylpyrido[4,3-a]phenazin-10-amine (6d) as a red powder, 20 mg (0.07 mmol, 3.5%); DSC = 204.68 °C; 1H-NMR (CDCl3; 500.2 MHz) δ = 3.26 (s, 6H, NCH3), 7.25 (d, 3JH,H = 2.9 Hz, 1H, aromatic), 7.65 (dd, 3JH,H = 9.5 Hz, 4JH,H = 2.8 Hz, 1H, aromatic), 7.93 (dd, 3JH,H = 9.2 Hz, 4JH,H = 0.7 Hz, 1H, aromatic), 8.03 (d, 3JH,H = 9.2 Hz, 1H, aromatic), 8.12 (d, 3JH,H = 9.5 Hz,1H, aromatic), 8.89 (d, 3JH,H = 5.5 Hz, 1H, aromatic), 9.10 (dt, 3JH,H = 5.5 Hz, 4JH,H = 0.8 Hz, 1H, aromatic), 9.28 (d, 4JH,H = 1.0 Hz, 1H, aromatic); 13C{1H}-NMR (CDCl3; 125.8 MHz) δ = 40.7, 103.9, 118.2, 122.8, 126.9, 128.4, 129.4, 130.0, 136.4, 139.3, 141.16, 141.25, 144.7, 146.1, 150.5, 151.5; IR (KBr): ν = 3427, 2962, 1627, 1501, 1414, 1356, 1262, 1219, 1097, 1028, 805, 703 cm−1;HRMS (TOF-ES+): m/z Calcd for C17H15N4 (M + H)+ = 275.1297, found 275.1296.
3.9. Synthesis of 6e and 7
The solution of N1,N1-dimethylbenzene-1,4-diamine (286 mg, 2.10 mmol) in toluene (50 mL) was brought to a gentle reflux, and silver carbonate on celite (2.5 g, 50% w/w, 4.50 mmol Ag2CO3) was added in small portions over 30 min. The reaction mixture was stirred under reflux for 4 h and filtered under reduced pressure without cooling. The solid residue was flushed with ca. 50 mL of ethyl acetate until a clean, colorless filtrate was observed. The obtained solution was concentrated and purified by column chromatography on silica gel using chloroform and/or ethyl acetate/hexane eluent system to afford:
N2,N2,N7,N7-tetramethylphenazine-2,7-diamine (6e) as a red powder, 90 mg (0.34 mmol, 33.8%); DSC = 255.69–255.86 °C; 1H-NMR (CDCl3; 500.2 MHz) δ = 3.15 (s, 12H, NCH3), 7.07 (d, 3JH,H = 2.8 Hz, 2H, aromatic), 7.52 (dd, 3JH,H = 9.6 Hz, 4JH,H = 2.8 Hz, 2H, aromatic), 7.93 (dd, 3JH,H = 9.5 Hz, 2H, aromatic); 13C{1H}-NMR (CDCl3; 125.8 MHz) δ = 40.8, 104.3, 122.5, 129.1, 139.6, 143.2, 149.8; IR (KBr): ν = 2956, 1586, 1472, 1421, 1372 cm−1; HRMS (TOF-ES+): m/z Calcd for C16H19N4 (M + H)+ = 267.1610, found 267.1609.
(E)-4,4′-(diazene-1,2-diyl)bis(N,N-dimethylaniline) (7) as a brown powder, 92 mg (0.34 mmol, 34.3%); DSC = 257.68 °C [59]; 1H-NMR (CDCl3; 500.2 MHz) δ = 3.06 (s, 12H, NCH3), 6.76 (d, 3JH,H = 9.1 Hz, 4H, aromatic), 7.80 (d, 3JH,H = 9.0 Hz, 4H, aromatic); 13C{1H}-NMR (CDCl3; 100.6 MHz) δ = 29.9, 40.6, 111.9, 124.1, 144.3, 151.7.
3.10. Hirshfeld Surface Calculations
Hirshfeld surfaces were calculated using Crystal Explorer 21.5 [31] to analyze the close contacts in the phenothiazine and phenazine compounds. The normalized contact distance (dnorm) values are mapped with red-blue-white colors, where the closer contacts with dnorm < 0 are represented in red areas, while the blue region indicates longer contacts with dnorm > 0, and white areas correspond to contacts with distances equal to the van der Waals radii (dnorm = 0). The shape index visualizes complementary molecules in the crystal with red (hollows) and blue (bumps) colors, where molecular surfaces touch one another. Curvedness maps show planar regions on the surface in green, which is separated by blue edges corresponding to large positive curvature.
3.11. Density Functional Theory (DFT) Computations
The molecular structures of the phenothiazines and phenazines in the ground singlet state (S0) were optimized with the DFT/B3LYP/6-31G(d,p) approach [60,61,62,63]. For the calculated molecules, all vibrational wavenumbers were found to be real, indicating the location of the true minimum on the hypersurface of the total energy. The first excited singlet (S1) and triplet (T1) states, as well as vertical S0-Sn transitions, were calculated within the time-dependent (TD) DFT approximation [64] with the same B3LYP/6-31G(d,p) method [61,62,63]. The solvent effects on the absorption and fluorescence spectra were taken into account using the polarized-continuum model [65]. The Gaussian 16 program package [66] was used for all the DFT and TDDFT calculations.
3.12. X-ray Diffraction Experiments
The data for 1, 2a, 3, 5, and 6a were collected using a SuperNova diffractometer (Agilent Technologies, currently Rigaku Oxford Diffraction). Accurate cell parameters were determined and refined using the CrysAlisPro program [67]. Additionally, integration of the collected data was performed with this program. The data for 2b were collected using an Xcaliburdiffractometer (Oxford Diffraction, currently Rigaku Oxford Diffraction). Determination and refinement of the cell parameters, as well as the integration of the collected data, were performed using the CrysAlisPro program [68]. All the structures were solved using direct methods with the SHELXS97 program and then refined using the SHELXL-2018/3 program [69]. Nonhydrogen atoms were refined with anisotropic displacement parameters. The hydrogen atoms were fixed at calculated distances and allowed to ride on the parent atoms.
4. Conclusions
A series of N-heteroaromatics have been strategically designed and synthesized to compare their spectroscopic properties and rationalize them on the basis of theoretical calculations (Scheme 1). We described and analyzed the crystal structure of the N-benzoylated phenothiazine-phenazine hybrid molecule 1. Compounds of this type possess rich future chemistry potential for photoluminescent materials [27] and polymerization photoinitiating systems [23]. The presented synthesis protocols allowed obtaining the symmetrical phenazines with good yields (molecules 5 and 6e) and with low yields for unsymmetrical representatives 6a–d, giving six novel compounds. The crystal structures of phenazines 5 and 6a were determined by single-crystal X-ray diffraction measurements. Interestingly, molecules 6a–d are structurally related to the well-known biological stain neutral red, the analogs of which exhibit promising photosensitizing [70] and electrochemical properties [71].
Our results show that phosphorylation of phenothiazine proceeds exclusively to one new product 2a, with a newly formed P-N bond together with rearrangement leading to a sulfurylated thiophosphoric acid 2b. The S1 and T1 excited states of compound 2a possess hybridized local and charge-transfer characters and show excellent luminescent properties. This benefit makes molecule 2a a potentially highly luminous molecule. The N-phosphoryl moiety does not participate directly in the low-energy excitations of molecule 2a (it does not contribute to frontier molecular orbitals, Figure 8b) but provides a hypsochromic shift. According to the energy gap law [72], this blue shift leads to hindered non-radiative quenching of the T1 state and to sustainable phosphorescence.
Comparing the molecular orbitals involved in excitations for 2a and 3, it should be noted that for both phenothiazines, these MOs are localized in the phenothiazine ring, but their shapes and charge transfer nature differ significantly. The presence of phosphorus in molecule 2a provides a strong stabilizing hyperconjugation effect, and the charge is transferred through the nitrogen atom to the thiazine-type ring. Additionally, the absence of π–π stacking interactions in molecule 2a and the strong charge-transfer character of the T1 state provide efficient σ-π orbital mixing responsible for the increase in spin-orbit coupling and intersystem crossing in this compound, which contributes to high phosphorescence. Phenothiazine 3 has no stabilizing hyperconjugation effect; the first S0→S1 transition possesses a charge transfer character mainly from sulfur to nitro groups. In contrast to molecule 2a, compound 3 does not exhibit phosphorescence at room temperature, which is typical for nitroaromatic compounds.
Supplementary Materials
The following supporting information can be downloaded at https://www.mdpi.com/article/10.3390/molecules27217519/s1: Tables S1–S16 and Figures S1–S18. The following are available online: CCDC 2177507 for 1, CCDC 2177508 for 2a, CCDC 2178722 for 2b, CCDC 2177509 for 3, CCDC 2177510 for 5 and CCDC 2177511 for 6a contains the supplementary crystallographic data for this paper, accessed on 13 June 2022. These data can be obtained free of charge from http://www.ccdc.cam.ac.uk/conts/retrieving.html (accessed on 13 June 2022) or from the Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK; Fax: (+44) 1223-336-033; or e-mail: deposit@ccdc.cam.ac.uk. Calculations have been carried out at the Wroclaw Centre for Networking and Supercomputing (http://www.wcss.wroc.pl (accessed on 13 June 2022)).
Author Contributions
J.E.N.: Conceptualization, D.S. and J.E.N. Methodology; R.P. Photophysical studies; N.K.-K. and B.M. provided Hirshfeld surface analysis and DFT studies. Funding acquisition; M.K. and J.K.: Single-crystal X-ray diffraction measurements and analysis; all authors: Investigation, Visualization, and Writing manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
The research activities were co-financed by the funds granted under the Research Excellence Initiative of the University of Silesia in Katowice. The study was supported by the Ministry of Education and Science of Ukraine (projects no. 0122U000760 and 0121U107533).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data set presented in this study is available in this article.
Conflicts of Interest
The authors declare no conflict of interest.
Sample Availability
Samples of the compounds are available on request from the corresponding authors.
Abbreviations
| DFT | density-functional theory |
| LC-MS | liquid chromatography-mass spectrometry |
| DMF | dimethylformamide |
| THF | tetrahydrofuran |
| DMSO | dimethyl sulfoxide |
| MeCN | acetonitrile |
| TDDFT | time-dependent density-functional theory |
| HRMS | high-resolution mass spectrometry |
| ESI | electrospray ionisation |
| iPrOH | 2-propanol |
| Et2O | diethyl ether |
References
- Padnya, P.L.; Khadieva, A.I.; Stoikov, I.I. Current achievements and perspectives in synthesis and applications of 3,7-disubstituted phenothiazines as Methylene Blue analogues. Dye. Pigment. 2022, 208, 110806. [Google Scholar] [CrossRef]
- Che, Y.-X.; Qi, X.-N.; Lin, Q.; Yao, H.; Qu, W.-J.; Shi, B.; Zhang, Y.-M.; Wei, T.-B. Design strategies and applications of novel functionalized phenazine derivatives: A review. J. Mater. Chem. C 2022, 10, 11119–11174. [Google Scholar] [CrossRef]
- Posso, M.C.; Domingues, F.C.; Ferreira, S.; Silvestre, S. Development of Phenothiazine Hybrids with Potential Medicinal Interest: A Review. Molecules 2022, 27, 276. [Google Scholar] [CrossRef] [PubMed]
- Laursen, J.B.; Nielsen, J. Phenazine Natural Products: Biosynthesis, Synthetic Analogues, and Biological Activity. Chem. Rev. 2004, 104, 1663–1685. [Google Scholar] [CrossRef]
- Mavrodi, D.M.; Bonsall, R.F.; Delaney, S.M.; Soule, M.J.; Phillips, G.; Thomashow, L.S. Functional Analysis of Genes for Biosynthesis of Pyocyanin and Phenazine-1-Carboxamide from Pseudomonas aeruginosa PAO1. J. Bacteriol. 2001, 183, 6454–6465. [Google Scholar] [CrossRef]
- Dietz, K.; Keller, H.J.; Nóthe, D.; Wehe, D. Charge Transfer and Addition Products between Alkylated Phenazine Donors and Ethanetetracarbonitrile (TCNE): Crystal and Molecular Structure of the Dye 2,2’-(2,3,5,10-Tetrahydro-5,10-dimethylphenazine-2,3-diylidene)bis(propanedinitrile) (TMPP). J. Am. Chem. Soc. 1982, 104, 7581–7585. [Google Scholar] [CrossRef]
- Mosnaim, A.D.; Ranade, V.V.; Wolf, M.E.; Puente, J.; Antonieta, V.M. Phenothiazine molecule provides the basic chemical structure for various classes of pharmacotherapeutic agents. Am. J. Ther. 2006, 13, 261–273. [Google Scholar] [CrossRef]
- Franz, A.W.; Rominger, F.; Muller, T.J.J. Synthesis and electronic properties of sterically demanding N-arylphenothiazines and unexpected Buchwald-Hartwig aminations. J. Org. Chem. 2008, 73, 1795–1802. [Google Scholar] [CrossRef]
- Okafor, C.O. Studies in the heterocyclic series: The chemistry and biological activity of new aza and thia-phenothiazines, and related dibenzothiazepines. Phosphorus Sulfur. 1978, 4, 79–80. [Google Scholar] [CrossRef]
- Sailer, M.; Nonnenmacher, M.; Oeser, T.; Muller, T.J.J. Synthesis and electronic properties of 3-acceptor-substituted and 3,7-bisacceptor-substituted phenothiazines. Eur. J. Org. Chem. 2006, 2006, 423435. [Google Scholar] [CrossRef]
- Pluta, K.; Morak-Miodawska, B.; Jelen, M. Recent progress in biological activities of synthesized phenothiazines. Eur. J. Med. Chem. 2011, 46, 3179–3189. [Google Scholar] [CrossRef] [PubMed]
- Kramer, C.S.; Zeitler, K.; Muller, T.J.J. First synthesis and electronic properties of (hetero) aryl bridged and directly linked redox active phenothiazinyl dyads and triads. Tetrahedron Lett. 2001, 42, 8619–8624. [Google Scholar] [CrossRef]
- Chen, M.-C.; Lee, Y.-L.; Huang, Z.-X.; Chen, D.-G.; Chou, P.-T. Tuning Electron-Withdrawing Strength on Phenothiazine Derivatives: Achieving 100% Photoluminescence Quantum Yield by NO2 Substitution. Chem. Eur. J. 2020, 26, 7124–7130. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, T.; dos Santos, C.G.; Riposati, A.; Barbosa, L.R.; Di Mascio, P.; Itri, R.; Baptista, M.S.; Nascimento, O.R.; Nantes, I.L. Photochemically Generated Stable Cation Radical of Phenothiazine Aggregates in Mildly Acid Buffered Solutions. J. Phys. Chem. B 2006, 110, 12257–12265. [Google Scholar] [CrossRef] [PubMed]
- Upadhyay, K.; Asthana, A.; Tamrakar, R.K. Sensitive spectrophotometric method for determination of some phenothiazine drugs. Res. Chem. Intermed. 2015, 41, 7481–7495. [Google Scholar] [CrossRef]
- Murphy, C.M.; Ravner, H.; Smith, N.L. Mode of Action of Phenothiazine-Type Antioxidants. Ind. Eng. Chem. 1950, 42, 2479–2489. [Google Scholar] [CrossRef]
- Fukuzumi, K.; Ikeda, N.; Egawa, M. Phenothiazine derivatives as new antioxidants for the autoxidation of methyl linoleate and their reaction mechanisms. J. Am. Oil Chem. Soc. 1976, 53, 623–627. [Google Scholar] [CrossRef]
- Yamamura, T.; Suzuchi, K.; Yamaguchi, T.; Nishiyama, T. Synthetic Studies on Polypropionate-Derived 4-Pyrone-Containing Marine Natural Products. Bull. Chem. Soc. Jpn. 1997, 70, 413–419. [Google Scholar] [CrossRef]
- Cini, M.; Fariello, R.G.; Bianchetti, A.; Moretti, A. Studies on lipid peroxidation in the rat brain. Neurochem. Res. 1994, 19, 283–288. [Google Scholar] [CrossRef]
- Slater, T.F. The inhibitory effects in vitro of phenothiazines and other drugs on lipid-peroxidation systems in rat liver microsomes, and their relationship to the liver necrosis produced by carbon tetrachloride. Biochem. J. 1968, 106, 155–160. [Google Scholar] [CrossRef]
- Levy, L.B. Inhibition of acrylic acid polymerization by phenothiazine and p-methoxyphenol. J. Polym. Sci. 1985, 23, 1505–1515. [Google Scholar] [CrossRef]
- Nicolson, A. The Effect of Oxygen Concentration on Methylacrylic Acid Stability. Plant Oper. Prog. 1991, 10, 171–183. [Google Scholar] [CrossRef]
- Podsiadły, R.; Strzelczyk, R. N-substituted quinoxalinobenzothiazine/iodonium salt systems as visible photoinitiators for hybrid polymerization. Dye. Pigment. 2013, 97, 462–468. [Google Scholar] [CrossRef]
- Jin, R.; Bub, C.L.; Patureau, F.W. Phenothiazinimides: Atom-Efficient Electrophilic Amination Reagents. Org. Lett. 2018, 20, 2884–2887. [Google Scholar] [CrossRef]
- Milen, M.; Ábrányi-Balogh, P.; Balogh, G.; Drahos, L.; Keglevich, G. A study on the phosphorylation of indole, imidazole, carbazole, and phenothiazine derivatives. Phosphorus Sulfur Silicon Relat. Elem. 2012, 187, 1091–1100. [Google Scholar] [CrossRef]
- Prinz, H.; Chamasmani, B.; Vogel, K.; Böhm, K.J.; Aicher, B.; Gerlach, M.; Günther, E.G.; Amon, P.; Ivanov, I.; Müller, K. N-Benzoylated Phenoxazines and Phenothiazines: Synthesis, Antiproliferative Activity, and Inhibition of Tubulin Polymerization. J. Med. Chem. 2011, 54, 4247–4263. [Google Scholar] [CrossRef]
- Tian, S.; Ma, H.; Wang, X.; Lv, A.; Shi, H.; Geng, Y.; Li, J.; Liang, F.; Su, Z.-M.; An, Z.; et al. Utilizing d–pπ Bonds for Ultralong Organic Phosphorescence. Angew. Chem. Int. Ed. 2019, 58, 6645–6649. [Google Scholar] [CrossRef]
- Nycz, J.E.; Małecki, J.G.; Pajchel, Ł.; Ponikiewski, Ł.; Wiącek, M. One-pot synthesis of selected P-vinylbenzyls under solvent-free conditions. ChemistrySelect 2022, 7, e202200568. [Google Scholar] [CrossRef]
- Nycz, J.E. The synthesis of hypodiphosphoric acid and derivatives with P-P bond, including esters and diphosphine dioxides: A Review. Molecules 2021, 26, 7286. [Google Scholar] [CrossRef]
- Spackman, M.A.; Jayatilakaa, D. Hirshfeld surface analysis. Cryst. Eng. Comm. 2009, 11, 19–32. [Google Scholar] [CrossRef]
- Spackman, P.R.; Turner, M.J.; McKinnon, J.J.; Wolff, S.K.; Grimwood, D.J.; Jayatilaka, D.; Spackman, M.A. CrystalExplorer: A program for Hirshfeld surface analysis, visualization and quantitative analysis of molecular crystals. J. Appl. Crystallogr. 2021, 54, 1006–1011. [Google Scholar] [CrossRef] [PubMed]
- Nycz, J.E.; Szala, M.; Malecki, G.J.; Nowak, M.; Kusz, J. Synthesis, spectroscopy and computational studies of selected hydroxyquinolines and their analogues. Spectrochim. Acta A 2014, 117, 351–359. [Google Scholar] [CrossRef] [PubMed]
- Malecki, G.; Nycz, J.E.; Ryrych, E.; Ponikiewski, L.; Nowak, M.; Kusz, J.; Pikies, J. Synthesis, spectroscopy and computational studies of some biologically important hydroxyhaloquinolines and their novel derivatives. J. Mol. Struct. 2010, 969, 130–138. [Google Scholar] [CrossRef]
- Nycz, J.E.; Malecki, G.; Ponikiewski, L.; Leboschka, M.; Nowak, M.; Kusz, J. Synthesis, spectroscopy and computational studies of some novel phosphorylated derivatives of quinoline-5,8-diones. J. Mol. Struct. 2011, 986, 39–48. [Google Scholar] [CrossRef]
- Wantulok, J.; Swoboda, D.; Nycz, J.E.; Książek, M.; Kusz, J.; Malecki, J.G.; Kubíček, V. Direct amination of nitroquinoline derivatives via nucleophilic displacement of aromatic hydrogen. Molecules 2021, 26, 1857. [Google Scholar] [CrossRef]
- Staab, A.H.; Elbl, K. Tetrakis(dialkylamino)aromatics and Their Charge Transfer Complexes. US Patent 4,999,441, 12 March 1991. [Google Scholar]
- Seth, K.; Roy, S.R.; Chakraborti, A.K. Synchronous double C–N bond formation via C–H activation for a novel synthetic route to phenazine. Chem. Commun. 2016, 52, 922–925. [Google Scholar] [CrossRef]
- Seth, K.; Roy, S.R.; Kumar, A.; Chakraborti, A.K. The palladium and copper contrast: A twist to products of different chemotypes and altered mechanistic pathways. Catal. Sci. Technol. 2016, 6, 2892–2896. [Google Scholar] [CrossRef]
- Thompson, D.C.; Eling, T.E. Reactive Intermediates Formed during the Peroxidative Oxidation of Anisidine Isomers. Chem. Res. Toxicol. 1991, 4, 474–481. [Google Scholar] [CrossRef]
- Stiborová, M.; Mikšanová, M.; Havlíček, V.; Schmeiser, H.H.; Frei, E. Mechanism of peroxidse-mediated oxidation of carcinogenic o-anisidine and its binding to DNA. Mutat. Res. 2002, 500, 49–66. [Google Scholar] [CrossRef]
- Mortzfeld, F.B.; Pietruszka, J.; Baxendale, I.R. A Simple and Efficient Flow Preparation of Pyocyanin a Virulence Factor of Pseudomonas aeruginosa. Eur. J. Org. Chem. 2019, 2019, 5424–5433. [Google Scholar] [CrossRef]
- Edward, J.T. Stability of glycosides to acid hydrolysis. Chem. Ind. 1955, 36, 1102–1104. [Google Scholar]
- Lemieux, R.U.; Chu, P. 133rd National Meeting of the American Chemical Society 31N; American Chemical Society: Washington, DC, USA, 1958. [Google Scholar]
- Gold, V.; Loening, K.L.; McNaught, A.D.; Sehmi, P. International Union of Pure and Applied Chemistry Compendium of Chemical Terminology IUPAC Recommendations; Blackwell Scientific Publications Limited: Malden, MA, USA, 1987. [Google Scholar]
- Bentrude, W.G.; Setzer, W.N.; Sopchik, A.E.; Bajwa, G.S.; Burright, D.D.; Hutchinson, J.P. Conformations of saturated six-membered-ring phosphorus heterocycles related to cyclophosphamide. NMR, x-ray, and infrared studies of 2-methoxy-2-oxo-1,3,2-oxazaphosphorinane and 2-thio-1,3,2-oxazaphosphorinane. J. Am. Chem. Soc. 1986, 108, 6669–6675. [Google Scholar] [CrossRef]
- Bentrude, W.G.; Hargis, J.H. Conformations of six-membered ring phosphorus heterocycles. I. Ring conformations and phosphorus configurations of isomeric six-membered ring phosphites. J. Am. Chem. Soc. 1970, 92, 7136–7144. [Google Scholar] [CrossRef]
- Cramer, C.J.; Truhlar, D.G.; French, A.D. Exo-anomeric effects on energies and geometries of different conformations of glucose and related systems in the gas phase and aqueous solution. Carbohyd. Res. 1997, 298, 105. [Google Scholar] [CrossRef]
- Manju, T.; Manoj, N.; Braunc, A.M.; Oliveros, E. Self sensitized photooxidation of N-methyl phenothiazine: Acidity control of the competition between electron and energy transfer mechanisms. Photochem. Photobiol. Sci. 2012, 11, 1744–1755. [Google Scholar] [CrossRef]
- García, C.; Oyola, R.; Piñero, L.E.; Arce, R.; Silva, J.; Sánchez, V. Substitution and Solvent Effects on the Photophysical Properties of Several Series of 10-Alkylated Phenothiazine Derivatives. J. Phys. Chem. A 2005, 109, 3360–3371. [Google Scholar] [CrossRef] [PubMed]
- Dilelio, M.C.; Kaufman, T.S.; Iglesias, B.A.; Silveira, C.C. Synthesis and evaluation of photophysical and electrochemical properties of vinyl chalcogenide derivatives of phenothiazines. Dye. Pigment. 2022, 198, 109982. [Google Scholar] [CrossRef]
- Schmidt, M.; Esser, B. Cavity-promotion by pillar[5]arenes expedites organic photoredox-catalysed reductive dehalogenations. Chem. Commun. 2021, 57, 9582–9585. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Huang, R.; Batsanov, A.S.; Pander, P.; Hsu, Y.-T.; Chi, Z.; Dias, F.B.; Martin, R.; Bryce, M.R. Intramolecular Charge Transfer Controls Switching Between Room Temperature Phosphorescence and Thermally Activated Delayed Fluorescence. Angew. Chem. Int. Ed. 2018, 57, 16407. [Google Scholar] [CrossRef]
- Yao, L.; Zhang, S.; Wang, R.; Li, W.; Shen, F.; Yang, B.; Ma, Y. Highly Efficient Near-Infrared Organic Light-Emitting Diode Based on a Butterfly-Shaped Donor–Acceptor Chromophore with Strong Solid-State Fluorescence and a Large Proportion of Radiative Excitons. Angew. Chem. Int. Ed. 2014, 53, 2119–2123. [Google Scholar] [CrossRef]
- Minaev, B.F.; Terpugova, A.F. Spin-orbit interaction in charge-transfer complexes. Sov. Phys. J. 1968, 12, 1260–1263. [Google Scholar] [CrossRef]
- Mitchell, S.C.; Kestell, P.; Steventon, G.B.; Waring, R.H. Fate of the anthelmintic, phenothiazine, in man. Xenobiotica 2002, 32, 771–782. [Google Scholar] [CrossRef]
- Hayashi, H.; Koizumi, T. Preparation and electrochemical behavior of N-substituted phenothiazine oxide. Heterocycles 2016, 92, 1441–1449. [Google Scholar] [CrossRef]
- Mellinger, T.J.; Keeler, C.E. Spectrofluorometric identification of phenothiazine drugs. Anal. Chem. 1963, 35, 554–558. [Google Scholar] [CrossRef]
- Nycz, J.E.; Wantulok, J.; Sokolova, R.; Pajchel, L.; Stankevič, M.; Szala, M.; Malecki, J.G.; Swoboda, D. Synthesis and electrochemical and spectroscopic characterization of 4,7-diamino-1,10-phenanthrolines and their precursors. Molecules 2019, 24, 4102. [Google Scholar] [CrossRef]
- Hu, L.; Cao, X.; Shi, L.; Qi, F.; Guo, Z.; Lu, J.; Gu, H. A Highly Active Nano-Palladium Catalyst for the Preparation of Aromatic Azos under Mild Conditions. Org. Lett. 2011, 13, 5640–5643. [Google Scholar] [CrossRef]
- Mardirossian, N.; Head-Gordon, M. Thirty years of density functional theory in computational chemistry: An overview and extensive assessment of 200 density functional. Mol. Phys. 2017, 115, 2315–2372. [Google Scholar] [CrossRef]
- Becke, A.D. Perspective: Fifty years of density-functional theory in chemical physics. J. Chem. Phys. 2014, 140, 18A301. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Francl, M.M.; Petro, W.J.; Hehre, W.J.; Binkley, J.S.; Gordon, M.S.; DeFrees, D.J.; Pople, J.A. Self-consistent molecular orbital methods. XXIII. A polarization-type basis set for second-row elements. J. Chem. Phys. 1982, 77, 3654–3666. [Google Scholar] [CrossRef]
- Runge, E.; Gross, E.K.U. Density-Functional Theory for Time-Dependent Systems. Phys. Rev. Lett. 1984, 52, 997–1000. [Google Scholar] [CrossRef]
- Miertuš, S.; Scrocco, E.; Tomasi, J. Electrostatic Interaction of a Solute with a Continuum. A Direct Utilizaion of Ab Initio Molecular Potentials for the Prevision of Solvent Effects. Chem. Phys. 1981, 55, 117–129. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision A.03; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- CrysAlisPro, Version 1.171.38.41q. Rigaku Oxford Diffraction. Applied Rigaku Technologies, Inc.: Austin, TX, USA, 2015.
- CrysAlisPro, Version 1.171.39.15e. Rigaku Oxford Diffraction. Applied Rigaku Technologies, Inc.: Austin, TX, USA, 2015.
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. A 2008, 64, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Wainwright, M. Photosensitisers in Biomedicine; John Wiley & Sons, Ltd.: Chichester, UK, 2009; pp. 54–58. [Google Scholar]
- Cheema, S.; Zhang, M.; Labine-Romain, M.; Lal, B.; Lavania, M.; Lee, M.; Li, X.; Lauro, F.M.; Beckmann, S.; Manefield, M. Neutral Red: The Synthetic Phenazine Full of Electrochemical Surprises. In Encyclopedia of Interfacial Chemistry; Elsevier: Amsterdam, The Netherlands, 2018; pp. 382–391. [Google Scholar]
- Lindgren, M.; Glimsdal, R.; Vestberg, R. Electronic states and phosphorescence of dendron functionalized platinum (II) acetylides. J. Lumin. 2007, 124, 302–310. [Google Scholar] [CrossRef]






















Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).