
Atom Economical Multi-Substituted Pyrrole Synthesis from Aziridine

Abstract
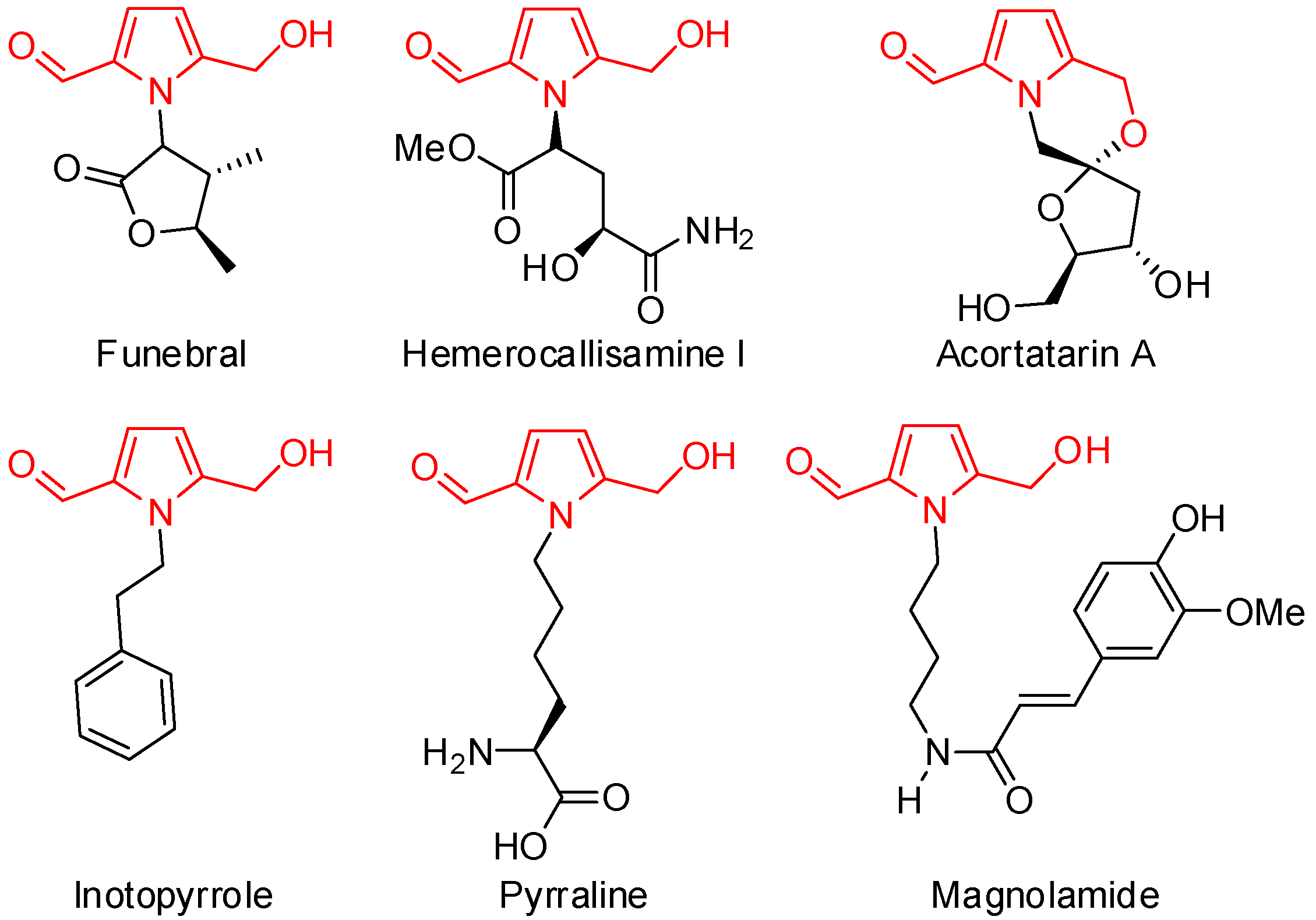
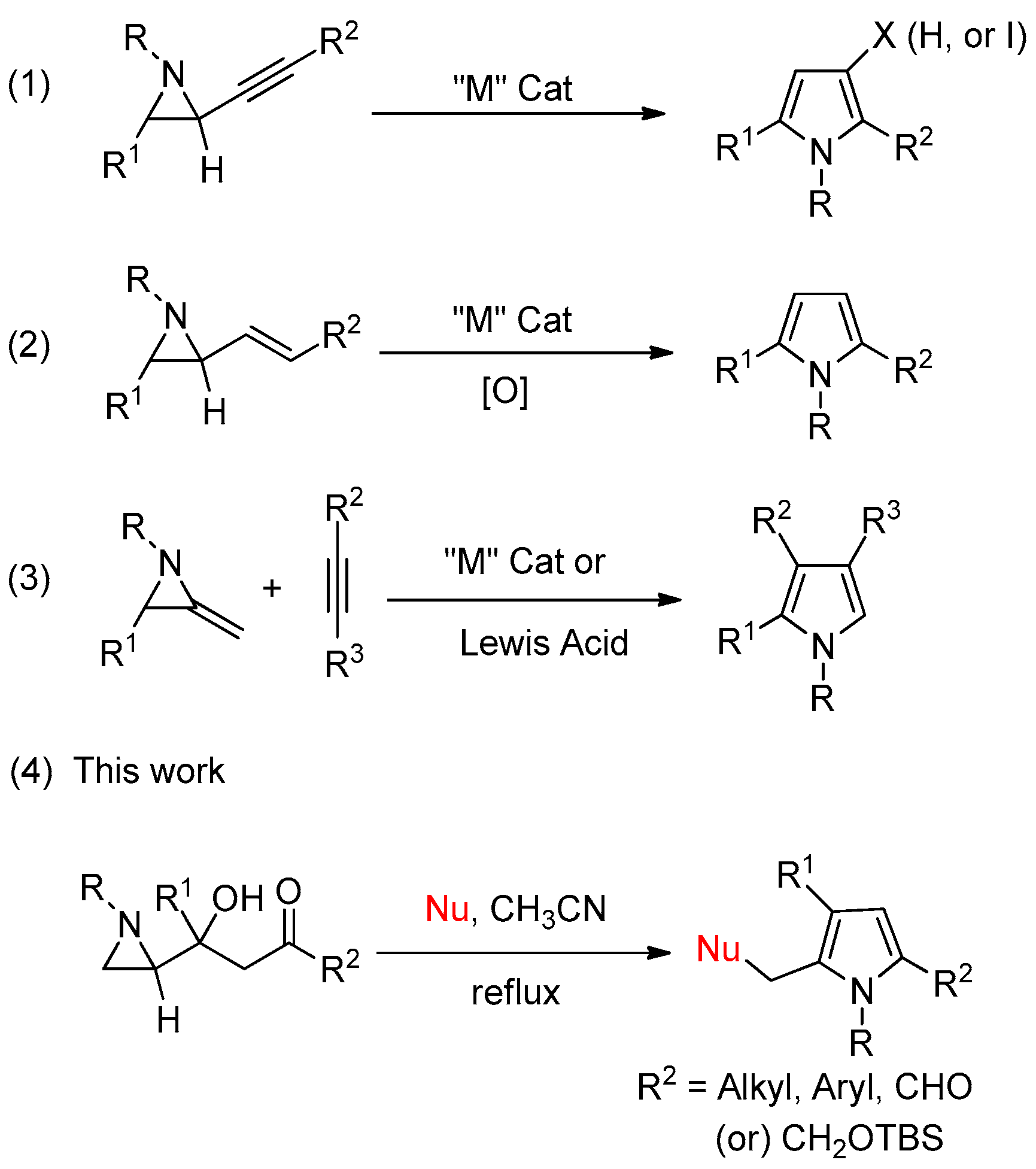
1. Introduction
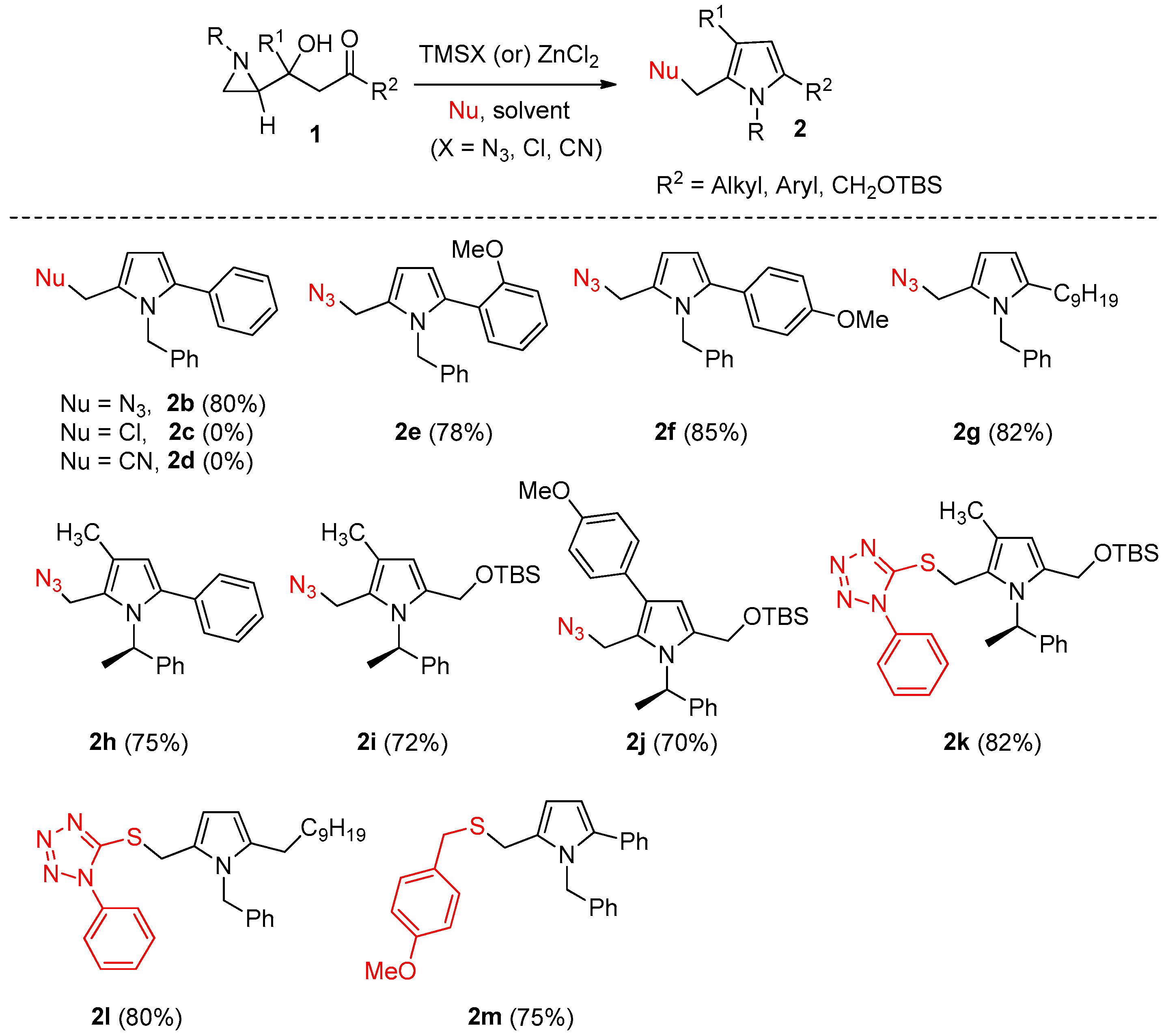
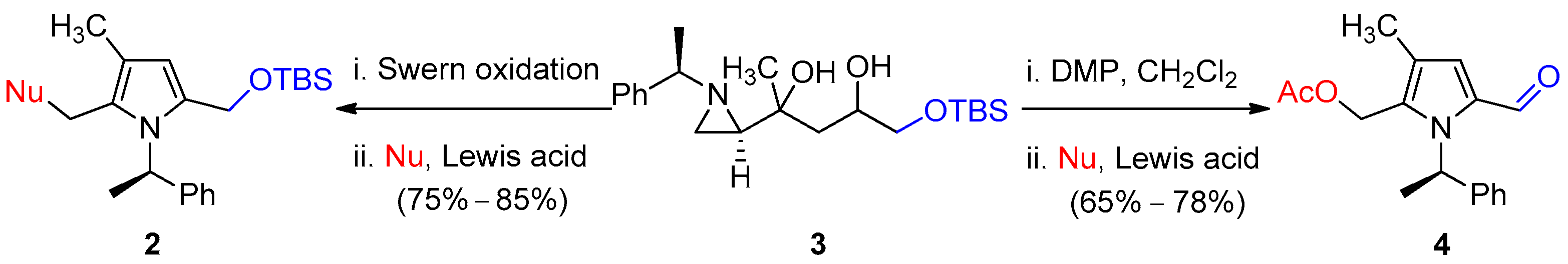
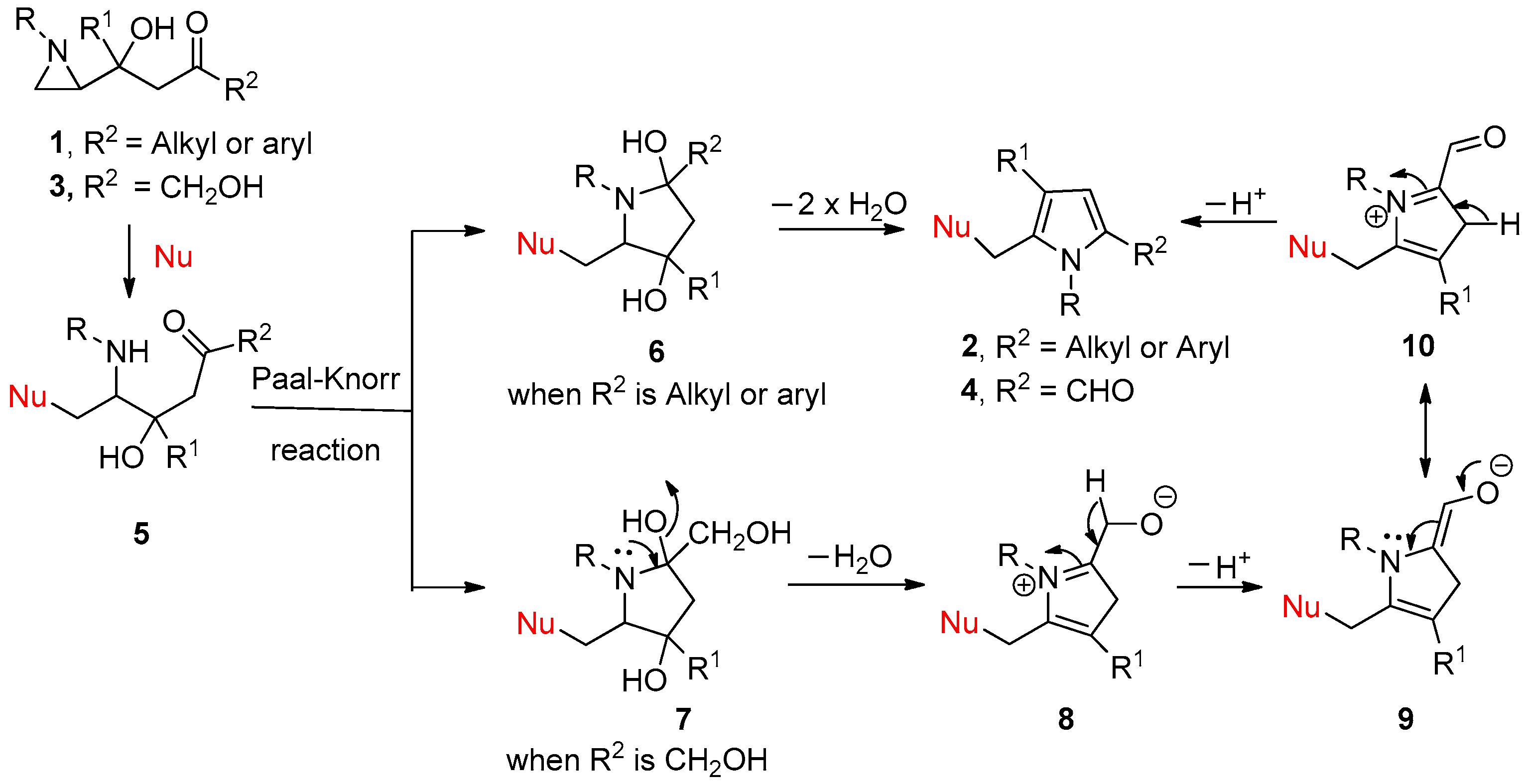
2. Results and Discussion
3. Materials and Methods
3.1. General Information
3.2. General Procedure for the Synthesis of Pyrroles
(R)-2-(Azidomethyl)-1-(1-phenylethyl)-5-propyl-1H-pyrrole (2a)
2-(Azidomethyl)-1-benzyl-5-phenyl-1H-pyrrole (2b)
2-(Azidomethyl)-1-benzyl-5-(2-methoxyphenyl)-1H-pyrrole (2e)
2-(Azidomethyl)-1-benzyl-5-(4-methoxyphenyl)-1H-pyrrole (2f)
2-(Azidomethyl)-1-benzyl-5-nonyl-1H-pyrrole (2g)
(R)-2-(Azidomethyl)-3-methyl-5-phenyl-1-(1-phenylethyl)-1H-pyrrole (2h)
(R)-2-(Azidomethyl)-5-(((tert-butyldimethylsilyl)oxy)methyl)-3-methyl-1-(1-phenylethyl)-1H-pyrrole (2i)
(R)-2-(Azidomethyl)-5-(((tert-butyldimethylsilyl)oxy)methyl)-3-(4-methoxyphenyl)-1-(1-phenylethyl)-1H-pyrrole (2j)
(R)-5-(((5-(((tert-Butyldimethylsilyl)oxy)methyl)-3-methyl-1-(1-phenylethyl)-1H-pyrrol-2-yl)methyl)thio)-1-phenyl-1H-tetrazole (2k)
5-(((1-Benzyl-5-nonyl-1H-pyrrol-2-yl)methyl)thio)-1-phenyl-1H-tetrazole (2l)
1-Benzyl-2-(((4-methoxybenzyl)thio)methyl)-5-phenyl-1H-pyrrole (2m)
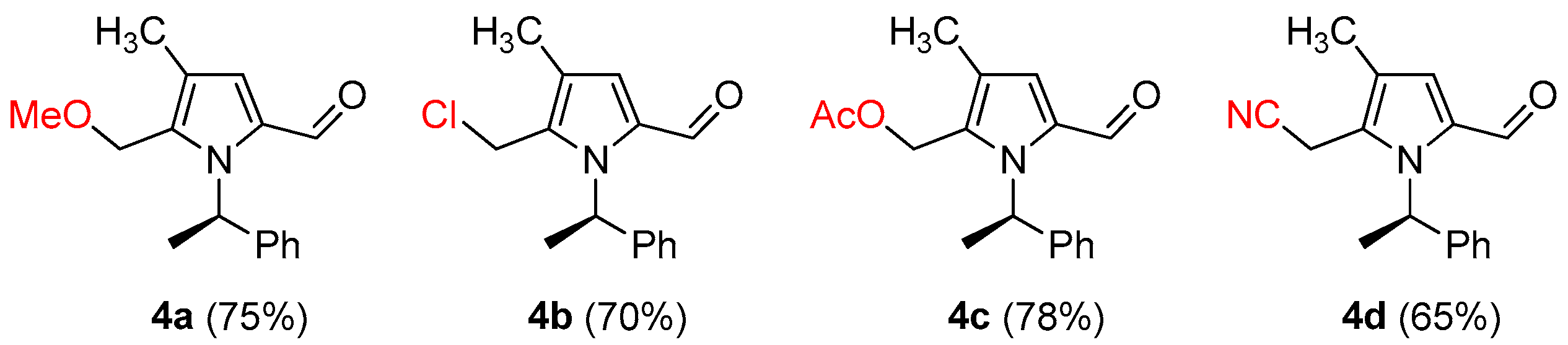
(R)-5-(methoxymethyl)-4-methyl-1-(1-phenylethyl)-1H-pyrrole-2-carbaldehyde (4a)
(R)-5-(Chloromethyl)-4-methyl-1-(1-phenylethyl)-1H-pyrrole-2-carbaldehyde (4b)
(R)-(5-Formyl-3-methyl-1-(1-phenylethyl)-1H-pyrrol-2-yl)methyl acetate (4c)
(R)-2-(5-Formyl-3-methyl-1-(1-phenylethyl)-1H-pyrrol-2-yl)acetonitrile (4d)
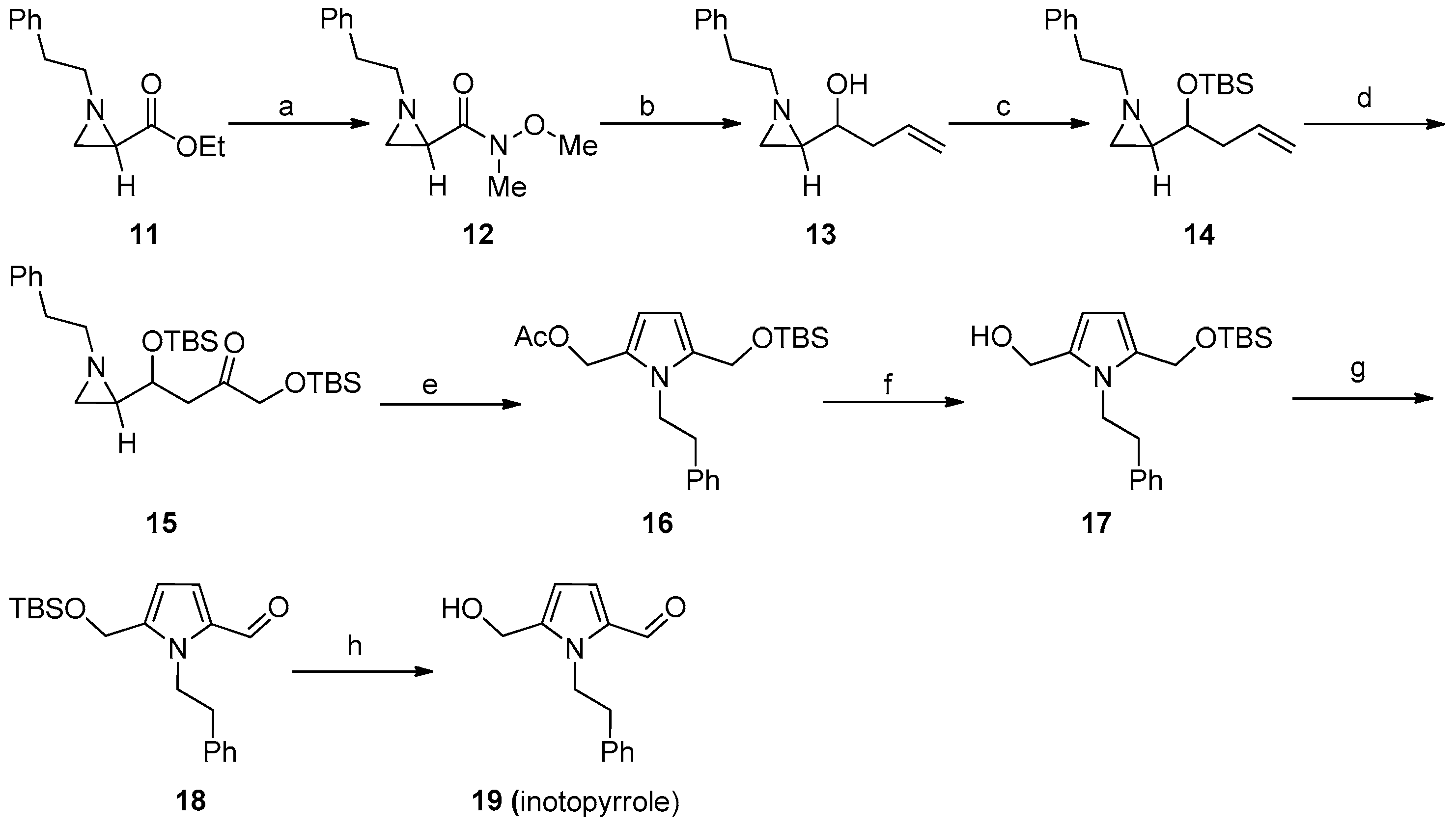
Ethyl 1-phenethylaziridine-2-carboxylate (11)
N-Methoxy-N-methyl-1-phenethylaziridine-2-carboxamide (12)
1-(1-Phenethylaziridin-2-yl)but-3-en-1-ol (13)
2-(1-((tert-Butyldimethylsilyl)oxy)but-3-en-1-yl)-1-phenethylaziridine (14)
Octamethyl-8-(1-phenethylaziridin-2-yl)-4,9-dioxa-3,10-disiladodecan-6-one (15)
(5-(((tert-Butyldimethylsilyl)oxy)methyl)-1-phenethyl-1H-pyrrol-2-yl)methyl acetate (16)
(5-(((tert-Butyldimethylsilyl)oxy)methyl)-1-phenethyl-1H-pyrrol-2-yl)methanol (17)
5-(((tert-Butyldimethylsilyl)oxy)methyl)-1-phenethyl-1H-pyrrole-2-carbaldehyde (18)
5-(Hydroxymethyl)-1-phenethyl-1H-pyrrole-2-carbaldehyde (19)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Gholap, S.S. Pyrrole: An emerging scaffold for construction of valuable therapeutic agents. Eur. J. Med. Chem. 2016, 110, 13–31. [Google Scholar] [CrossRef]
- Estévez, V.; Villacampa, M.; Menéndez, J.C. Multicomponent reactions for the synthesis of pyrroles. Chem. Soc. Rev. 2010, 39, 4402–4421. [Google Scholar] [CrossRef]
- Zhang, L.-Y.; Bai, H.-B.; Shan, W.-G.; Zhan, Z.-J. A new Alkaloid from the Mycelium of Inonotus Obliquus. J. Chem. Res. 2014, 38, 245–246. [Google Scholar] [CrossRef]
- Wood, J.M.; Furkert, D.P.; Brimble, M.A. 2-Formylpyrrole natural products: Origin, structural diversity, bioactivity and synthesis. Nat. Prod. Rep. 2019, 36, 289–306. [Google Scholar] [CrossRef]
- Mal, D.; Shome, B.; Dinda, B.K. Pyrrole and Its Derivatives. In Heterocycles in Natural Product Synthesis; Majumdar, K.C., Chattopadhyay, S.K., Eds.; Wiley: Hoboken, NJ, USA, 2011; Chapter 6; p. 187. [Google Scholar]
- Park, Y.M.; Won, J.H.; Kim, Y.H.; Choi, J.W.; Park, H.J.; Lee, K.L. In vivo and in vitro anti-inflammatory and anti-nociceptive effects of the methanol extract of Inonotus obliquus. J. Ethnopharmacol. 2005, 101, 120–128. [Google Scholar] [CrossRef]
- Lee, I.K.; Kim, Y.S.; Jang, Y.W.; Jung, J.Y.; Yun, B.S. New antioxidant polyphenols from the medicinal mushroom Inonotus obliquus. Bioorg Med Chem Lett. 2007, 17, 6678–6681. [Google Scholar] [CrossRef]
- Szychowski, K.A.; Skora, B.; Pomianek, T.; Gminski, J. Inonotus obliquus - from folk medicine to clinical use. J. Tradit. Complement. Med. 2021, 11, 293–302. [Google Scholar] [CrossRef]
- Zheng, W.; Zhang, M.; Zhao, Y.; Wang, Y.; Miao, K.; Wei, Z. Accumulation of antioxidant phenolic constituents in submerged cultures of Inonotus obliquus. Bioresour. Technol. 2009, 100, 1327–1335. [Google Scholar] [CrossRef]
- Cui, Y.; Kim, D.-S.; Park, K.-C. Antioxidant effect of Inonotus obliquus. J. Ethnopharmacol. 2005, 96, 79–85. [Google Scholar] [CrossRef]
- Kim, Y.O.; Han, S.B.; Lee, H.W.; Ahn, H.J.; Yoon, Y.D.; Jung, J.K.; Shin, C.S. Immuno-stimulating effect of the endo-polysaccharide produced by submerged culture of Inonotus obliquus. Life Sci. 2005, 77, 2438–2456. [Google Scholar] [CrossRef]
- Cha, J.-Y.; Jun, B.-S.; Yoo, K.-S.; Hahm, J.-R.; Cho, Y.-S. Fermented Chaga Mushroom (Inonotus obliquus) Effects on Hypolipidemia and Hepatoprotection in Otsuka Long-Evans Tokushima Fatty (OLETF) Rats. Food Sci. Biotechnol. 2006, 15, 122–127. [Google Scholar]
- Paal, C. Synthese von Thiophen-und Pyrrolderivaten. Ber. Dtsch. Chem. Ges. 1885, 18, 367. [Google Scholar] [CrossRef]
- Knorr, L. Synthesis of pyrroline-derivatives II. Chem. Ber. 1884, 17, 1635. [Google Scholar] [CrossRef]
- Hantzsch, A. Neue Bildungsweise von Pyrrolderivaten. Ber. Dtsch. Chem. Ges. 1890, 23, 1474–1476. [Google Scholar] [CrossRef]
- Gulevich, A.V.; Dudnik, A.S.; Chernyak, N.; Gevorgyan, V. Transition Metal-Mediated Synthesis of Monocyclic Aromatic Heterocycles. Chem. Rev. 2013, 113, 3084–3213. [Google Scholar] [CrossRef] [PubMed]
- Doyle, M.P.; Dong, K.; Humeidi, A.; Griffith, W.; Arman, H.; Xu, X. AgI-Catalyzed Reaction of Enol Diazoacetates and Imino Ethers: Synthesis of Highly Functionalized Pyrroles. Angew. Chem., Int. Ed. 2021, 60, 13394–13400. [Google Scholar]
- Stankovic, S.; D’hooghe, M.; Catak, S.; Eum, H.; Waroquier, M.; Van Speybroeck, V.; De Kimpe, N.; Ha, H.-J. Regioselectivity in the ring-opening of non-activated aziridines. Chem. Soc. Rev. 2012, 41, 643–665. [Google Scholar] [CrossRef]
- Choi, J.; Yu, T.; Ha, H.-J. Alkylative Aziridine Ring-Opening Reactions. Molecules 2021, 26, 1703. [Google Scholar] [CrossRef]
- Ranjith, J.; Ha, H.-J. Synthetic Applications of Aziridinium Ions. Molecules 2021, 26, 1774. [Google Scholar] [CrossRef]
- Chen, J.R.; Hu, X.Q.; Lu, L.Q.; Xiao, W.J. Exploration of Visible-Light Photocatalysis in Heterocycle Synthesis and Functionalization: Reaction Design and Beyond. Acc. Chem. Res. 2016, 49, 1911–1923. [Google Scholar] [CrossRef]
- Estevez, V.; Villacampa, M.; Menendez, J.C. Recent advances in the synthesis of pyrroles by multicomponent reactions. Chem. Soc. Rev. 2014, 43, 4633–4657. [Google Scholar] [CrossRef] [PubMed]
- Yadav, N.N.; Jeong, H.; Ha, H.-J. Atom-Economical and Metal-Free Synthesis of Multisubstituted Furans from Intramolecular Aziridine Ring Opening. ACS Omega 2017, 2, 7525–7531. [Google Scholar] [CrossRef] [PubMed]
- Macha, L.; D’hooghe, M.; Ha, H.J. Deployment of Aziridines for the Synthesis of Alkaloids and Their Derivatives. Synthesis 2019, 51, 1491–1515. [Google Scholar]
- Kim, J.H.; Lee, S.B.; Lee, W.K.; Yoo, D.-H.; Ha, H.-J. Synthesis of 1,2,5- and 1,2,3,5-substituted pyrroles from substituted aziridines via Ag(I)-catalyzed intramolecular cyclization. Tetrahedron 2011, 67, 3553–3558. [Google Scholar] [CrossRef]
- Lee, H.; Kim, J.H.; Lee, W.K.; Jung, J.-H.; Ha, H.-J. Synthesis of 2,5-Disubstituted 6-Azaindoles from Substituted Aziridines via Intramolecular Cyclization. Org. Lett. 2012, 14, 3120–3122. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | |||||
|---|---|---|---|---|---|
| Entry a | Nucleophile | Solvent | Temp | Time (h) | Yield b (%) |
| 1 | TMSN3 | THF | 85 | 4 | 0 |
| 2 | TMSN3 | Dioxane | 85 | 4 | 0 |
| 3 | TMSN3 | CH2Cl2 | 50 | 4 | 70 |
| 4 | TMSN3 | CH3CN | 85 | 4 | 85 |
| 5 | BF3.OEt2/NaN3 | CH2Cl2 | rt | 4 | 0 |
| 6 | FeCl3/NaN3 | CH2Cl2 | rt | 4 | 0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Macha, L.; Jala, R.; Na, S.-Y.; Ha, H.-J. Atom Economical Multi-Substituted Pyrrole Synthesis from Aziridine. Molecules 2022, 27, 6869. https://doi.org/10.3390/molecules27206869
Macha L, Jala R, Na S-Y, Ha H-J. Atom Economical Multi-Substituted Pyrrole Synthesis from Aziridine. Molecules. 2022; 27(20):6869. https://doi.org/10.3390/molecules27206869
Chicago/Turabian StyleMacha, Lingamurthy, Ranjith Jala, Sang-Yun Na, and Hyun-Joon Ha. 2022. "Atom Economical Multi-Substituted Pyrrole Synthesis from Aziridine" Molecules 27, no. 20: 6869. https://doi.org/10.3390/molecules27206869
APA StyleMacha, L., Jala, R., Na, S.-Y., & Ha, H.-J. (2022). Atom Economical Multi-Substituted Pyrrole Synthesis from Aziridine. Molecules, 27(20), 6869. https://doi.org/10.3390/molecules27206869