


Folic Acid Antimetabolites (Antifolates): A Brief Review on Synthetic Strategies and Application Opportunities

 and
and 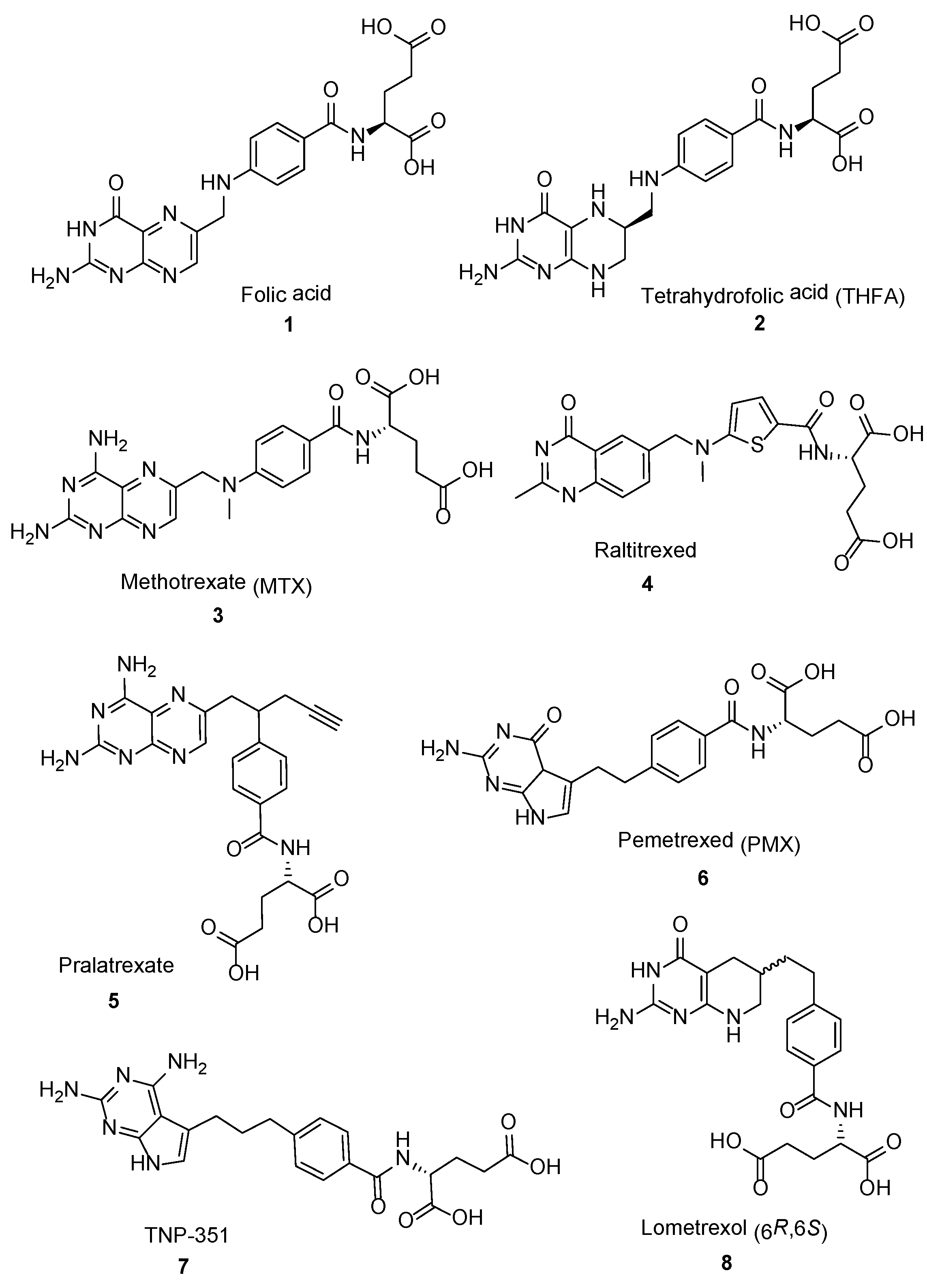{kind=link}
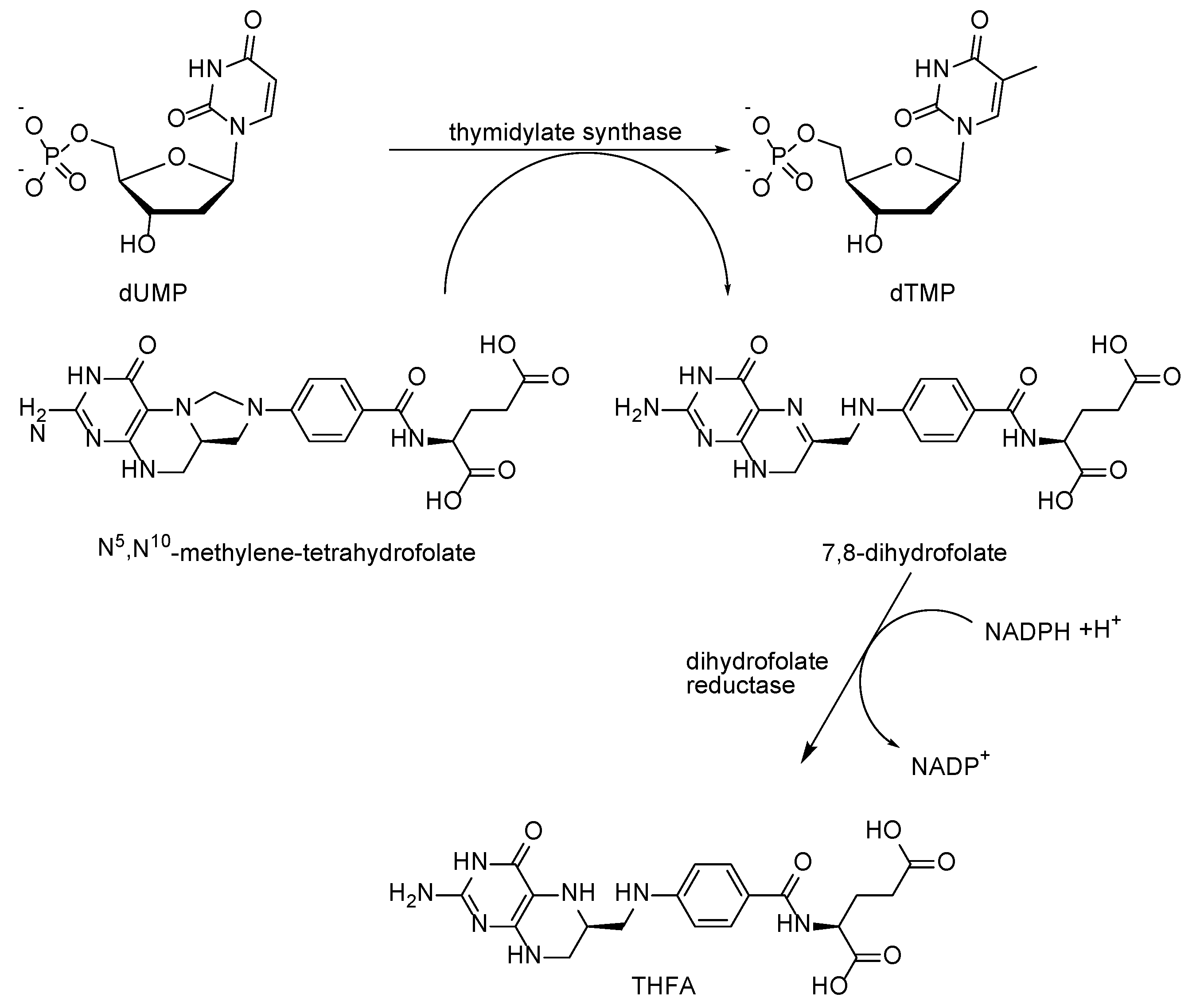{kind=link}
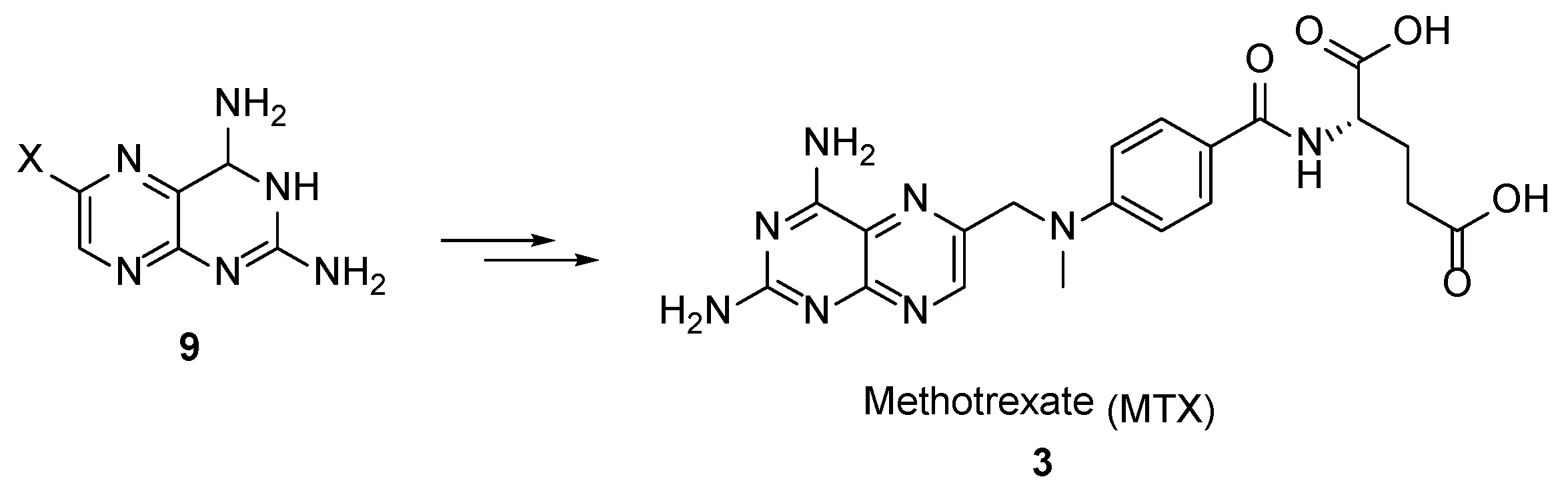{kind=link}
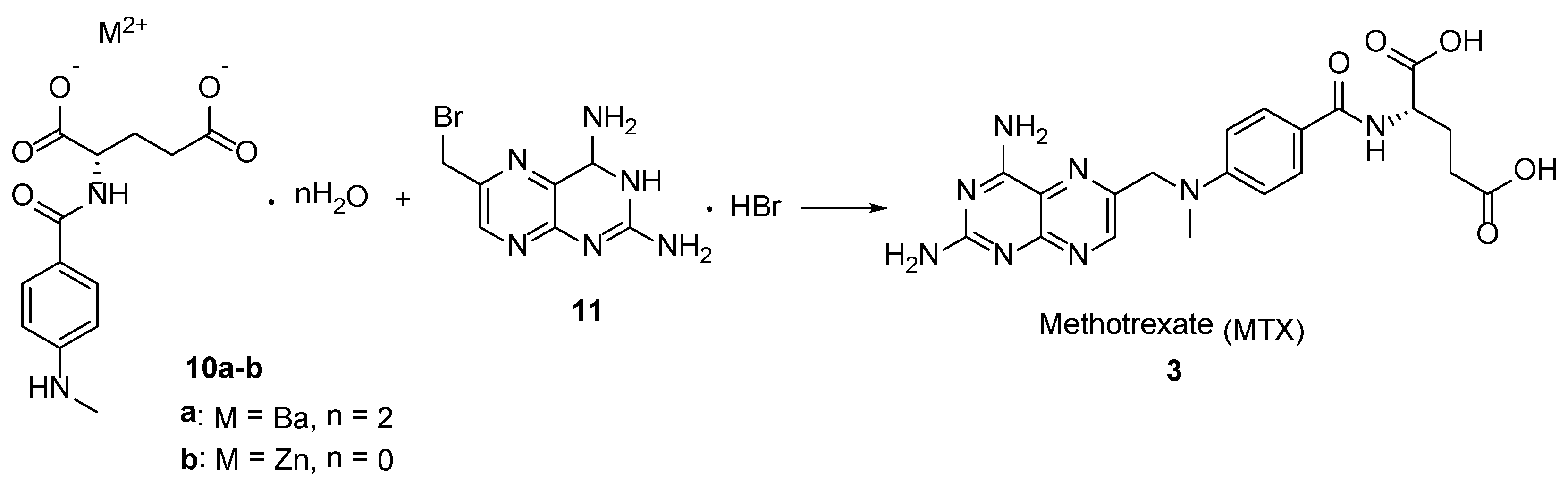{kind=link}
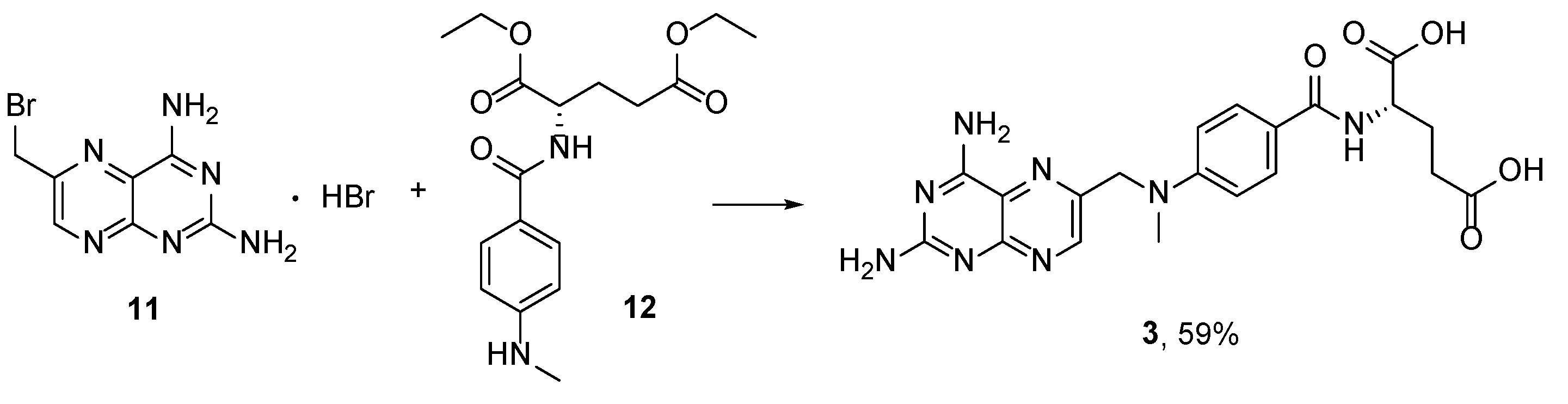{kind=link}
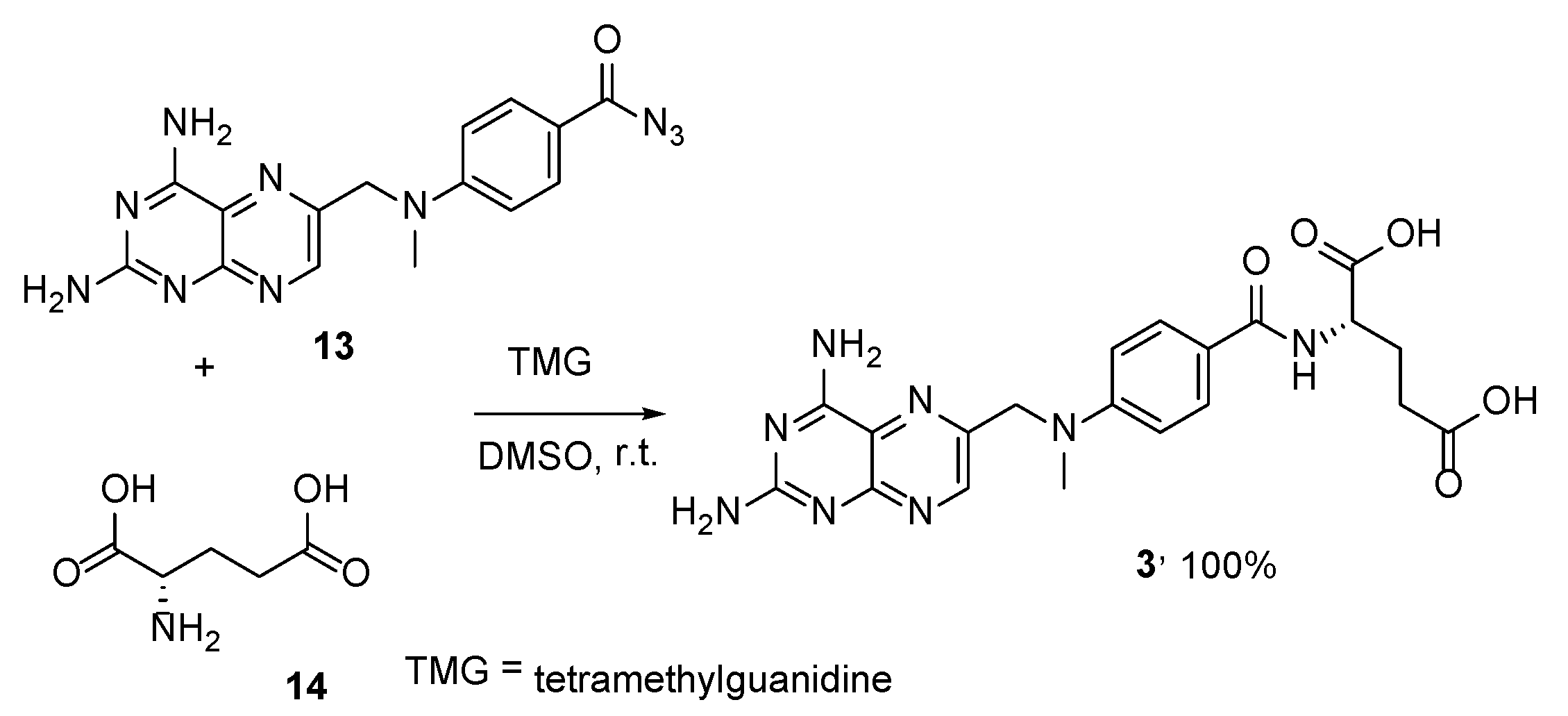{kind=link}
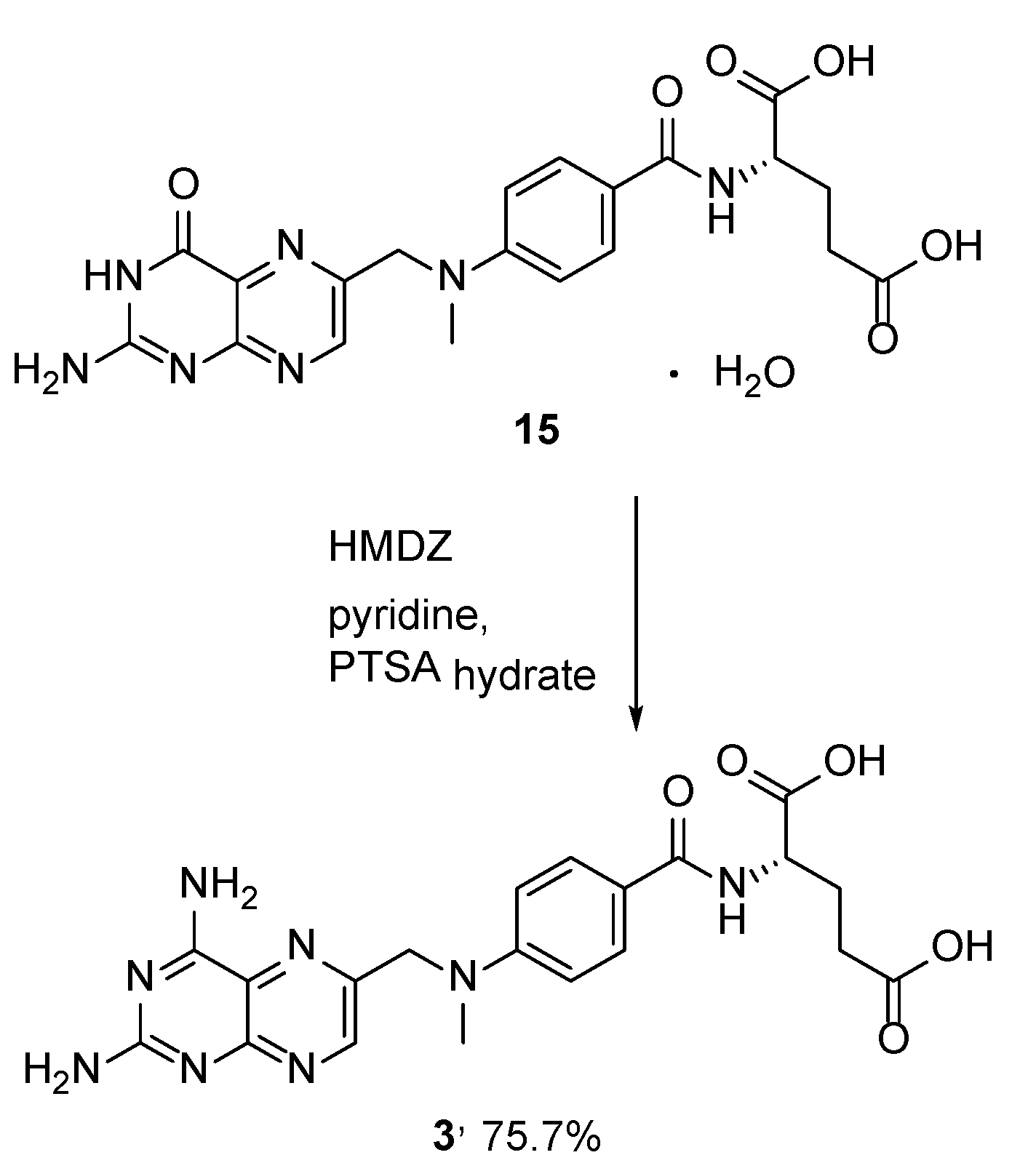{kind=link}
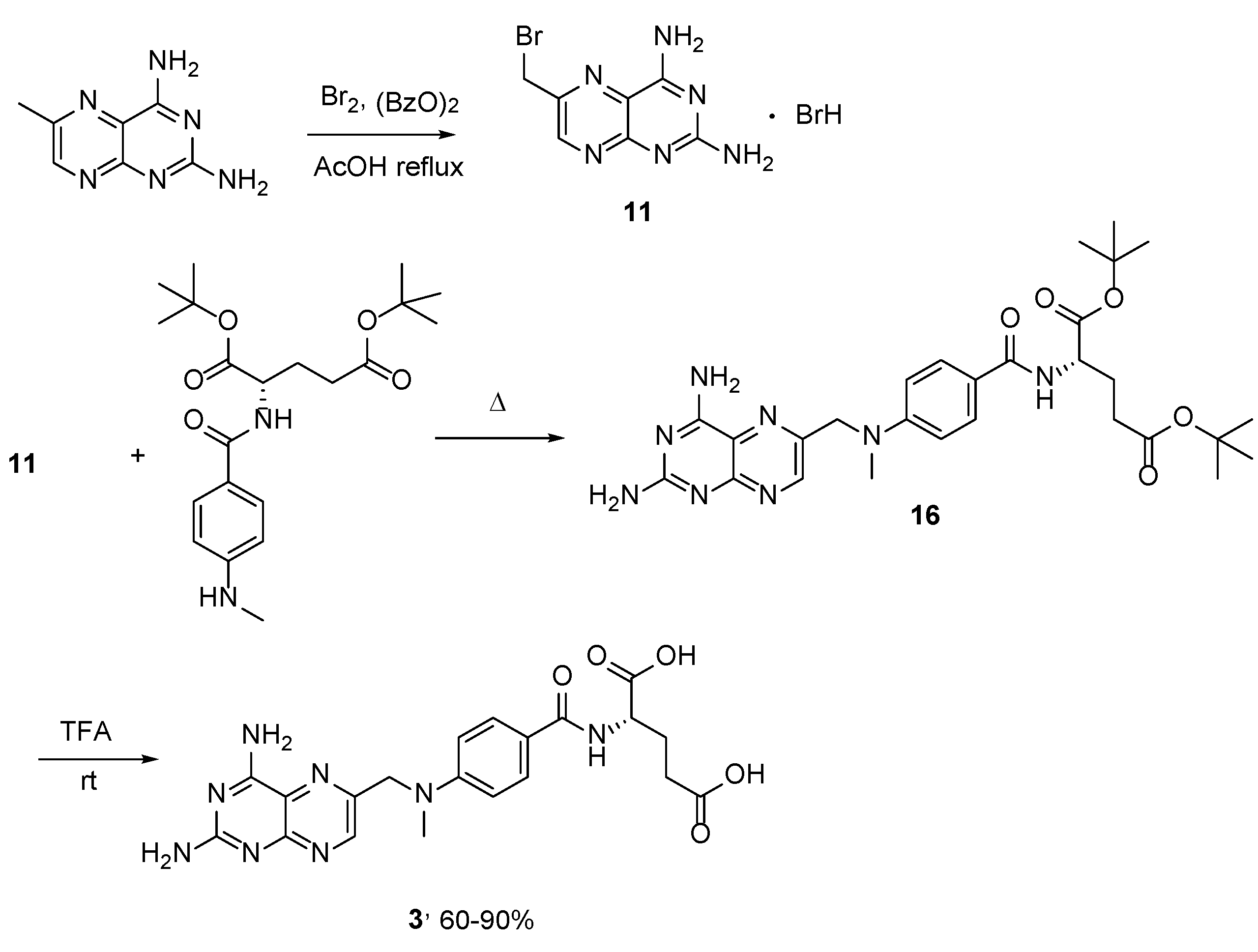{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
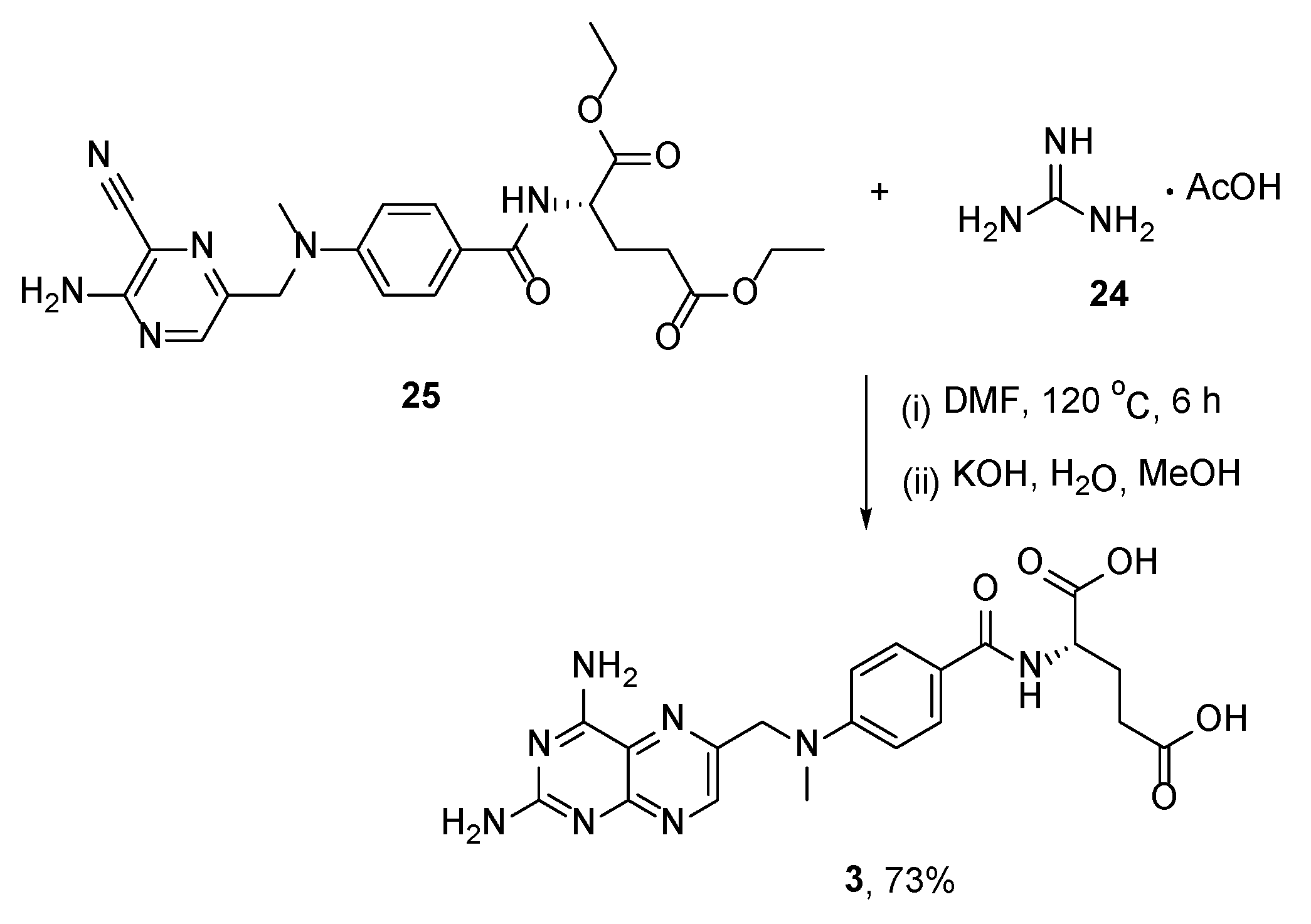{kind=link}
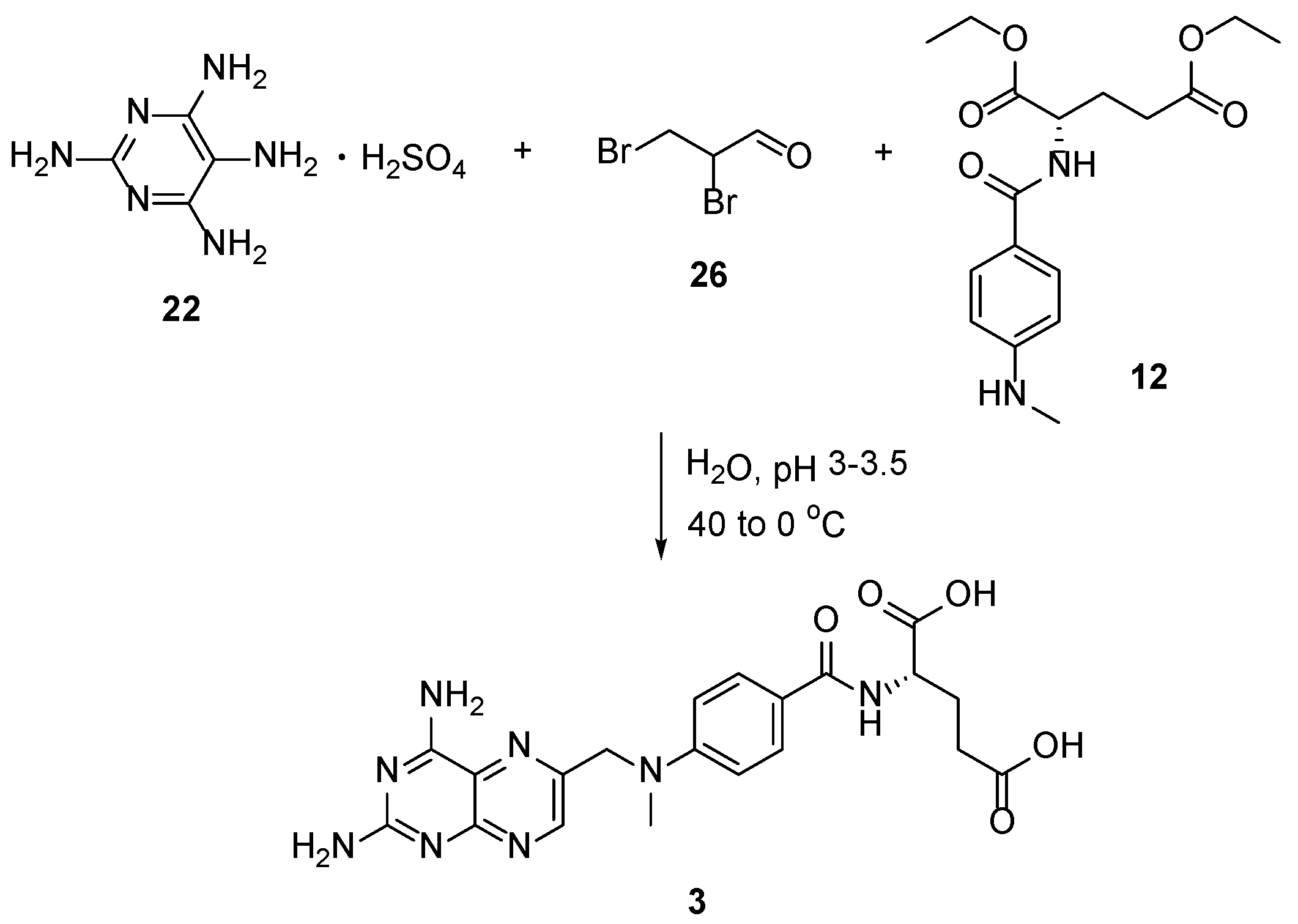{kind=link}
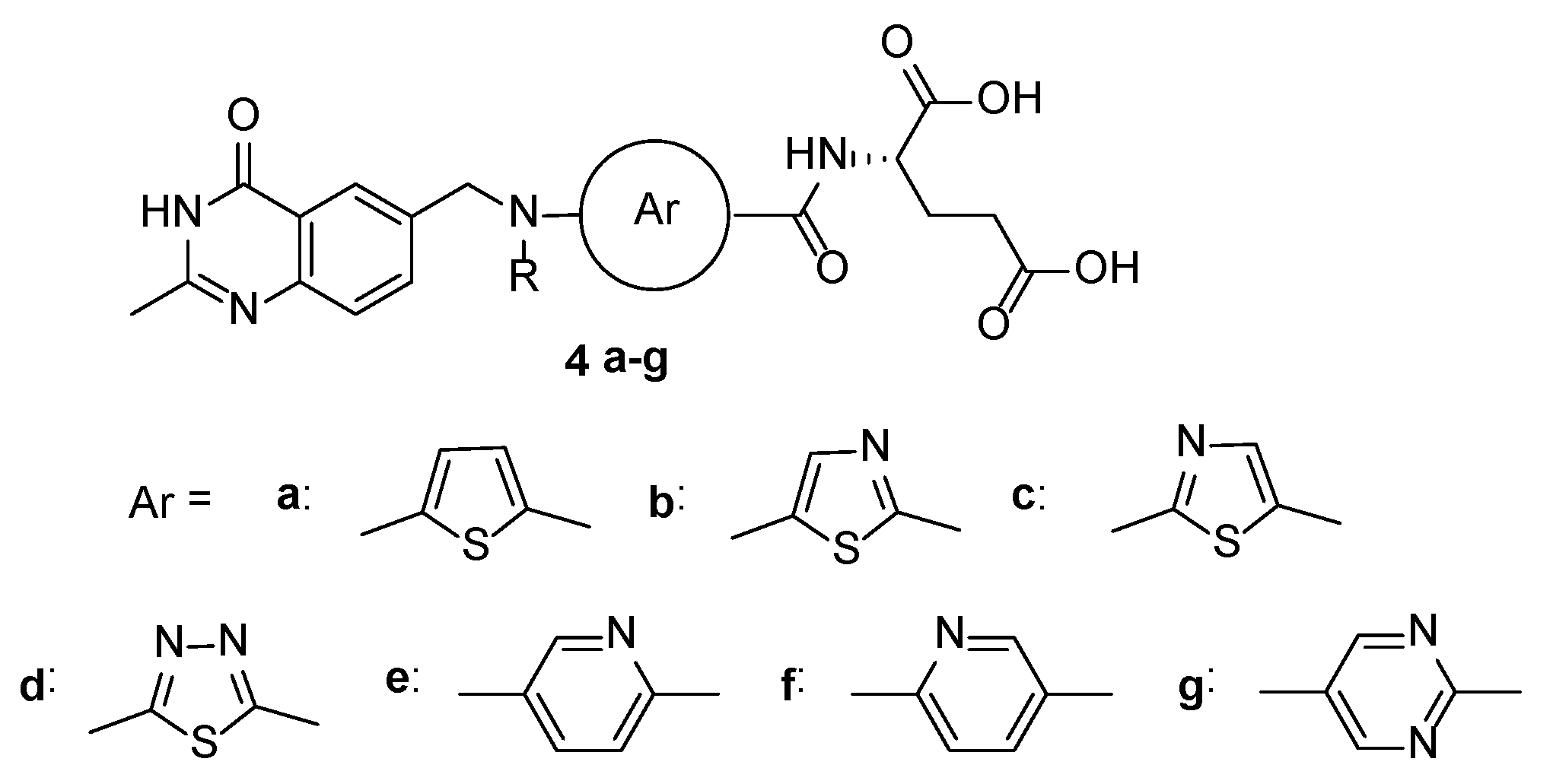{kind=link}
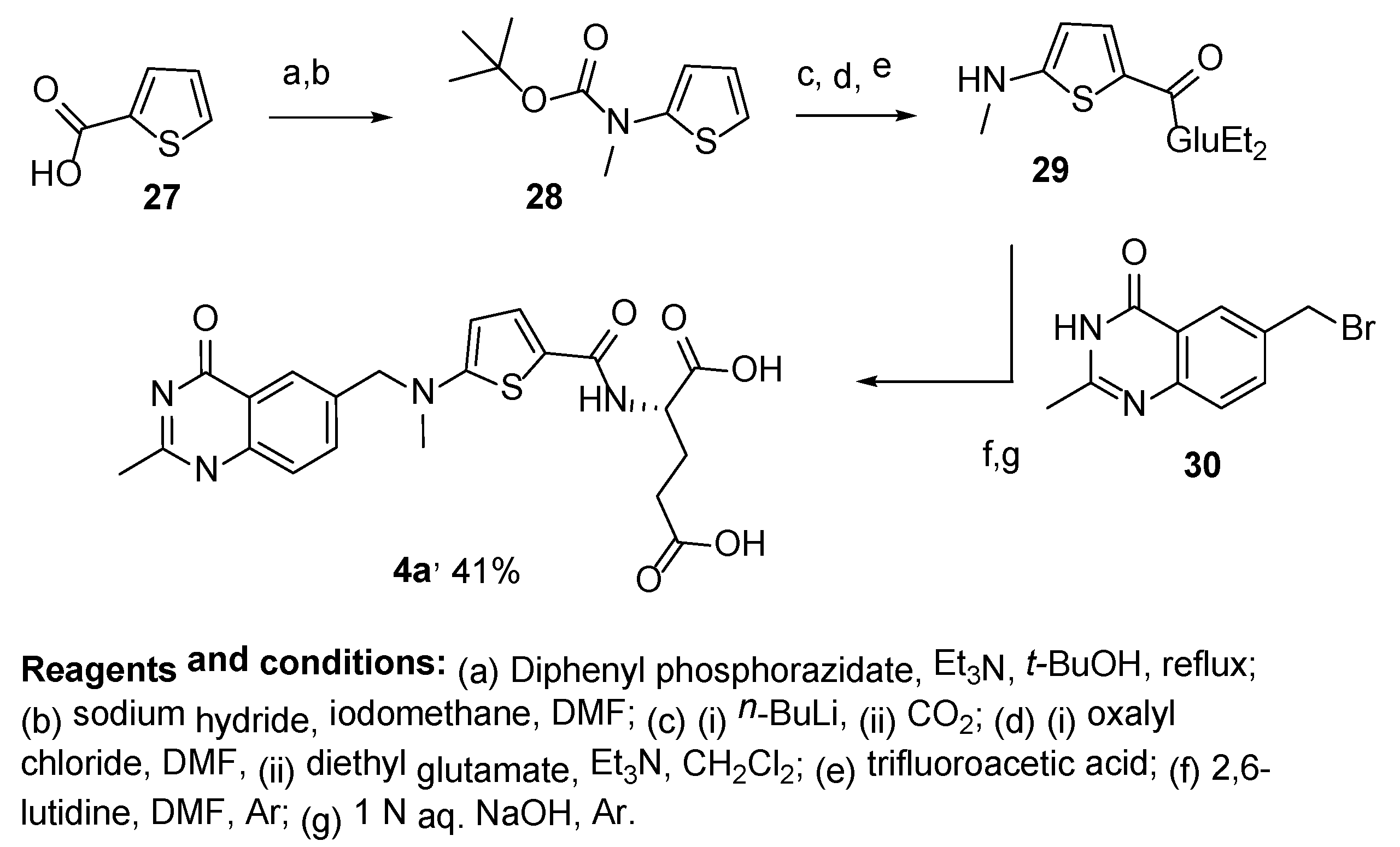{kind=link}
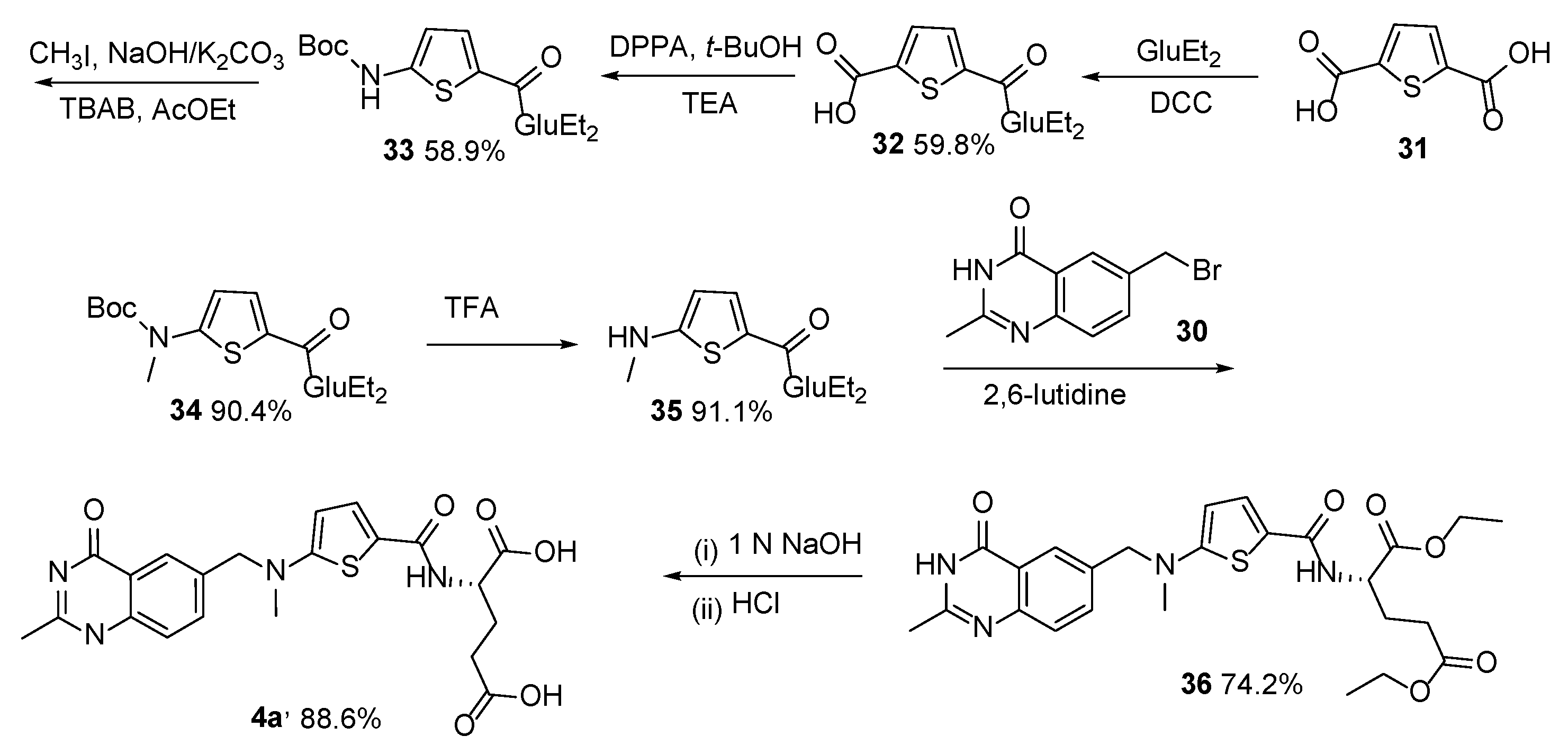{kind=link}
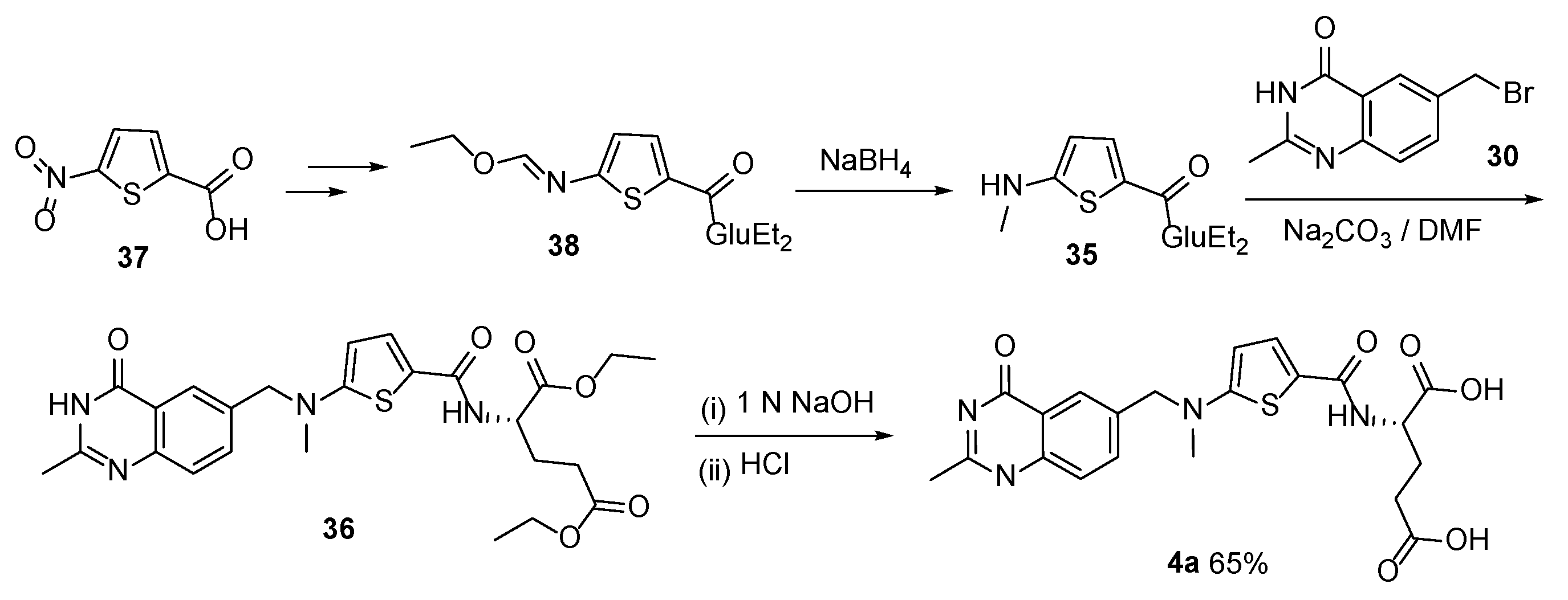{kind=link}
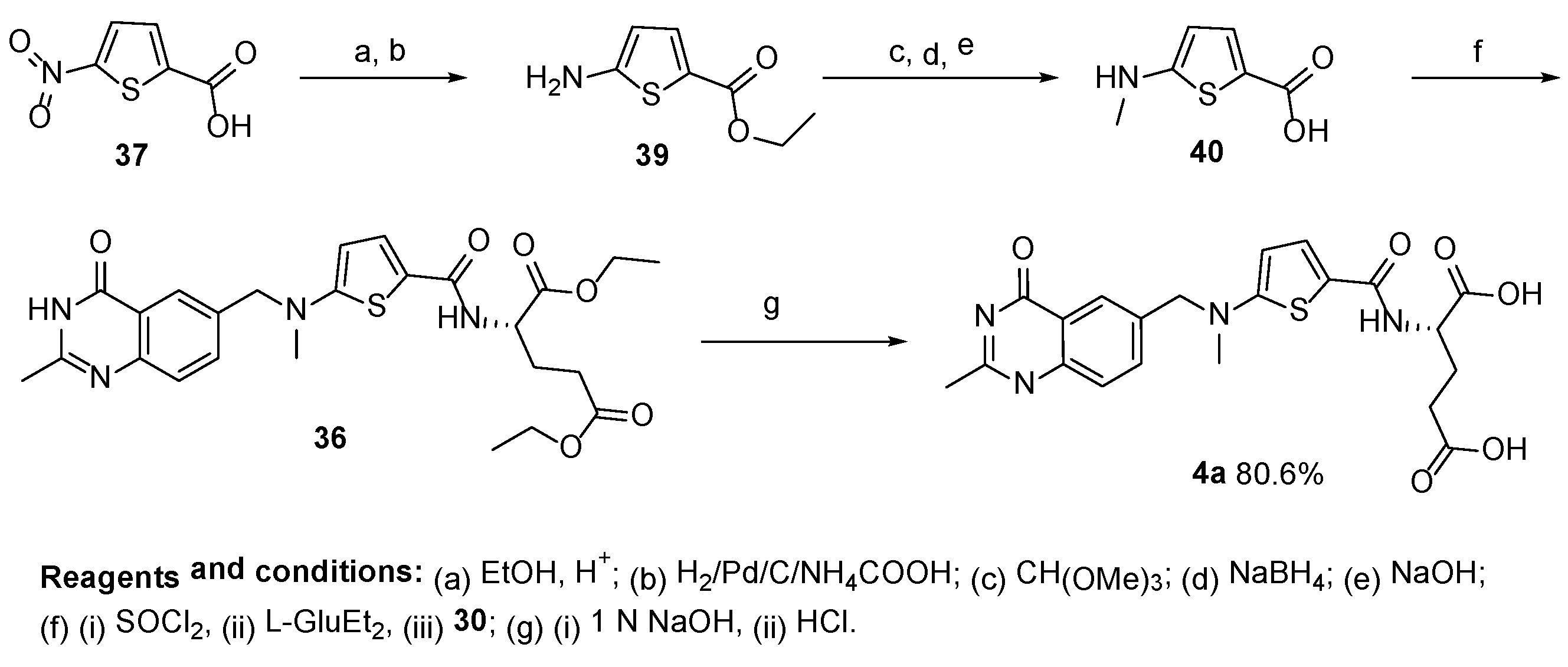{kind=link}
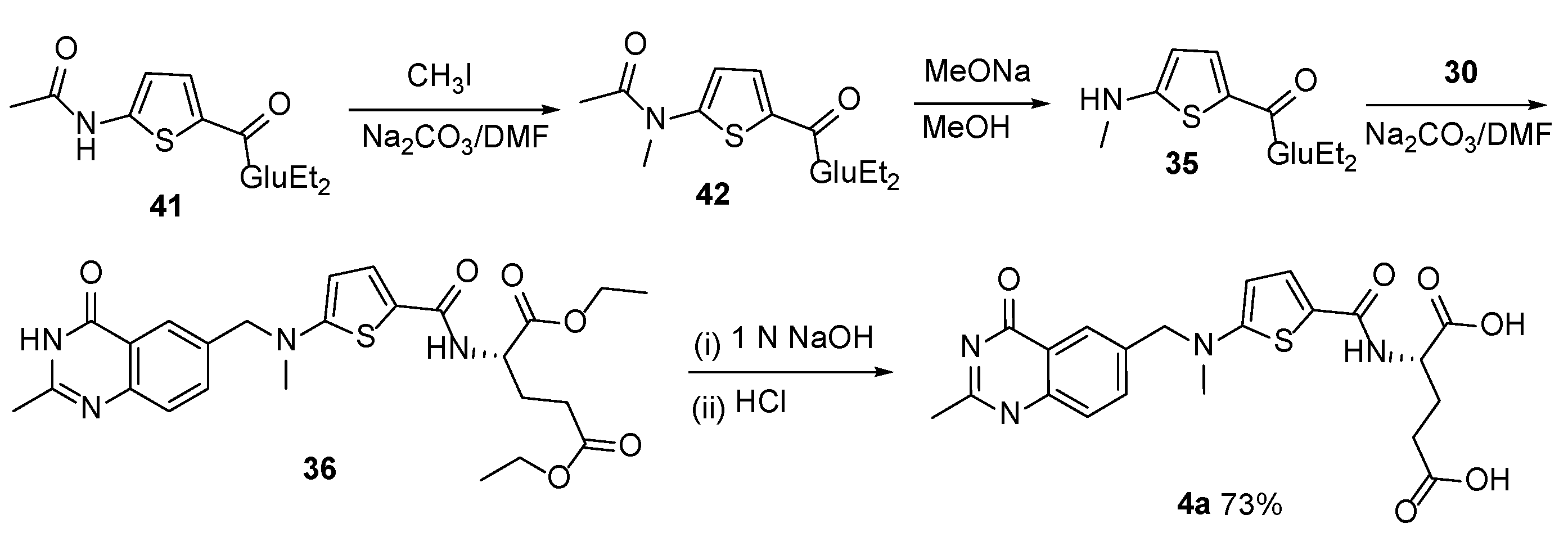{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Discussion
2.1. Mechanism of Antifolates Action
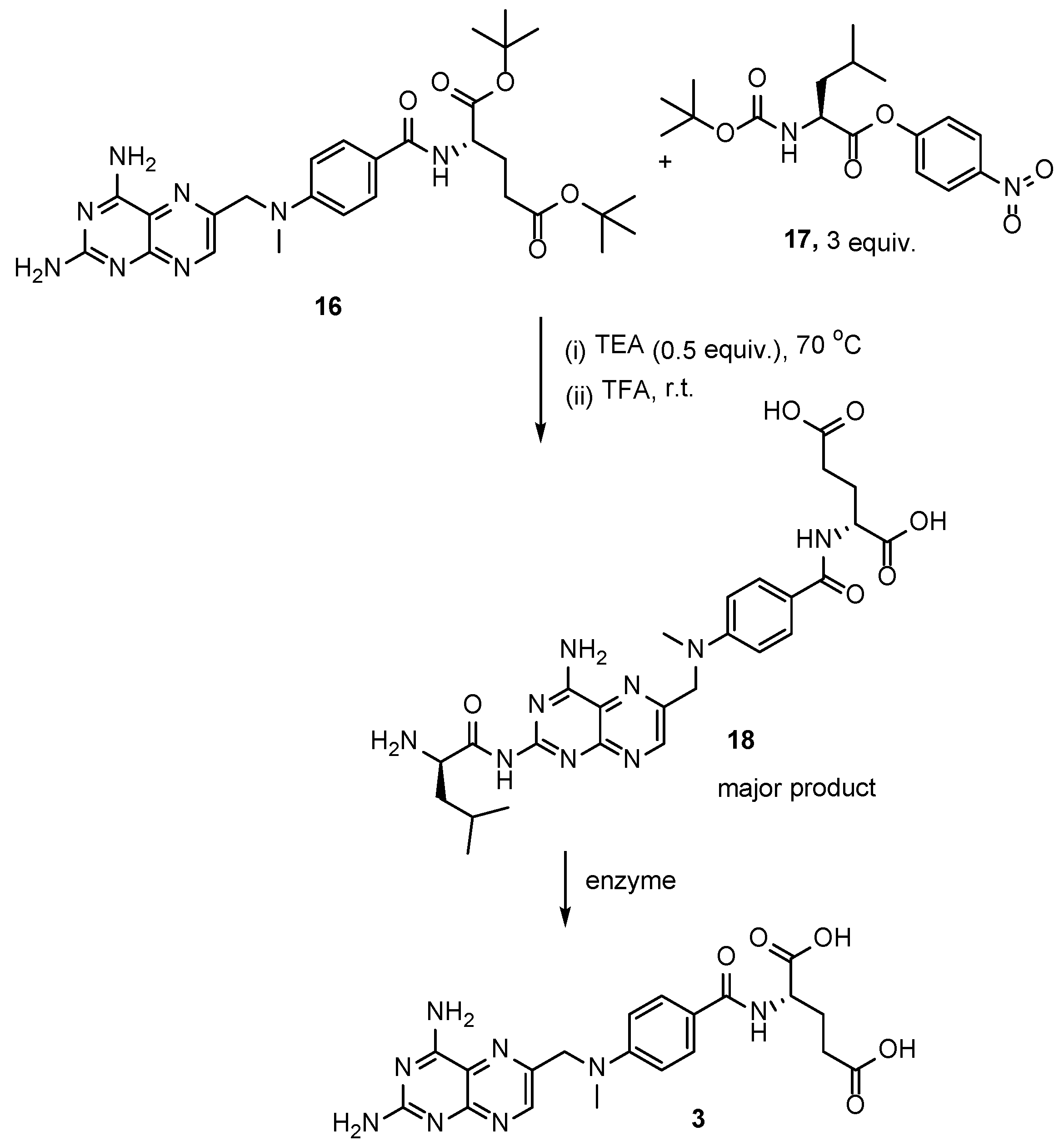
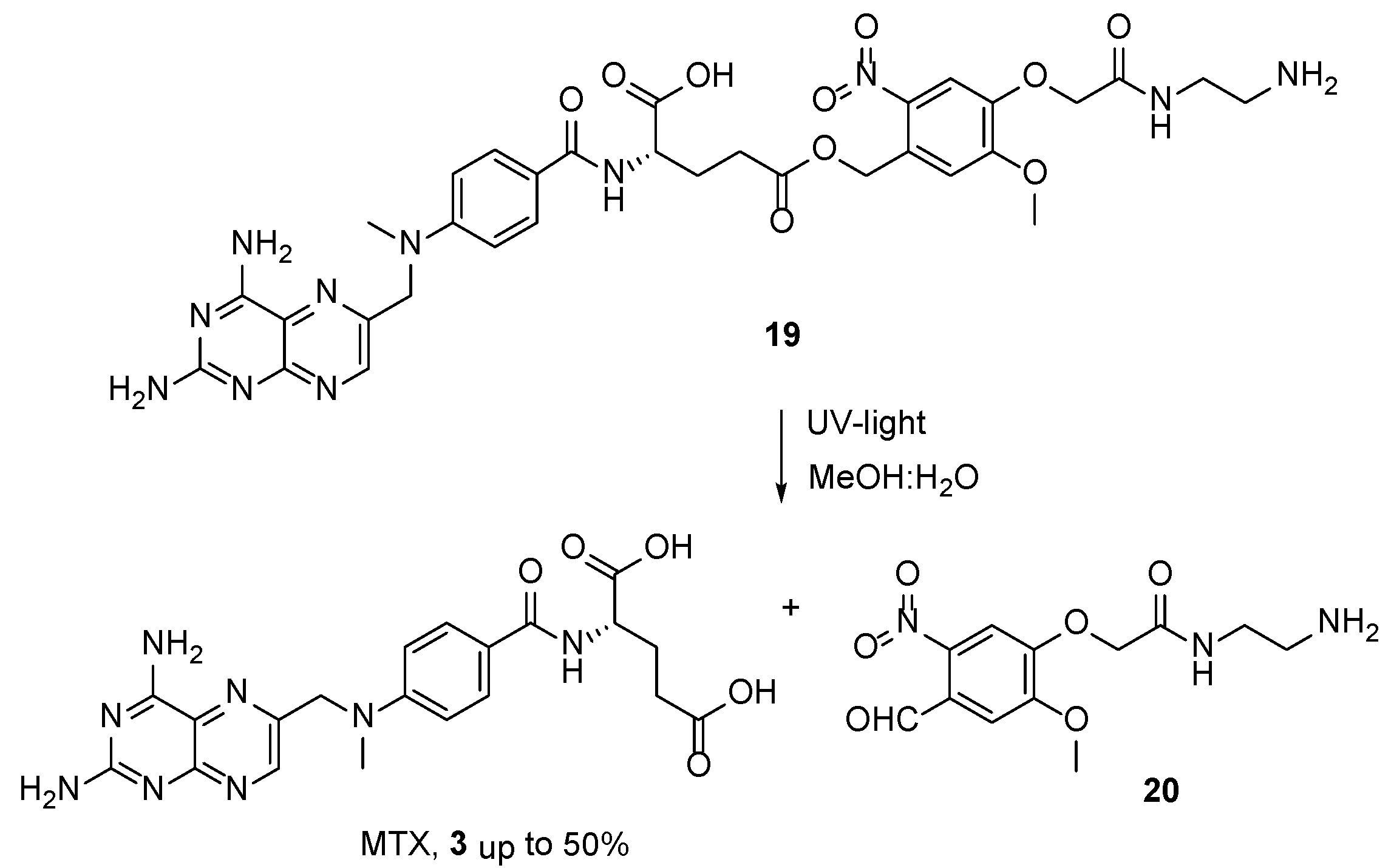
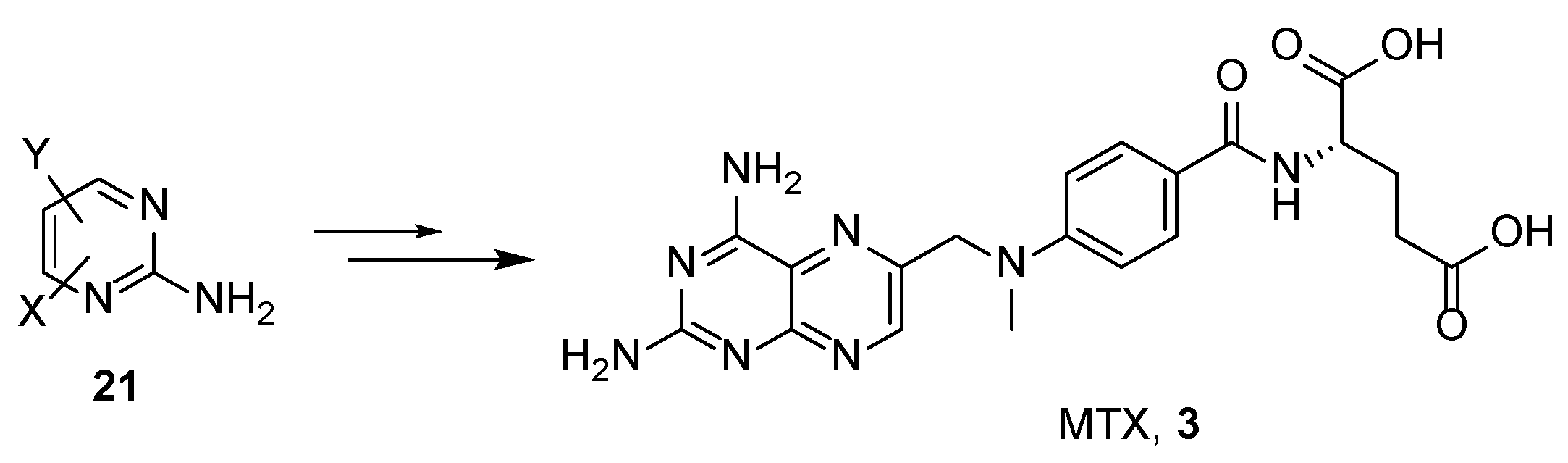
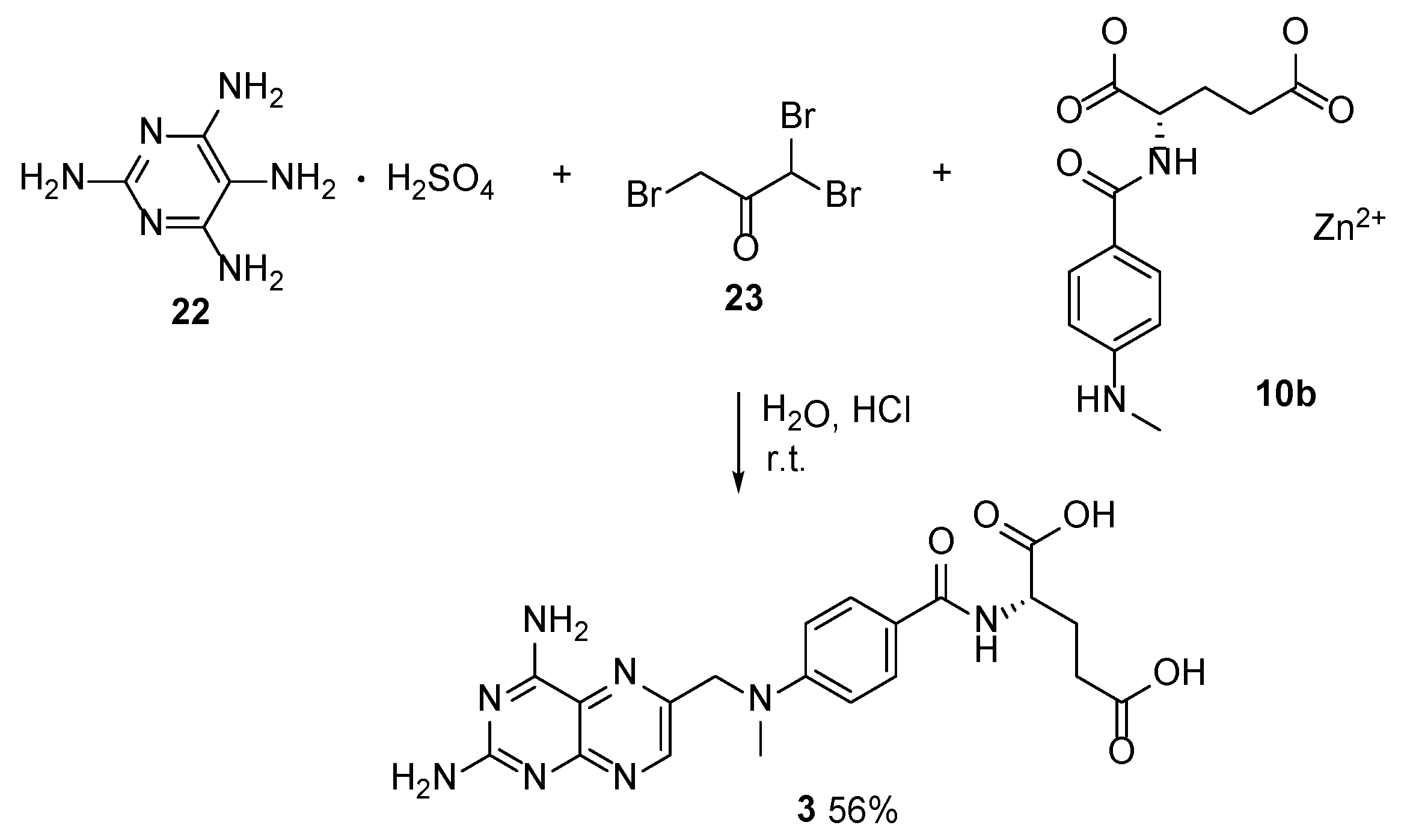
2.2. Methotrexate: (S)-2-(4-(((2,4-Diaminopteridin-6-yl)Methyl)(Methyl)Amino)benzamido) Pentanedioic Acid (MTX, Rheumatrex, Amethopterin, Abitrexate, Trexall, Methylaminopterin, Mexate, Metatrexan)
2.3. Raltitrexed: (2S)-2-[[5-[Methyl-[(2-Methyl-4-oxo-3H-Quinazolin-6-yl)Methyl]Amino] Thiophene-2-Carbonyl]Amino]Pentanedioic Acid (Tomudex, ZD1694)
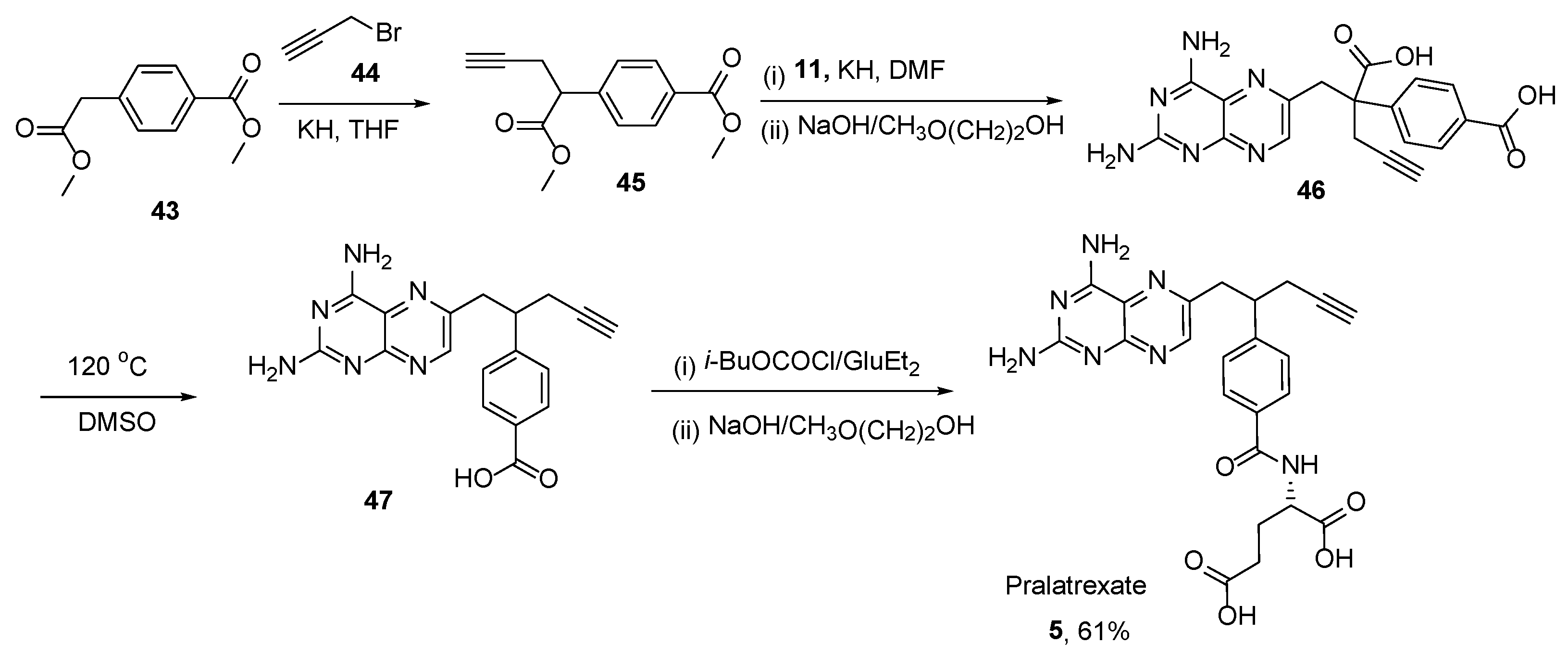
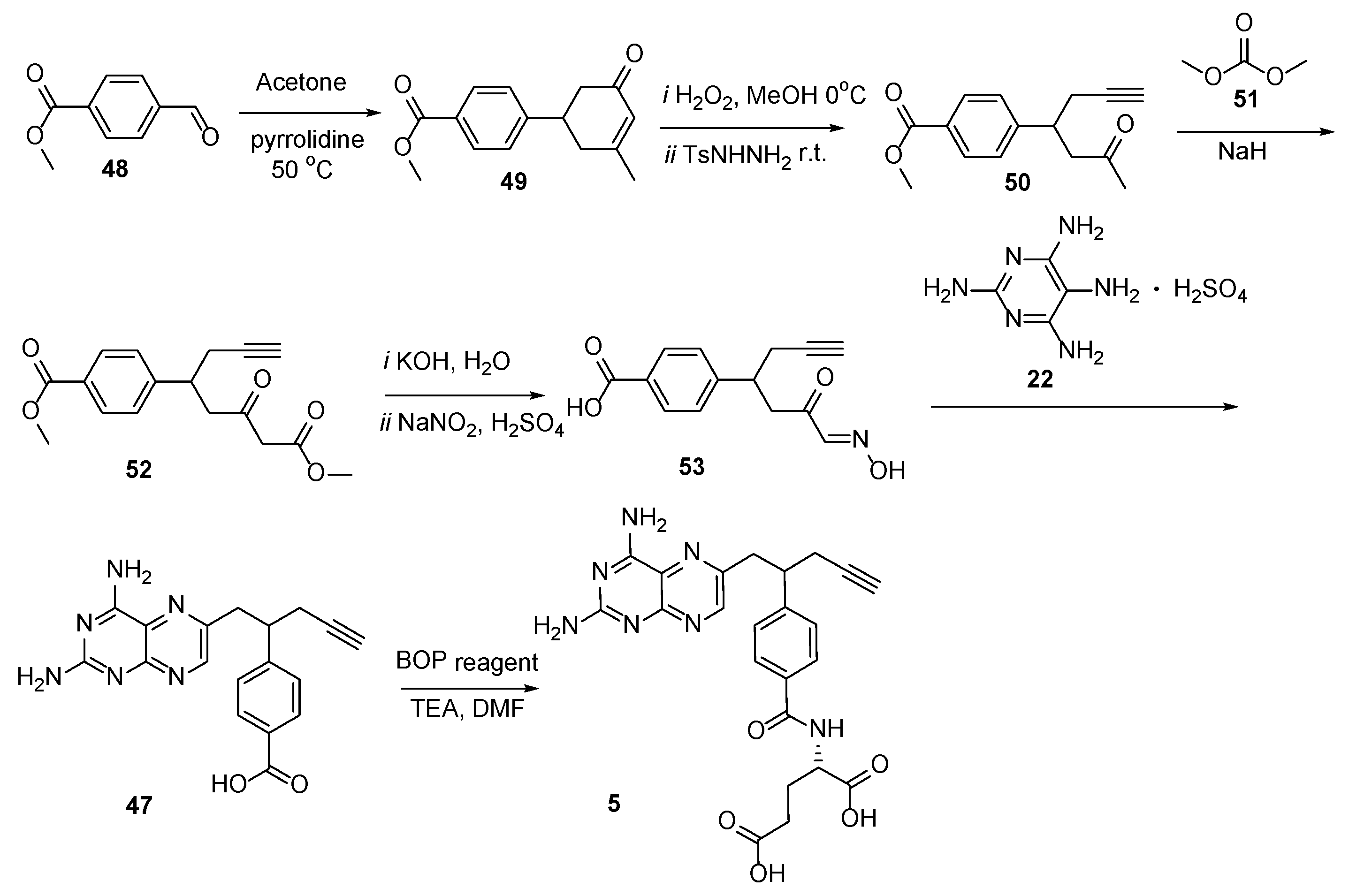
2.4. Pralatrexate (Folotyn): N-4-[1-(2,4-Diaminopteridin-6-yl)Pent-4-yn-2-yl]Benzoyl-L-Glutamic Acid
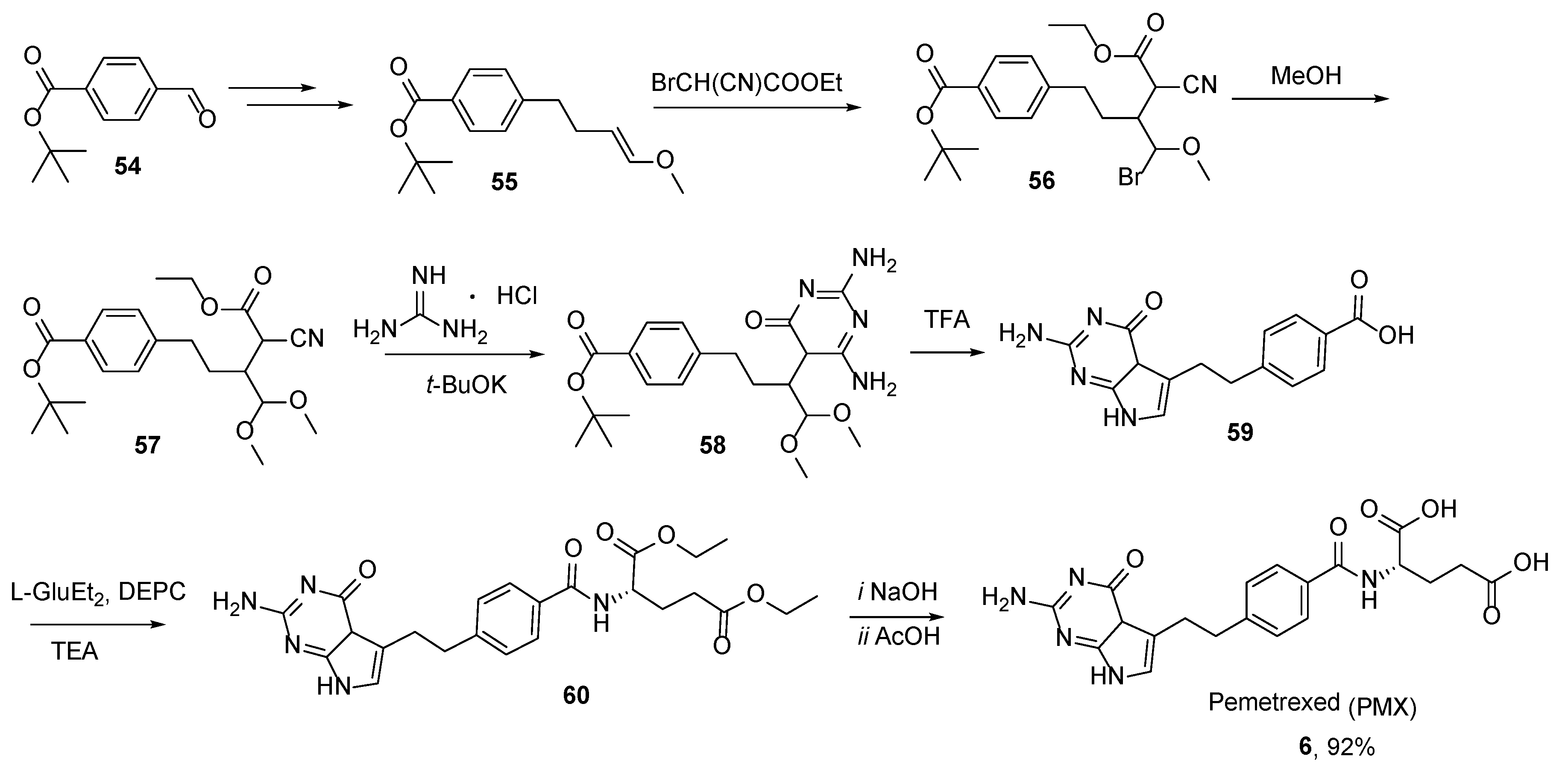
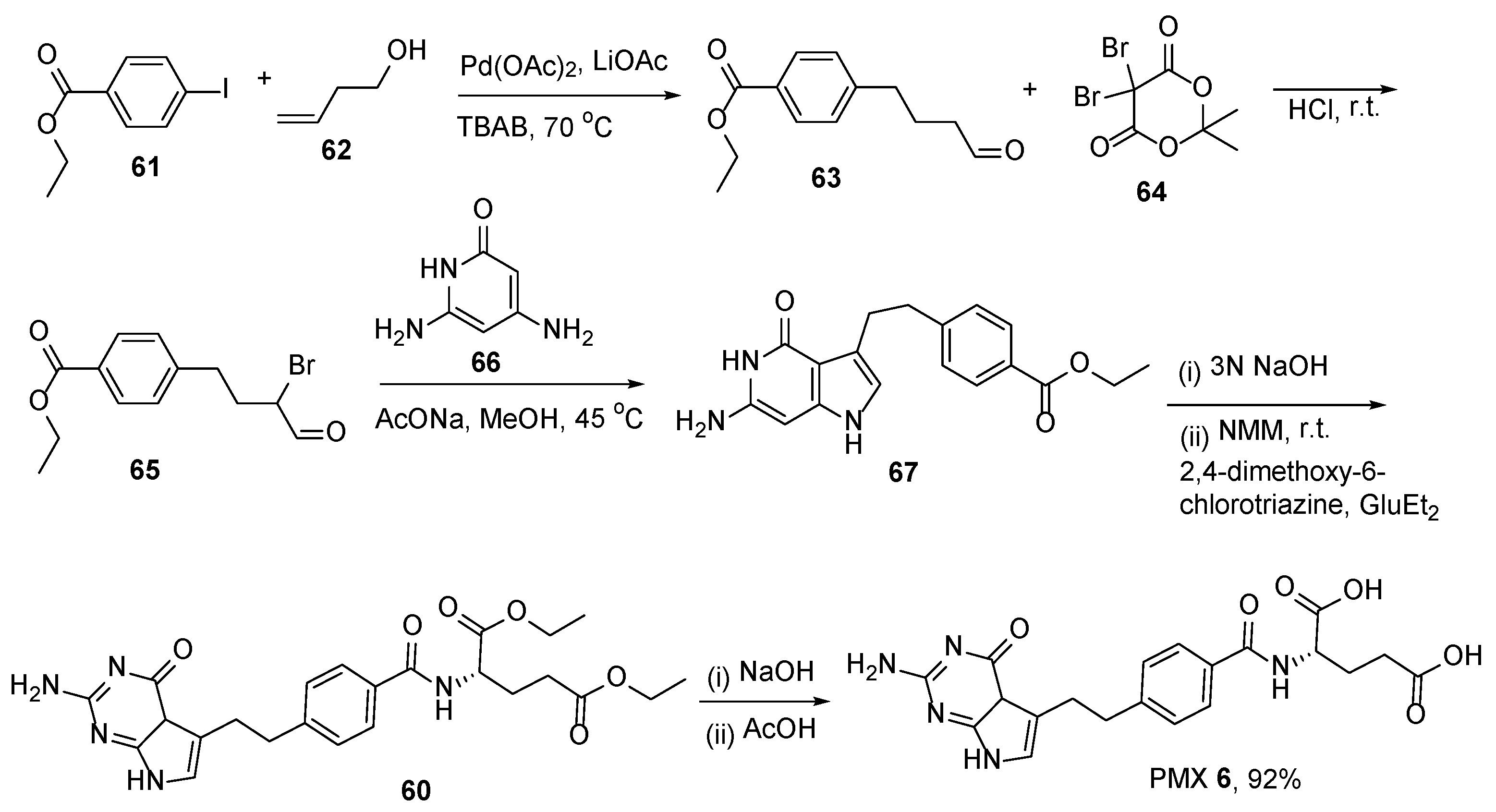
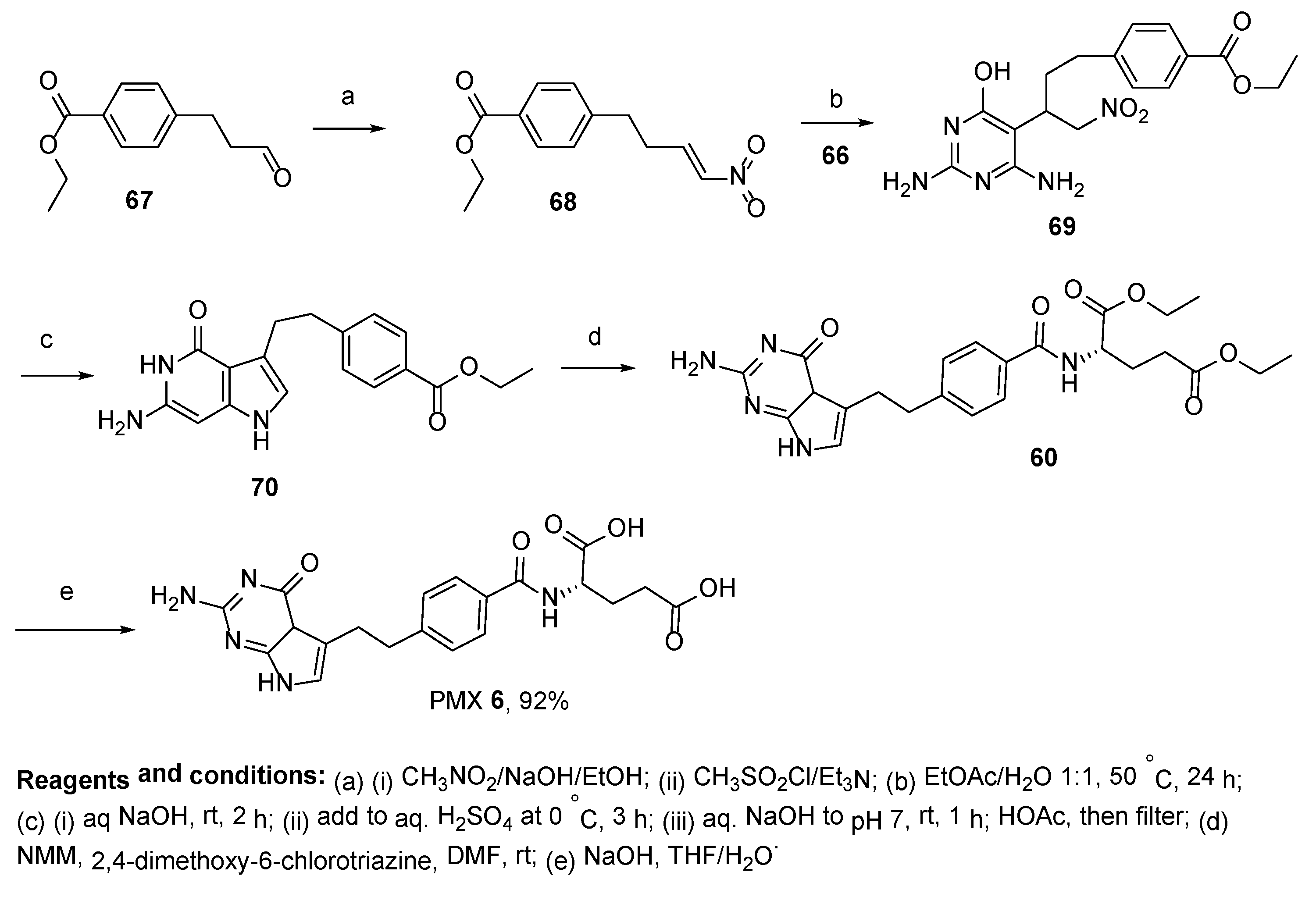
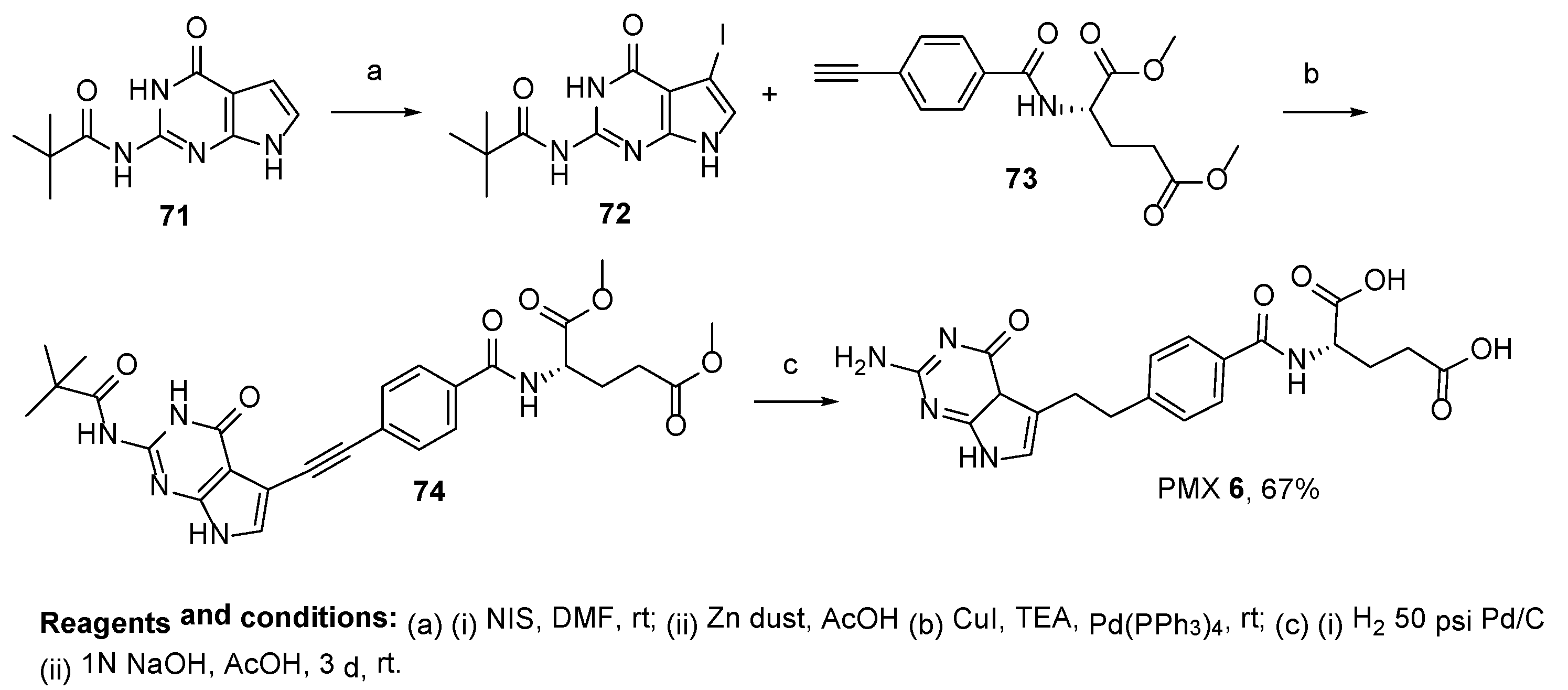
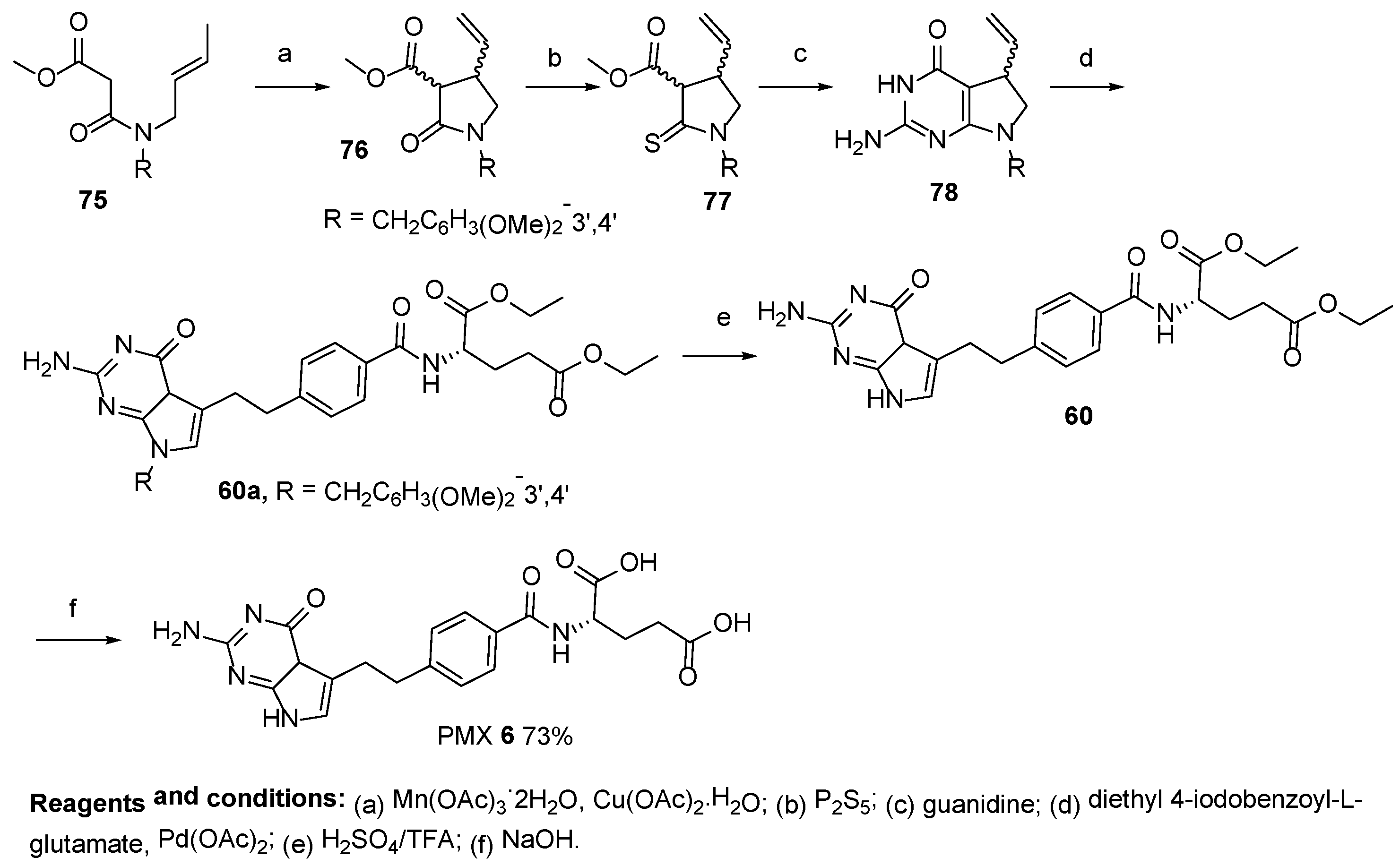
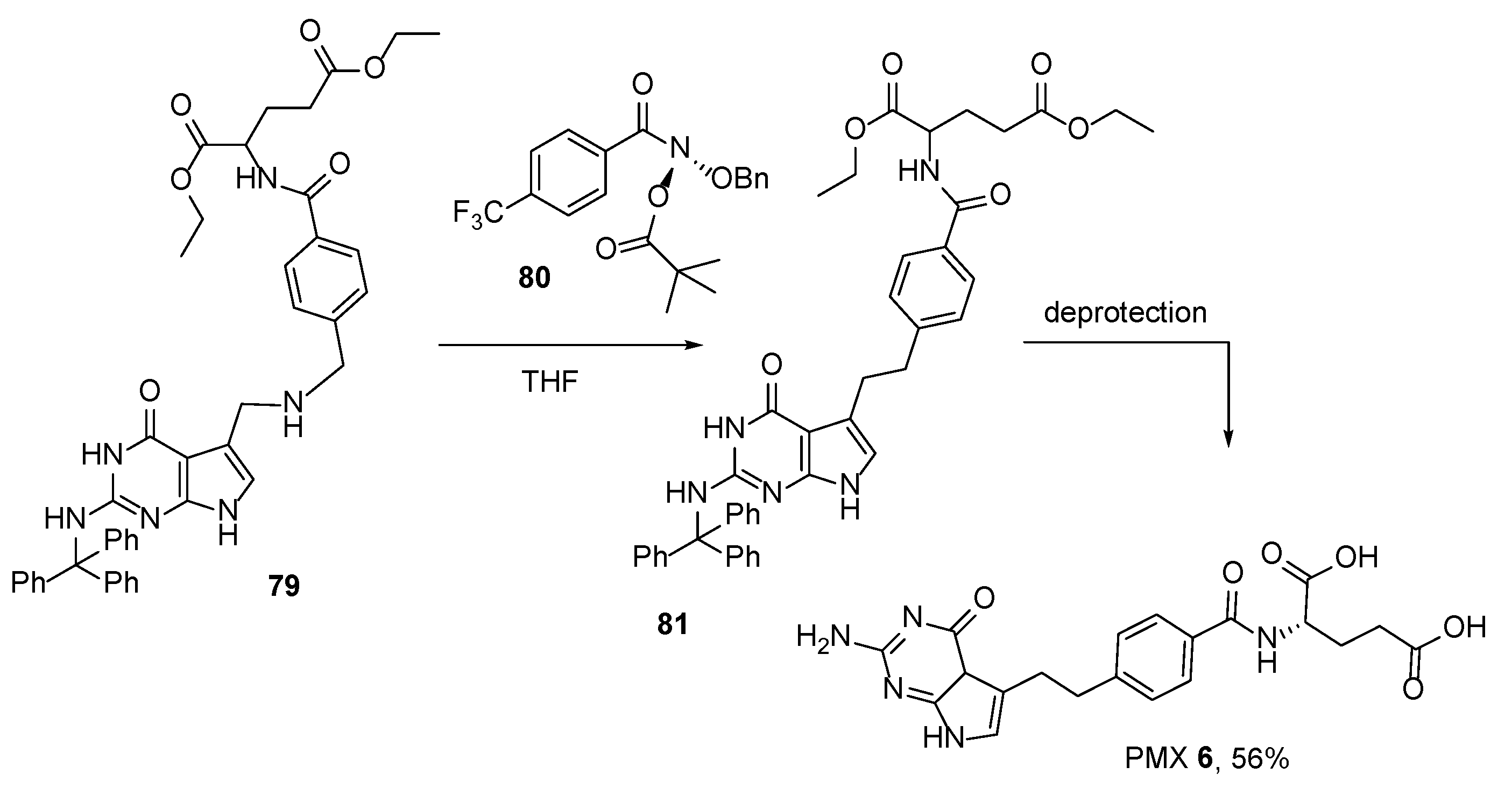
2.5. Pemetrexed: (S)-2-(4-(2-(2-Amino-4-oxo-4,7-Dihydro-1H-Pyrrolo[2,3-d]Pyrimidin-5-yl)Ethyl)Benzamido)Pentanedioic Acid (PMX, ALIMTA, LY231514, MTA)
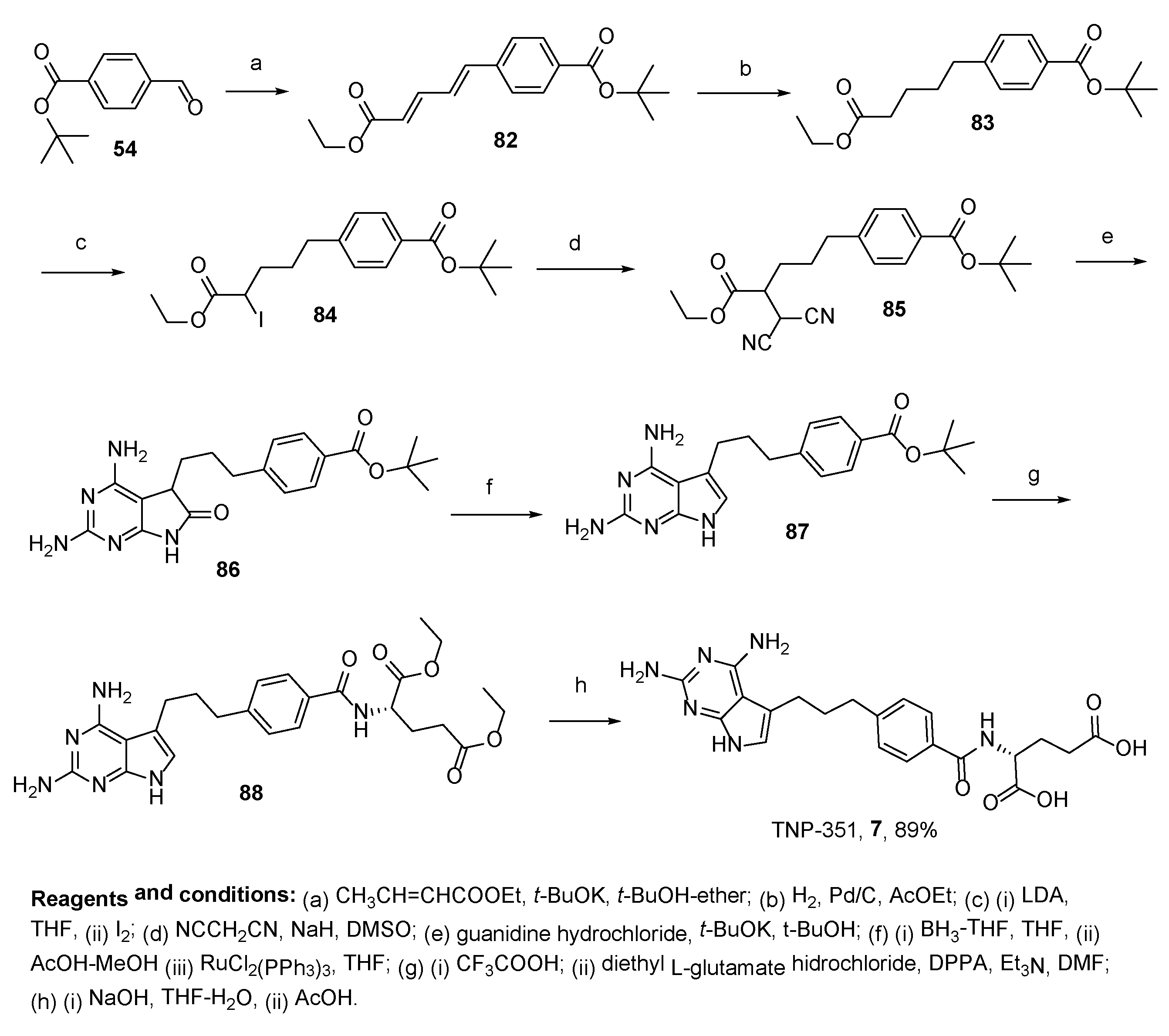
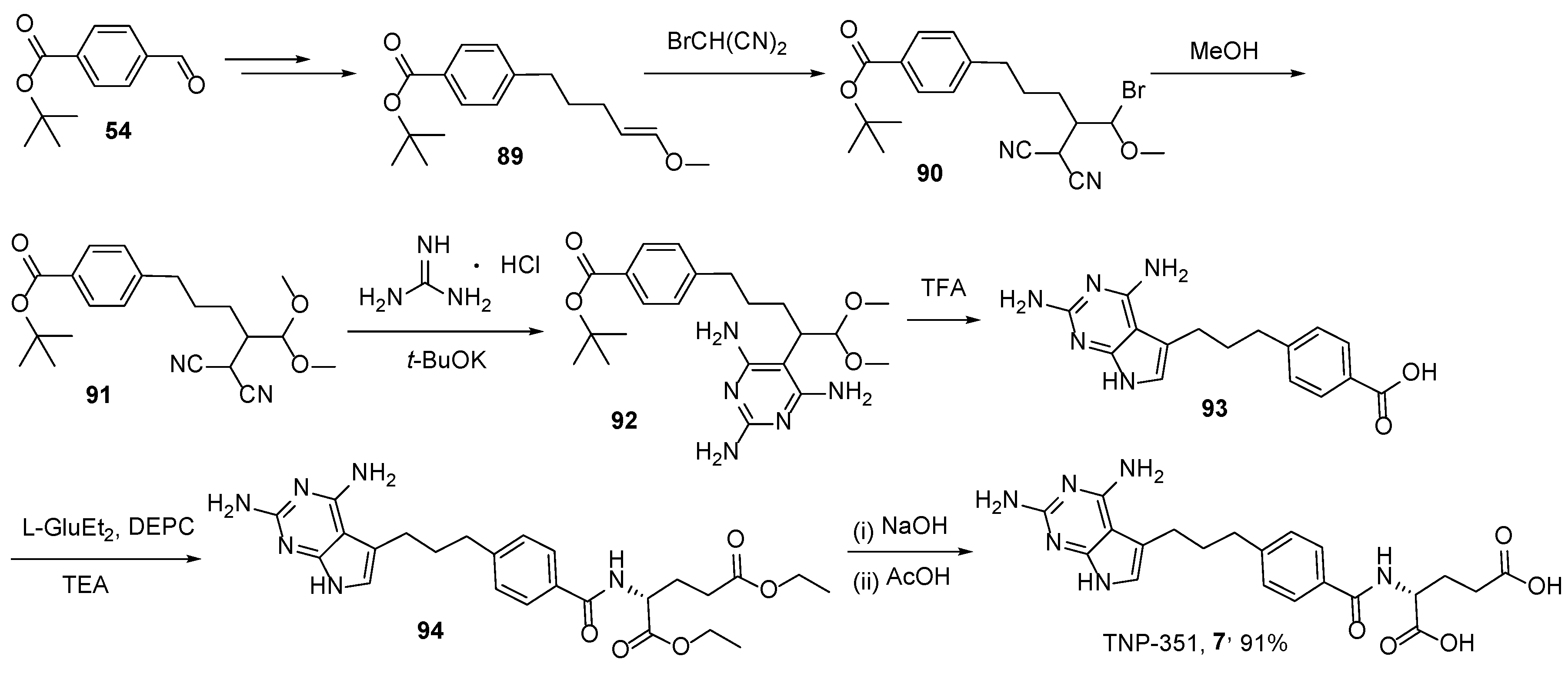
2.6. TNP-351: (2S)-2-[[4-[3-(2,4-Diamino-7H-Pyrrolo[2,3-d]Pyrimidin-5-yl)Propyl]benzoyl]Amino]Pentanedioic Acid (HY-19095)
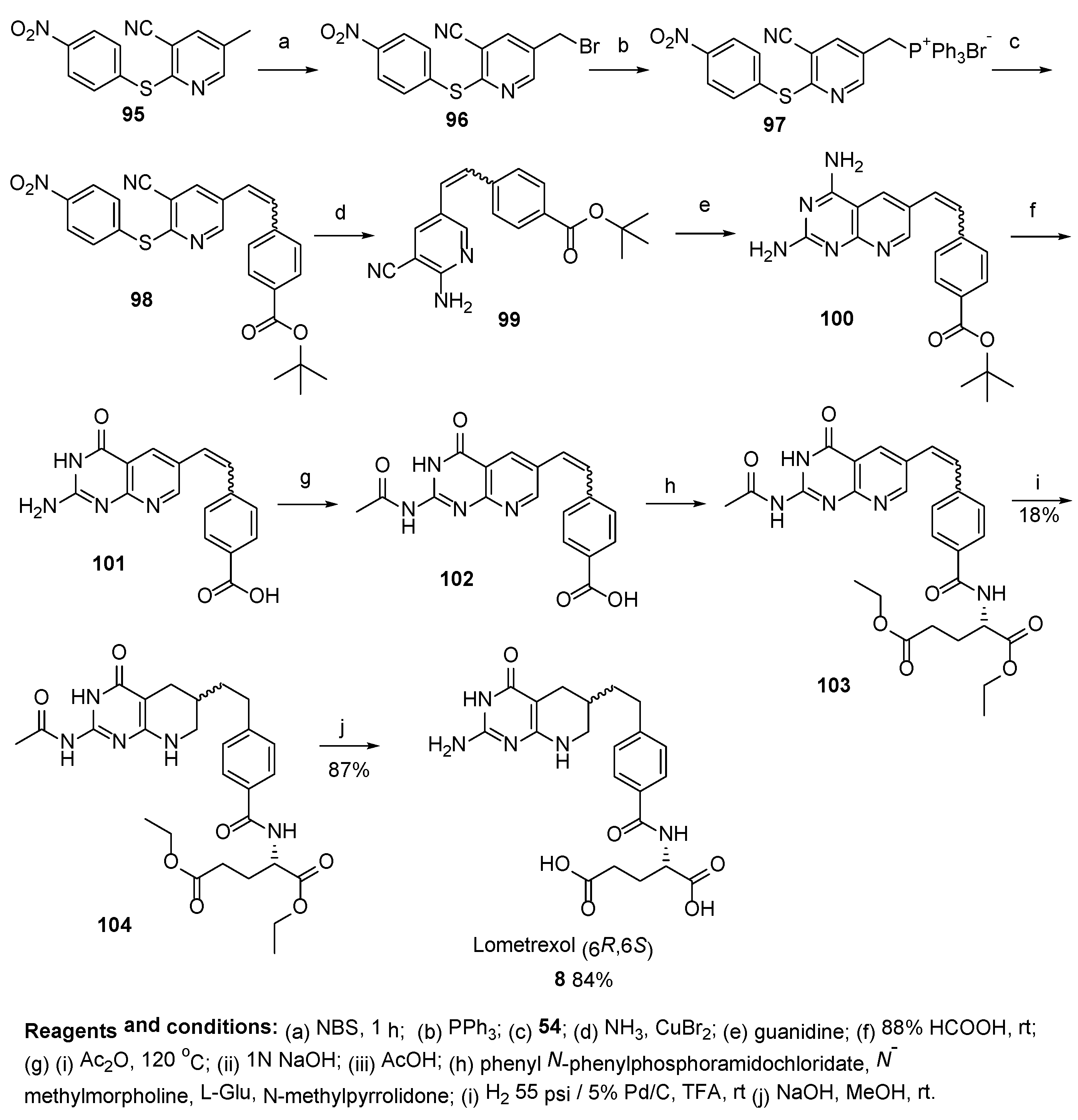
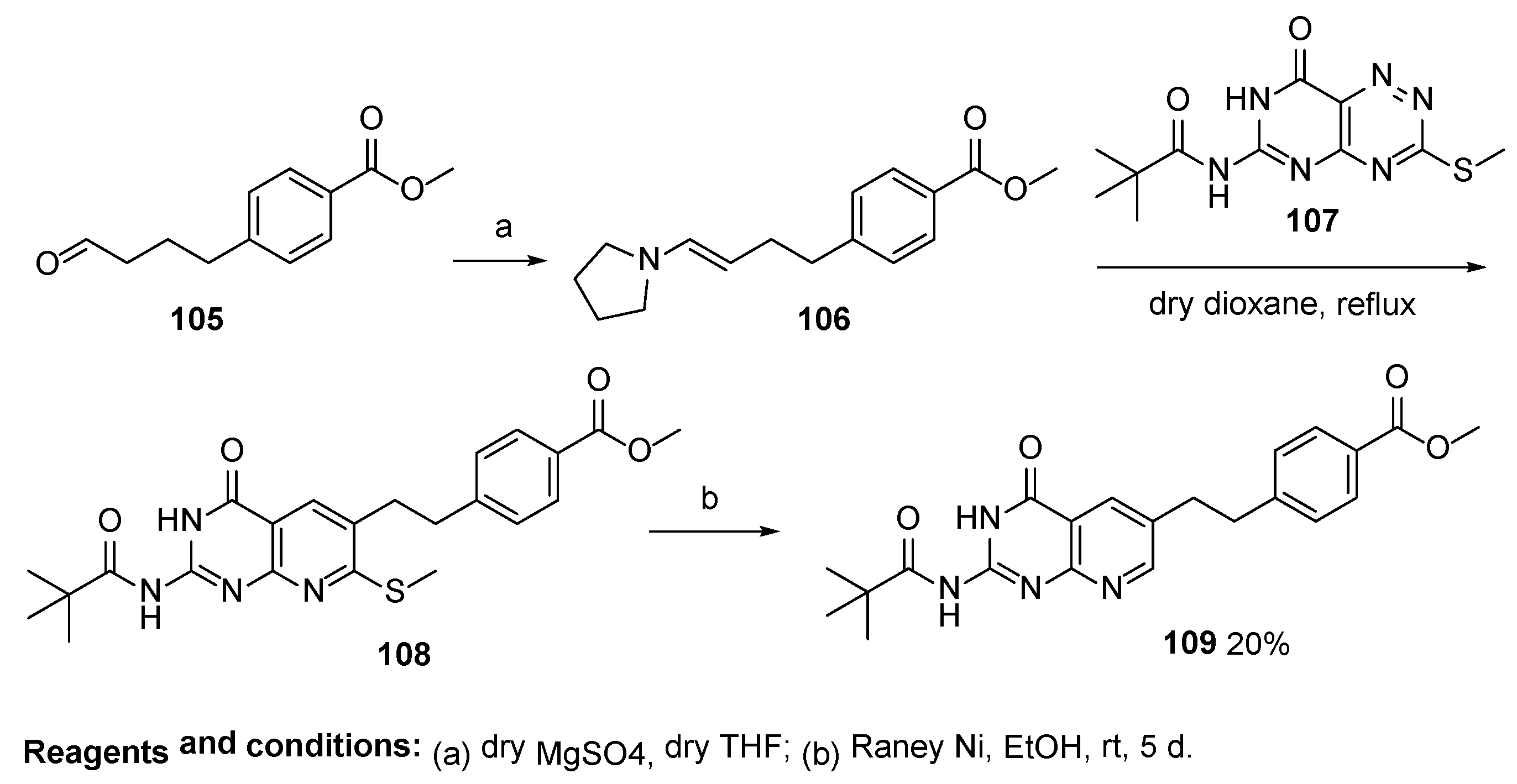
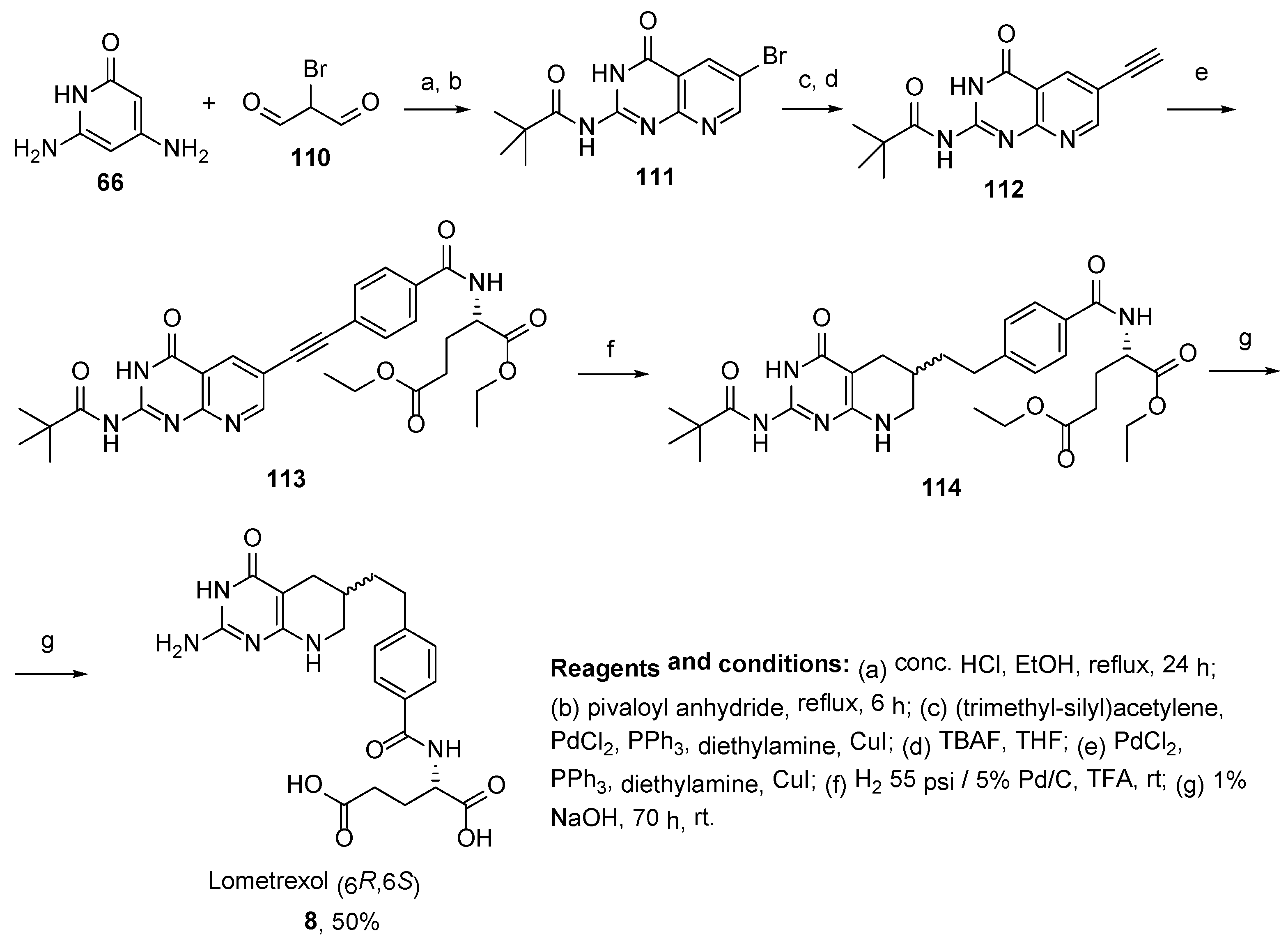
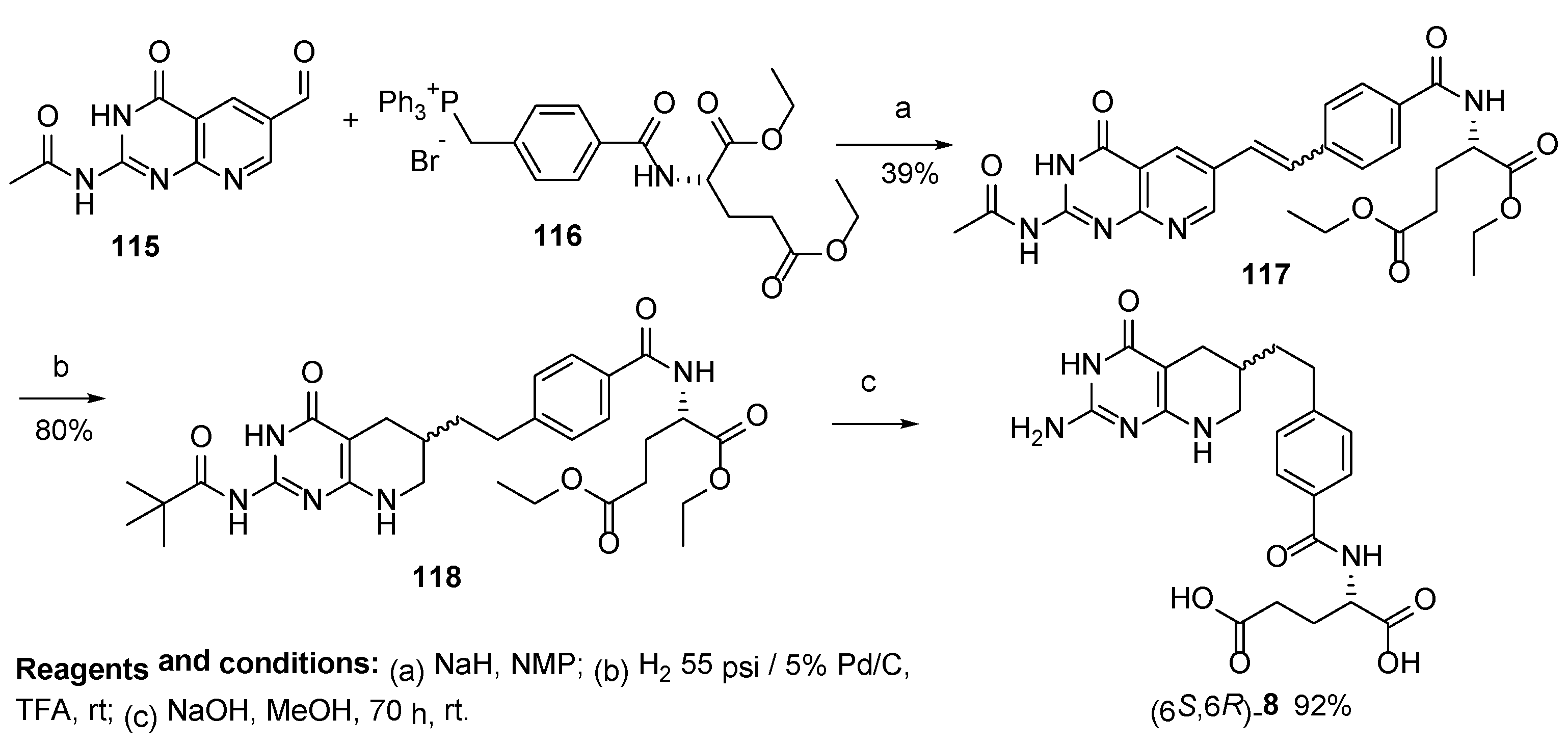
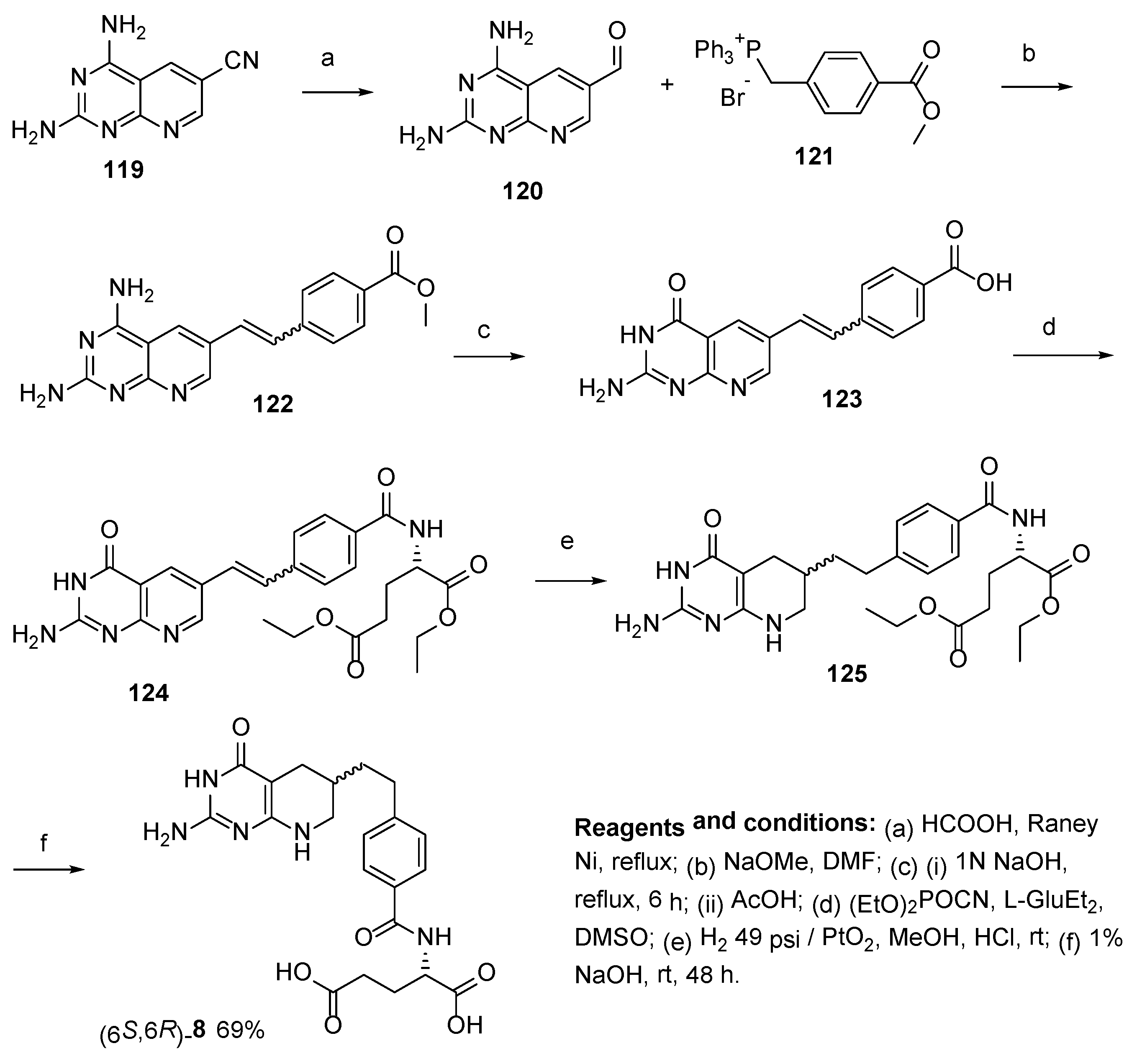
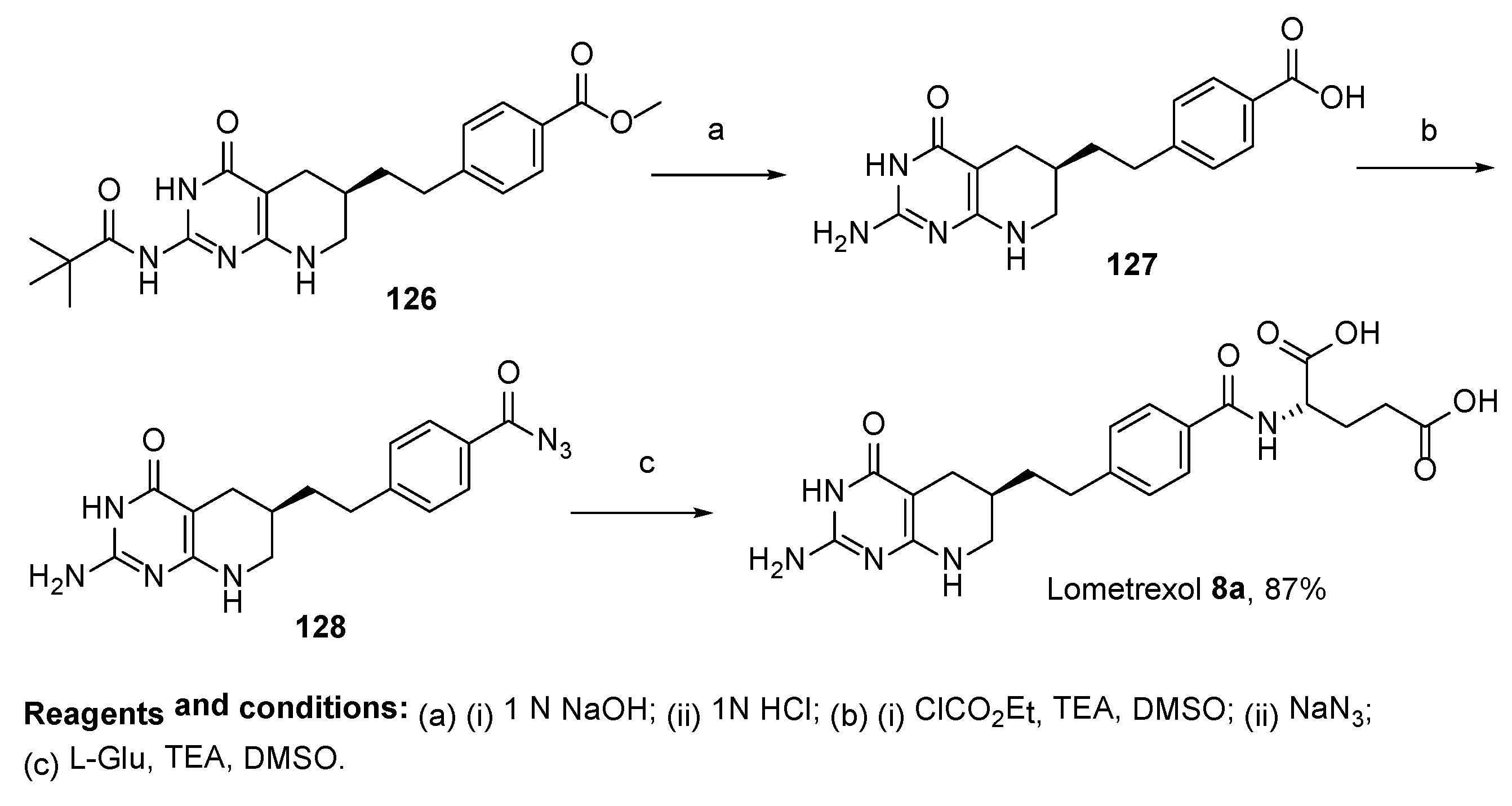
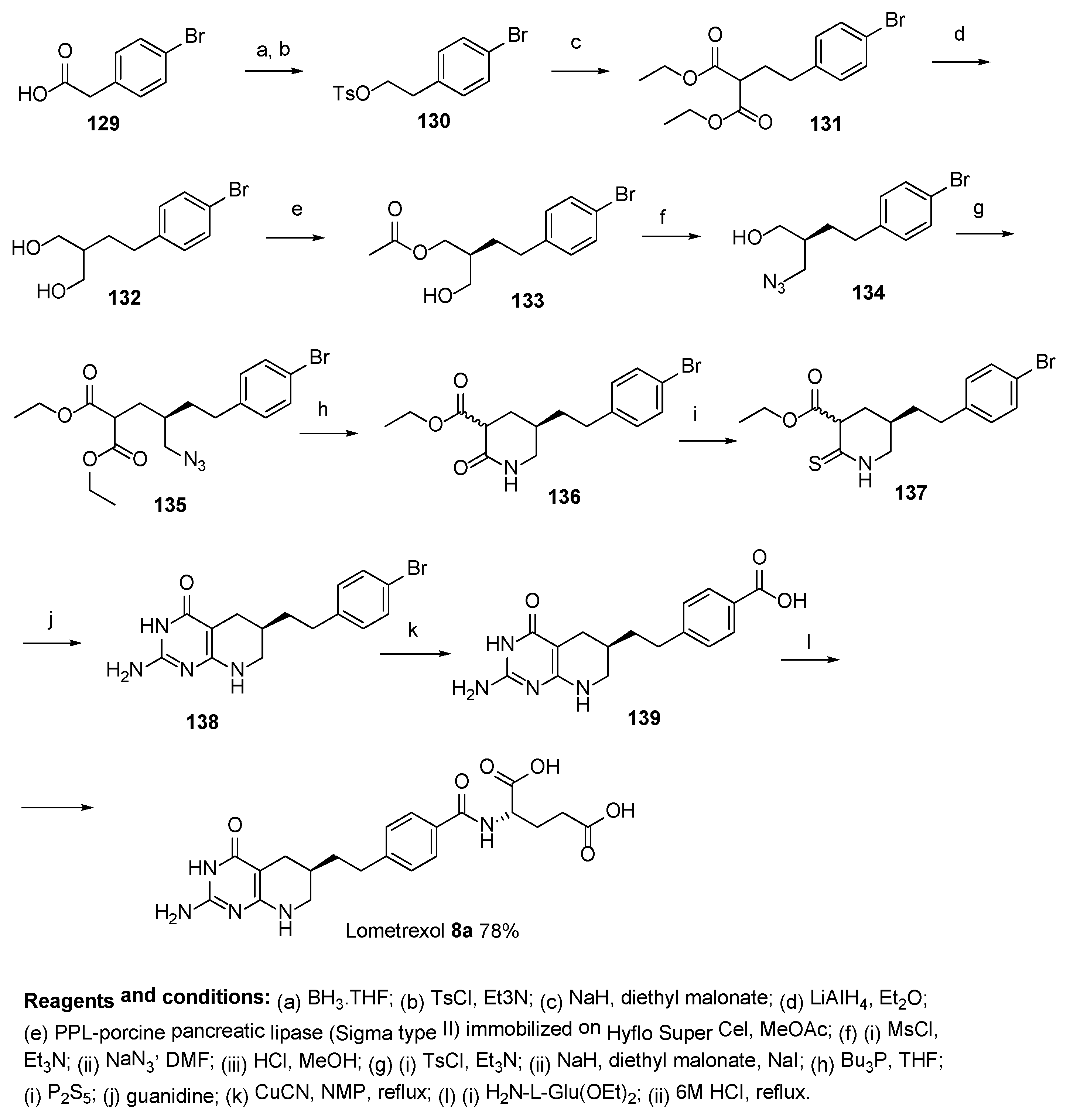
2.7. Lometrexol: (2S)-2-[[4-[2-[(6R)-2-Amino-4-oxo-5,6,7,8-Tetrahydro-1H-Pyrido[2,3-d]Pyrimidin-6-yl]Ethyl]Benzoyl]Amino]Pentanedioic Acid (LY 264618, DDATHF-B, Lometrexolum)
3. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ahluwalia, G.S.; Grem, J.L.; Hao, Z.; Cooney, D.A. Metabolism and action of amino acid analog anti-cancer agents. Pharmacol. Ther. 1990, 46, 243–271. [Google Scholar] [CrossRef]
- Xie, M.; Liu, D.; Yang, Y. Anti-cancer peptides: Classification, mechanism of action, reconstruction and modification. Open Biol. 2020, 10, 200004. [Google Scholar] [CrossRef]
- Chiangjong, W.; Chutipongtanate, S.; Hongeng, S. Anticancer peptide: Physicochemical property, functional aspect and trend in clinical application (Review). Int. J. Oncol. 2020, 57, 678–696. [Google Scholar] [CrossRef]
- Mandal, P.K.; Gao, F.; Lu, Z.; Ren, Z.; Ramesh, R.; Birtwistle, J.S.; Kaluarachchi, K.K.; Chen, X.; Bast, R.C., Jr.; Liao, W.S.; et al. Potent and selective phosphopeptide mimetic prodrugs targeted to the Src homology 2 (SH2) domain of signal transducer and activator of transcription 3. J. Med. Chem. 2011, 54, 3549–3563. [Google Scholar] [CrossRef] [PubMed]
- Gut, J. Aza Analogs of Pyrimidine and Purine Bases of Nucleic Acids. Adv. Heterocycl. Chem. 1963, 1, 189–251. [Google Scholar] [CrossRef]
- Gojkovic, Z.; Karlsson, A. Purine and Pyrimidine-Based Analogs and Suicide Gene Therapy. In Deoxynucleoside Analogs in Cancer Therapy; Humana Press: Totowa, NJ, USA, 2007; pp. 403–439. [Google Scholar] [CrossRef]
- Zelder, F.; Sonnay, M.; Prieto, L. Antivitamins for medicinal applications. ChemBioChem 2015, 16, 1264–1278. [Google Scholar] [CrossRef] [PubMed]
- Howell, A. The Role of Antihormones; CRC Press: Boca Raton, FL, USA, 1990. [Google Scholar]
- Albert, A. Antimetabolites: Antagonistic analogues of coenzymes and enzyme substrates. In Selective Toxicity; Albert, A., Ed.; Springer: Dordrecht, The Netherlands, 1985; pp. 323–378. [Google Scholar]
- Lansiaux, A. Les antimétabolites. Bull. Cancer 2011, 98, 1263–1274. [Google Scholar] [CrossRef] [PubMed]
- Petering, H.G. Folic acid antagonists. Physiol. Rev. 1952, 32, 197–213. [Google Scholar] [CrossRef]
- Ellison, R.R. Treating Cancer with Antimetabolites. Am. J. Nurs. 1962, 62, 79. [Google Scholar] [CrossRef]
- Panderi, I.; Koufopantelis, P. Methotrexate, an antimetabolite of folic acid, a brief throwback. Pharmakeftiki 2014, 26, 45–56. [Google Scholar]
- Chu, E.; Drake, J.C.; Boarman, D.; Baram, J.; Allegra, C.J. Mechanism of thymidylate synthase inhibition by methotrexate in human neoplastic cell lines and normal human myeloid progenitor cells. J. Biol. Chem. 1990, 265, 8470–8478. [Google Scholar] [CrossRef]
- Messmann, R.; Allegra, C. Antifolates. In Cancer Chemotherapy and Biotherapy; Chabner, B., Longo, D., Eds.; L. Williams & W: Philadelphia, PA, USA, 2001; pp. 139–184. [Google Scholar]
- Shih, C.; Chen, V.J.; Gossett, L.S.; Gates, S.B.; MacKellar, W.C.; Habeck, L.L.; Shackelford, K.A.; Mendelsohn, L.G.; Soose, D.J.; Patel, V.F.; et al. Ly231514, a pyrrolo [2,3-d]pyrimidine-based antifolate that inhibits multiple folate-requiring enzymes. Cancer Res. 1997, 57, 1116–1123. [Google Scholar] [PubMed]
- Avendaño, C.; Menéndez, J.C. (Eds.) Antimetabolites. In Medicinal Chemistry of Anticancer Drugs; Elsevier: Amsterdam, The Netherlands, 2008; pp. 9–52. [Google Scholar]
- Moran, R.G. Folate antimetabolites inhibitory to de novo purine synthesis. In New Drugs, Concepts and Results in Cancer Chemotherapy. Cancer Treatment and Research; Muggia, F.M., Ed.; Springer: Boston, MA, USA, 1991; Volume 58, pp. 65–87. [Google Scholar]
- Catalucci, E. Process for the Production of Methotrexate. U.S. Patent US4224446A, 23 September 1991. [Google Scholar]
- Bin, Y.; Shubin, W.; Zhichao, M.; Quansheng, S.; Yanjiao, X.; Lilian, L. A Kind of Preparation Method of Methotrexate (MTX). U.S. Patent CN109553619A, 2 April 2019. [Google Scholar]
- Piper, J.R.; Montgomery, J.A. 6-(Bromomethyl)-2,4-diaminopteridine Hydrobromide. U.S. Patent US4077957A, 11 April 1978. [Google Scholar]
- Luo, J.; Smith, M.D.; Lantrip, D.A.; Wang, S.; Fuchs, P.L. Efficient syntheses of pyrofolic acid and pteroyl azide, reagents for the production of carboxyl-differentiated derivatives of folic acid. J. Am. Chem. Soc. 1997, 119, 10004–10013. [Google Scholar] [CrossRef]
- Attoline, E.; Michieletti, M.; Rossi, D.; Allegrini, P. Process for the Preparation of Pteridine Derivatives. U.S. Patent US4767859, 17 November 1988. [Google Scholar]
- Cheung, A.H.T.; Smal, M.; Chau, D.D. Synthesis of multi-13C-enriched methotrexate for NMR studies of drug-Enzyme interactions. Heterocycles 1987, 25, 507–514. [Google Scholar] [CrossRef]
- Cheung, A.H.T.; Boadle, D.K.; Tran, T.Q. N-(L-α-aminoacyl) derivatives of methotrexate. Heterocycles 1989, 28, 751–758. [Google Scholar] [CrossRef]
- Choi, S.K.; Verma, M.; Silpe, J.; Moody, R.E.; Tang, K.; Hanson, J.J.; Baker, J.R., Jr. A photochemical approach for controlled drug release in targeted drug delivery. Bioorg. Med. Chem. 2012, 20, 1281–1290. [Google Scholar] [CrossRef]
- Lei, T.; Yi, Z.; Jianta, W.; Gaofeng, Z.; Xing, C. Synthesis Process of Methotrexate. U.S. Patent CN112851676A, 28 May 2021. [Google Scholar]
- Wiecko, J. Process for Preparing Methotrexate or an N-Substituted Derivative Thereof and/or a di (lower) Alkyl Ester Thereof and Precursor Therefor. U.S. Patent US4057548A, 30 March 1976. [Google Scholar]
- Seeger, D.R.; Cosulich, D.B.; Smith, J.M.; Hultquist, M.E. Analogs of Pteroylglutamic Acid. III. 4-Amino Derivatives. J. Am. Chem. Soc. 1949, 71, 1753–1758. [Google Scholar] [CrossRef]
- Widemann, B.C.; Balis, F.M.; Godwin, K.S.; McCully, C.; Adamson, P.C. The plasma pharmacokinetics and cerebrospinal fluid penetration of the thymidylate synthase inhibitor raltitrexed (Tomudex(TM)) in a nonhuman primate model. Cancer Chemother. Pharmacol. 1999, 44, 439–443. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Wu, W.; Hong, W.; Sun, X.; Wu, J.; Huang, Q. Raltitrexed-based chemotherapy for advanced colorectal cancer. Clin. Res. Hepatol. Gastroenterol. 2014, 38, 219–225. [Google Scholar] [CrossRef]
- Cocconi, G.; Cunningham, D.; Van Cutsem, E.; Francois, E.; Gustavsson, B.; van HazelD Kerr, G.; Possinger, K.; Hietschold, S.M. Open, randomized, multicenter trial of raltitrexed versus fluorouracil plus high-dose leucovorin in patients with advanced colorectal cancer. Tomudex Colorectal Cancer Study Group. J. Clin. Oncol. 2016, 16, 2943–2952. [Google Scholar] [CrossRef]
- Marsham, P.R.; Hughes, L.R.; Jackman, A.L.; Hayter, A.J.; Oldfield, J.; Wardleworth, J.M.; Bishop, J.A.M.; O’Connor, B.M.; Calvert, A.H. Quinazoline Antifolate Thymidylate Synthase Inhibitors: Heterocyclic Benzoyl Ring Modifications. J. Med. Chem. 1991, 34, 1594–1605. [Google Scholar] [CrossRef] [PubMed]
- Cao, S.L.; Wan, R.; Feng, Y.P. New synthesis of thymidylate synthase inhibitor raltitrexed. Synth. Commun. 2003, 33, 3519–3526. [Google Scholar] [CrossRef]
- Yao, T.; Zubing, W.; Shuwang, G.; Jian, W.; Yuzhu, C.; Feng, L.; Dan, X.; Chunxia, Z.; Zhoushan, T. The Formoxyl of 5-((Alkoxy methylene)amino)thienyl-2-formyl Group)-L-glutamic Acid Dialkyl Ester and Preparation Method Thereof. U.S. Patent CN106957296A, 18 July 2017. [Google Scholar]
- Xiqun, J.; Wei, W.; Zhoushan, T.; Jun, H.; Jing, W.; Yuzhu, C.; Huaping, W.; Dan, X.; Chunxia, Z.; Chuanjun, W. A kind of Pharmaceutical Composition of Raltitrexed Pharmaceutical Composition and Preparation Method Thereof. U.S. Patent CN107616976A, 23 January 2018. [Google Scholar]
- Shaojie, H.; Wei, S.; Zhaobai, Z.; Xu, S. Preparation Method of Raltitrexed. U.S. Patent CN110551114A, 10 December 2019. [Google Scholar]
- O’Connor, O.A.; Pro, B.; Pinter-Brown, L.; Bartlett, N.; Popplewell, L.; Coiffier, B.; Lechowicz, M.J.; Savage, K.J.; Shustov, A.R.; Gisselbrecht, C.; et al. Pralatrexate in patients with relapsed or refractory peripheral T-cell lymphoma: Results from the pivotal PROPEL study. J. Clin. Oncol. 2011, 29, 1182–1189. [Google Scholar] [CrossRef] [PubMed]
- Amengual, J.E.; Lichtenstein, R.; Lue, J.; Sawas, A.; Deng, C.; Lichtenstein, E.; Khan, K.; Atkins, L.; Rada, A.; Kim, H.A.; et al. A phase 1 study of romidepsin and pralatrexate reveals marked activity in relapsed and refractory T-cell lymphoma. Blood 2018, 131, 397–407. [Google Scholar] [CrossRef] [PubMed]
- Parker, T.; Barbarotta, L.; Foss, F. Pralatrexate: Treatment of T-cell non-Hodgkin’s lymphoma. Future Oncol. 2012, 9, 21–29. [Google Scholar] [CrossRef] [PubMed]
- DeGraw, J.I.; Colwell, W.T.; Piper, J.R.; Sirotnak, F.M. Synthesis and Antitumor Activity of 10-Propargyl-10-deazaaminopterin. J. Med. Chem. 1993, 36, 2228–2231. [Google Scholar] [CrossRef]
- Guimin, Z.; Chengfu, C.; Chuanbing, W. A Kind of Preparation Method of Pralatrexate. U.S. Patent CN108069970, 02 June 2020. [Google Scholar]
- Giust, W.; Burton, R.; Gorin, B.; Clayton, J. Process for Preparation of an Antifolate Agent. U.S. Patent WO2013/177713A1, 5 December 2013. [Google Scholar]
- O’Connor, O.A.; Sirotnak, F.M. Treatment of T-Cell Lymphoma Using 10-Propargyl-10-Deazaaminopterin. U.S. Patent US2005/267117A1, 1 December 2005. [Google Scholar]
- Lahiri, S.; Gupta, N.; Singh, H.K.; Panda, N.; Handa, V.; Abul, A.; Gupta, C.K.; Sanghani, S.; Sonavane, G.M. Process for the Preparation of Pralatrexate. U.S. Patent US9440979B2, 13 September 2016. [Google Scholar]
- Lahiri, S.; Gupta, N.; Singh, H.K. Salts of Pralatrexate. U.S. Patent WO2014/20553A1, 6 February 2014. [Google Scholar]
- Lahiri, S.; Gupta, N.; Singh, H.K.; Panda, N.; Handa, V.; Abul, A.; Gupta, C.K.; Sanghani, S.; Sonavane, G.M. Improved Process for the Preparation of Pralatrexate. U.S. Patent WO2014016740A2, 30 January 2014. [Google Scholar]
- Tiseni, P.S.; Galluzzo, C.; Canavesi, A.; Biljan, T. Processes and Intermediates for Preparing Pralatrexate. U.S. Patent EP2794610B1, 27 June 2013. [Google Scholar]
- Pronk, G.J. Optically Pure Diastereomers of 10-Propargyl-10-Deazaaminopterin and Methods of Using Same. U.S. Patent US8835433B2, 4 August 2011. [Google Scholar]
- Alla, R.R.V.; Ramarao, C.; Michel, P.T.; Nitlikar, L.H.; Kalam, B.R.; Duduka, R. A Process for Preparing Intermediates of 10-propargyl-10-deazaaminopterin (pralatrexate) Synthesis and the Intermediates Thereof. U.S. Patent WO2013164856A1, 7 November 2013. [Google Scholar]
- Cohen, M.H.; Justice, R.; Pazdur, R. Approval Summary: Pemetrexed in the Initial Treatment of Advanced/Metastatic Non-Small Cell Lung Cancer. Oncologist 2009, 14, 930–935. [Google Scholar] [CrossRef] [PubMed]
- Rossi, A.; Ricciardi, S.; Maione, P.; de Marinis, F.; Gridelli, C. Pemetrexed in the treatment of advanced non-squamous lung cancer. Lung Cancer 2009, 66, 141–149. [Google Scholar] [CrossRef]
- Gandhi, L.; Rodríguez-Abreu, D.; Gadgeel, S.; Esteban, E.; Felip, E.; De Angelis, F.; Domine, M.; Clingan, P.; Hochmair, M.J.; Powell, S.F.; et al. Pembrolizumab plus Chemotherapy in Metastatic Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2018, 378, 2078–2092. [Google Scholar] [CrossRef] [PubMed]
- Azzoli, C.G.; Kris, M.G.; Pfister, D.G. Cisplatin versus carboplatin for patients with metastatic non-small-cell lung cancer–an old rivalry renewed. J. Natl. Cancer Inst. 2007, 99, 828–829. [Google Scholar] [CrossRef]
- Manegold, C. Pemetrexed (alimta, MTA, multitargeted antifolate, LY231514) for malignant pleural mesothelioma. Semin. Oncol. 2003, 30, 32–36. [Google Scholar] [CrossRef] [PubMed]
- McLeod, H.L.; Cassidy, J.; Powrie, R.H.; Priest, D.G.; Zorbas, M.A.; Synold, T.W.; Shibata, S.; Spicer, D.; Bissett, D.; Pithavala, Y.K.; et al. Pharmacokinetic and Pharmacodynamic Evaluation of the Glycinamide Ribonucleotide Formyltransferase Inhibitor AG20341. Clin. Cancer Res. 2000, 6, 2677–2684. [Google Scholar] [PubMed]
- Miwa, T.; Hitaka, T.; Akimoto, H. A Novel Synthetic Approach to Pyrrolo [2,3-d]pyrimidine Antifolates. J. Org. Chem. 1993, 58, 1696–1701. [Google Scholar] [CrossRef]
- Mitchell-Ryan, S.; Wang, Y.; Raghavan, S.; Ravindra, M.P.; Hales, E.; Orr, S.; Cherian, C.; Hou, Z.; Matherly, L.H.; Gangjee, A. Discovery of 5-substituted pyrrolo[2,3-d]pyrimidine antifolates as dual-acting inhibitors of glycinamide ribonucleotide formyltransferase and 5-aminoimidazole-4-carboxamide ribonucleotide formyltransferase in de novo purine nucleotide biosynthesis. J. Med. Chem. 2013, 56, 10016–10032. [Google Scholar] [CrossRef]
- Bonaccorsi, F.; Calvani, F.; Pasqui, F. Process for the Preparation of Pemetrexed and Lysin Salt Thereof. U.S. Patent EP2882753B1, 13 February 2014. [Google Scholar]
- Michalak, O.; Gruza, M.M.; Witkowska, A.; Bujak, I.; Cmoch, P. Synthesis and physicochemical characterization of the impurities of pemetrexed disodium, an anticancer drug. Molecules 2015, 20, 10004–10031. [Google Scholar] [CrossRef]
- Taylor, E.C.; Liu, B. A simple and concise synthesis of LY231514 (MTA). Tetrahedron Lett. 1999, 40, 4023–4026. [Google Scholar] [CrossRef]
- Taylor, E.C.; Liu, B. A New and Efficient Synthesis of Pyrrolo[2,3-d]pyrimidine Anticancer Agents: Alimta (LY231514, MTA), Homo-Alimta, TNP-351, and Some Aryl 5-Substituted Pyrrolo[2,3-d]pyrimidines. J. Org. Chem. 2003, 68, 9938–9947. [Google Scholar] [CrossRef]
- Taylor, E.C.; Liu, B. Process for the Preparation of pyrrolo[2,3-d]pyrimidines. U.S. Patent US6066732 A, 23 May 2000. [Google Scholar]
- Taylor, E.C.; Kuhnt, D.; Shih, C.; Rinzel, S.M.; Grindey, G.B.; Barredo, J.; Jannatipour, M.; Moran, R.G. A Dideazatetrahydrofolate Analogue Lacking a Chiral Center at C-6, N-[4-[2-(2-Amino-3,4-dihydro-4-oxo-7H-pyrrolo[2,3-d]pyrimidin-5yl)ethyl[benzoyl]-l-glutamic Acid is an Inhibitor of Thymidylate Synthase. J. Med. Chem. 1992, 35, 4450–4454. [Google Scholar] [CrossRef]
- Taylor, E.C.; Liu, B. A novel synthetic route to 7-substituted derivatives of the antitumor agent LY231514 (MTA). Tetrahedron Lett. 1999, 40, 5291–5294. [Google Scholar] [CrossRef]
- Kennedy, S.H.; Dherange, B.D.; Berger, K.J.; Levin, M.D. Skeletal editing through direct nitrogen deletion of secondary amines. Nature 2021, 593, 223–227. [Google Scholar] [CrossRef]
- Itoh, F.; Russello, O.; Akimoto, H.; Beardsley, G.P. Novel pyrrolo[2,3-d]pyrimidine antifolate TNP-351: Cytotoxic effect on methotrexate-resistant CCRF-CEM cells and inhibition of transformylases of de novo purine biosynthesis. Cancer Chemother. Pharmacol. 1994, 34, 273–279. [Google Scholar] [CrossRef] [PubMed]
- Miwa, T.; Hitaka, T.; Akimoto, H.; Nomura, H.J. Novel pyrrolo [2, 3-d] pyrimidine antifolates: Synthesis and antitumor activities. Med. Chem. 1991, 34, 555–560. [Google Scholar]
- Adams, J.; Elliott, P.J. New agents in cancer clinical trials. Oncogene 2000, 19, 6687–6692. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Newell, D.R. Clinical pharmacokinetics of antitumor antifolates. Semin. Oncol. 1999, 26, 74–81. [Google Scholar]
- Bronder, J.L.; Moran, R.G. Antifolates targeting purine synthesis allow entry of tumor cells into S phase regardless of p53 function. Cancer Res. 2002, 62, 5236–5241. [Google Scholar] [PubMed]
- Taylor, E.C.; Harrington, P.J.; Fletcher, S.R.; Beardsley, G.P.; Moran, R.G. Synthesis of the Antileukemic Agents 5, 10-Dideazaaminopterin and 5, 10-Dideaza-5, 6, 7, 8-tetrahydroaminopterin. J. Med. Chem. 1985, 28, 914–921. [Google Scholar] [CrossRef] [PubMed]
- Taylor, E.C.; Harrington, P.M.; Warner, J.C. Diels-Alder reactions of 6-azapterins. An alternative strategy for the synthesis of 5,10-dideaza-5,6,7,8-tetrahydrofolic acid (DDATHF). Heterocycles 1988, 27, 1925–1928. [Google Scholar] [CrossRef]
- Taylor, E.C.; Wong, G.S.K. Convergent and Efficient Palladium-Effected Synthesis of 5,10-Dideaza-5,6,7,8-tetrahydrofolic Acid (DDATHF). J. Org. Chem. 1989, 54, 3618–3624. [Google Scholar] [CrossRef]
- Boschelli, D.H.; Webber, S.; Whiteley, J.M.; Oronsky, A.L.; Kerwar, S.S. Synthesis and biological properties of 5,10-dideaza-5,6,7,8-tetrahydrofolic acid. Arch. Biochem. Biophys. 1988, 265, 43–49. [Google Scholar] [CrossRef]
- Piper, J.R.; McCaleb, G.S.; Montgomery, J.A.; Kisliuk, R.L.; Gaumont, Y.; Thorndike, J.; Sirotnak, F.M. Synthesis and Antifolate Activity of 5-Methyl-5,10-dideaza Analogues of Aminopterin and Folic Acid and an Alternative Synthesis of 5,10-Dideazatetrahydrofolic Acid, a Potent Inhibitor of Glycinamide Ribonucleotide Formyltransferase. J. Med. Chem. 1988, 31, 2164–2169. [Google Scholar] [CrossRef] [PubMed]
- Taylor, E.C.; Chaudhari, R.; Lee, K. A simplified and efficient synthesis of 5,10-dideaza-5,6,7,8-tetrahydrofolic acid (DDATHF). Investig. New Drugs 1996, 14, 281–285. [Google Scholar] [CrossRef] [PubMed]
- Tomsho, J.W.; McGuire, J.J.; Coward, J.K. Synthesis of (6R)- and (6S)-5,10-dideazatetrahydrofolate oligo-γ-glutamates: Kinetics of multiple glutamate ligations catalyzed by folylpoly-γ-glutamate synthetase. Org. Biomol. Chem. 2005, 3, 3388–3398. [Google Scholar] [CrossRef]
- Barnett, C.J.; Wilson, T.M. Asymmetric synthesis and absolute configuration of 5,10-dideaza-5,6,7,8-tetrahydropteroic acid and 5,10-dideaza-5,6,7,8-tetrahydrofolic acid (DDATHF). Tetrahedron Lett. 1989, 30, 6291–6294. [Google Scholar] [CrossRef]
- Barnett, C.J.; Wilson, T.M.; Wendel, S.R.; Winningham, M.J.; Deeter, J.B. Asymmetric Synthesis of Lometrexol ((6R)-5,10-Dideaza-5,6,7,8-tetrahydrofolic Acid). J. Org. Chem. 1994, 59, 7038–7045. [Google Scholar] [CrossRef]
- Sirichaiwat, C.; Intaraudom, C.; Kamchonwongpaisan, S.; Vanichtanankul, J.; Thebtaranonth, Y.; Yuthavong, Y. Target Guided Synthesis of 5-Benzyl-2,4-diamonopyrimidines: Their Antimalarial Activities and Binding Affinities to Wild Type and Mutant Dihydrofolate Reductases from Plasmodium falciparum. J. Med. Chem. 2004, 47, 345–354. [Google Scholar] [CrossRef] [PubMed]
- Kimmitt, P.T.; Harwood, C.R.; Barer, M.R. Toxin gene expression by Shiga toxin-producing Escherichia coli: The role of antibiotics and the bacterial SOS response. Emerg. Infect. Dis. 2000, 6, 458–465. [Google Scholar] [CrossRef] [PubMed]
- Heaslet, H.; Harris, M.; Fahnoe, K.; Sarver, R.; Putz, H.; Chang, J.; Subramanyam, C.; Barreiro, G.; Miller, J.R. Structural comparison of chromosomal and exogenous dihydrofolate reductase from Staphylococcus aureus in complex with the potent inhibitor trimethoprim. Proteins Struct. Funct. Bioinforma. 2009, 76, 706–717. [Google Scholar] [CrossRef]
- Brogden, R.N.; Carmine, A.A.; Heel, R.C.; Speight, T.M.; Avery, G.S. Trimethoprim: A Review of its Antibacterial Activity, Pharmacokinetics and Therapeutic Use in Urinary Tract Infections. Drugs 1982, 23, 405–430. [Google Scholar] [CrossRef]
- Topless, R.K.; Green, R.; Morgan, S.L.; Robinson, P.C.; Merriman, T.R.; Gaffo, A.L. Folic acid and methotrexate use and their association with COVID-19 diagnosis and mortality: An analysis from the UK Biobank. medRxiv 2022. [Google Scholar] [CrossRef]
- Sheybani, Z.; Dokoohaki, M.H.; Negahdaripour, M.; Dehdashti, M.; Zolghadr, H.; Moghadami, M.; Masoompour, S.M.; Zolghadr, A.R. The Role of Folic Acid in the Management of Respiratory Disease Caused by COVID-19. ChemRxiv Camb. Camb. Open Engag. 2020. [Google Scholar] [CrossRef]
- Meisel, E.; Efros, O.; Bleier, J.; Halevi, T.B.; Segal, G.; Rahav, G.; Leibowitz, A.; Grossman, E. Folate Levels in Patients Hospitalized with Coronavirus Disease 2019. Nutrients 2021, 13, 812. [Google Scholar] [CrossRef] [PubMed]
- Stegmann, K.M.; Dickmanns, A.; Gerber, S.; Nikolova, V.; Klemke, L.; Manzini, V.; Schlösser, D.; Bierwirth, C.; Freund, J.; Sitte, M.; et al. The folate antagonist methotrexate diminishes replication of the coronavirus SARS-CoV-2 and enhances the antiviral efficacy of remdesivir in cell culture models. Virus Res. 2021, 302, 198469. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Guo, R.; Kim, S.H.; Shah, H.; Zhang, S.; Liang, J.H.; Fang, Y.; Gentili, M.; O’ Leary, C.N.; Elledge, S.J.; et al. SARS-CoV-2 hijacks folate and one-carbon metabolism for viral replication. Nat. Commun. 2021, 12, 1676. [Google Scholar] [CrossRef]
- Mujwar, S.; Tripathi, A. Repurposing benzbromarone as antifolate to develop novel antifungal therapy for Candida albicans. J. Mol. Model. 2022, 28, 193. [Google Scholar] [CrossRef] [PubMed]





































Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kovalev, I.S.; Zyryanov, G.V.; Santra, S.; Majee, A.; Varaksin, M.V.; Charushin, V.N. Folic Acid Antimetabolites (Antifolates): A Brief Review on Synthetic Strategies and Application Opportunities. Molecules 2022, 27, 6229. https://doi.org/10.3390/molecules27196229
Kovalev IS, Zyryanov GV, Santra S, Majee A, Varaksin MV, Charushin VN. Folic Acid Antimetabolites (Antifolates): A Brief Review on Synthetic Strategies and Application Opportunities. Molecules. 2022; 27(19):6229. https://doi.org/10.3390/molecules27196229
Chicago/Turabian StyleKovalev, Igor S., Grigory V. Zyryanov, Sougata Santra, Adinath Majee, Mikhail V. Varaksin, and Valery N. Charushin. 2022. "Folic Acid Antimetabolites (Antifolates): A Brief Review on Synthetic Strategies and Application Opportunities" Molecules 27, no. 19: 6229. https://doi.org/10.3390/molecules27196229
APA StyleKovalev, I. S., Zyryanov, G. V., Santra, S., Majee, A., Varaksin, M. V., & Charushin, V. N. (2022). Folic Acid Antimetabolites (Antifolates): A Brief Review on Synthetic Strategies and Application Opportunities. Molecules, 27(19), 6229. https://doi.org/10.3390/molecules27196229