
Two Ways to Achieve the Same Goal—Two Validated Quantitative NMR Strategies for a Low-Abundance Natural Product in Standardized Extracts: The Case of Hepatodamianol in Turnera diffusa

 , ,
, ,
Abstract

1. Introduction
2. Results and Discussion
2.1. Selection of Solvent and Signal for Quantitation
2.2. Purity Evaluation of the Isolated Hepatodamianol
2.3. Measurement of Longitudinal Relaxation Time
2.4. Method Validation
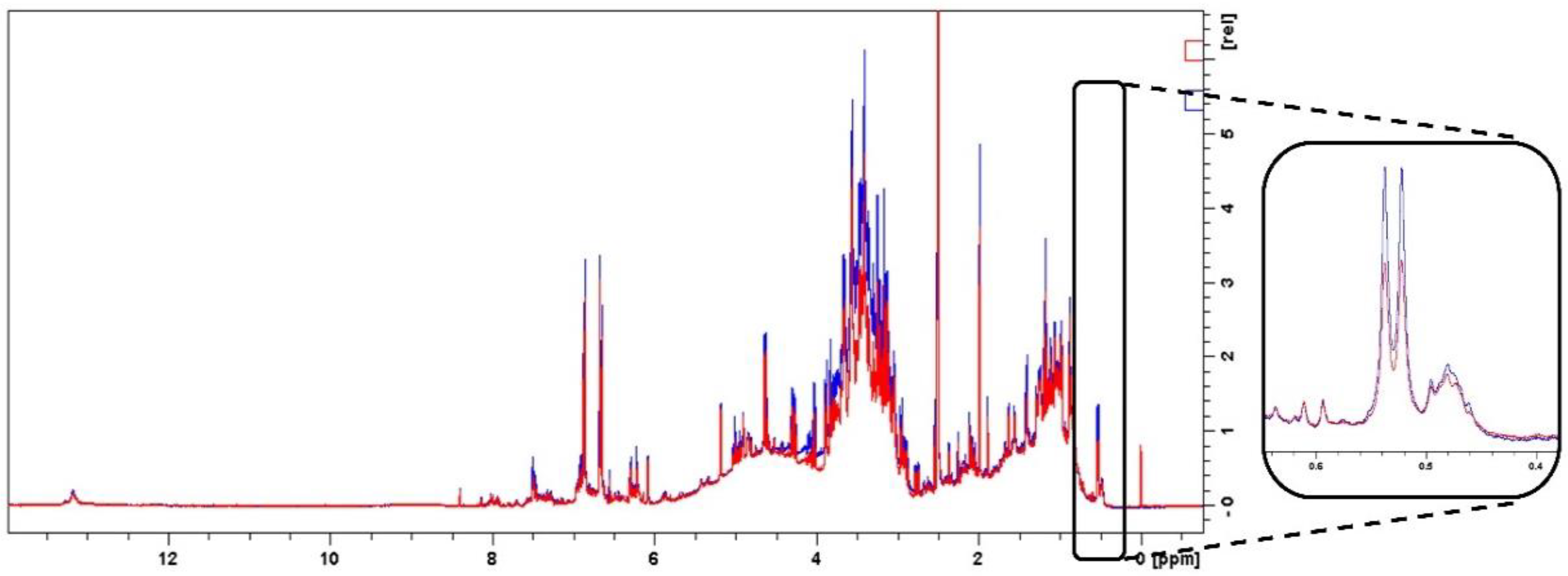
2.4.1. Specificity
2.4.2. Linearity
2.4.3. Precision
2.4.4. Accuracy
2.4.5. Limit of Detection
2.4.6. Limit of Quantitation
2.4.7. Robustness
2.5. Hepatodamianol Quantitation in Standardized Extract Samples
3. Experimental: Materials and Methods
3.1. Chemicals and Reagents
3.2. Plant Material
3.3. Sample Preparation
3.4. Evaluation of Purity of the Isolated Hepatodamianol Using High-Performance Liquid Chromatography with Diode Array Detection
3.5. Evaluation of the Purity of Isolated Hepatodamianol Using qNMR
3.6. Preparation of Standard Solutions
3.7. NMR Experiments
3.8. Method Validation
3.9. Comparison of Calibration Approaches
3.10. Statistical Analysis
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Szewczyk, K.; Zidorn, C. Ethnobotany, Phytochemistry, and Bioactivity of the Genus Turnera (Passifloraceae) with a Focus on Damiana—Turnera diffusa. J. Ethnopharmacol. 2014, 152, 424–443. [Google Scholar] [CrossRef] [PubMed]
- Ramírez-Durón, R.; Almaguer, L.C.; Salazar-Aranda, R.; Salazar-Cavazos, M.D.L.; de Torres, N.W. Evaluation of Thin-Layer Chromatography Methods for Quality Control of Commercial Products containing Aesculus hippocastanum, Turnera diffusa, Matricaria recutita, Passiflora incarnata, and Tilia occidentalis. J. AOAC Int. 2007, 90, 920–924. [Google Scholar] [CrossRef] [PubMed]
- Godoi, A.F.L.; Vilegas, W.; Godoi, R.H.M.; van Vaeck, L.; van Grieken, R. Application of Low-Pressure Gas Chromatography-Ion-Trap Mass Spectrometry to the Analysis of the Essential Oil of Turnera diffusa (Ward.) Urb. J. Chromatogr. A 2004, 1027, 127–130. [Google Scholar] [CrossRef]
- Lucio-Gutiérrez, J.R.; Garza-Juárez, A.; Coello, J.; Maspoch, S.; Salazar-Cavazos, M.L.; Salazar-Aranda, R.; Waksman de Torres, N. Multi-Wavelength High-Performance Liquid Chromatographic Fingerprints and Chemometrics to Predict the Antioxidant Activity of Turnera diffusa as Part of Its Quality Control. J. Chromatogr. A 2012, 1235, 68–76. [Google Scholar] [CrossRef] [PubMed]
- Garza-Juárez, A.; Salazar-Cavazos, M.D.L.L.; Salazar-Aranda, R.; Pérez-Meseguer, J.; de Torres, N.W. Correlation Between Chromatographic Fingerprint and Antioxidant Activity of Turnera diffusa (Damiana). Planta Med. 2011, 77, 958–963. [Google Scholar] [CrossRef] [PubMed]
- Camargo, E.E.S.; Vilegas, W. Controle de Qualidade Dos Extratos Polares de Turnera diffusa Willd. ex Schult., Turneraceae. Braz. J. Pharmacogn. 2010, 20, 228–232. [Google Scholar] [CrossRef][Green Version]
- Pérez-Meseguer, J.; Garza-Juárez, A.; Salazar-Aranda, R.; Salazar-Cavazos, M.L.; de la Torre, R.Y.C.; Rivas-Galindo, V.; de Torres, N.W. Development and Validation of an HPLC-DAD Analytical Procedure for Quality Control of Damiana (Turnera diffusa), Using an Antioxidant Marker Isolated from the Plant. J. AOAC Int. 2010, 93, 1161–1168. [Google Scholar] [CrossRef]
- Lucio-Gutiérrez, J.R.; Montemayor, C.D.; Coello-Bonilla, J.; Minsky, N.W.; Saucedo, A.L. Selective 1D-TOCSY and Chemometrics to Evaluate Authenticity of Turnera diffusa and Related Botanical Extracts. Phytochem. Lett. 2019, 30, 62–68. [Google Scholar] [CrossRef]
- Willer, J.; Jöhrer, K.; Greil, R.; Zidorn, C.; Çiçek, S.S. Cytotoxic Properties of Damiana (Turnera diffusa) Extracts and Constituents and A Validated Quantitative UHPLC-DAD Assay. Molecules 2019, 24, 855. [Google Scholar] [CrossRef]
- Estrada-Reyes, R.; Carro-Juárez, M.; Martínez-Mota, L. Pro-Sexual Effects of Turnera diffusa Wild (Turneraceae) in Male Rats Involves the Nitric Oxide Pathway. J. Ethnopharmacol. 2013, 146, 164–172. [Google Scholar] [CrossRef]
- Naranjo, A.P.; Delgado-Montemayor, C.; Fraga-López, A.; Castañeda-Corral, G.; Salazar-Aranda, R.; Acevedo-Fernández, J.J.; Waksman, N. Acute Hypoglycemic and Antidiabetic Effect of Teuhetenone A Isolated from Turnera diffusa. Molecules 2017, 22, 599. [Google Scholar] [CrossRef] [PubMed]
- Delgado-Montemayor, C.; Cordero-Pérez, P.; Torres-González, L.; Salazar-Cavazos, M.D.L.L.; Saucedo, A.L.; Paniagua-Vega, D.; Waksman-Minsky, N.H. Development of a Hepatoprotective Herbal Drug from Turnera diffusa. Evid. Based Complement. Altern. Med. 2022, 2022, 5114948. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez, D.R.; Lozano-Sepulveda, S.A.; Delgado-Montemayor, C.; Waksman, N.; Cordero-Perez, P.; Rivas-Estilla, A.M. Turnera diffusa extract attenuates profibrotic, extracellular matrix and mitochondrial markers in activated human hepatic stellate cells (HSC). Ann. Hepatol. 2021, 22, 100281. [Google Scholar] [CrossRef] [PubMed]
- Delgado-Montemayor, C.; Salazar-Aranda, R.; Cordero-Pérez, P.; Torres-González, L.; Perez-Meseguer, J.; de Torres, N.W. Uso de Un Compuesto Derivado de Luteolina de Una Planta Del Género Turnera Como Agente Hepatoprotector. Patent 2014008805, 21 January 2016. [Google Scholar]
- Nishizaki, Y.C.; Lankin, D.; Chen, S.-N.; Pauli, G. Accurate and Precise External Calibration Enhances the Versatility of Quantitative NMR (QNMR). Anal. Chem. 2021, 93, 2733–2741. [Google Scholar] [CrossRef]
- Simmler, C.; Napolitano, J.G.; McAlpine, J.B.; Chen, S.-N.; Pauli, G.F. Universal Quantitative NMR Analysis of Complex Natural Samples. Curr. Opin. Biotechnol. 2014, 25, 51–59. [Google Scholar] [CrossRef]
- Pauli, G.F.; Jaki, B.U.; Lankin, D.C. Quantitative HNMR: Development and Potential of a Method for Natural Products Analysis. J. Nat. Prod. 2005, 68, 133–149. [Google Scholar] [CrossRef]
- Bharti, S.K.; Roy, R. Quantitative 1H NMR Spectroscopy. TrAC Trends Anal. Chem. 2012, 35, 5–26. [Google Scholar] [CrossRef]
- Liu, Y.; Chen, S.N.; McAlpine, J.B.; Klein, L.L.; Friesen, J.B.; Lankin, D.C.; Pauli, G.F. Quantification of a Botanical Negative Marker without an Identical Standard: Ginkgotoxin in Ginkgo biloba. J. Nat. Prod. 2014, 77, 611–617. [Google Scholar] [CrossRef]
- Wider, G.; Dreier, L. Measuring Protein Concentrations by NMR Spectroscopy. J. Anal. Chem. Soc. 2006, 128, 2571–2576. [Google Scholar] [CrossRef]
- Monakhova, Y.B.; Kohl-Himmelseher, M.; Kuballa, T.; Lachenmeier, D.W. Determination of the Purity of Pharmaceutical Reference Materials by 1H NMR Using the Standardless PULCON Methodology. J. Pharm. Biomed. Anal. 2014, 100, 381–386. [Google Scholar] [CrossRef]
- Jung, Y.S.; Hyeon, J.S.; Hwang, G.S. Software-Assisted Serum Metabolite Quantification Using NMR. Anal. Chim. Acta 2016, 934, 194–202. [Google Scholar] [CrossRef] [PubMed]
- Goldoni, L.; Beringhelli, T.; Rocchia, W.; Realini, N.; Piomelli, D. A Simple and Accurate Protocol for Absolute Polar Metabolite Quantification in Cell Cultures Using Quantitative Nuclear Magnetic Resonance. Anal. Biochem. 2016, 501, 26–34. [Google Scholar] [CrossRef] [PubMed]
- Hohmann, M.; Felbinger, C.; Christoph, N.; Wachter, H.; Wiest, J.; Holzgrabe, U. Quantification of Taurine in Energy Drinks Using 1H NMR. J. Pharm. Biomed. Anal. 2014, 93, 156–160. [Google Scholar] [CrossRef] [PubMed]
- Williamson, K.; Hatzakis, E. Evaluating the Effect of Roasting on Coffee Lipids Using a Hybrid Targeted-Untargeted NMR Approach in Combination with MRI. Food Chem. 2019, 299, 125039. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, R.; Sugai, C.; Yamazaki, T.; Matsushima, R.; Uchida, H.; Matsumiya, M.; Takatsu, A.; Suzuki, T. Quantitative Nuclear Magnetic Resonance Spectroscopy Based on PULCON Methodology: Application to Quantification of Invaluable Marine Toxin, Okadaic Acid. Toxins 2016, 8, 294. [Google Scholar] [CrossRef] [PubMed]
- Ramos, A.S.; Mar, J.M.; da Silva, L.S.; Acho, L.D.R.; Silva, B.J.P.; Lima, E.S.; Campelo, P.H.; Sanches, E.A.; Bezerra, J.A.; Chaves, F.C.M. Pedra-Ume Caá Fruit: An Amazon Cherry Rich in Phenolic Compounds with Antiglycant and Antioxidant Properties. Food Res. Int. 2019, 123, 674–683. [Google Scholar] [CrossRef]
- Hsieh, L.Y.; Chan, H.H.; Kuo, P.C.; Hung, H.Y.; Li, Y.C.; Kuo, C.L.; Peng, Y.; Zhao, Z.Z.; Kuo, D.H.; Sun, I.W.; et al. A Feasible and Practical 1H NMR Analytical Method for the Quality Control and Quantification of Bioactive Principles in Lycii Fructus. J. Food Drug Anal. 2018, 26, 1105–1112. [Google Scholar] [CrossRef]
- Pauli, G.F.; Gödecke, T.; Jaki, B.U.; Lankin, D.C. Quantitative 1H NMR. Development and Potential of an Analytical Method: An update. J. Nat. Prod. 2012, 75, 834–851. [Google Scholar] [CrossRef]
- Pauli, G.F.; Chen, S.N.; Simmler, C.; Lankin, D.C.; Gödecke, T.; Jaki, B.U.; Friesen, J.B.; McAlpine, J.B.; Napolitano, J.G. Importance of Purity Evaluation and the Potential of Quantitative 1H NMR as a Purity Assay: Miniperspective. J. Med. Chem. 2014, 57, 9220–9231. [Google Scholar] [CrossRef]
- Huang, B.; Xiao, S.; Chen, T.; Xie, Y.; Luo, P. Purity Assessment of Ginsenoside Rg1 Using Quantitative 1H Nuclear Magnetic Resonance. J. Pharm. Biomed. Anal. 2017, 139, 193–204. [Google Scholar] [CrossRef]
- Schönberger, T.; Monakhova, Y.B.; Lachenmeier, D.W.; Kuballa, T. Guide to NMR Method Development and Validation. Part I: Identification and Quantification; Eurolab: Berlin, Germany, 2014. [Google Scholar]
- Malz, F.; Jancke, H. Validation of Quantitative NMR. J. Pharm. Biomed. Anal. 2005, 38, 813–823. [Google Scholar] [CrossRef] [PubMed]
- Schoenberger, T. Guideline for QNMR Analysis; European Network of Forensic Science Institutes: Wiesbaden, Germany, 2019. [Google Scholar]
- Benedito, L.E.C.; Maldaner, A.O.; Oliveira, A.L. An External Reference 1H qNMR Method (PULCON) for Characterization of High Purity Cocaine Seizures. Anal. Methods 2018, 10, 489–495. [Google Scholar] [CrossRef]
- Fernandez-Pastor, I.; Luque-Muñoz, A.; Rivas, F.; O’Donnell, M.; Martinez, A.; Gonzalez-Maldonado, R.; Haidour, A.; Parra, A. Quantitative NMR Analysis of L-Dopa in Seeds from Two Varieties of Mucuna pruriens. Phytochem. Anal. 2019, 30, 89–94. [Google Scholar] [CrossRef]
- Duangdee, N.; Chamboonchu, N.; Kongkiatpaiboon, S.; Prateeptongkum, S. Quantitative 1HNMR Spectroscopy for the Determination of Oxyresveratrol in Artocarpus lacucha Heartwood. Phytochem. Anal. 2019, 30, 617–622. [Google Scholar] [CrossRef]
- Bekiroglu, S.; Myrberg, O.; Östman, K.; Ek, M.; Arvidsson, T.; Rundlöf, T.; Hakkarainen, B. Validation of a Quantitative NMR Method for Suspected Counterfeit Products Exemplified on Determination of Benzethonium Chloride in Grapefruit Seed Extracts. J. Pharm. Biomed. Anal. 2008, 47, 958–961. [Google Scholar] [CrossRef] [PubMed]
- Monakhova, Y.B.; Diehl, B.W.K. Practical Guide for Selection of 1H qNMR Acquisition and Processing Parameters Confirmed by Automated Spectra Evaluation. Magn. Reson. Chem. 2017, 55, 996–1005. [Google Scholar] [CrossRef]
- Li, Z.Y.; Welbeck, E.; Wang, R.F.; Liu, Q.; Yang, Y.B.; Chou, G.X.; Bi, K.S.; Wang, Z.T. A Universal Quantitative 1H Nuclear Magnetic Resonance (qNMR) Method for Assessing the Purity of Dammarane-Type Ginsenosides. Phytochem. Anal. 2015, 26, 8–14. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Quantitation Method | % Purity | %RSD |
|---|---|---|
| Chromatography | 56.98 | 0.42 |
| qNMR ERETIC2 | 32.82 | 2.38 |
| 1H Atom (Substance) | T1 (s) |
|---|---|
| CH3-6‴ (rutin) | 0.43 |
| CH3-6‴ (hepatodamianol) | 0.53 |
| Calibration Method | Curve Equation | Determination Coefficient (r2) |
|---|---|---|
| ERETIC2 | 4,208,618.33 x − 455.71 | 0.99999 |
| External standard calibration with rutin | 4,094,508.77 x + 32,601.21 | 0.99989 |
| Theoretical Concentration, mM | ESC a Concentration, mM | % Error | %RSD | ERETIC2 Concentration, mM | % Error | %RSD b |
|---|---|---|---|---|---|---|
| 0.250 | 0.240 | –4.012 | 0.585 | 0.241 | –3.467 | 0.633 |
| 0.500 | 0.497 | –0.636 | 0.284 | 0.491 | –1.733 | 0.311 |
| 1.000 | 1.010 | 0.961 | 0.889 | 0.990 | −1.000 | 0.898 |
| 2.000 | 2.031 | 1.528 | 1.214 | 1.983 | –0.833 | 1.198 |
| 3.000 | 3.012 | 0.397 | 0.609 | 2.938 | –2.067 | 0.621 |
| 4.000 | 3.988 | –0.302 | 0.619 | 3.888 | –2.808 | 0.616 |
| 5.000 | 4.998 | –0.049 | 0.222 | 4.870 | –2.600 | 0.229 |
| Concentration, mM | %RSD a ESC b | %RSD ERETIC2 |
|---|---|---|
| 0.50 | 1.104 | 0.311 |
| 2.00 | 1.504 | 1.198 |
| 5.00 | 0.933 | 0.229 |
| ESC Quantitation | ERETIC2 Quantitation | |||||
|---|---|---|---|---|---|---|
| Sample (mg) | Unspiked Sample (mg/mL) | Spiked Sample (mg/mL) | Recovery (%) | Unspiked Sample (mg/mL) | Spiked Sample (mg/mL) | Recovery (%) |
| Average | 1.05 | 1.42 | 99.94 | 1.03 | 1.39 | 97.22 |
| %RSD | 2.61 | 0.52 | 6.36 | 2.60 | 0.53 | 6.32 |
| Concentration (mM) | %RSD | % Error | ||
|---|---|---|---|---|
| ESC with Rutin | ERETIC2 | ESC with Rutin | ERETIC2 | |
| 0.125 | 1.85 | 2.01 | –12.00 | –8.27 |
| 0.250 | 0.58 | 0.63 | –4.10 | –3.47 |
| ESC (Rutin) | ERETIC2 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Parameter | Concentration (mM) | Value | Average Concentration (mM) | %RSD | p-Value | Robustness | Average Concentration (mM) | %RSD | p-Value | Robustness |
| Processing * | 0.5 | A1 | 0.49 | 0.05 | 0.281 | Yes | 0.49 | 0.00 | 0.139 | Yes |
| A2 | 0.50 | 0.28 | 0.49 | 0.31 | ||||||
| A3 | 0.50 | 0.37 | 0.49 | 0.31 | ||||||
| 2.0 | A1 | 2.03 | 1.36 | 0.996 | Yes | 1.99 | 1.35 | 0.996 | Yes | |
| A2 | 2.03 | 1.21 | 1.98 | 1.20 | ||||||
| A3 | 2.03 | 1.12 | 1.98 | 1.11 | ||||||
| 5.0 | A1 | 5.14 | 4.44 | 0.392 | Yes | 5.01 | 4.43 | 0.391 | Yes | |
| A2 | 5.00 | 0.22 | 4.87 | 0.23 | ||||||
| A3 | 5.00 | 0.24 | 4.87 | 0.24 | ||||||
| LB value | 0.5 | 0.05 | 0.49 | 0.21 | 0.097 | Yes | 0.49 | 0.24 | 0.139 | Yes |
| 0.10 | 0.50 | 0.28 | 0.49 | 0.31 | ||||||
| 0.15 | 0.50 | 0.59 | 0.49 | 0.65 | ||||||
| 2.0 | 0.05 | 2.03 | 1.29 | 0.998 | Yes | 1.98 | 1.27 | 0.998 | Yes | |
| 0.10 | 2.03 | 1.21 | 1.98 | 1.20 | ||||||
| 0.15 | 2.03 | 1.20 | 1.98 | 1.20 | ||||||
| 5.0 | 0.05 | 5.00 | 0.25 | 0.709 | Yes | 4.87 | 0.24 | 0.708 | Yes | |
| 0.10 | 5.00 | 0.22 | 4.87 | 0.23 | ||||||
| 0.15 | 4.99 | 0.21 | 4.87 | 0.21 | ||||||
| Baseline correction algorithm | 0.5 | ABS | 0.50 | 0.20 | 0.002 | No | 0.49 | 0.20 | 0.002 | No |
| ABSN | 0.50 | 0.28 | 0.49 | 0.31 | ||||||
| ABSD | 0.49 | 0.25 | 0.49 | 0.24 | ||||||
| 2.0 | ABS | 2.03 | 1.31 | 0.875 | Yes | 1.98 | 1.30 | 0.873 | Yes | |
| ABSN | 2.03 | 1.21 | 1.98 | 1.20 | ||||||
| ABSD | 2.02 | 1.30 | 1.97 | 1.31 | ||||||
| 5.0 | ABS | 5.00 | 0.26 | 0.092 | Yes | 4.87 | 0.26 | 0.092 | Yes | |
| ABSN | 5.00 | 0.22 | 4.87 | 0.23 | ||||||
| ABSD | 4.97 | 0.29 | 4.85 | 0.28 | ||||||
| Hepatodamianol (mg)/Sample (g) (%RSD) | ||
|---|---|---|
| T. diffusa Sample | ESC | ERETIC2 |
| 1 | 36.13 ± 1.17 | 35.33 ± 3.20 |
| 2 | 52.44 ± 1.37 | 51.25 ± 2.60 |
| 3 | 28.69 ± 0.39 | 28.08 ± 1.36 |
| 4 | 36.16 ± 0.84 | 35.37 ± 2.29 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Parra-Naranjo, A.; Delgado-Montemayor, C.; Salazar-Aranda, R.; Castro-Ríos, R.; Saucedo, A.L.; Waksman-Minsky, N. Two Ways to Achieve the Same Goal—Two Validated Quantitative NMR Strategies for a Low-Abundance Natural Product in Standardized Extracts: The Case of Hepatodamianol in Turnera diffusa. Molecules 2022, 27, 6593. https://doi.org/10.3390/molecules27196593
Parra-Naranjo A, Delgado-Montemayor C, Salazar-Aranda R, Castro-Ríos R, Saucedo AL, Waksman-Minsky N. Two Ways to Achieve the Same Goal—Two Validated Quantitative NMR Strategies for a Low-Abundance Natural Product in Standardized Extracts: The Case of Hepatodamianol in Turnera diffusa. Molecules. 2022; 27(19):6593. https://doi.org/10.3390/molecules27196593
Chicago/Turabian StyleParra-Naranjo, Aída, Cecilia Delgado-Montemayor, Ricardo Salazar-Aranda, Rocío Castro-Ríos, Alma L. Saucedo, and Noemí Waksman-Minsky. 2022. "Two Ways to Achieve the Same Goal—Two Validated Quantitative NMR Strategies for a Low-Abundance Natural Product in Standardized Extracts: The Case of Hepatodamianol in Turnera diffusa" Molecules 27, no. 19: 6593. https://doi.org/10.3390/molecules27196593
APA StyleParra-Naranjo, A., Delgado-Montemayor, C., Salazar-Aranda, R., Castro-Ríos, R., Saucedo, A. L., & Waksman-Minsky, N. (2022). Two Ways to Achieve the Same Goal—Two Validated Quantitative NMR Strategies for a Low-Abundance Natural Product in Standardized Extracts: The Case of Hepatodamianol in Turnera diffusa. Molecules, 27(19), 6593. https://doi.org/10.3390/molecules27196593