
New Triazinoindole Bearing Benzimidazole/Benzoxazole Hybrids Analogs as Potent Inhibitors of Urease: Synthesis, In Vitro Analysis and Molecular Docking Studies


 , , , ,
, , , ,  , and
, and
Abstract
:1. Introduction
2. Results and Discussion
2.1. Chemistry
2.1.1. Synthesis of 7-Nitro-5H-[1,2,4]triazino[5,6-b]indole-3-thiol (III)
2.1.2. Synthesis of 2-(Chloromethyl)-1H-benzo[d]imidazole (VIb)
2.1.3. Synthesis of 2-(Chloromethyl)benzo[d]oxazole (VIb)
2.1.4. Synthesis of 3-(((1H-Benzo[d]imidazol-2-yl)methyl)thio)-8-nitro-5H-[1,2,4]triazino[5,6-b]indole (V)
2.1.5. Synthesis of 2-(((8-Nitro-5H-[1,2,4]triazino[5,6-b]indol-3-yl)thio)methyl)benzo[d]oxazole (VI)
2.1.6. Synthesis of Substituted 2-(((8-Nitro-5H-[1,2,4]triazino[5,6-b]indol-3-yl)thio)methyl)benzo[d]oxazole (1–12)
2.1.7. Synthesis of Substituted (3-((Benzo[d]oxazol-2-ylmethyl)thio)-8-nitro-5H-[1,2,4]triazino[5,6-b]indol-5-yl)(phenyl)methanone (13–25)
2.2. In Vitro Urease Inhibitory Activity
2.2.1. Structure-Activity Relationship (SAR) for Urease Inhibitory Activity
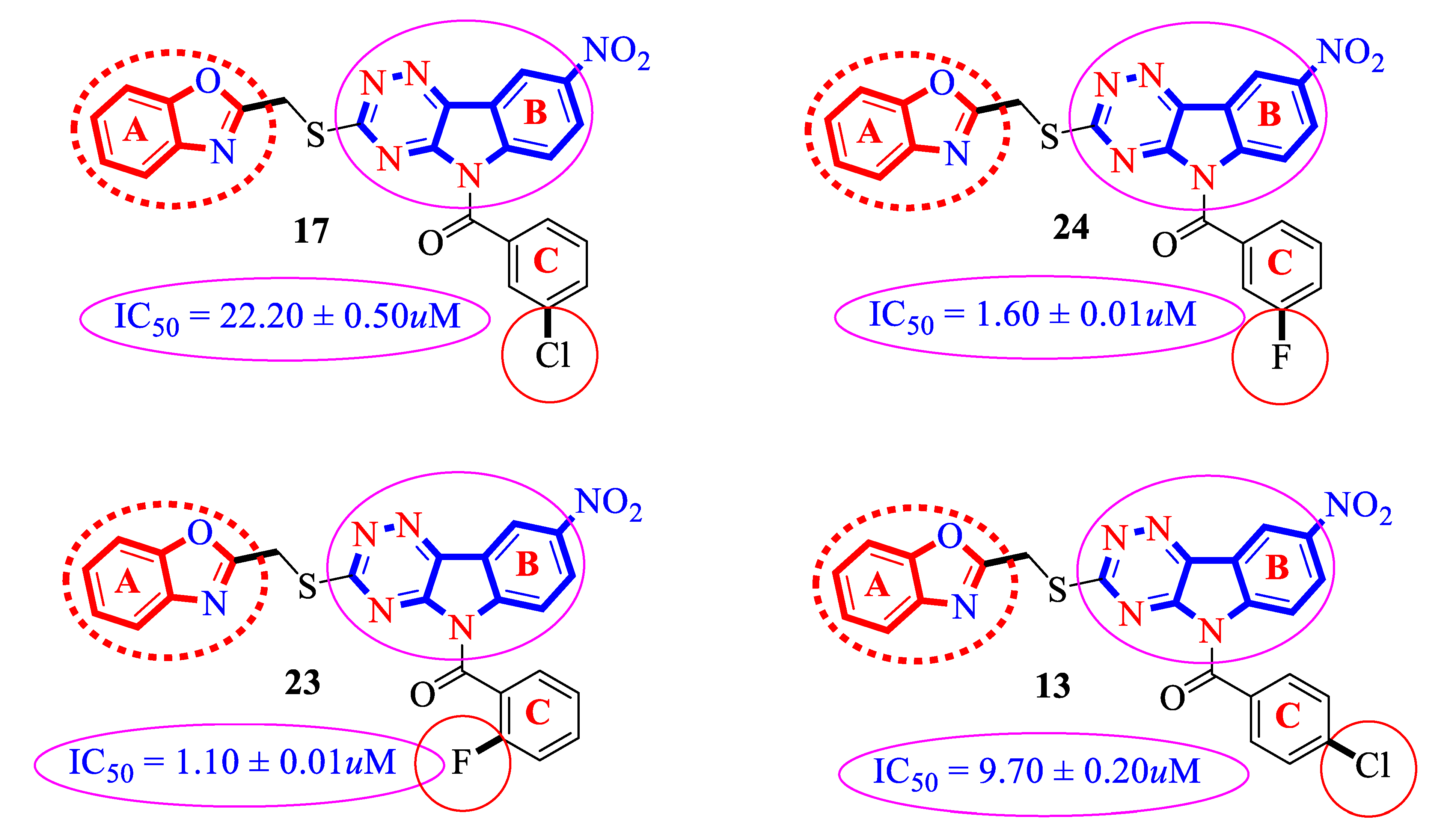
2.2.2. Category “A”
2.2.3. Category “B”
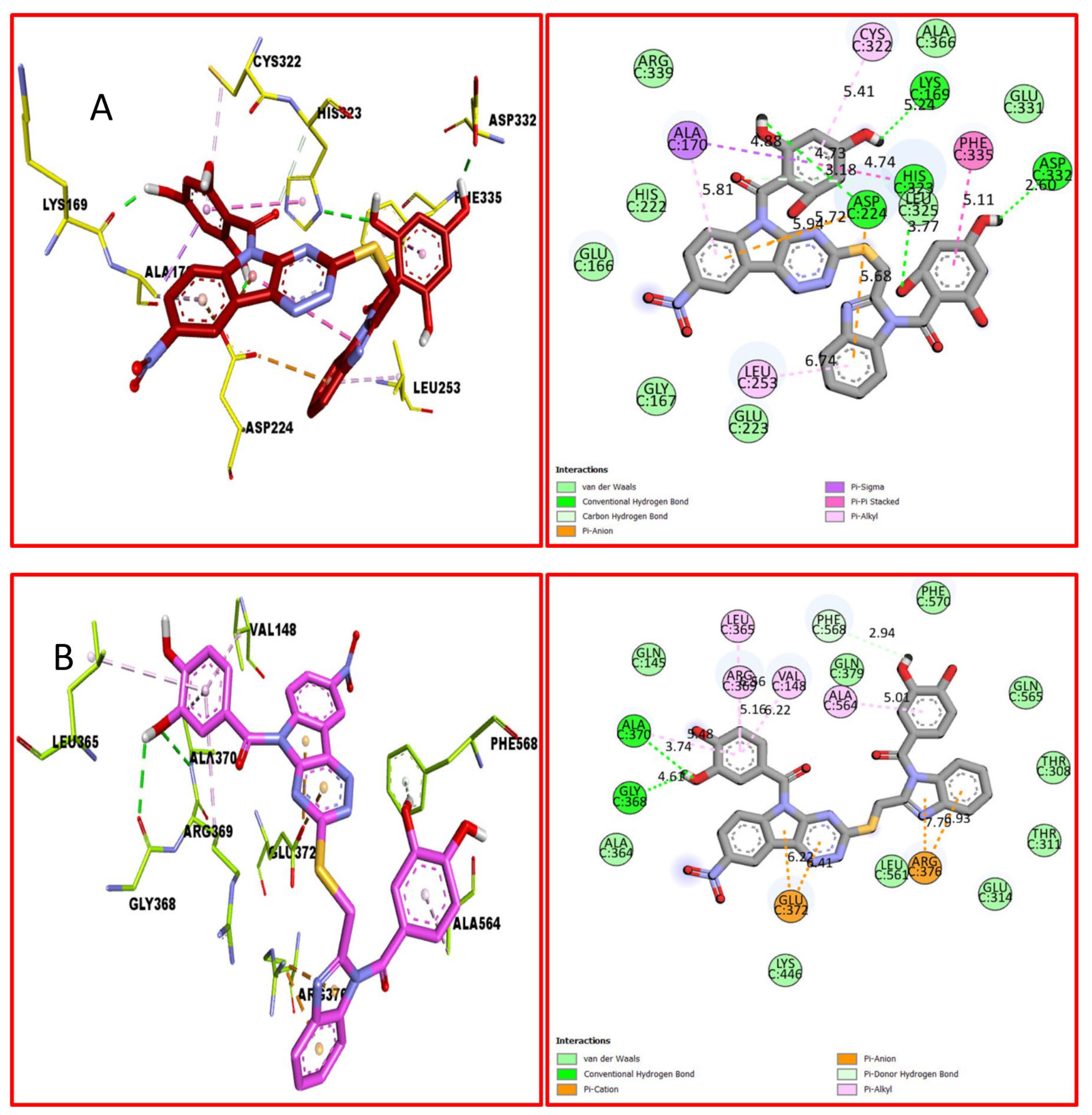
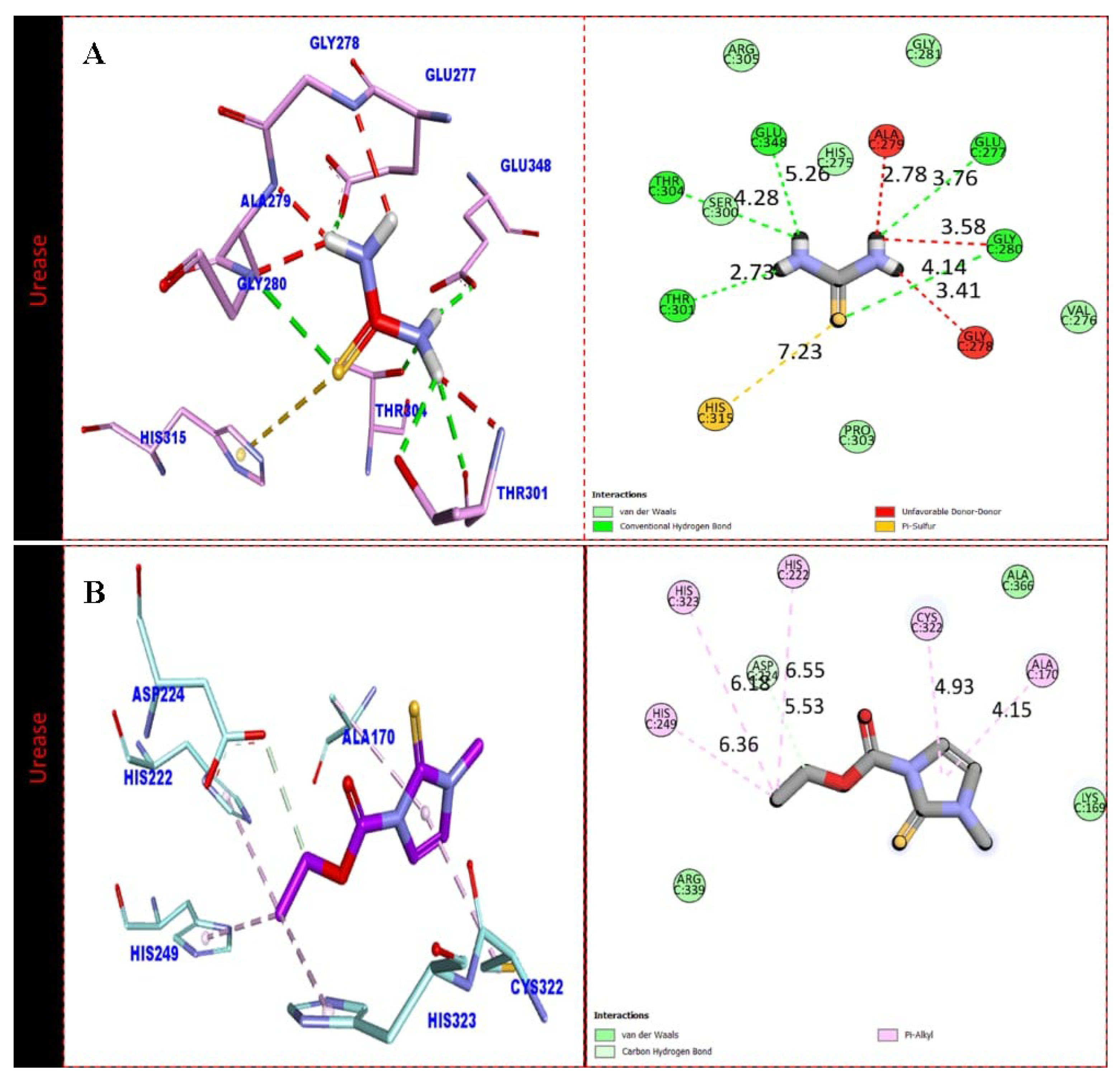
2.3. Docking Study
3. Experimental
3.1. General Information
3.2. General Procedure for the Formation of Triazinoindole-Based Benzimidazole/Benzoxazole Derivatives (1–25)
3.3. Urease Inhibition Assay
3.4. Assay Protocol for Docking Study
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Zambelli, B.; Musiani, F.; Benini, S.; Ciurli, S. Chemistry of Ni2+ in urease: Sensing, trafficking, and catalysis. Acc. Chem. Res. 2011, 44, 520–530. [Google Scholar] [CrossRef] [PubMed]
- Mobley, H.L.; Hausinger, R.P. Microbial ureases: Significance, regulation, and molecular characterization. Microbiol. Rev. 1989, 53, 85–108. [Google Scholar] [CrossRef] [PubMed]
- Lodhi, M.A.; Nawaz, S.A.; Iqbal, S.; Khan, K.M.; Rode, B.M.; Choudhary, M.I. 3D-QSAR CoMFA studies on bis-coumarine analogues as urease inhibitors: A strategic design in anti-urease agents. Bioorg. Med. Chem. 2008, 16, 3456–3461. [Google Scholar]
- Tanaka, T.; Kawase, M.; Tani, S. Urease inhibitory activity of simple α, β-unsaturated ketones. Life Sci. 2003, 73, 2985–2990. [Google Scholar] [CrossRef]
- Hanif, M.; Saleem, M.; Hussain, M.T.; Rama, N.H.; Zaib, S.; Aslam, M.A.M.; Jones, P.G.; Iqbal, J. Synthesis, urease inhibition, antioxidant and antibacterial studies of some 4-amino-5-aryl-3H-1, 2, 4-triazole-3-thiones and their 3, 6-disubstituted 1, 2, 4-triazolo [3, 4-b] 1, 3, 4-thiadiazole derivatives. J. Braz. Chem. Soc. 2012, 23, 854–860. [Google Scholar] [CrossRef] [Green Version]
- Saeed, A.; Rehman, S.; Channar, P.A.; Larik, F.A.; Abbas, Q.; Hassan, M.; Raza, H.; Flörke, U.; Seo, S. Synthesis, x-ray crystal structure elucidation and hirshfeld surface analysis of n-((4-(1h-benzo[d]imidazole-2-yl)phenyl)carbamothioyl)benzamide: Investigations for elastase inhibition, antioxidant and dna binding potentials for biological applications. J. Taiwan Inst. Chem. Eng. 2017, 4, 1–10. [Google Scholar]
- Arora, R.; Issar, U.; Kakkar, R. In Silico study of the active site of Helicobacter pylori urease and its inhibition by hydroxamic acids. J. Mol. Graph. Model. 2018, 83, 64–73. [Google Scholar] [CrossRef]
- Amtul, Z.; Siddiqui, R.A.; Choudhary, M.I. Chemistry and mechanism of urease inhibition. Curr. Med. Chem. 2002, 9, 1323–1348. [Google Scholar] [CrossRef]
- Abid, O.U.R.; Babar, T.M.; Ali, F.I.; Ahmed, S.; Wadood, A.; Rama, N.H.; Uddin, R.; Khan, A.; Choudhary, M.I. Identification of novel urease inhibitors by high-throughput virtual and in vitro screening. Med. Chem. Lett. 2010, 1, 145–149. [Google Scholar] [CrossRef] [Green Version]
- Pervez, H.; Chohan, Z.H.; Ramzan, M.; Nasim, F.U.H.; Khan, K.M. Synthesis and biological evaluation of some new N4-substituted isatin-3-thiosemicarbazones. J. Enzyme Inhib. Med. Chem. 2009, 24, 437–446. [Google Scholar] [CrossRef]
- Aslam, M.A.S.; Mahmood, S.U.; Shahid, M.; Saeed, A.; Iqbal, J. Synthesis, biological assay in vitro and molecular docking studies of new Schiff base derivatives as potential urease inhibitors. Eur. J. Med. Chem. 2011, 46, 5473–5479. [Google Scholar] [CrossRef] [PubMed]
- Perveen, S.; Khan, K.M.; Lodhi, M.A.; Choudhary, M.I.; Voelter, W. Urease and α-chymotrypsin inhibitory effects of selected urea derivatives. Lett Drug Des. Discov. 2008, 5, 401–405. [Google Scholar] [CrossRef]
- Hanif, M.; Shoaib, K.; Saleem, M.; Hasan Rama, N.; Zaib, S.; Iqbal, J. Synthesis, urease inhibition, antioxidant, antibacterial, and molecular docking studies of 1, 3, 4-oxadiazole derivatives. Int. Sch. Res. Not. 2012, 2012, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shamim, S.; Khan, K.M.; Salar, U.; Ali, F.; Lodhi, M.A.; Taha, M.; Khan, F.A.; Ashraf, S.; Ul-Haq, Z.; Ali, M.; et al. 5-Acetyl-6-methyl-4-aryl-3, 4-dihydropyrimidin-2 (1H)-ones: As potent urease inhibitors; synthesis, in vitro screening, and molecular modeling study. Bioorg. Chem. 2018, 76, 37–52. [Google Scholar] [CrossRef] [PubMed]
- Taha, M.; Wadood, A. Synthesis and molecular docking study of piperazine derivatives as potent urease inhibitors. Bioorg. Chem. 2018, 78, 411–417. [Google Scholar] [CrossRef] [PubMed]
- Fadel, H.H.; Lotfy, S.N.; Asker, M.M.; Mahmoud, M.G.; Al-Okbi, S.Y. Nutty-like flavor production by Corynbacterium glutamicum 1220T from enzymatic soybean hydrolysate. Effect of encapsulation and storage on the nutty flavoring quality. J. Adv. Res. 2018, 10, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Shelke, S.M.; Bhosale, S.H. Synthesis, antidepressant evaluation and QSAR studies of novel 2-(5H-[1,2,4]triazino [5,6-b]indol-3-ylthio)-N-(substituted phenyl)acetamides. Bioorg. Med. Chem. Lett. 2010, 20, 4661–4664. [Google Scholar] [CrossRef]
- Monge, A.; Palop, J.; Ramierz, C.; Font, M.; Fernandez-Alveraz, E. New 5H-1,2,4-triazino [5,6-b]indole and aminoindole derivatives. Synthesis and studies as inhibitors of blood platelet aggregation, anti-hypertensive agents and thromboxane synthetase inhibitorsNouveaux dérivés 5H-1,2,4-triazino [5,6-b]indole et de l’aminoindole. Synthèse et évaluation comme inhibiteurs de l’agrégation plaquettaire, agents anti-hypertenseurs et inhibiteurs de la thromboxane synthétase. Eur. J. Med. Chem. 1991, 26, 179. [Google Scholar]
- Aswar, U.M.; Kalshetti, P.P.; Shelke, S.M.; Bhosale, S.H.; Bodhankar, S.L. Effect of newly synthesized 1,2,4-triazino [5,6-b]indole-3-thione derivatives on olfactory bulbectomy induced depression in rats. Asian Pac. J. Trop. Biomed. 2012, 2, 992. [Google Scholar] [CrossRef] [Green Version]
- Tomchin, A.B.; Uryupov, O.Y.; Zhukova, T.I.; Kuznetsova, T.A.; Kostycheva, M.V.; Smirnov, A.V. Thiourea and Thiosemicarbazide Derivatives: Structure, Transformations, and Pharmacological Activity. Part II. Antihypoxic Activity of 1,2,4-triazino [5,6-b]indole Derivatives. Pharmaceut. Chem. J. 1997, 31, 125–133. [Google Scholar] [CrossRef]
- Kgokong, J.L.; Smith, P.P.; Matsabisa, G.M. 1,2,4-Triazino-[5,6b]indole derivatives: Effects of the trifluoromethyl group on in vitro antimalarial activity. Bioorg. Med. Chem. 2005, 13, 2935–2942. [Google Scholar] [CrossRef] [PubMed]
- Gladych, J.M.Z.; Hunt, J.H.; Jack, D.; Haff, R.F.; Boyle, J.J.; Stewart, R.C.; Ferlanto, R.J. Inhibition of Rhinovirus by Isatin Thiosemicarbazone Analogues. Nature 1969, 221, 286. [Google Scholar] [CrossRef] [PubMed]
- Boyle, J.J.; Raupp, W.G.; Stanfield, F.J.; Haff, R.F.; Dick, E.C.; Alessio, D.D.; Dick, C.R. Progress in Rhinovirus Chenotherapy. Ann. N. Y. Acad. Sci. 1970, 173, 477. [Google Scholar] [CrossRef]
- Gwaltney, J.M. Rhinovirus inhibition by 3-substituted triazinoindoles. Proc. Soc. Exp. Biol. Med. 1970, 133, 1148. [Google Scholar] [CrossRef]
- Haff, R.F.; Flagg, W.B.; Gallo, J.J.; Hoover, J.R.E.; Miller, J.A.; Pinto, C.A.; Pagano, J.F. The in vitro antiviral activity of a triazinoindole (SK & F 40491). Proc. Soc. Exp. Biol. Med. 1972, 141, 475–478. [Google Scholar]
- Taha, M.; Ismail, N.H.; Jamil, W.; Rashwan, H.; Kashif, S.M.; Sain, A.A.; IlhamAdenan, M.; Anouar, E.H.; Ali, M.; Rahim, F.; et al. Synthesis of novel derivatives of 4- methylbenzimidazole and evaluation of their biological activities. Eur. J. Med. Chem. 2014, 84, 731–738. [Google Scholar] [CrossRef] [PubMed]
- Xiang, P.; Zhou, T.; Wang, L.; Sun, C.Y.; Hu, J.; Zhao, Y.L.; Yang, L. Novel benzothiazole benzimidazole and benzoxazole derivatives as potential antitumor agents: Synthesis and preliminary in vitro biological evaluation. Molecules 2012, 17, 873–883. [Google Scholar] [CrossRef] [Green Version]
- Valdez-Padilla, D.; Rodríguez-Morales, S.; Campos, A.H.; Luis, F.H.; Mulia, L.Y..; Contreras, A.T.; Castillo, R. Synthesis and antiprotozoal activity of novel 1-methylbenzimidazole derivatives. Bioorg. Med. Chem. 2009, 17, 1724–1730. [Google Scholar] [CrossRef]
- Ishikawa, M.; Nonoshita, K.; Ogino, Y.; Nagae, Y.; Tsukahara, D.; Hosaka, H.; Hosaka, H.; Maruki, H.; Ohyama, S.; Yoshimoto, R.; et al. Discovery of novel 2-(pyridine-2-yl)-1H-benzimidazole derivatives as potent glucokinase activators. Bioorg. Med. Chem. Lett. 2009, 19, 4450–4454. [Google Scholar] [CrossRef]
- Hamaguchi, W.; Masuda, N.; Isomura, M.; Miyamoto, S.; Kikuchi, S.; Amano, Y.; Hanbou, K.; Mahira, T.; Watanabe, T. Design and synthesis of novel benzimidazole derivatives as phosphodiesterase 10A inhibitors with reduced CYP1A2 inhibition. Bioorg. Med. Chem. 2013, 21, 7612–7623. [Google Scholar] [CrossRef]
- Luo, Y.; Yao, J.P.; Yang, L.; Feng, C.L.; Tang, W.; Wang, G.F.; Lu, W. Design and synthesis of novel benzimidazole derivatives as inhibitors of hepatitis B virus. Bioorg. Med. Chem. 2010, 18, 5048–5055. [Google Scholar] [CrossRef] [PubMed]
- Padmavathi, V.; Venkatesh, B.C.; Muralikrishna, A.; Padmaja, A. The Reactivity of Gem Cyanoester Ketene Dithiolates towards the Development of Potent Antioxidant Heterocycles. Chem. Pharm. Bull. 2012, 60, 449–4458. [Google Scholar] [CrossRef] [Green Version]
- Vasquez, R.J.; Howell, B.; Yvon, A.M.; Wadsworth, P.; Cassimeris, L. Nanomolar concentrations of nocodazole alter microtubule dynamic instability in vivo and in vitro. Mol. Biol. Cell. 1997, 8, 973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knauf, W.U.; Lissichkov, T.; Aldaoud, A. Phase III randomized study of bendamustine compared with chlorambucil in previously untreated patients with chronic lymphocytic leukemia. J. Clin. Oncol. 2009, 27, 4378. [Google Scholar] [CrossRef]
- Jenkins, T.C. Targeting multi-stranded DNA structures. Curr. Med. Chem. 2000, 7, 99. [Google Scholar] [CrossRef] [PubMed]
- Baldisserotto, A.; Demurtas, M.; Lampronti, I.; Tacchini, M.; Moi, D.; Balboni, G.; Pacifico, S.; Vertuani, S.; Manfredini, S.; Onnis, V. Synthesis and evaluation of antioxidant and antiproliferative activity of 2-arylbenzimidazoles. Bioorg. Chem. 2019, 94, 103396. [Google Scholar] [CrossRef]
- Sirgamalla, R.; Kommakula, A.; Konduru, S.; Ponakanti, R.; Devaram, J.; Boda, S. Cupper-catalyzed an efficient synthesis, characterization of 2-substituted benzoxazoles, 2-substituted benzothiazoles derivatives and their anti-fungal activity. Chem. Data Collec. 2020, 27, 100362. [Google Scholar] [CrossRef]
- Aswathy, V.V.; Alper-Hayta, S.; Yalcin, G.; Mary, Y.S.; Panicker, C.Y.; Jojo, P.J.; Kaynak-Onurdag, F.; Armaković, S.; Armaković, S.J.; Yildiz, I.; et al. Modification of benzoxazole derivative by bromine-spectroscopic, antibacterial and reactivity study using experimental and theoretical procedures. J. Mol. Structu. 2017, 1141, 495–511. [Google Scholar] [CrossRef]
- Tariq, S.; Kamboj, P.; Alam, O.; Amir, M. 1, 2, 4-Triazole-based benzothiazole/benzoxazole derivatives: Design, synthesis, p38α MAP kinase inhibition, anti-inflammatory activity and molecular docking studies. Bioorg. Chem. 2018, 81, 630–641. [Google Scholar] [CrossRef]
- Demmer, C.S.; Bunch, L. Benzoxazoles and oxazolopyridines in medicinal chemistry studies. Eur. J. Med. Chem. 2015, 97, 778–785. [Google Scholar] [CrossRef]
- Kakkar, S.; Tahlan, S.; Lim, S.M.; Ramasamy, K.; Mani, V.; Shah, S.A.A.; Narasimhan, B. Benzoxazole derivatives: Design, synthesis and biological evaluation. Chem. Cent. J. 2018, 12, 92. [Google Scholar] [CrossRef] [PubMed]
- Guzow, K.; Mulkiewicz, E.; Obuchowski, M.; Wiczk, W. Biological activity of 3-(2-benzoxazol-5-yl) alanine derivatives. Amino Acids 2021, 53, 1257–1268. [Google Scholar] [CrossRef] [PubMed]
- Taha, M.; Alshamrani, F.J.; Rahim, F.; Hayat, S.; Ullah, H.; Zaman, K.; Imran, S.; Khan, K.M.; Naz, F. Synthesis of novel triazinoindole-based thiourea hybrid: A study on α-glucosidase inhibitors and their molecular docking. Molecules 2019, 24, 3819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahim, F.; Tariq, S.; Taha, M.; Ullah, H.; Zaman, K.; Uddin, I.; Wadood, A.; Khan, A.A.; Rehman, A.U.; Uddin, N.; et al. New triazinoindole bearing thiazole/oxazole analogues: Synthesis, α-amylase inhibitory potential and molecular docking study. Bioorg. Chem. 2019, 92, 103284. [Google Scholar] [CrossRef]
- Balogun, M.M.; Shamim, S.; Khan, K.M.; Salar, U.; Oladosu, I.A.; Lateef, M.; Wadood, A.; Taha, M.; Moronkola, D.O.; Rehman, A.U.; et al. 2-Mercapto Benzoxazole Derivatives as Novel Leads: Urease Inhibition, In Vitro and In Silico Studies. Chem. Sel. 2021, 6, 8490–8498. [Google Scholar] [CrossRef]
- Zaman, K.; Rahim, F.; Taha, M.; Ullah, H.; Wadood, A.; Nawaz, M.; Khan, F.; Wahab, Z.; Shah, S.A.A.; Rehman, A.U.; et al. Synthesis, in vitro urease inhibitory potential and molecular docking study of Benzimidazole analogues. Bioorg. Chem. 2019, 89, 103024. [Google Scholar] [CrossRef]
- Hussain, R.; Ullah, H.; Rahim, F.; Sarfraz, M.; Taha, M.; Iqbal, R.; Abdelaziz, M.A. Multipotent Cholinesterase Inhibitors for the Treatment of Alzheimer’s Disease: Synthesis, Biological Analysis and Molecular Docking Study of Benzimidazole-Based Thiazole Derivatives. Molecules 2022, 27, 6087. [Google Scholar] [CrossRef]
- Khan, S.; Iqbal, S.; Khan, M.; Rehman, W.; Shah, M.; Hussain, R.; Farouk, A.E. Design, Synthesis, In Silico Testing, and In Vitro Evaluation of Thiazolidinone-Based Benzothiazole Derivatives as Inhibitors of α-Amylase and α-Glucosidase. Pharmaceuticals 2022, 15, 1164. [Google Scholar] [CrossRef]
- Weatherburn, M.W. Phenol-hypochlorite reaction for determination of ammonia. Anal. Chem. 1967, 39, 971–974. [Google Scholar] [CrossRef]
- Khan, S.; Ullah, H.; Rahim, F.; Nawaz, M.; Hussain, R.; Rasheed, L. Synthesis, in vitro α-amylase, α-glucosidase activities and molecular docking study of new benzimidazole bearing thiazolidinone derivatives. J. Mol. Structu. 2022, 1269, 133812. [Google Scholar] [CrossRef]
- Doğan, Ş.D.; Gündüz, M.G.; Doğan, H.; Krishna, V.S.; Lherbet, C.; Sriram, D. Design and synthesis of thiourea-based derivatives as Mycobacterium tuberculosis growth and enoyl acyl carrier protein reductase (InhA) inhibitors. Eur. J. Med. Chem. 2020, 199, 112402. [Google Scholar] [CrossRef] [PubMed]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | |||||
| S.NO | R | IC50 ± SEM (μM) | S.NO | R | IC50 ± SE (μM) |
| 1 | 4-nitro | 17.60 ± 0.30 | 2 | 2,4-dihydroxy | 1.90 ± 0.10 |
| 3 | 3,5-dihydroxy | 2.10 ± 0.10 | 4 | 2-methoxy | 32.20 ± 0.30 |
| 5 | 3,4-dihydroxy | 0.90 ± 0.01 | 6 | 2,5-dihydroxy | 6.70 ± 0.20 |
| 7 | 3-hydroxy | 18.90 ± 0.40 | 8 | 2,4,6-trihydroxy | 0.20 ± 0.01 |
| 9 | 4-dimethylamino | 31.10 ± 0.30 | 10 | 2-nitro | 19.30 ± 0.40 |
| 11 | 3-nitro | 26.60 ± 0.50 | 12 | 4-hydroxy | 14.60 ± 0.30 |
| Standard Thiourea | 21.86 ± 0.40 μM | ||||
 | |||||
| 13 | 4-chloro | 9.70 ± 0.20 | 14 | 4-methyl ethanoate | 23.90 ± 0.50 |
| 15 | 2-methyl | 36.20 ± 0.70 | 16 | 4-methyl | 28.20 ± 0.50 |
| 17 | 3-chloro | 22.20 ± 0.50 | 18 | 2-hydroxy | 3.80 ± 0.10 |
| 19 | 3-methyl | 34.90 ± 0.70 | 20 | 3-hydroxy-4-methoxy | 2.70 ± 0.10 |
| 21 | 2,4-dimethoxy | 31.76 ± 0.50 | 22 | 2,3-dihydroxy | 1.70 ± 0.10 |
| 23 | 2-fluoro | 1.10 ± 0.01 | 24 | 3-fluoro | 1.60 ± 0.01 |
| 25 | 3-nitro | 21.20 ± 0.40 | - | - | - |
| Standard Thiourea | 21.86 ± 0.40 μM | ||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mumtaz, S.; Iqbal, S.; Shah, M.; Hussain, R.; Rahim, F.; Rehman, W.; Khan, S.; Abid, O.-u.-R.; Rasheed, L.; Dera, A.A.; et al. New Triazinoindole Bearing Benzimidazole/Benzoxazole Hybrids Analogs as Potent Inhibitors of Urease: Synthesis, In Vitro Analysis and Molecular Docking Studies. Molecules 2022, 27, 6580. https://doi.org/10.3390/molecules27196580
Mumtaz S, Iqbal S, Shah M, Hussain R, Rahim F, Rehman W, Khan S, Abid O-u-R, Rasheed L, Dera AA, et al. New Triazinoindole Bearing Benzimidazole/Benzoxazole Hybrids Analogs as Potent Inhibitors of Urease: Synthesis, In Vitro Analysis and Molecular Docking Studies. Molecules. 2022; 27(19):6580. https://doi.org/10.3390/molecules27196580
Chicago/Turabian StyleMumtaz, Sundas, Shahid Iqbal, Mazloom Shah, Rafaqat Hussain, Fazal Rahim, Wajid Rehman, Shoaib Khan, Obaid-ur-Rahman Abid, Liaqat Rasheed, Ayed A. Dera, and et al. 2022. "New Triazinoindole Bearing Benzimidazole/Benzoxazole Hybrids Analogs as Potent Inhibitors of Urease: Synthesis, In Vitro Analysis and Molecular Docking Studies" Molecules 27, no. 19: 6580. https://doi.org/10.3390/molecules27196580
APA StyleMumtaz, S., Iqbal, S., Shah, M., Hussain, R., Rahim, F., Rehman, W., Khan, S., Abid, O.-u.-R., Rasheed, L., Dera, A. A., Al-ghulikah, H. A., Kehili, S., Elkaeed, E. B., Alrbyawi, H., & Alahmdi, M. I. (2022). New Triazinoindole Bearing Benzimidazole/Benzoxazole Hybrids Analogs as Potent Inhibitors of Urease: Synthesis, In Vitro Analysis and Molecular Docking Studies. Molecules, 27(19), 6580. https://doi.org/10.3390/molecules27196580