
Synthesis, In Vitro Anti-Microbial Analysis and Molecular Docking Study of Aliphatic Hydrazide-Based Benzene Sulphonamide Derivatives as Potent Inhibitors of α-Glucosidase and Urease


 , , ,
, , ,
Abstract
1. Introduction
2. Material and Methods
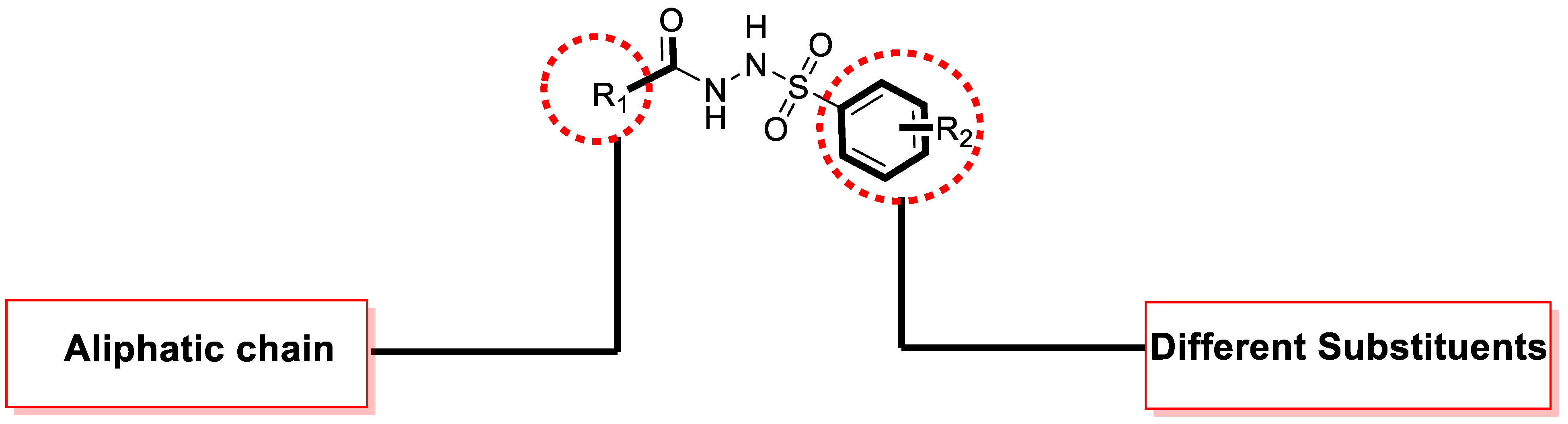
General Procedure of Aliphatic Hydrazide-Based Benzene Sulphonamide Derivatives (1–15)
3. Results and Discussion
Chemistry
4. Spectral Analysis
5. Biological Profile
5.1. α-Glucosidase Inhibitory Activity
5.2. Anti-Urease Inhibitory Activity
5.3. Anti-Microbial Activity
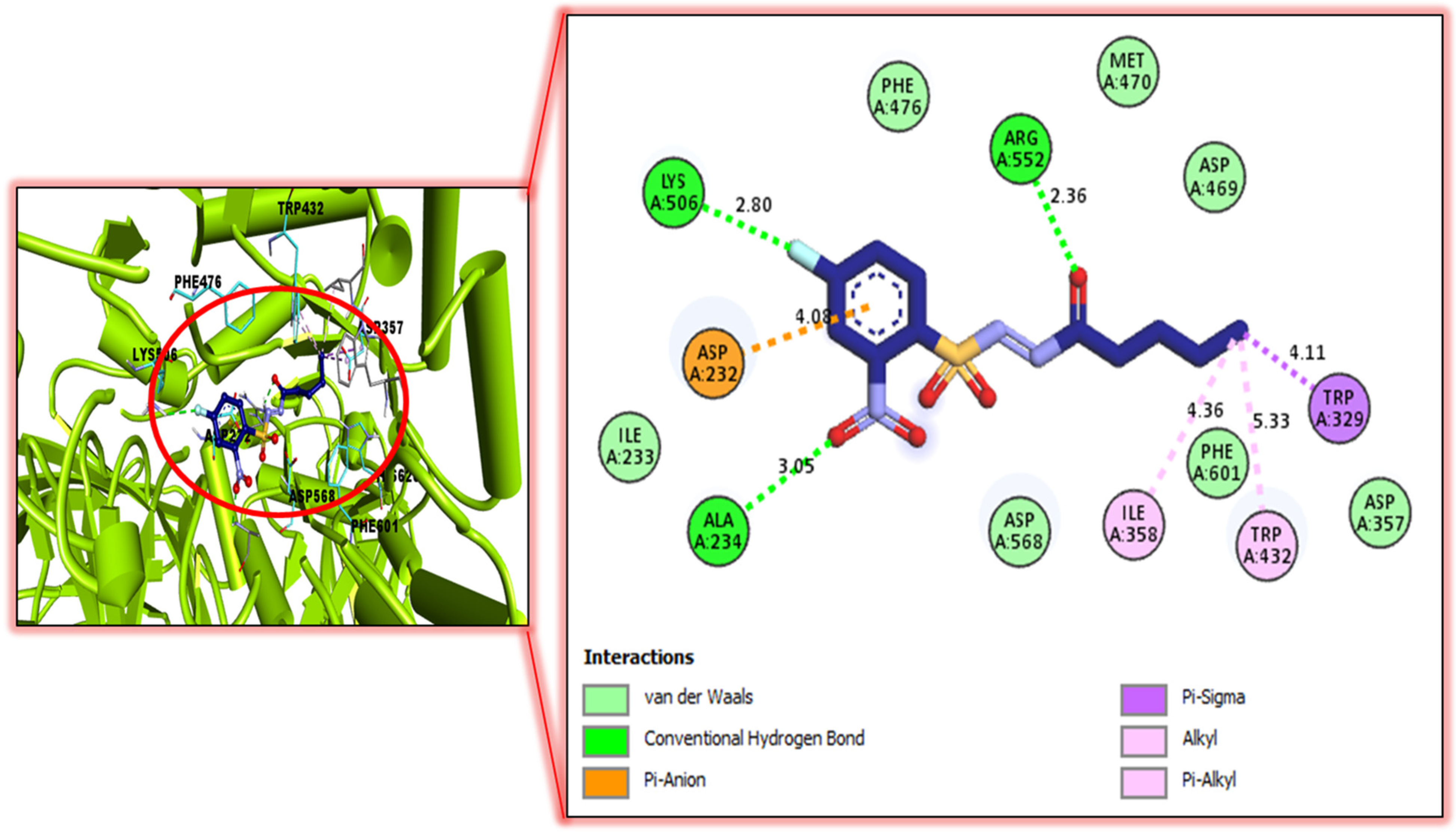
5.4. Molecular Docking Studies
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Whiting, D.R.; Guariguata, L.; Weil, C.; Shaw, J. IDF diabetes atlas: Global estimates of the prevalence of diabetes for 2011 and 2030. Diabetes Res. Clin. Pract. 2011, 94, 311–321. [Google Scholar] [CrossRef] [PubMed]
- Taylor, S.I.; Accili, D.; Imai, Y. Insulin resistance or insulin deficiency: Which is the primary cause of NIDDM? Diabetes 1994, 43, 735–740. [Google Scholar] [CrossRef] [PubMed]
- Porte, D., Jr. β-cells in typeII diabetes mellitus. Diabetes 1991, 40, 166–180. [Google Scholar] [CrossRef]
- Butler, A.E.; Janson, J.; Bonner-Weir, S.; Ritzel, R.; Rizza, R.A.; Butler, P.C. β-cell deficit and increased β-cell apoptosis in humans with type 2 diabetes. Diabetes 2003, 52, 102–110. [Google Scholar] [CrossRef]
- Tang, P.C.; Lin, Z.G.; Wang, Y.; Yang, F.L.; Wang, Q.; Fu, J.H.; Zhang, L.; Gong, A.S.; Luo, J.J.; Dai, J.; et al. Design and synthesis of DPP-4 inhibitor for the treatment of type 2 diabetes. Chin. Chem. Lett. 2010, 21, 253–256. [Google Scholar] [CrossRef]
- Wu, Y.H. Synthesis of (S)‐2‐Ethoxy‐3‐Phenylpropanoic Acid Derivatives and Their Insulin‐Sensitizing Activity. Chin. J. Chem. 2007, 25, 265–267. [Google Scholar] [CrossRef]
- Salar, U.; Khan, K.M.; Chigurupati, S.; Taha, M.; Wadood, A.; Vijayabalan, S.; Ghufran, M.; Perveen, S. New hybrid hydrazinyl thiazole substituted chromones: As potential α-amylase inhibitors and radical (DPPH & ABTS) scavengers. Sci. Rep. 2017, 7, 16980. [Google Scholar]
- Sun, H.; Wang, D.; Song, X.; Zhang, Y.; Ding, W.; Peng, X.; Zhang, X.; Li, Y.; Ma, Y.; Wang, R.; et al. Natural prenylchalconaringenins and prenylnaringenins as antidiabetic agents: α-glucosidase and α-amylase inhibition and in vivo antihyperglycemic and antihyperlipidemic effects. J. Agric. Food Chem. 2017, 65, 1574–1581. [Google Scholar] [CrossRef] [PubMed]
- Campos, C. Chronic hyperglycemia and glucose toxicity: Pathology and clinical sequelae. Postgrad. Med. 2012, 124, 90–97. [Google Scholar] [CrossRef]
- Rask-Madsen, C.; King, G.L. Vascular complications of diabetes: Mechanisms of injury and protective factors. CellMetab. 2013, 17, 20–33. [Google Scholar]
- Miller, B.R.; Nguyen, H.; Hu, C.J.H.; Lin, C.; Nguyen, Q.T. New and emerging drugs and targets for type 2 diabetes: Reviewing the evidence. Am. Health Drug Benefits 2014, 7, 452. [Google Scholar]
- Raghu, C.; Arjun, H.A.; Anantharaman, P. In vitro and in silico inhibition properties of fucoidan against α-amylase and α-D-glucosidase with relevance to type 2 diabetes mellitus. Carbohydr. Polym. 2019, 209, 350–355. [Google Scholar]
- Henry, R.J. The mode of action of sulfonamides. Bacteriol. Rev. 1943, 7, 175–262. [Google Scholar] [CrossRef] [PubMed]
- Wegst-Uhrich, S.R.; Navarro, D.A.; Zimmerman, L.; Aga, D.S. Assessing antibiotic sorption in soil: A literature review and new case studies on sulfonamides and macrolides. Chem. Cent. J. 2014, 8, 5. [Google Scholar] [CrossRef] [PubMed]
- Kosak, U.; Brus, B.; Knez, D.; Zakelj, S.; Trontelj, J.; Pislar, A.; Sink, R.; Jukic, M.; Zivin, M.; Podkowa, A.; et al. The magic of crystal structure-based inhibitor optimization: Development of a butyrylcholinesterase inhibitor with picomolar affinity and in vivo activity. J. Med. Chem. 2018, 61, 119–139. [Google Scholar] [PubMed]
- Košak, U.; Brus, B.; Knez, D.; Šink, R.; Žakelj, S.; Trontelj, J.; Pišlar, A.; Šlenc, J.; Gobec, M.; Živin, M.; et al. Development of an in-vivo active reversible butyrylcholinesterase inhibitor. Sci. Rep. 2016, 6, 39495. [Google Scholar] [CrossRef] [PubMed]
- Riaz, S.; Khan, I.U.; Bajda, M.; Ashraf, M.; Shaukat, A.; Rehman, T.U.; Mutahir, S.; Hussain, S.; Mustafa, G.; Yar, M. Pyridine sulfonamide as a small key organic molecule for the potential treatment of type-II diabetes mellitus and Alzheimer’s disease: In vitro studies against yeast α-glucosidase, acetylcholinesterase and butyrylcholinesterase. Bioorg. Chem. 2015, 63, 64–71. [Google Scholar] [CrossRef]
- Markowicz-Piasecka, M.; Huttunen, K.M.; Broncel, M.; Sikora, J. Sulfenamide and sulfonamide Derivatives of Metformin—A New option to Improve endothelial Function and plasma Haemostasis. Sci. Rep. 2019, 9, 6573. [Google Scholar] [CrossRef]
- Zajdel, P.; Partyka, A.; Marciniec, K.; Bojarski, A.J.; Pawlowski, M.; Wesolowska, A. Quinoline-and isoquinoline-sulfonamide analogs of aripiprazole: Novel antipsychotic agents? Future Med. Chem. 2014, 6, 57–75. [Google Scholar] [CrossRef]
- Mutahir, S.; Jończyk, J.; Bajda, M.; Khan, I.U.; Khan, M.A.; Ullah, N.; Ashraf, M.; Riaz, S.; Hussain, S.; Yar, M. Novel biphenyl bis-sulfonamides as acetyl and butyrylcholinesterase inhibitors: Synthesis, biological evaluation and mo-lecular modeling studies. Bioorg. Chem. 2016, 64, 13–20. [Google Scholar] [CrossRef]
- Apiraksattayakul, S.; Pingaew, R.; Prachayasittikul, V.; Ruankham, W.; Jongwachirachai, P.; Songtawee, N.; Phopin, K. Neuroprotective Properties of Bis-Sulfonamide Derivatives Against 6-OHDA-Induced Parkinson's Model via Sirtuin 1 Ac-tivity and in silico Pharmacokinetic Properties. Front. Mol. Neurosci. 2022, 15, 890838. [Google Scholar] [CrossRef] [PubMed]
- Morris, M.; Knudsen, G.M.; Maeda, S.; Trinidad, J.C.; Ioanoviciu, A.; Burlingame, A.L.; Mucke, L. Tau post-translational modifications in wild-type and human amyloid precursor protein transgenic mice. Nat. Neurosci. 2015, 18, 1183–1189. [Google Scholar] [CrossRef] [PubMed]
- Hu, L.; Li, Z.R.; Li, Y.; Qu, J.; Ling, Y.H.; Jiang, J.D.; Boykin, D.W. Synthesis and structure—Activity relationships of carbazole sulfonamides as a novel class of antimitotic agents against solid tumors. J. Med. Chem. 2006, 49, 6273–6282. [Google Scholar] [CrossRef] [PubMed]
- ShoaibAhmadShah, S.; Rivera, G.; Ashfaq, M. Recent advances in medicinal chemistry of sulfonamides: Rational design as anti-tumoral, anti-bacterial and an-ti-inflammatory agents. MiniRev. Med. Chem. 2013, 13, 70–86. [Google Scholar]
- Takagi, M.; Honmura, T.; Watanabe, S.; Yamaguchi, R.; Nogawa, M.; Nishimura, I.; Katoh, F.; Matsuda, M.; Hidaka, H. In vivo antitumor activity of a novel sulfonamide, HMN-214, against human tumor xenografts in mice and the spectrum of cytotoxicity of its active metabolite, HMN-176. Investig. New Drugs 2003, 21, 387–399. [Google Scholar]
- Owa, T.; Yoshino, H.; Okauchi, T.; Yoshimatsu, K.; Ozawa, Y.; Sugi, N.H.; Nagasu, T.; Koyanagi, N.; Kitoh, K. Discovery of novel antitumor sulfonamides targeting G1 phase of the cell cycle. J. Med. Chem. 1999, 42, 3789–3799. [Google Scholar] [CrossRef] [PubMed]
- Gul, H.I.; Yamali, C.; Sakagami, H.; Angeli, A.; Leitans, J.; Kazaks, A.; Tars, K.; Ozgun, D.O.; Supuran, C.T. New anticancer drug candidate’s sulfonamides as selective hCA IX or hCA XII inhibitors. Bioorg. Chem. 2018, 77, 411–419. [Google Scholar] [CrossRef] [PubMed]
- Wan, Y.; Fang, G.; Chen, H.; Deng, X.; Tang, Z. Sulfonamide derivatives as potential anti-cancer agents and their SARs elucidation. Eur. J. Med. Chem. 2021, 226, 113837. [Google Scholar] [CrossRef] [PubMed]
- Mahajan, A.; Singh, H.; Singh, A.; Agrawal, D.K.; Arora, A.; Chundawat, T.S. Trifluoromethylated Quinolone-Hydantoin Hybrids: Synthesis and Antibacterial Evaluation. Science 2022, 4, 30. [Google Scholar] [CrossRef]
- Kharb, M.; Jat, R.K.; Parjapati, G.; Gupta, A. Introduction to molecular docking software technique in medicinal chemistry. Int. J. Drug Res. Tech. 2012, 2, 189–197. [Google Scholar]
- Li, Z.; Gu, J.; Zhuang, H.; Kang, L.; Zhao, X.; Guo, Q. Adaptive molecular docking method based on information entropy genetic algorithm. Appl. Soft Comput. 2015, 26, 299–302. [Google Scholar]
- Rao, C.M.M.P.; Naidu, N.; Priya, J.; Rao, K.P.C.; Ranjith, K.; Shobha, S.; Siddiraju, S. Molecular docking and dynamic simulations of benzimidazoles with beta-tubulins. Bioinformation 2021, 17, 404. [Google Scholar] [PubMed]
- Khan, S.; Ullah, H.; Rahim, F.; Nawaz, M.; Hussain, R.; Rasheed, L. Synthesis, in vitro α-amylase, α-glucosidase activities and molecular docking study of new benzimidazole bearing thiazolidinone derivatives. J. Mol. Struct. 2022, 1269, 133812. [Google Scholar] [CrossRef]
- Khan, Y.; Iqbal, S.; Shah, M.; Maalik, A.; Hussain, R.; Khan, S.; Khan, I.; Pashameah, R.A.; Alzahrani, E.; Farouk, A.E. New Quinoline-based triazole hybrid analogues as effective inhibitors of α-amylase and αglucosidase: Synthesis, in vitro evaluation and molecular docking along with in silico study. Front. Chem. 2022, 10, 1099. [Google Scholar]
- Khan, Y.; Rehman, W.; Hussain, R.; Khan, S.; Malik, A.; Khan, M.; Liaqat, A.; Rasheed, L.; Begum, F.; Fazil, S.; et al. New biologically potent benzimidazole‐based‐triazole derivatives as acetylcholinesterase and butyrylcholinesterase inhib-itors along with molecular docking study. J. Hetercyc. Chem. 2022, in press. [Google Scholar]
- Weatherburn, M.W. Phenol-hypochlorite reaction for determination of ammonia. Anal. Chem. 1967, 39, 971–974. [Google Scholar] [CrossRef]
- Srinivasa, M.G.; Aggarwal, N.N.; Gatpoh, B.F.D.; Shankar, M.K.; Byadarahalli Ravindranath, K.; Gurubasavaraj Veeranna, P.; Bistuvalli Chandrashekarappa, R. Identification of benzothiazole‐rhodanine derivatives as α-amylase and α-glucosidase inhibitors: Design, synthesis, in silico, and in vitro analysis. J. Mol. Recognit. 2022, 35, 2959. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| C.No | R1 | R2 | Alpha-Glucosidase IC50 (µM ± SEM) | Anti-Urease IC50 (µM ± SEM) |
|---|---|---|---|---|
| 1 |  |  | 8.80 ± 0.20 | 17.60 ± 0.50 |
| 2 |  |  | 9.20 ± 0.30 | 21.20 ± 0.70 |
| 3 |  |  | 8.60 ± 0.20 | 12.30 ± 0.30 |
| 4 |  |  | 5.70 ± 0.70 | 8.80 ± 0.10 |
| 5 |  |  | 3.20 ± 0.40 | 2.10 ± 0.10 |
| 6 |  |  | 2.50 ± 0.40 | 5.30 ± 0.20 |
| 7 |  |  | 4.50 ± 0.80 | 9.80 ± 0.10 |
| 8 |  |  | 6.70 ± 0.80 | 13.10 ± 0.20 |
| 9 |  |  | 7.40 ± 0.10 | 11.10 ± 0.20 |
| 10 |  |  | 27.60 ± 0.20 | 23.40 ± 0.20 |
| 11 |  |  | 29.85 ± 0.20 | 25.30 ± 0.30 |
| 12 |  |  | 23.10 ± 0.30 | 16.30 ± 0.10 |
| 13 |  |  | 22.50 ± 0.20 | 18.40 ± 0.30 |
| 14 |  |  | 6.50 ± 0.70 | 7.40 ± 0.40 |
| 15 |  |  | 7.90 ± 0.80 | 8.60 ± 0.20 |
| Standard drug Acarbose and thiourea | IC50 = 8.24 ± 0.08 | IC50 = 7.80 ± 0.30 | ||
| Compound Analog-5 against Urease | Receptor | Interaction | Distance | Docking Score |
| LYS-A-506 H-B 2.80A ARG-A-552 H-B 2.36A TRP-A-329 Pi-Sigma 4.11A TRP-A-432 R 5.33A ILE-A-358 Pi-R 4.36A ALA-A-234 H-B 3.05A ASP-A-232 Pi-anion 4.08A | −9.80 | |||
| Analog-6 against α-Glucosidase | HIS-A-626 H-B 2.57A PHE-A-601 R 2.36A ASP-A-568 Pi-anion 3.36A TRP-A-432 Pi-Sigma 4.84A ASP-A-232 Attractive charges 2.99A LYS-A-506 Pi-R 3.57A PHE-A-476 Pi-R 4.97A ASP-A-357 H-F 3.41A | −10.70 | ||
| Acarbose drug | LYS-A-506 H-B 2.80A ASP-A-630 H-B 4.98A ALA-A-329 V.W Interaction 4.92A GLU-A-603 H-B 4.08A SER-A-505 H-B 3.72A SER-A-505 H-B 3.53A ASN-A-496 H-B 4.81A LYS-A-506 H-B 3.71A ASP-A-232 H-B 4.50A | −9.30 | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khan, S.; Iqbal, S.; Shah, M.; Rehman, W.; Hussain, R.; Rasheed, L.; Alrbyawi, H.; Dera, A.A.; Alahmdi, M.I.; Pashameah, R.A.; et al. Synthesis, In Vitro Anti-Microbial Analysis and Molecular Docking Study of Aliphatic Hydrazide-Based Benzene Sulphonamide Derivatives as Potent Inhibitors of α-Glucosidase and Urease. Molecules 2022, 27, 7129. https://doi.org/10.3390/molecules27207129
Khan S, Iqbal S, Shah M, Rehman W, Hussain R, Rasheed L, Alrbyawi H, Dera AA, Alahmdi MI, Pashameah RA, et al. Synthesis, In Vitro Anti-Microbial Analysis and Molecular Docking Study of Aliphatic Hydrazide-Based Benzene Sulphonamide Derivatives as Potent Inhibitors of α-Glucosidase and Urease. Molecules. 2022; 27(20):7129. https://doi.org/10.3390/molecules27207129
Chicago/Turabian StyleKhan, Shoaib, Shahid Iqbal, Mazloom Shah, Wajid Rehman, Rafaqat Hussain, Liaqat Rasheed, Hamad Alrbyawi, Ayed A. Dera, Mohammed Issa Alahmdi, Rami Adel Pashameah, and et al. 2022. "Synthesis, In Vitro Anti-Microbial Analysis and Molecular Docking Study of Aliphatic Hydrazide-Based Benzene Sulphonamide Derivatives as Potent Inhibitors of α-Glucosidase and Urease" Molecules 27, no. 20: 7129. https://doi.org/10.3390/molecules27207129
APA StyleKhan, S., Iqbal, S., Shah, M., Rehman, W., Hussain, R., Rasheed, L., Alrbyawi, H., Dera, A. A., Alahmdi, M. I., Pashameah, R. A., Alzahrani, E., & Farouk, A.-E. (2022). Synthesis, In Vitro Anti-Microbial Analysis and Molecular Docking Study of Aliphatic Hydrazide-Based Benzene Sulphonamide Derivatives as Potent Inhibitors of α-Glucosidase and Urease. Molecules, 27(20), 7129. https://doi.org/10.3390/molecules27207129