2.1. Prediction of the Numerical Values of the Parameter k7 Using the GUSAR2019 Program
According to the consensus approach implemented in the GUSAR2019 program, six consensus QSPR models M1–M6 were built to predict the numerical values of logk
7 for the phenolic type antioxidants, namely the sulfur-containing alkylphenols, the natural phenols, the chromane derivatives, the betulonic and betulinic acids, and 20-hydroxyecdysone. These models differ in the type of descriptors they contain and the number of partial regression relationships. The descriptive power characteristics of the M1–M6 consensus models, calculated automatically in the GUSAR2019 program by comparing the experimental logk
7 values with those predicted by these six models are presented in
Table 1. Note that the determination coefficients, the standard deviations, and Fisher’s criterion values presented in
Table 1 are the average values obtained taking into account all partial regression models included in the consensus model Mi (i = 1–6).
For a QSPR model to be adequate, that is, to be acceptable for use within its reach, its results must correctly describe and predict the target property. According to the recommendations of the QSAR/QSPR modeling experts, the validation is the most important concept in the development and application of the QSPR models. The validation of the developed QSPR models based on the external test set structures is the “gold standard” that validates the reliability of these models along with the acceptability of each step during their development: the assessment of the input data quality, the diversity of the data sets, the predictivity, the domain of applicability, and interpretability.
In this regard, to objectively characterize the descriptive and predictive powers of the M1–M6 consensus models, we performed the prediction of the logk
7 values for the antioxidant structures contained in test sets TR1 and TR2. As in our previous studies [
67,
68,
69,
70], in addition to the parameters calculated in the GUSAR2019 program (average R
2, average Q
2, average F), we used metrics based on the R
2 determination factors (R
2, R
20, R
2’0, average R
2m, ΔR
2m Q
2F1, Q
2F2, CCC); and the metrics designed to estimate the prediction errors of the logk
7 values (RMSE, MAE, SD) [
34,
35,
36,
37]. The metrics based on the prediction error estimates were used to determine the true prediction quality index for the parameter logk
7 for the compounds of both test sets. Their calculation was performed using the Xternal Validation Plus 1.2 program [
71]. The same program was used to check the models for systematic errors.
The statistical criteria measuring the descriptive and predictive powers of the M1–M6 QSPR models, which were estimated for 95% of the structures of the training and test sets TR1, TR2 and TS1, TS2, respectively, are presented in
Table 2,
Table 3.
Tables S2–S8 (
Supplementary Material) present a complete set of the statistical parameters calculated using the Xternal Validation Plus 1.2 software for the training and test sets TR1, TR2 and TS1, TS2, taking into account both 100% and 95% of the antioxidant structures they contain.
The analysis of the statistical characteristics of the M1-M6 consensus models, summarized in
Table 2,
Table 3 showed that almost all of the models successfully reproduced the experimental data contained in both training sets (the condition is satisfied for 100% and 95% of the data). Thus, the values of the determination coefficients R
2, R
20, R
2’0, average R
2m, and CCC, evaluated by comparing the values of logk
7pred and logk
7exp fully met all of the the requirements, corresponding to the models with a high descriptive power listed in part 2.3. The M6 model (100% and 95% of the data) had the highest descriptive power in a number of determination coefficients (R
2, R
20, CCC). At the same time, other criteria indicated the best reproducibility of the experimental data of test sets TS1 and TS2 (100% and 95% data) using the models M3 (R
2’0, ∆R
2m), M4 (average R
2m), and M5 (R
2’0, average R
2m). The M3 and M6 models were characterized by the lowest values of the prediction errors of the logk
7 value for the structures of both training sets (RMSE, MAE, SD) at 100% and 95% of the data contained in them. At the same time, the best characteristics were in the M3 model (
Table 2). The minimum SD value at 100% of the data in both training sets was shown by the M4 model. In the case of 95% of the data in these training sets, the best result was observed for the M6 model. Since the numerical values of the MAE for all of the models were in the range of 0.0599–0.0679, which is significantly lower than 0.706 (10% of the range of the simulated logk
7 values), and simultaneously, the numerical values of the MAE+3SD criterion were also significantly smaller than 0.706, we can conclude that almost all of the models had a high descriptive power.
However, the same models were characterized by the rather low values of the different determination coefficients for the comparison of the experimental and predicted logk7 values for the 100% antioxidant structures contained in test sets TS1 and TS2. Thus, the coefficient of determination R2 and its analogs (R20, R2’0) were in the range of 0.4500–0.6882, the CCC criterion ranged from 0.6483 to 0.8086, which allowed us to characterize the prognostic ability of these models as low. At the same time, the most successful predictions, if we focus on these criteria, were observed for the structures of test set TS2. Meanwhile, a more reliable estimate of the predictive power of the M1–M6 models, taking into account 100% of the data in the test sets, can be obtained by analyzing the criteria based on the logk7 prediction errors for the same antioxidant structures. Specifically, the MAE and MAE+3SD criteria ranged from 0.3472 (M6, TS2) to 0.4696 (M5, TS1) and from 1.4586 (M4, TS2) to 2.1112 (M5, TS1). According to these criteria, the models with moderate predictive powers are M1 (TS1), M3 (TS1), and M4–M6 (TS2). Thus, the analysis of prediction errors for antioxidant structures contained in test sets TS1 (100% data) and TS2 (100% data) did not remove the uncertainty factor in assessing the predictive power of the M1–M6 models.
The removal of 5% of the structures from both test sets led to a significant increase in the numerical values of the various types of determination coefficients and a decrease in the logk7 prediction errors for the structures contained in TS1 and TS2.
The numerical value of the R
2 criterion increased approximately by 30% and ranged from 0.7289 to 0.8204. The coefficient of determination R
20 increased in parallel and was almost in the same range: 0.7263–0.8115. The maximum values of these criteria were found in both cases when the M1 model was used for the prediction tasks in the series of antioxidants contained in test set TS1 (95% of the data). According to the criteria mentioned in Part 2.3, the M5 and M6 models were insignificantly inferior in their predictive power. This fact was established in the prediction of logk
7 for the antioxidants included in test set TS2 (95% data). From the analysis of the numerical values of all of the other types of determination coefficients, which are presented in
Table 3, we can conclude that in some cases, the M5 model demonstrated the greatest prognostic ability. We reached this conclusion by analyzing CCC, R
2’0, and average R
2m values for the compounds of test set TS2 (95% data). The highest values of the criteria Q
2F1, Q
2F2 differed in the results of the prediction of logk
7 for the structures of the same test set performed using the M6 model (
Table 3). When evaluating the prognostic ability of the M1-M6 models, taking into account the prediction errors of the logk
7 values for 95% of the data in test sets TS1, TS2, the most successful predictions were also observed for the test set TS2 structures. The M6 model showed the lowest values of the RMSEP error, the SD standard deviation, and the MAE+3SD criterion. On the same dataset, the M5 model showed the minimum MAE error.
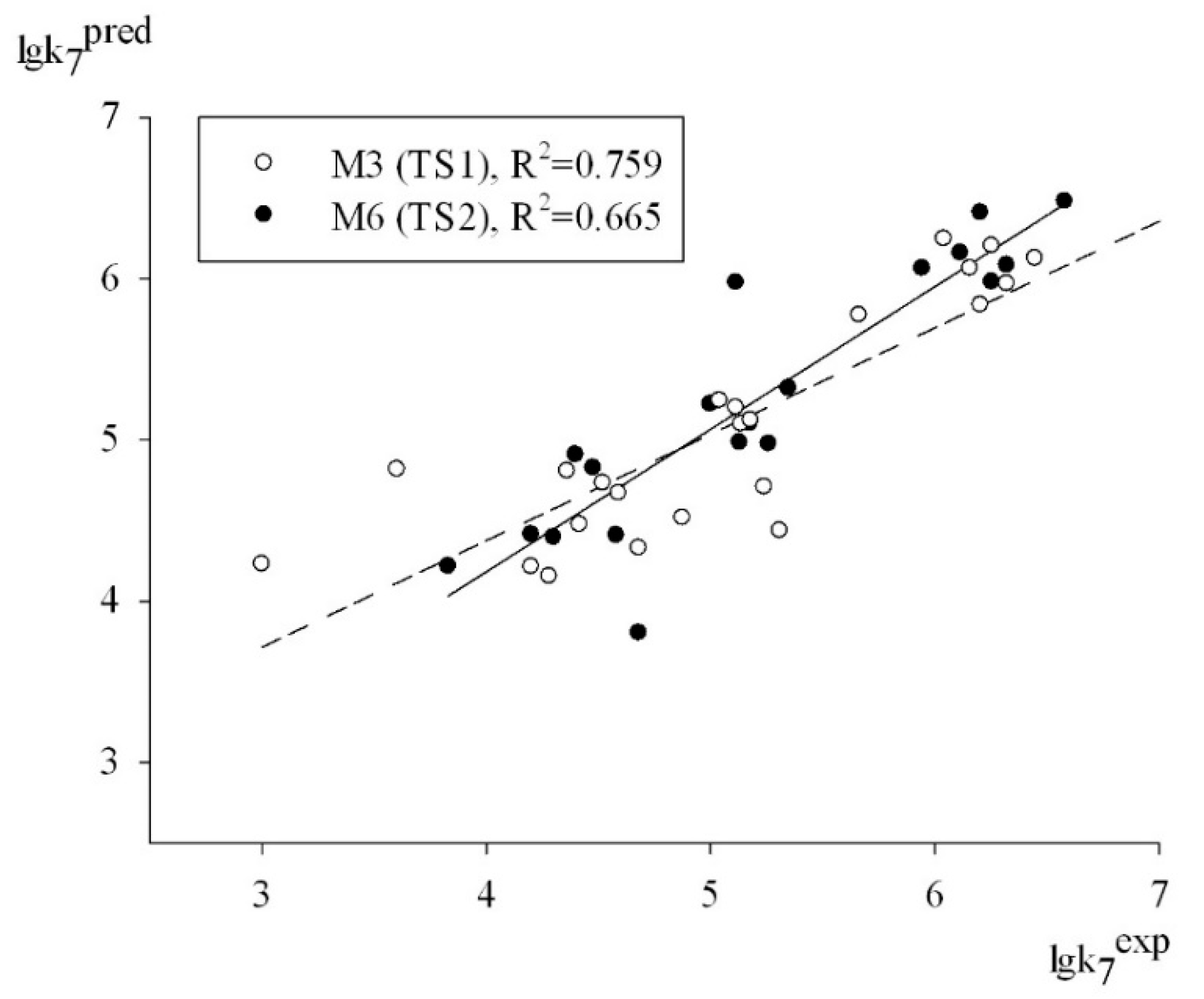
Thus, relying on the set of criteria summarized in
Table 3, we can conclude that all of the models had a moderate predictive power in predicting the logk
7 values for the antioxidant structures contained in test sets TS1 and TS2. An obvious proof of this fact is the plot depicted in
Figure 2, which shows a satisfactory correlation between the experimental and predicted values of logk
7 for the structures of test sets TS1 and TS2 (95% data).
The insignificant difference between the numerical values of the various types of determination coefficients, in combination with the acceptable values of the MAE and MAE+3SD parameters, summarized in
Table 2,
Table 3, indicates that the valid QSPR models focused on predicting logk
7 values for the antioxidants can be constructed using either one particular type of descriptor (QNA or MNA descriptors) or a combination of the descriptors in a consensus approach.
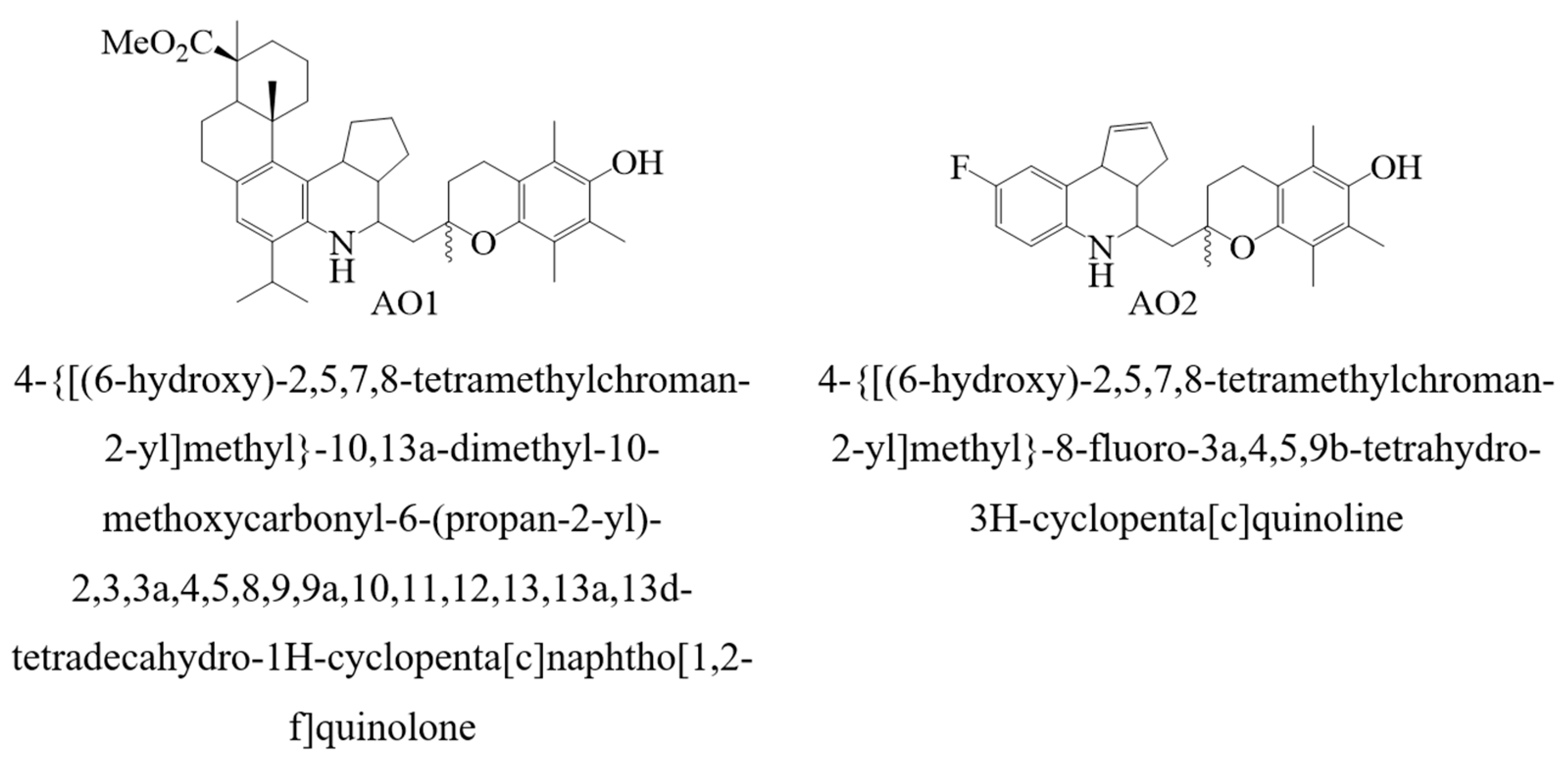
Subsequently, the M1–M6 consensus model was used to predict the numerical values of logk
7 for the antioxidants AO1 and AO2. The results of these calculations are summarized in
Table 4.
The approximate 95% confidence interval for predicting future data is ±2RMSE if the model is correct and the errors are normally distributed.
2.2. Experimental Determination of the Inhibition Rate Constants k7 for Compounds AO1 and AO2. Methods of the Kinetic Experiment to Determine the Antioxidant Activity of Compounds AO1 and AO2
The synthesis, the physico-chemical properties, and the antioxidant assays of compounds AO1 and AO2 (
Figure 3) were reported previously [
72]. In the present study, we describe the kinetics of the radical chain oxidation of an organic compound in the presence of additives AO1 and AO2.
The experimental logk
7 values for compounds AO1 and AO2 were determined by the manometric method using air oxygen absorption as a model liquid-phase oxidation of 1,4-dioxane, initiated by azobis(isobutyronitrile) (AIBN). The experiments were performed according to the standard technique described earlier [
72,
73,
74,
75,
76,
77,
78]. The model reaction was carried out in a thermostatically controlled glass reactor where the solutions of the initiator (AIBN) and the studied substances in 1,4-dioxane were loaded. The temperature of the reaction mixture was 348 K. The reaction mixture was maintained in the thermostat for 5 min. The kinetic curves was measured using a universal manometric differential unit, the design of which was reported earlier [
75,
76,
77,
78]. Subsequently, the initial rates of the oxidation of 1,4-dioxane were calculated from the initial sections of the kinetic curves recorded in the absence and in the presence of compounds AO1 and AO2 using the least-squares method. The numerical values of the effective inhibition rate constants for compounds AO1 and AO2 were calculated from the degree of the decrease in the initial oxygen uptake rate during the oxidation of 1,4-dioxane. The initiation rate of the oxidative process was constant and was V
i = 1 × 10
−7 mol·l
−1·s
−1. It was determined using the equation V
i = 2ek
p[AIBN], where k
p is the rate constant of the AIBN decay, e is the probability of the radical escape into the bulk). For k
p, the value measured in cyclohexanol was taken [
79]:
Since the reaction was performed according to the standard technique [
73,
74,
75,
76,
77,
78], we assumed that the initiated oxidation of 1,4-dioxane proceeded by the radical chain mechanism, which we schematically show in
Figure 3 [
1,
2,
3,
4,
5,
6,
7].
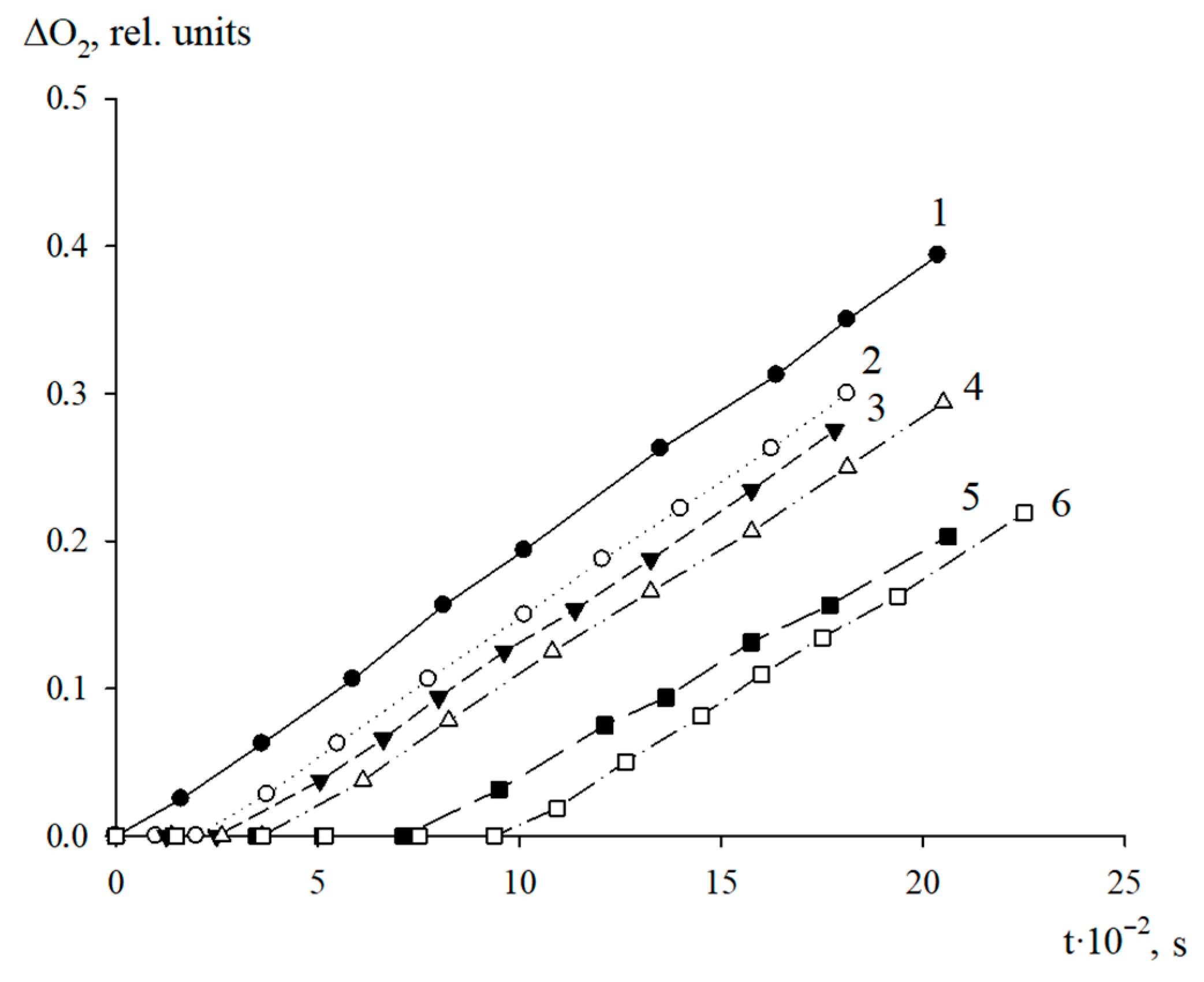
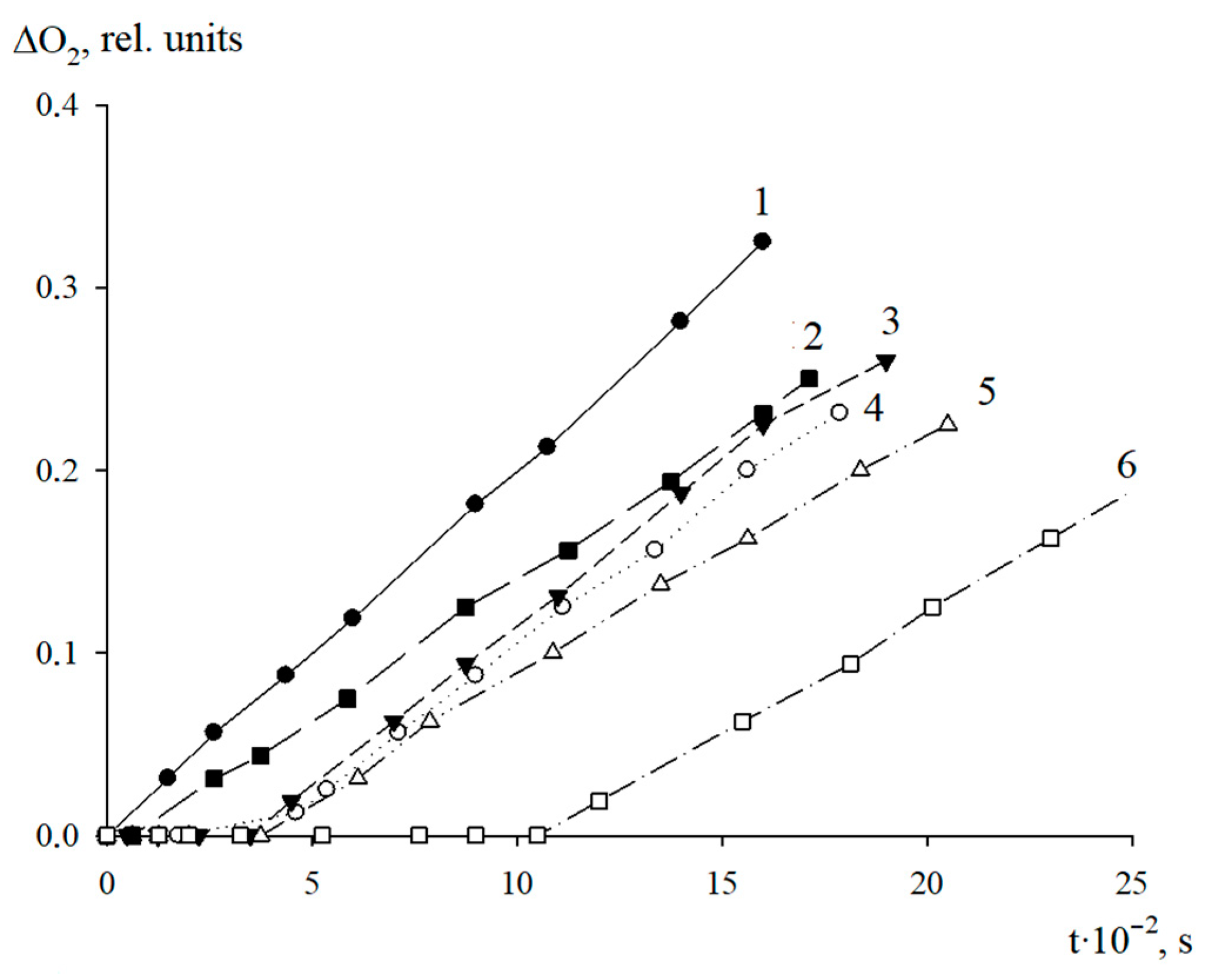
The antioxidant properties of AO1 and AO2 were studied in the AIBN-initiated radical chain oxidation of 1,4-dioxane in the kinetic regime at 348 K. The typical kinetic curves of the oxygen uptake in the presence of additives of AO1 and AO2 at different concentrations are shown in
Figure 4 and
Figure 5. In the absence of compounds AO1 and AO2, the kinetic curves of the oxygen uptake in the oxidation of 1,4-dioxane were straight lines, i.e., the reaction order with respect to oxygen was zero. Consequently, the oxidation of 1,4-dioxane proceeded in the kinetic regime. In this case, the chain propagation and termination reactions were run by peroxyl radicals.
As can be seen in
Figure 4 and
Figure 5, the introduction of the additives of AO1 and AO2 brings about a clear induction period in the kinetic curves of the oxygen uptake, indicating a pronounced antioxidant effect of the studied substances.
Using the Excel 2016 word-processor, we calculated the initial oxidation rates of the model substrate at different concentrations of the added substances. The resulting numerical values are presented in
Table 5. As can be seen, the introduction of compounds AO1 and AO2 separately in the concentration range of (0.44–3.13) × 10
−6 mol/L for AO1 or AO2, respectively, into 1,4-dioxane being oxidized, led to a decrease in the initial oxidation rate. Thus, the qualitative analysis allows us to conclude that we consider that both compounds effectively inhibit the oxidation process of the model substrate (
Figure 4 and
Figure 5,
Table 5).
The numerical values of the effective rate constants of inhibition fk
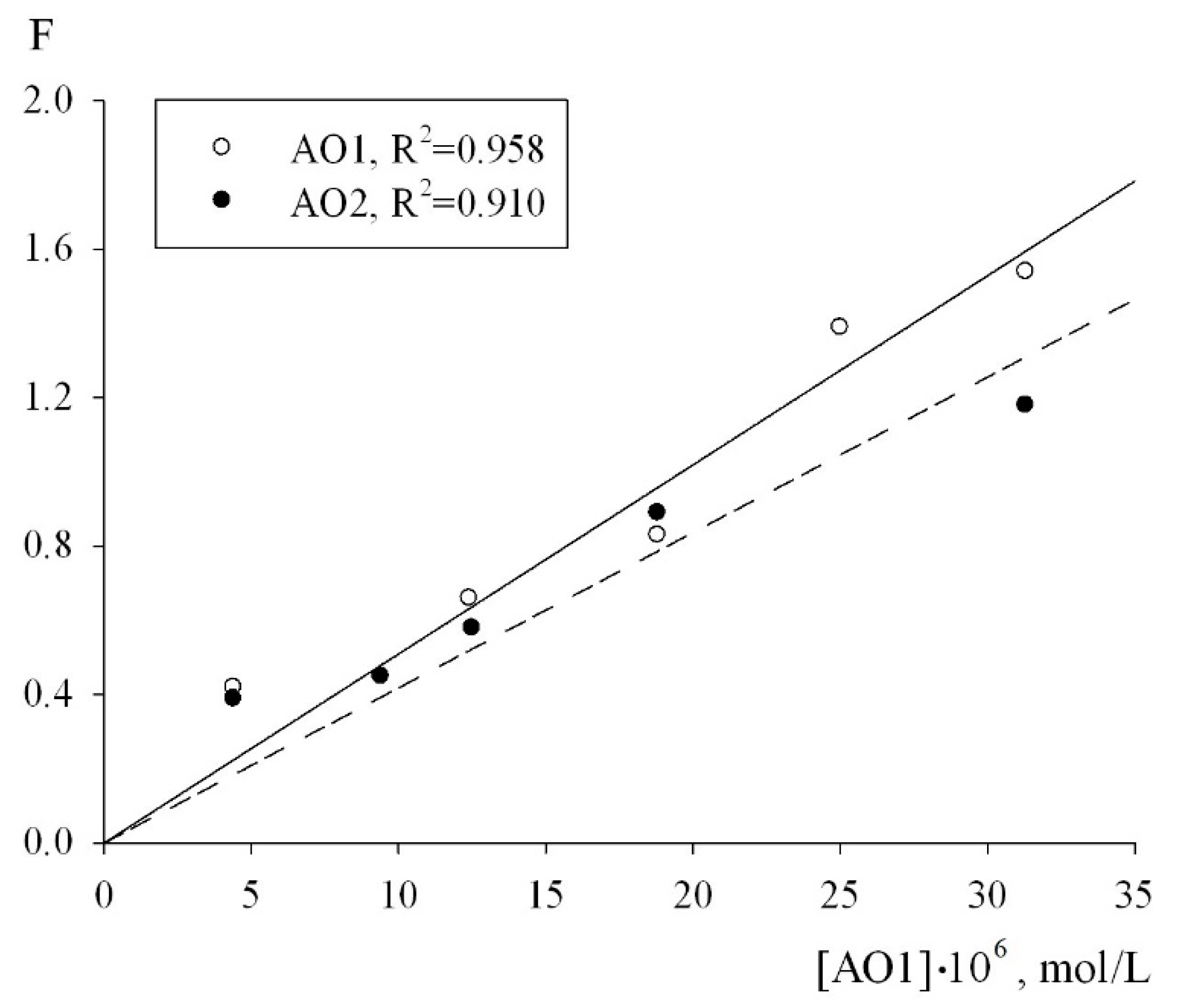
7 for each of the antioxidants were calculated using Equation (2). The condition for the applicability of this equation is a linear dependence of the inhibition parameter F on the concentration of the antioxidants. As can be seen from
Figure 6, in the oxidation chain regime in the (0.44–3.13) × 10
−6 mol/L concentration range of the AO1 and AO2 compounds, the inhibition parameter F, calculated from the initial rates of the inhibited oxidation of 1,4-dioxane by formula (2) actually followed a linear dependence on the AO1 and AO2 concentrations (
Figure 6):
where V
0 and V are the initial rates of the oxygen uptake during the oxidation of 1,4-dioxane in the absence and in the presence of each of the antioxidants taken separately, respectively, [AO] is the concentration of the added AO, k
7 and 2k
6 are the rate constants of the oxidation chain termination by the antioxidant and the quadratic chain termination via peroxyl radicals of the substrate, respectively [
1,
2,
3,
4,
5,
6,
7], [RH] is the concentration of 1,4-dioxane ([RH] = 11.75 mol/L), k
2 is the rate constant of the chain propagation for the oxidation of the model substrate (k
2 = 7.9 l·mol
−1·s
−1 [
2]). When calculating these values, we used the quadratic chain termination rate constant 2k
6 = 6.67 × 10
7 l·mol
−1·s
−1 known from the literature [
2]. The errors in determining the fk
7 and f values were calculated using the Excel 2016 word processor (Regression tab).
The adjustment of the experimental data in the coordinates of Equation (2), the effective inhibition constants for compounds AO1 and AO2 were determined to be f = (1.32 ± 0.3) × 10
6 M
−1s
−1 and f = (1.08 ± 0.2) × 10
6 M
−1s
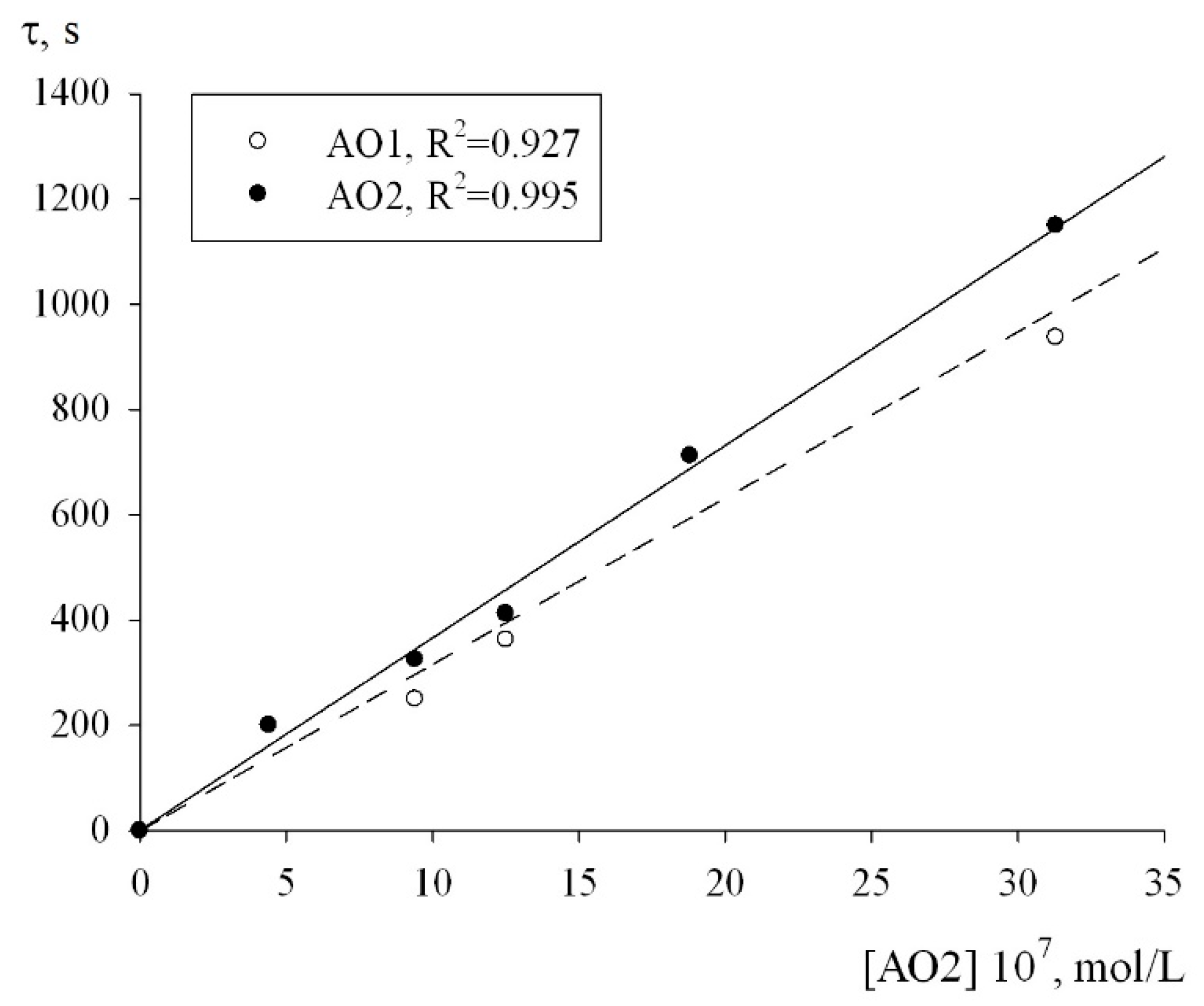
−1, respectively. In addition, to determine the numerical value of the stoichiometric inhibition coefficient, we studied the dependence of the induction period, which appeared on the kinetic curves of the oxygen uptake, on the concentrations of AO1 and AO2. As can be seen from
Figure 7, the dependence of the induction period τ on the concentrations of AO1 and AO2 is linear. In this case, it is correct to use Equation (3) to determine the stoichiometric inhibition coefficient f:
where τ is the induction period on the kinetic curves of the oxygen uptake during the oxidation of 1,4-dioxane inhibited by AO1 and AO2; V
i is the initiation rate of the oxidation. Conversion of the experimental data in the coordinates of Equation (3) gave the stoichiometric inhibition factors f for the antioxidants AO1 and AO2 to be 30 ± 4 and 40 ± 2, respectively.
The inhibition rate constant k
7exp for AO1 and AO2 was calculated by formula (4):
The numerical values of k7exp for the antioxidants AO1 and AO2 were k7exp = (4.3 ± 1.0) × 104 M−1s−1 and k7exp = (2.7 ± 0.5) × 104 M−1s−1, respectively.
The comparative analysis of the calculated logk
7pred and the experimental logk
7exp values for compounds AO1 and AO2 (
Table 4) suggests that the M1–M6 QSPR consensus model has a moderate predictive ability and can be applied to the search and development of new antioxidants. The difference between the predicted and experimentally determined logk
7 values for these antioxidants does not exceed the 2RMSEP range.
Thus, all M1–M6 QSPR consensus models are characterized by a high descriptive and moderate predictive power for comparing the experimental and predicted logk7 values for training set structures TR1 and TR2, the external and internal test set structures TS1 and TS2, and compounds AO1 and AO2. These models can be used for the screening of virtual libraries and databases in order to search for new antioxidants in the series of some sulfur-containing alkylphenols, natural phenols, chromane and lupanoic acids, betulonic and betulinic acids, and 20-hydroxyecdysone.
In general, the approach implemented in the GUSAR2019 program, which was previously used only for modeling the biological activity of low-molecular-weight compounds, allows a high degree of reliability in modeling the kinetic characteristics of antioxidants expressed as the k7 parameter. Thus, this program can be recommended as an additional tool in the search for new antioxidants.

 ,
, 










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
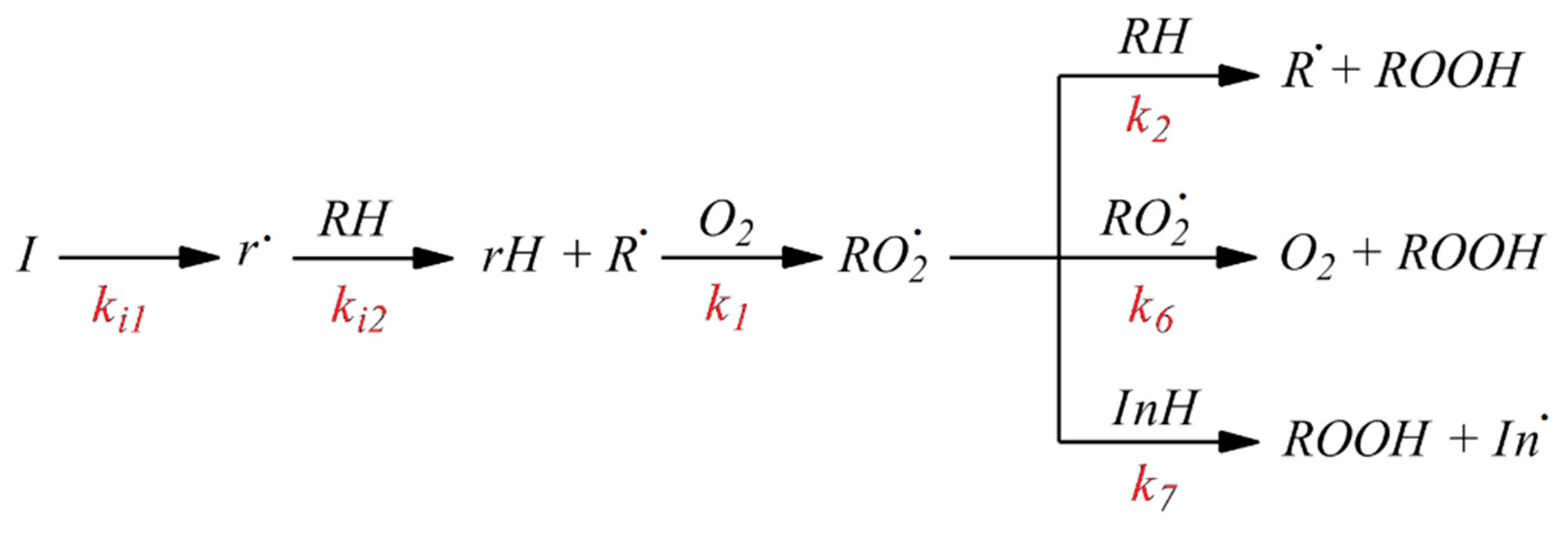{kind=link}