
Synthesis of Novel Benzimidazole-Based Thiazole Derivatives as Multipotent Inhibitors of α-Amylase and α-Glucosidase: In Vitro Evaluation along with Molecular Docking Study

 ,
,  ,
,  , ,
, ,  , ,
, ,  , and
, and 
Abstract
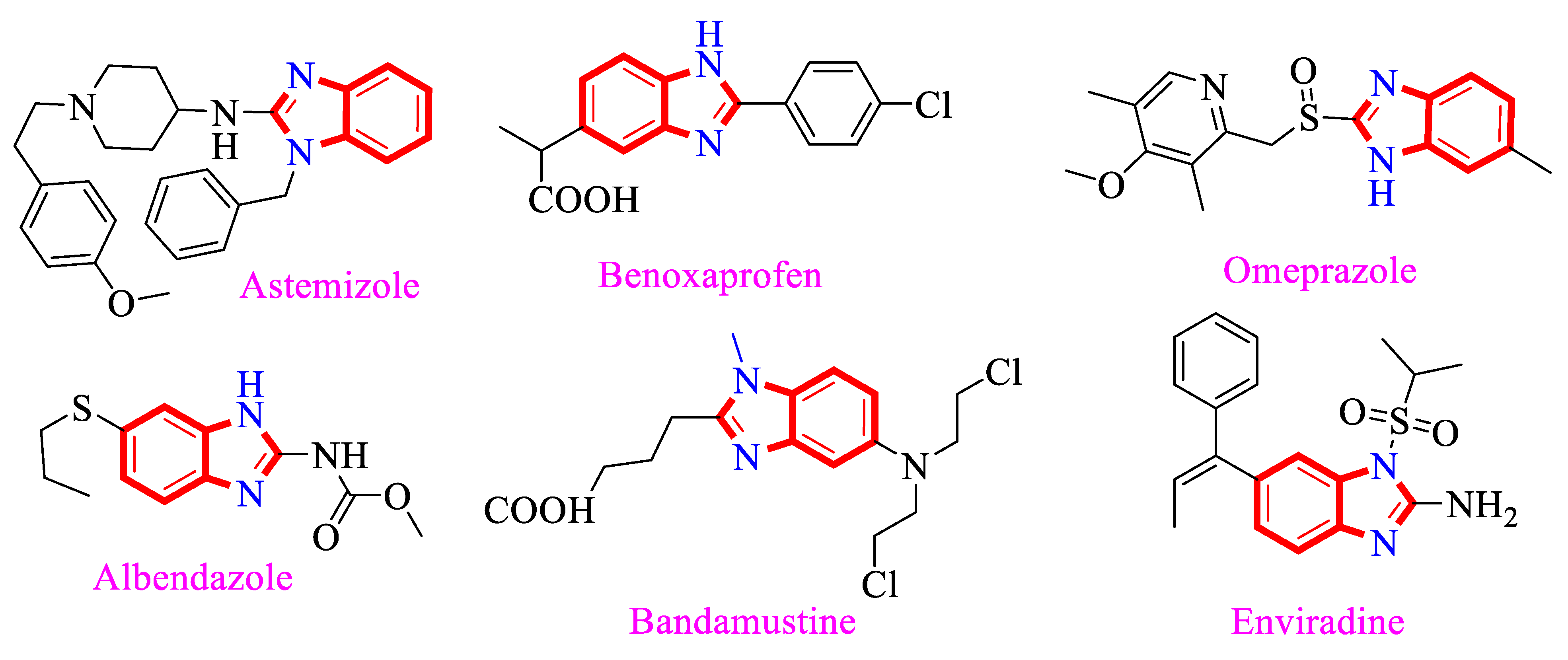
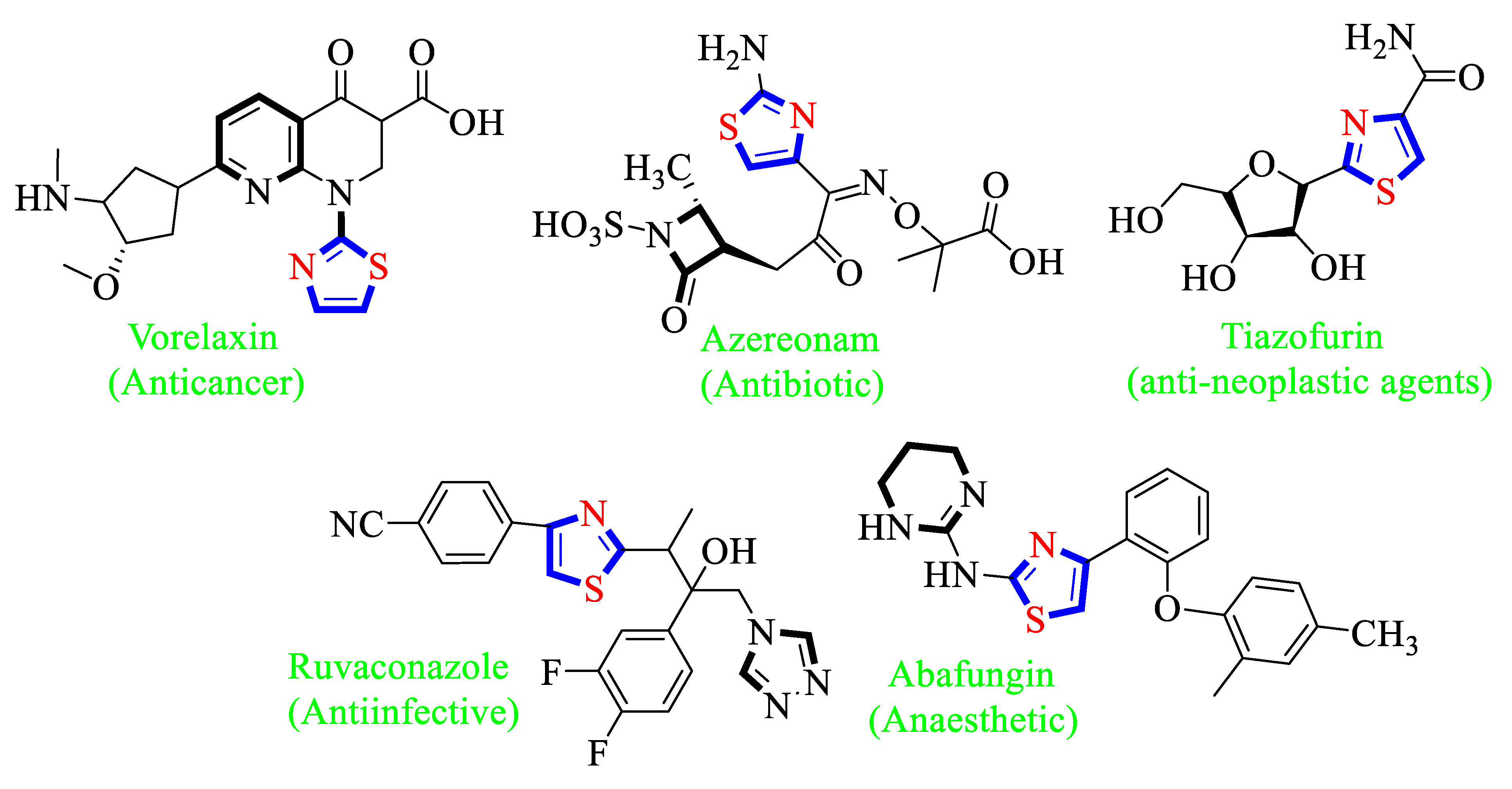
1. Introduction
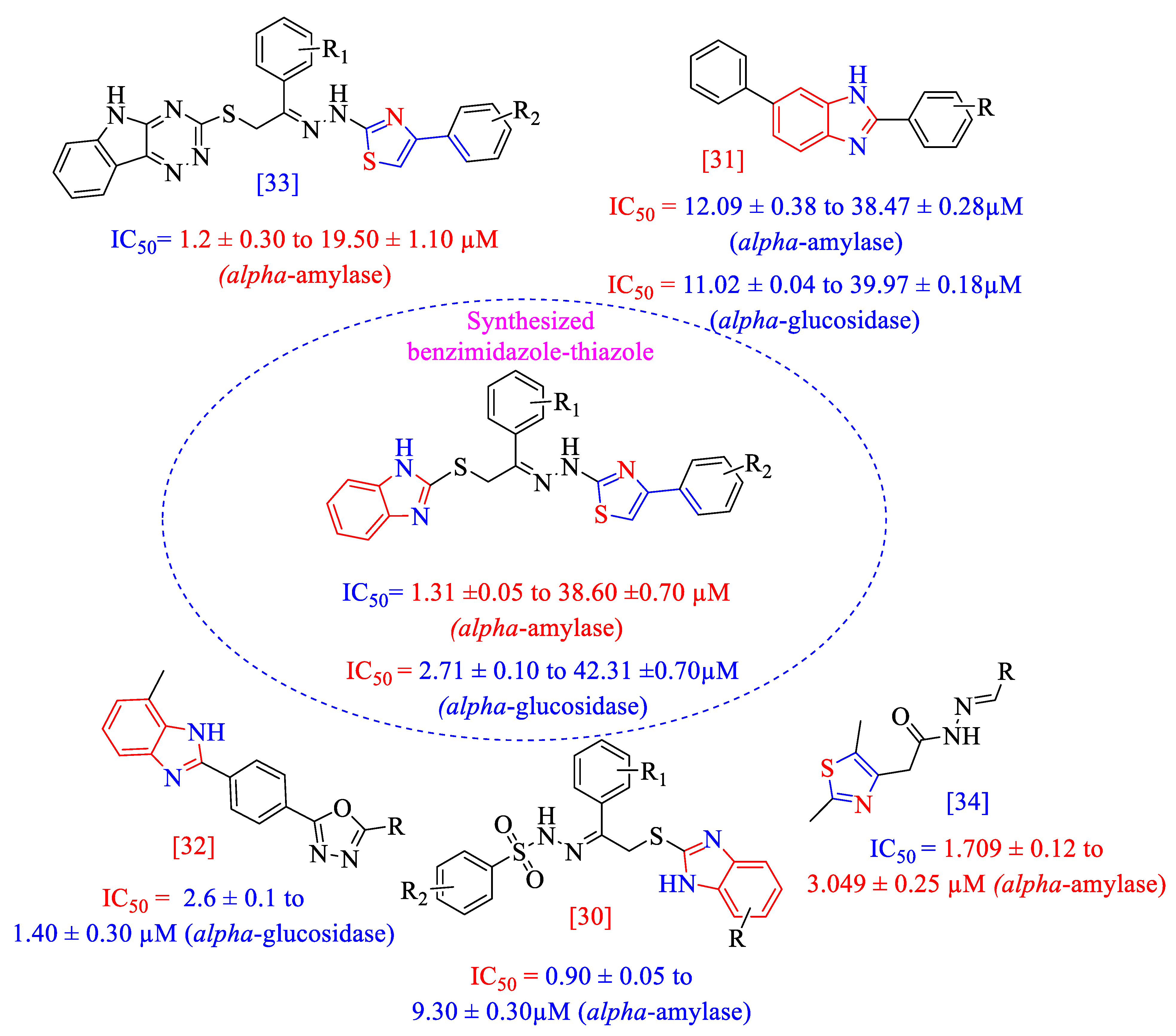
2. Results and Discussion
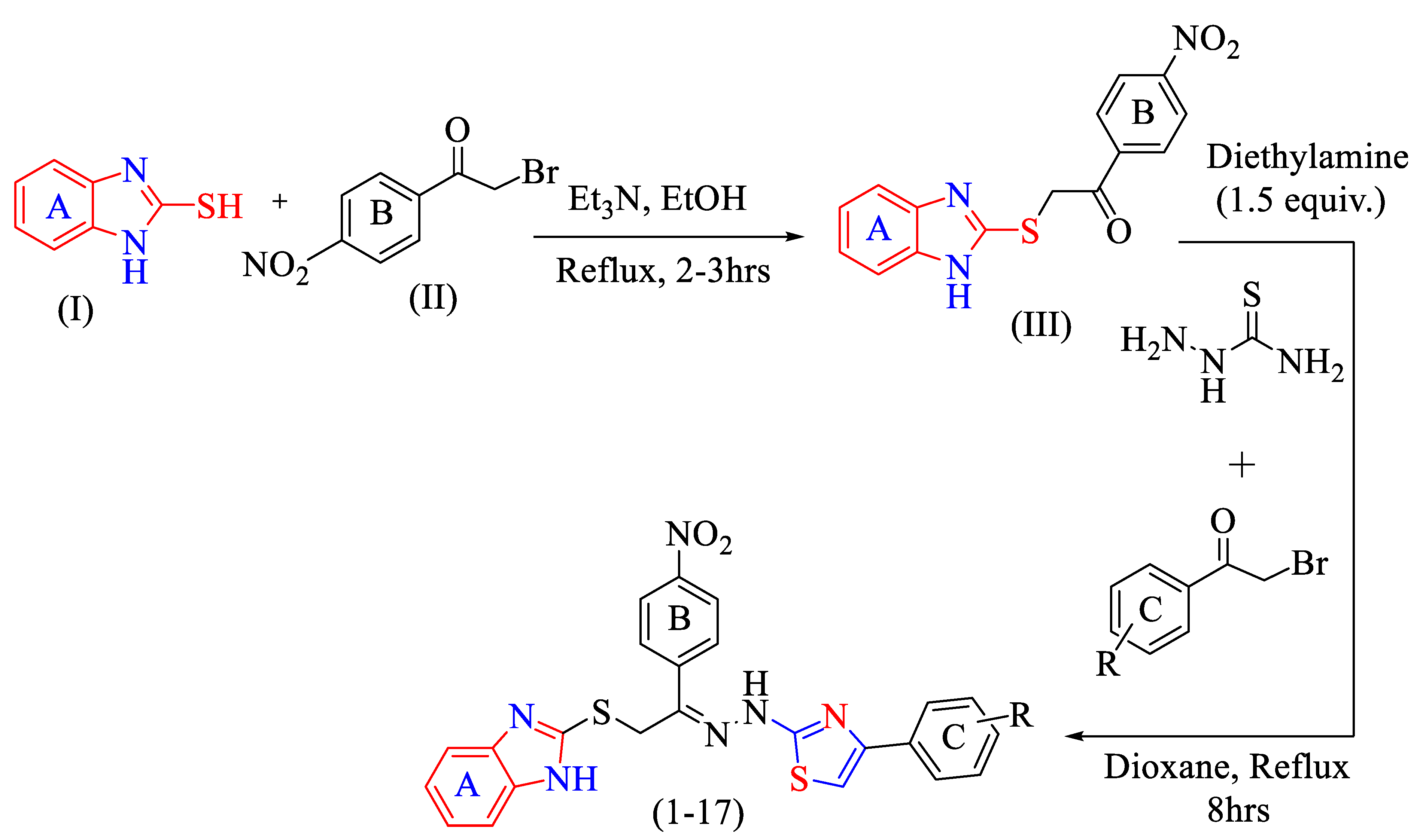
2.1. Chemistry
2.2. In Vitroα-Amylase and α-Glucosidase Inhibitory Activities (1–17)
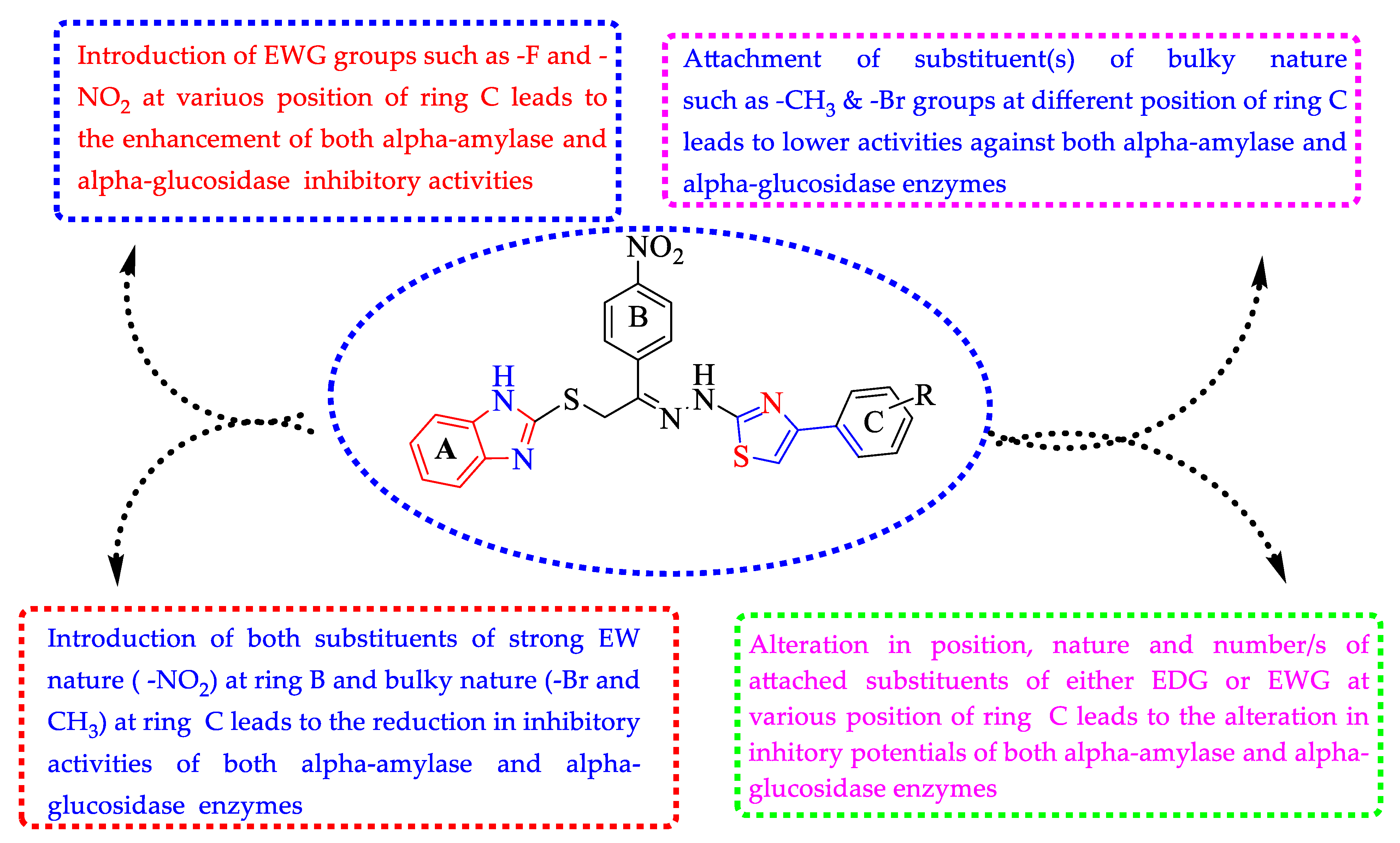
Structure–Activity Relationship (SAR) for Inhibitory Actions of α-Amylase and α-Glucosidase (1–17)
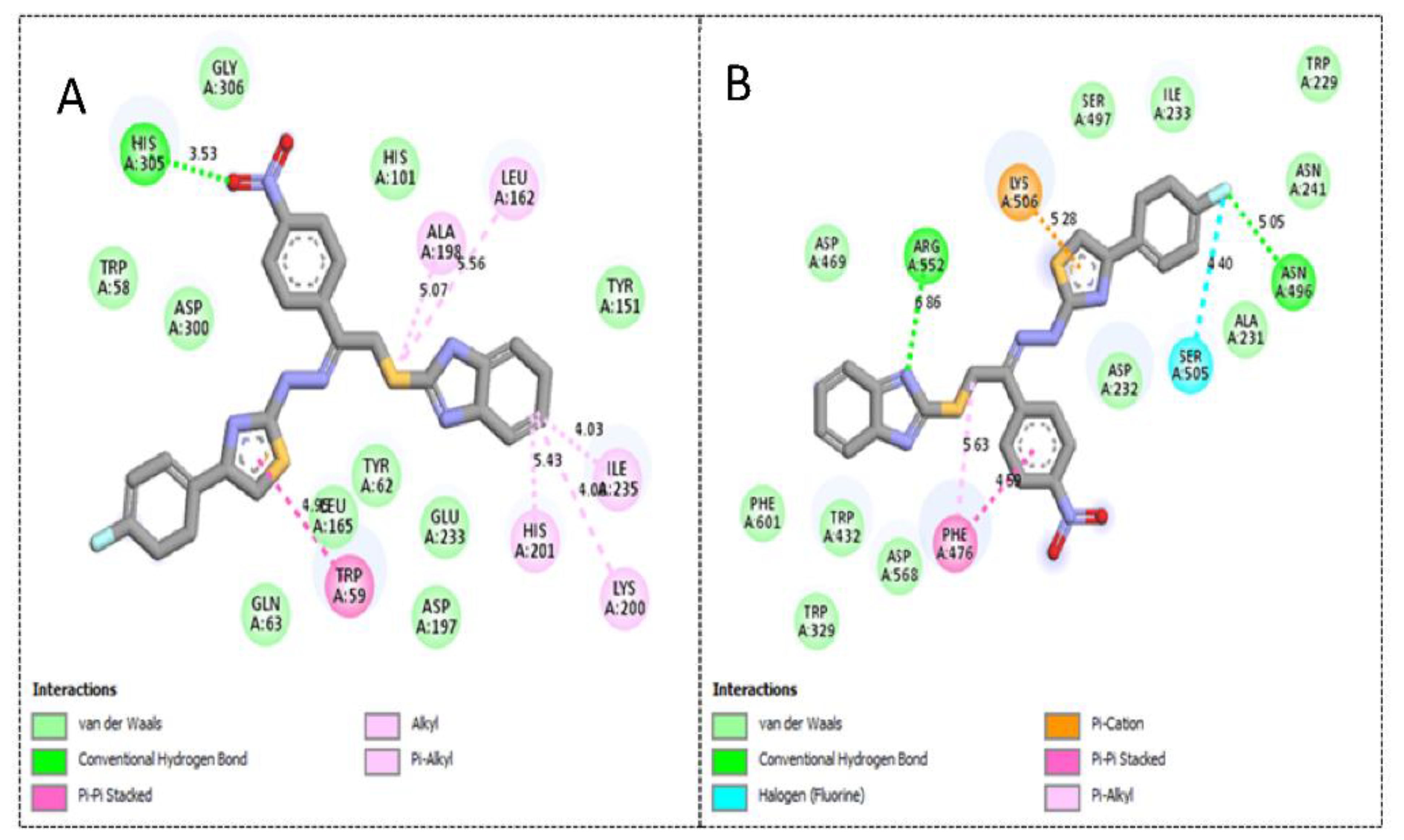
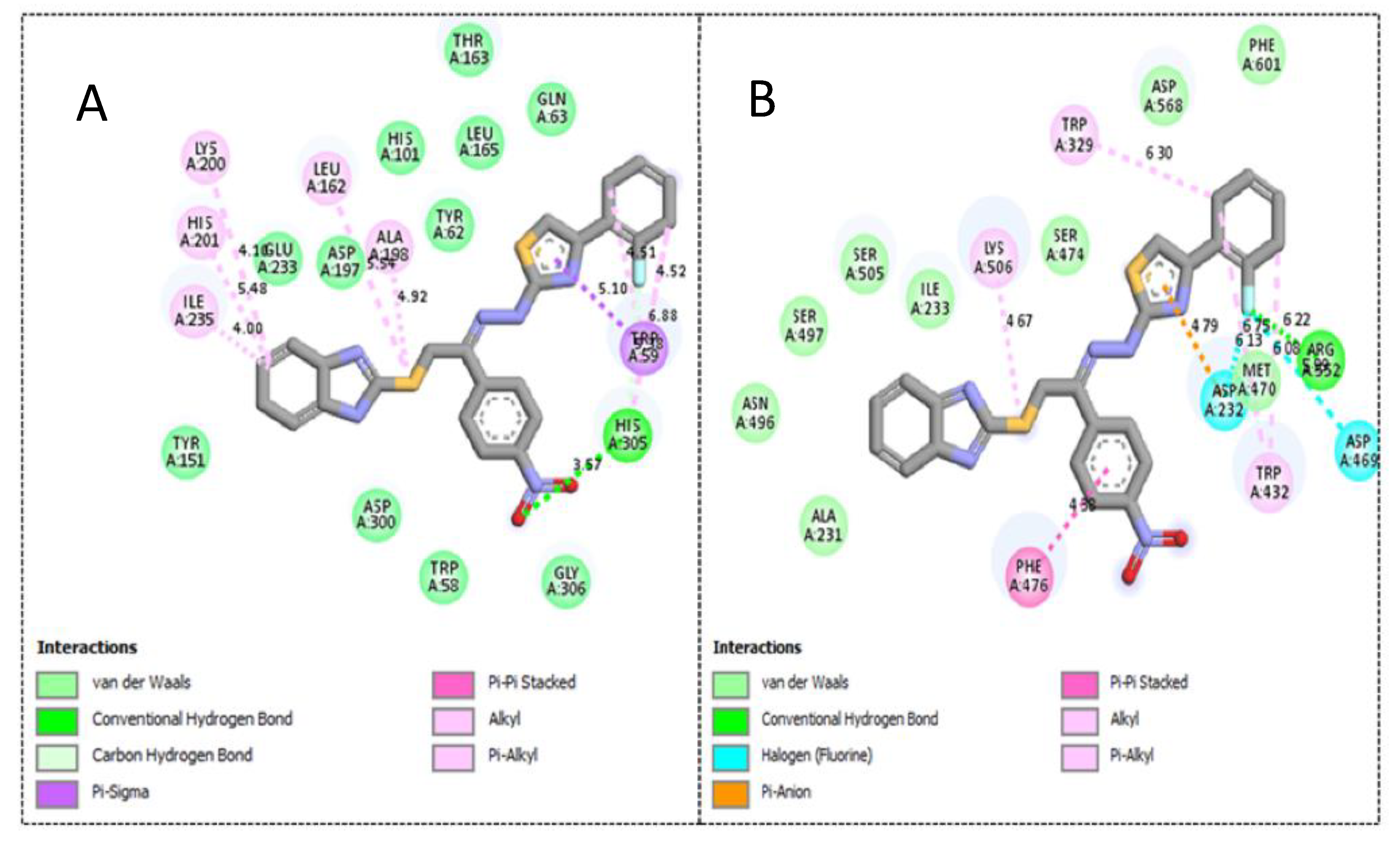
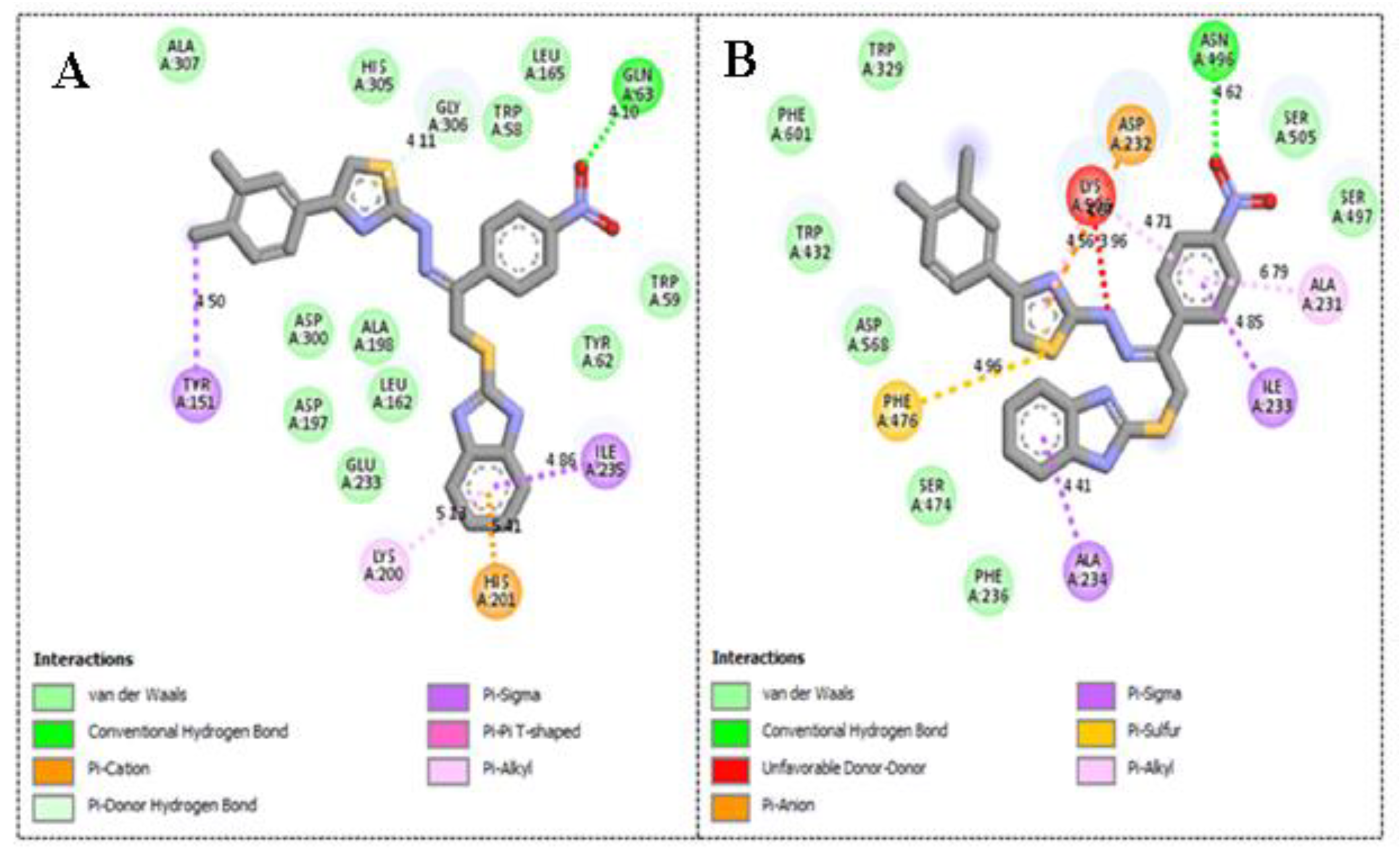
2.3. Molecular Docking Study
3. Experimental
3.1. General Information
3.2. General Method for the Production of Thiazole Scaffolds Based on Benzimidazole (1–17)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Chen, L.; Magliano, D.J.; Zimmet, P.Z. The worldwide epidemiology of type 2 diabetes mellitus—Present and future perspectives. Nat. Rev. Endocrinol. 2012, 8, 228–236. [Google Scholar] [CrossRef]
- Munhoz, A.; Frode, T.S. Isolated compounds from natural products with potential antidiabetic activity—A systematic review. Curr. Diabetes Rev. 2018, 14, 36–106. [Google Scholar] [CrossRef] [PubMed]
- Kahn, S.E.; Cooper, M.E.; Del Prato, S. Pathophysiology and treatment of type 2 diabetes: Perspectives on the past, present, and future. Lancet 2014, 383, 1068–1083. [Google Scholar] [CrossRef]
- Taha, M.; Baharudin, M.S.; Ismail, N.H.; Selvaraj, M.; Salar, U.; Alkadi, K.A.; Khan, K.M. Synthesis and in silico studies of novel sulfonamides having oxadiazole ring: As ß-glucuronidase inhibitors. Bioorganic Chem. 2017, 71, 86–96. [Google Scholar] [CrossRef] [PubMed]
- Qaisar, M.N.; Chaudhary, B.A.; Sajid, M.U.; Hussain, N. Evaluation of a-glucosidase inhibitory activity of dichloromethane and methanol extracts of Croton bonplandianumbaill. Trop. J. Pharm. Res. 2014, 13, 1833–1836. [Google Scholar] [CrossRef]
- Tundis, R.; Loizzo, M.; Menichini, F. Natural products as a-amylase and aglucosidase inhibitors and their hypoglycaemic potential in the treatment of diabetes: An update. Mini Rev. Med. Chem. 2010, 10, 315–331. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.; Alam, A.; Khan, K.M.; Salar, U.; Chigurupati, S.; Wadood, A.; Ali, F.; Mohammad, J.I.; Riaz, M.; Perveen, S. Flurbiprofen derivatives as novel a-amylase inhibitors: Biology-oriented drug synthesis (BIODS), in vitro, and in silico evaluation. Bioorganic Chem. 2018, 81, 157–167. [Google Scholar] [CrossRef]
- Okuyama, M.; Saburi, W.; Mori, H.; Kimura, A. a-glucosidases and a-1,4-glucan lyases: Structures, functions, and physiological actions. CellMol. Life Sci. 2016, 73, 2727–2751. [Google Scholar] [CrossRef]
- Taha, M.; Irshad, M.; Imran, S.; Chigurupati, S.; Selvaraj, M.; Rahim, F.; Ismail, N.H.; Nawaz, F.; Khan, K.M. Synthesis of piperazine sulfonamide analogs as diabetic-II inhibitors and their molecular docking study. Eur. J. Med. Chem. 2017, 141, 530–537. [Google Scholar] [CrossRef]
- Group, S.-N.T.R.; Chiasson, J.-L.; Josse, R.G.; Gomis, R.; Hanefeld, M.; Karasik, A.; Laakso, M. Acarbose for prevention of type 2 diabetes mellitus: The STOP-NIDDM randomised trial. Lancet 2002, 359, 2072–2077. [Google Scholar]
- Asgari, M.S.; Mohammadi-Khanaposhtani, M.; Kiani, M.; Ranjbar, P.R.; Zabihi, E.; Pourbagher, R.; Rahimi, R.; Faramarzi, M.A.; Biglar, M.; Larijani, B. Biscoumarin-1,2,3-triazole hybrids as novel anti-diabetic agents: Design, synthesis, in vitro aglucosidase inhibition, kinetic, and docking studies. Bioorganic Chem. 2019, 92, 103206. [Google Scholar] [CrossRef] [PubMed]
- Nissen, S.E.; Wolski, K. Effect of rosiglitazone on the risk of myocardial infarction and death from cardiovascular causes. N. Engl. J. Med. 2007, 356, 2457–2471. [Google Scholar] [CrossRef] [PubMed]
- Small, G.W.; Rabins, P.V.; Barry, P.P.; Buckholts, N.S.; Dekosky, S.T.; Ferris, S.H. Diagnosis and treatment of Alzheimer disease and related disorders. J. Am. Med. Assoc. 1997, 278, 1363–1371. [Google Scholar] [CrossRef]
- Spasov, A.; Yozhitsa, I.N.; Bugaeva, L.I.; Anisimova, V.A. Benzimidazole derivatives: Spectrum of pharmacological activity and toxicological properties (a review). Pharm. Chem. J. 1999, 33, 232–243. [Google Scholar] [CrossRef]
- Patil Ganguly, S.; Surana, S. A systematic review of benzimidazole derivatives as an antiulcer agent. Rasayan J Chem. 2008, 1, 447–460. [Google Scholar]
- Dubey, K.; Sanyal, P.K. Benzimidazoles in a wormy world. Vet Scan Online Vet. Med. J. 2010, 5, 63. [Google Scholar]
- Boiani, M.; González, M. Imidazole and benzimidazole derivatives as chemotherapeutic agents. Mini Rev. Med. Chem. 2005, 5, 409–424. [Google Scholar] [CrossRef]
- Narasimhan Sharma, D.; Kumar, P. Benzimidazole: A medicinally important heterocyclic moiety. Med. Chem. Res. 2012, 21, 269–283. [Google Scholar] [CrossRef]
- VishnuJi, R.; Arun, S.; Mahendra, N.; Ramendra, P. The Chemistry of Heterocycles: Nomenclature and Chemistry of Three-to-Five Membered Heterocycles; Elsevier: Amsterdam, The Netherlands, 2019; pp. 149–478. [Google Scholar]
- Bansal, Y.; Silakari, O.; Lapinsky, D.J.; Kusayanagi, T.; Tsukuda, S.; Shimura, S.; Manita, D.; Iwakiri, K.; Kamisuki, S.; Takakusagi, Y.; et al. The therapeutic journey of benzimidazoles: A review. Bioorganic Med. Chem. 2012, 20, 6208–6236. [Google Scholar] [CrossRef]
- Parekh, N.M.; Juddhawala, K.V.; Rawal, B.M. Antimicrobial activity of thiazolyl 3-benzenesulfonamide-condensed 2,4-thiazolidinediones derivatives. Med. Chem. Res. 2013, 22, 2737–2745. [Google Scholar] [CrossRef]
- Haroun, M.; Tratrat, C.; Tsolaki, E.; Geronikaki, A. Thiazole-Based Thiazolidinones as Potent Antimicrobial Agents. Design, Synthesis and Biological Evaluation. Comb. Chem. High Throughput Screen 2016, 19, 51–57. [Google Scholar] [CrossRef] [PubMed]
- Rostom, S.A.; El-Ashmawy, I.M.; Abd El Razik, H.A.; Badr, M.H.; Ashour, H.M. Design and Synthesis of Some Thiazolyl and Thiadiazolyl Derivatives of Antipyrine as Potential Non-Acidic Anti-Inflammatory, Analgesic and Antimicrobial Agents. BioorganicMed. Chem. 2009, 17, 882–895. [Google Scholar] [CrossRef]
- Zablotskaya, A.; Segal, I.; Germane, S.; Shestakova, I.; Domracheva, I.; Nesterova, A.; Geronikaki, A.; Lukevics, E. Silyl Modification of Biologically Active Compounds. 8. Trimethylsilyl Ethers of Hydroxyl-Containing Thiazole Derivatives. Chem. Heterocycl. Compd. 2002, 38, 859–866. [Google Scholar] [CrossRef]
- Luzina, E.L.; Popov, A.V. Synthesis and Anticancer Activity of N-Bis(Trifluoromethyl) Alkyl-N’-Thiazolyl and N-Bis(Trifluoromethyl)Alkyl-N’-BenzothiazolylUreas. Eur. J. Med. Chem. 2009, 44, 4944–4953. [Google Scholar] [CrossRef]
- Turan-Zitouni, G.; Chevallet, P.; Kilic, F.S.; Erol, K. Synthesis of Some ThiazolylPyrazoline Derivatives and Preliminary Investigation of Their Hypotensive Activity. Eur. J. Med. Chem. 2000, 35, 635–641. [Google Scholar] [CrossRef]
- Britschgi, M.; Greyerz, S.; Burkhart, C.; Pichler, W.J. Molecular Aspects of Drug Recognition by Specific T Cells. Curr. Drug Targets 2003, 4, 1–11. [Google Scholar] [CrossRef]
- Haroon, M.; Khalid, M.; Shahzadi, K.; Akhtar, T.; Saba, S.; Rafique, J.; Ali, S.; Irfan, M.; Alam, M.M.; Imran, M. Alkyl 2-(2-(arylidene) alkylhydrazinyl) thiazole-4-carboxylates: Synthesis, acetyl cholinesterase inhibition and docking studies. J. Mole. Struct. 2021, 1245, 131063. [Google Scholar] [CrossRef]
- Chhabria, M.; Patel, S.; Modi, P.; Brahmkshatriya, P. Thiazole: A review on chemistry, synthesis and therapeutic importance of its derivatives. Curr. Top. Med. Chem. 2016, 16, 2841–2862. [Google Scholar] [CrossRef] [PubMed]
- Hussain, S.; Taha, M.; Rahim, F.; Hayat, S.; Zaman, K.; Iqbal, N.; Selvaraj, M.; Sajid, M.; Bangesh, M.A.; Khan, F.; et al. Synthesis of benzimidazole derivatives as potent inhibitors for α-amylase and their molecular docking study in management of type-II diabetes. J. Mol. Struct. 2021, 1232, 130029. [Google Scholar] [CrossRef]
- Aroua, L.M.; Almuhaylan, H.R.; Alminderej, F.M.; Messaoudi, S.; Chigurupati, S.; Al-Mahmoud, S.; Mohammed, H.A. A facile approach synthesis of benzoylaryl benzimidazole as potential α-amylase and α-glucosidase inhibitor with antioxidant activity. Bioorganic Chem. 2021, 114, 105073. [Google Scholar] [CrossRef]
- Taha, M.; Rahim, F.; Zaman, K.; Selvaraj, M.; Uddin, N.; Farooq, R.K.; Nawaz, M.; Sajid, M.; Nawaz, F.; Ibrahim, M.; et al. Synthesis, α-glycosidase inhibitory potential and molecular docking study of benzimidazole derivatives. Bioorganic Chem. 2020, 95, 103555. [Google Scholar] [CrossRef] [PubMed]
- Rahim, F.; Tariq, S.; Taha, M.; Ullah, H.; Zaman, K.; Uddin, I.; Wadood, A.; Khan, A.A.; Rehman, A.U.; Uddin, N.; et al. New triazinoindole bearing thiazole/oxazole analogues: Synthesis, α-amylase inhibitory potential and molecular docking study. Bioorganic Chem. 2019, 92, 103284. [Google Scholar] [CrossRef] [PubMed]
- Taha, M.; Irshad, M.; Imran, S.; Rahim, F.; Selvaraj, M.; Almandil, N.B.; Mosaddik, A.; Chigurupati, S.; Nawaz, F.; Ismail, N.H.; et al. Thiazole based carbohydrazide derivatives as α-amylase inhibitor and their molecular docking study. Heteroat. Chem. 2019, 2019, 7502347. [Google Scholar] [CrossRef]
- Rahim, F.; Zaman, K.; Taha, M.; Ullah, H.; Ghufran, M.; Wadood, A.; Rehman, W.; Uddin, N.; Shah, S.A.A.; Sajid, M.; et al. Synthesis, in vitro alpha-glucosidase inhibitory potential of benzimidazole bearing bis-Schiff bases and their molecular docking study. Bioorganic Chem. 2020, 94, 103394. [Google Scholar] [CrossRef] [PubMed]
- Kharb, M.; Jat, R.K.; Parjapati, G.; Gupta, A. Introduction to molecular docking software technique in medicinal chemistry. Int. J. Drug Res. Technol. 2012, 2, 89–197. [Google Scholar]
- Li, Z.; Gu, J.; Zhuang, H.; Kang, L.; Zhao, X.; Guo, Q. Adaptive molecular docking method based on information entropy genetic algorithm. Appl. Soft Comput. 2015, 26, 299–302. [Google Scholar] [CrossRef]
- Rao, C.M.M.P.; Naidu, N.; Priya, J.; Rao, K.P.C.; Ranjith, K.; Shobha, S.; Chowdary, B.S.; Siddiraju, S. Molecular docking and dynamic simulations of benzimidazoles with beta-tubulins. Bioinformation 2021, 17, 404. [Google Scholar]
- Khan, S.; Ullah, H.; Rahim, F.; Nawaz, M.; Hussain, R.; Rasheed, L. Synthesis, in vitro α-amylase, α-glucosidase activities and molecular docking study of new benzimidazole bearing thiazolidinone derivatives. J. Mol. Struct. 2022, 1269, 133812. [Google Scholar] [CrossRef]
- Blum, C.; Roli, A.; Sampels, M. (Eds.) Hybrid Metaheuristics: An Emerging Approach to Optimization; Springer: Berlin, Germany, 2008; Volume 114. [Google Scholar]
- Baxter, J. Local optima avoidance in depot location. J. Oper. Res. Soc. 1981, 32, 815–819. [Google Scholar] [CrossRef]
- Azam, S.S.; Abbasi, S.W. Molecular docking studies for the identification of novel melatoninergic inhibitors for acetylserotonin-O-methyltransferase using different docking routines. Theor. Biol. Med. Model. 2013, 10, 63. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Synthesized Compounds | R | α-Amylase IC50 [µM] | α-Glucosidase IC50 [µM] |
|---|---|---|---|
| 1 | 3,4-dichloro | 2.20 ± 0.10 | 3.90 ± 0.20 |
| 2 | 3-fluoro | 4.10 ± 0.10 | 5.60 ± 0.10 |
| 3 | 4-fluoro | 1.30 ± 0.05 | 2.70 ± 0.10 |
| 4 | 2-fluoro | 1.90 ± 0.10 | 2.90 ± 0.10 |
| 5 | 4-hydroxy | 3.60 ± 0.20 | 4.30 ± 0.30 |
| 6 | 4-bromo | 18.50 ± 0.30 | 25.80 ± 0.50 |
| 7 | 3-bromo | 34.70 ± 0.70 | 36.40 ± 0.70 |
| 8 | 2-bromo | 20.30 ± 0.50 | 29.30 ± 0.60 |
| 9 | 4-nitro | 16.40 ± 0.30 | 19.60 ± 0.40 |
| 10 | 2-nitro | 7.20 ± 0.20 | 9.60 ± 0.20 |
| 11 | 4-methyl | 24.40 ± 0.30 | 25.90 ± 0.40 |
| 12 | 3-methyl | 38.60 ± 0.70 | 34.40 ± 0.70 |
| 13 | 2-methyl | 34.20 ± 0.60 | 37.50 ± 0.70 |
| 14 | 4-chloro | 16.40 ± 0.30 | 17.30 ± 0.30 |
| 15 | 2-chloro | 5.20 ± 0.10 | 6.30 ± 0.10 |
| 16 | 3-chloro | 12.50 ± 0.20 | 13.60 ± 0.20 |
| 17 | 3-nitro | 28.40 ± 0.40 | 29.60 ± 0.40 |
| Standard acarbose drug | 10.30 ± 0.20 | 9.80 ± 0.20 | |
| Active Derivatives | Name of Enzyme | IC50 [µM] | Free Binding Energy (kcal/mol) | Number of HBs | Number of Closest Residues | Interacting Residues |
|---|---|---|---|---|---|---|
| 3 | α-amylase | 1.30 ± 0.05 | −12.13 | 1 | 17 | His305, Trp59, Ala198, Leu162, Lys200, His201 and Ile235 |
| α-glucosidase | 2.70 ± 0.10 | −11.48 | 2 | 16 | Phe476, Asp232, Trp432, Met470, Asp469, Arg552, Trp329 and Lys506 | |
| 4 | α-amylase | 1.90 ± 0.10 | −10.87 | 1 | 17 | Trp59, His201, Lys200, Ile235, Leu162, Ala198 and His305 |
| α-glucosidase | 2.90 ± 0.10 | −10.19 | 1 | 16 | Phe476, Ser505, Asn496, Lys506 and Arg552 | |
| 1 | α-amylase | 2.20 ± 0.10 | −9.23 | 1 | 17 | Tyr151, Lys200, His201, Ile235 and Gln63 |
| α-glucosidase | 3.90 ± 0.20 | −8.98 | 1 | 15 | Phe476, Ala 234, Ile233, Ala231, Asn496, Asp232 and Lys506 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hussain, R.; Iqbal, S.; Shah, M.; Rehman, W.; Khan, S.; Rasheed, L.; Rahim, F.; Dera, A.A.; Kehili, S.; Elkaeed, E.B.; et al. Synthesis of Novel Benzimidazole-Based Thiazole Derivatives as Multipotent Inhibitors of α-Amylase and α-Glucosidase: In Vitro Evaluation along with Molecular Docking Study. Molecules 2022, 27, 6457. https://doi.org/10.3390/molecules27196457
Hussain R, Iqbal S, Shah M, Rehman W, Khan S, Rasheed L, Rahim F, Dera AA, Kehili S, Elkaeed EB, et al. Synthesis of Novel Benzimidazole-Based Thiazole Derivatives as Multipotent Inhibitors of α-Amylase and α-Glucosidase: In Vitro Evaluation along with Molecular Docking Study. Molecules. 2022; 27(19):6457. https://doi.org/10.3390/molecules27196457
Chicago/Turabian StyleHussain, Rafaqat, Shahid Iqbal, Mazloom Shah, Wajid Rehman, Shoaib Khan, Liaqat Rasheed, Fazal Rahim, Ayed A. Dera, Sana Kehili, Eslam B. Elkaeed, and et al. 2022. "Synthesis of Novel Benzimidazole-Based Thiazole Derivatives as Multipotent Inhibitors of α-Amylase and α-Glucosidase: In Vitro Evaluation along with Molecular Docking Study" Molecules 27, no. 19: 6457. https://doi.org/10.3390/molecules27196457
APA StyleHussain, R., Iqbal, S., Shah, M., Rehman, W., Khan, S., Rasheed, L., Rahim, F., Dera, A. A., Kehili, S., Elkaeed, E. B., Awwad, N. S., Bajaber, M. A., Alahmdi, M. I., Alrbyawi, H., & Alsaab, H. O. (2022). Synthesis of Novel Benzimidazole-Based Thiazole Derivatives as Multipotent Inhibitors of α-Amylase and α-Glucosidase: In Vitro Evaluation along with Molecular Docking Study. Molecules, 27(19), 6457. https://doi.org/10.3390/molecules27196457