
Collision Cross Section Prediction with Molecular Fingerprint Using Machine Learning



Abstract
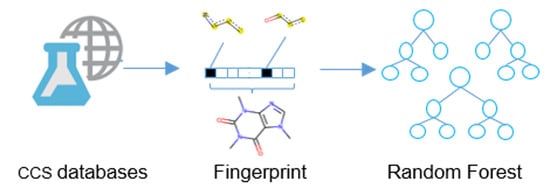
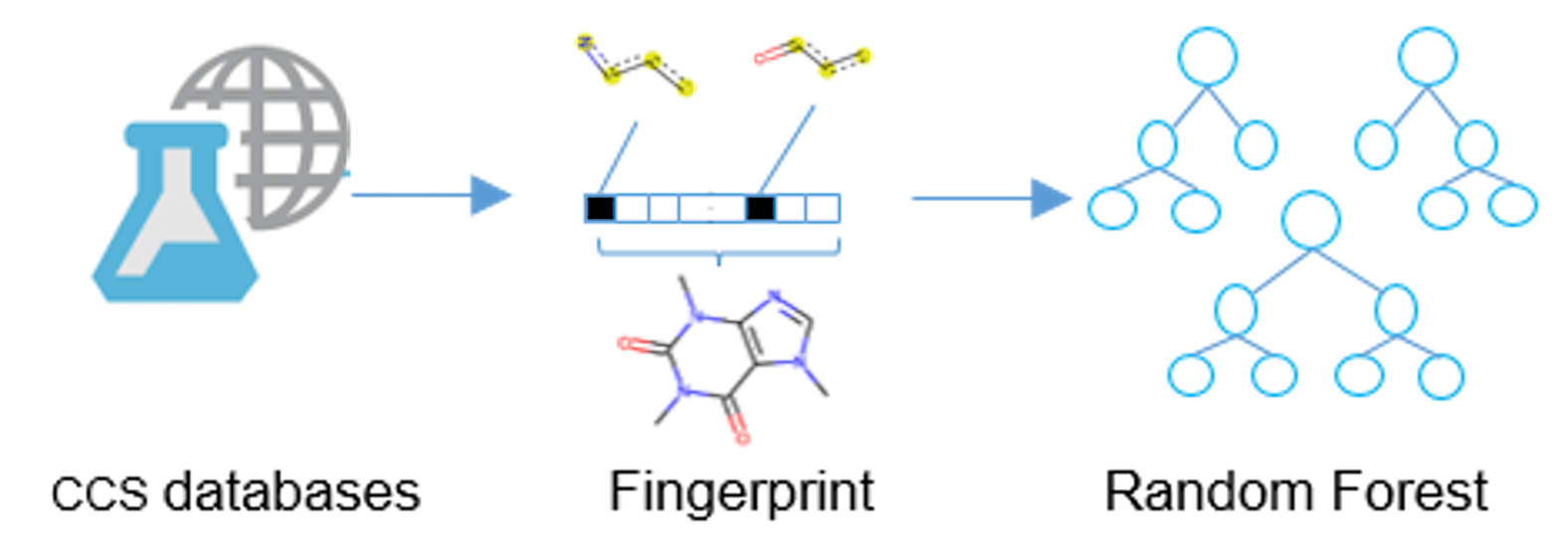
1. Introduction
2. Materials and Methods
2.1. Datasets
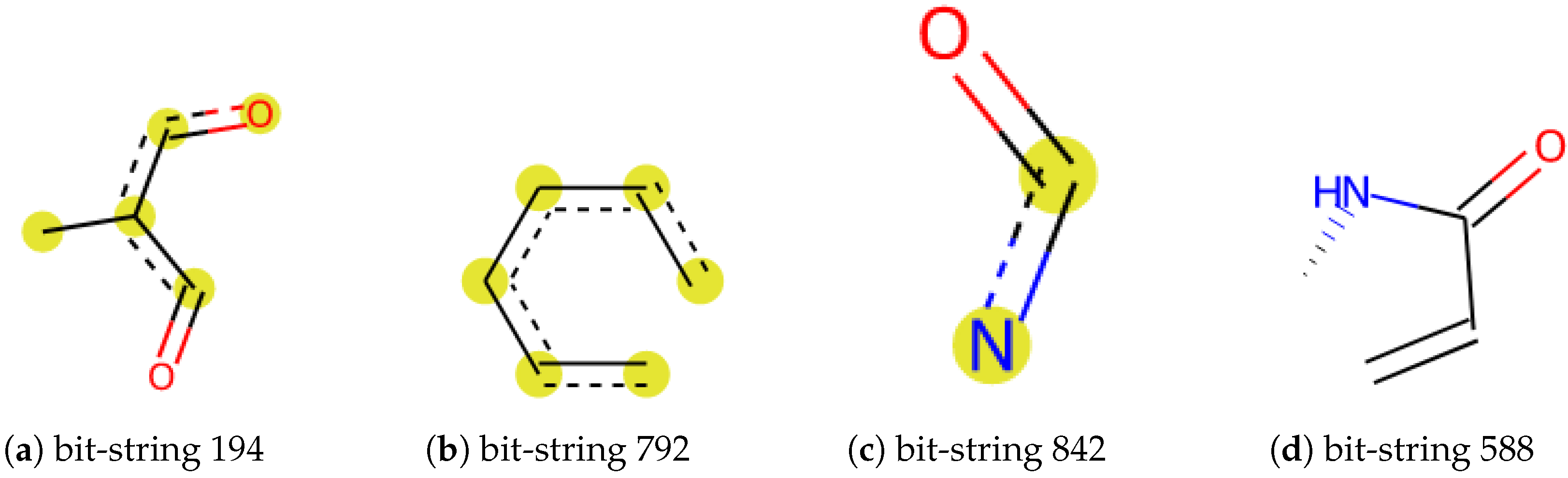
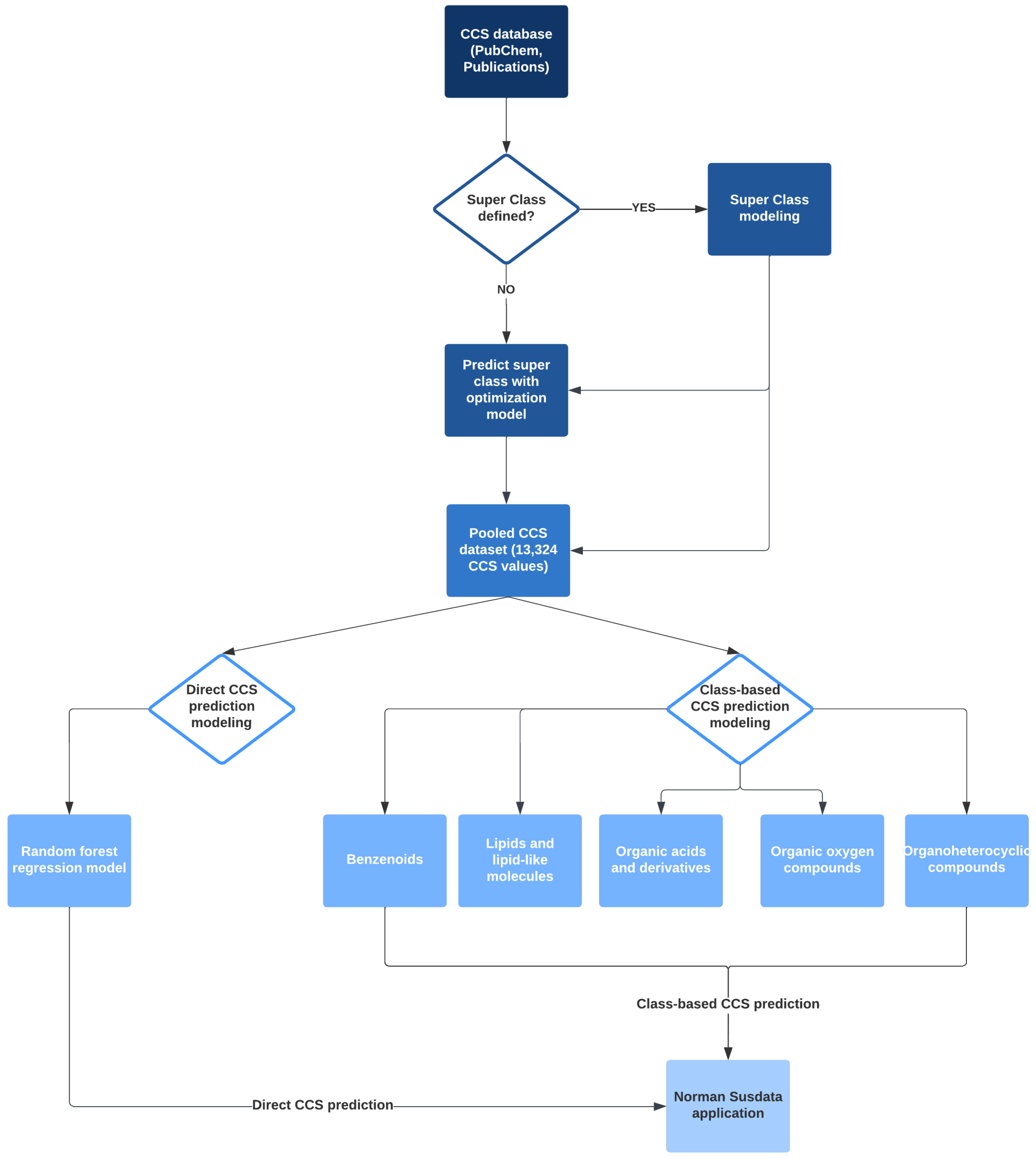
2.2. Overall Workflow
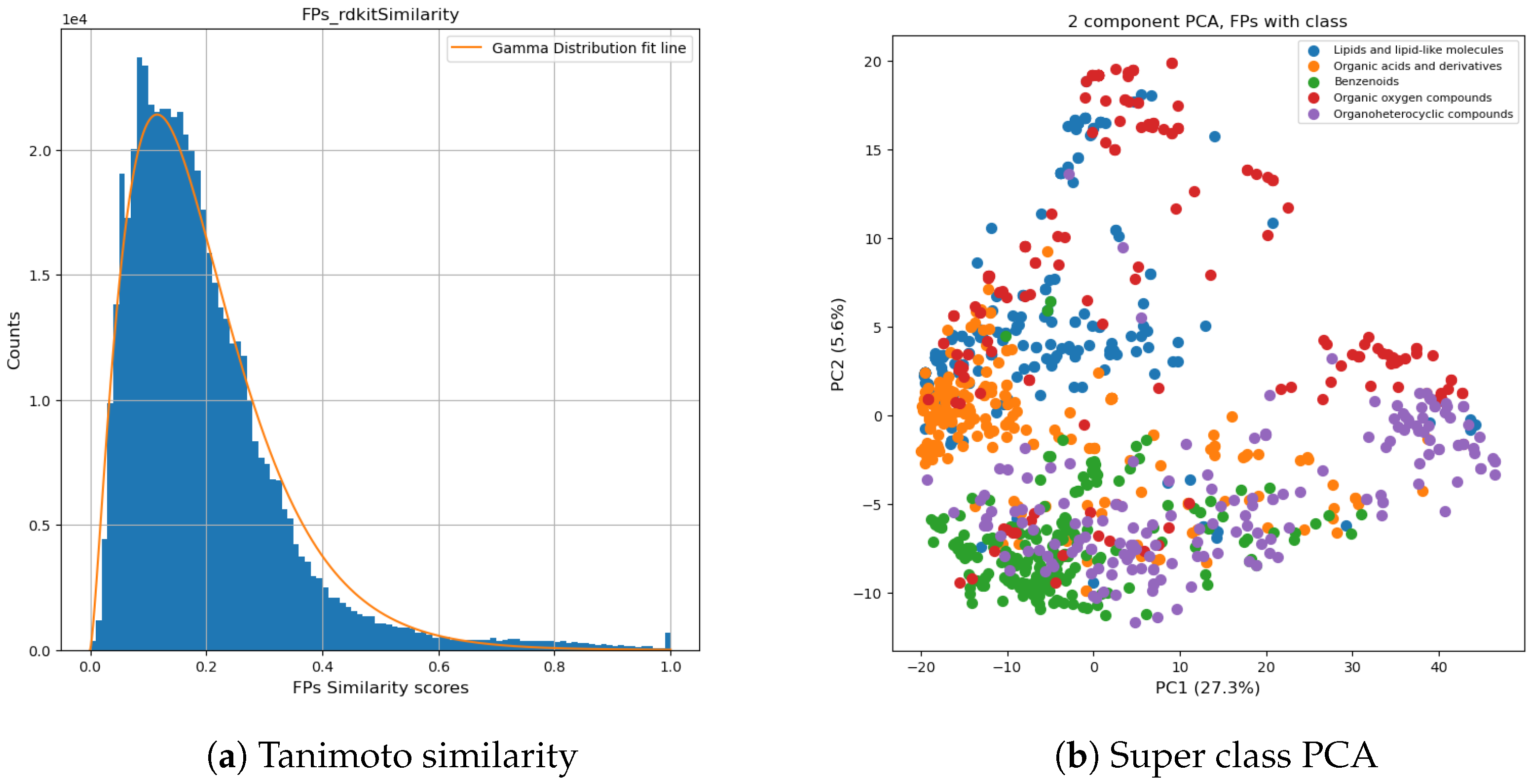
2.2.1. Dataset for Classification Model
2.2.2. Dataset for Regression

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reference | Number of Chemicals | Instrument * |
|---|---|---|
| Picache et al. [60] | 1195 | Agilent 6560 IM-QTOF MS |
| Hines et al. [64] | 1304 | Waters Synapt G2-Si HDMS |
| Celma et al. [40] | 631 | Waters VION IMS-QTOF MS |
| Zheng et al. [61,62] | 891 | Agilent 6560 IM-QTOF MS |
| Belova et al. [65] | 145 | Agilent 6560 IM-QTOF MS |
| Bijlsma et al. [51] | 193 | Waters VION IMS-QTOF MS |
| PubChem [66] | 8965 |
2.3. Modeling
2.3.1. Class Prediction
2.3.2. Class-Based CCS Regression
2.3.3. Direct CCS Regression
3. Results
3.1. Random Forest Classifier and Regression Prediction Model
3.2. Evaluation of Classification Model
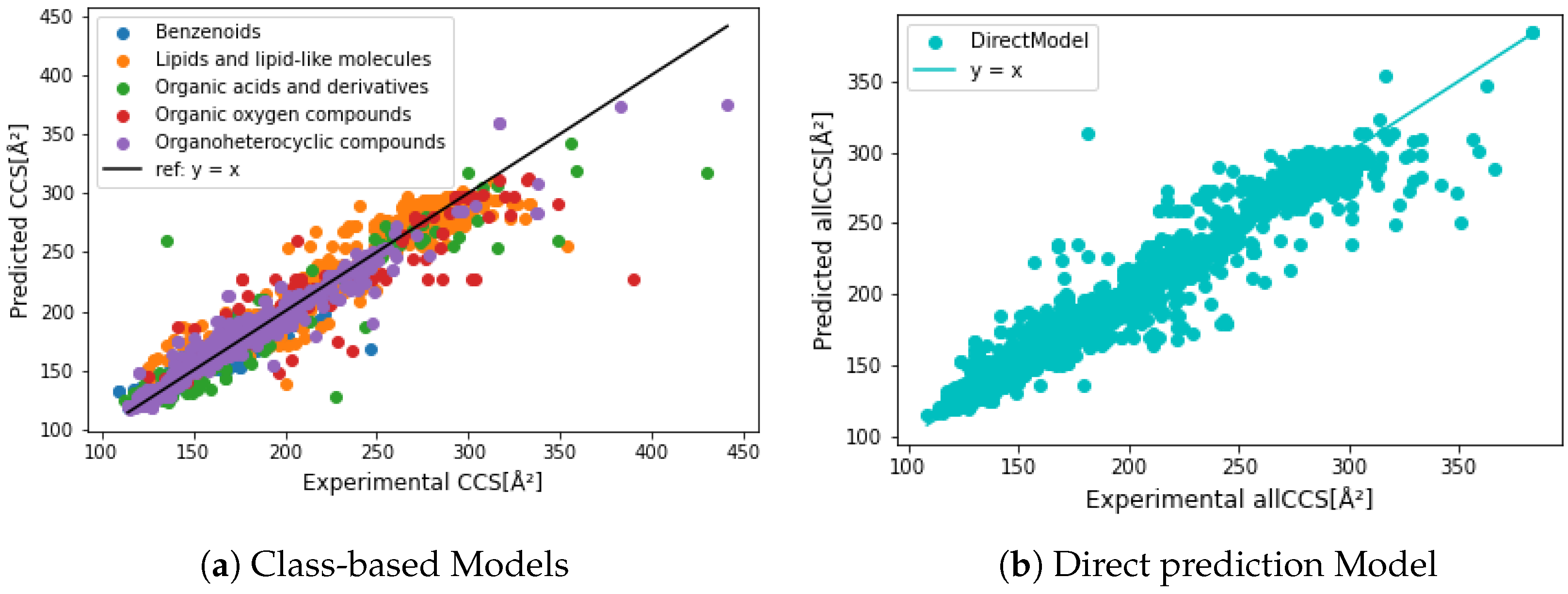
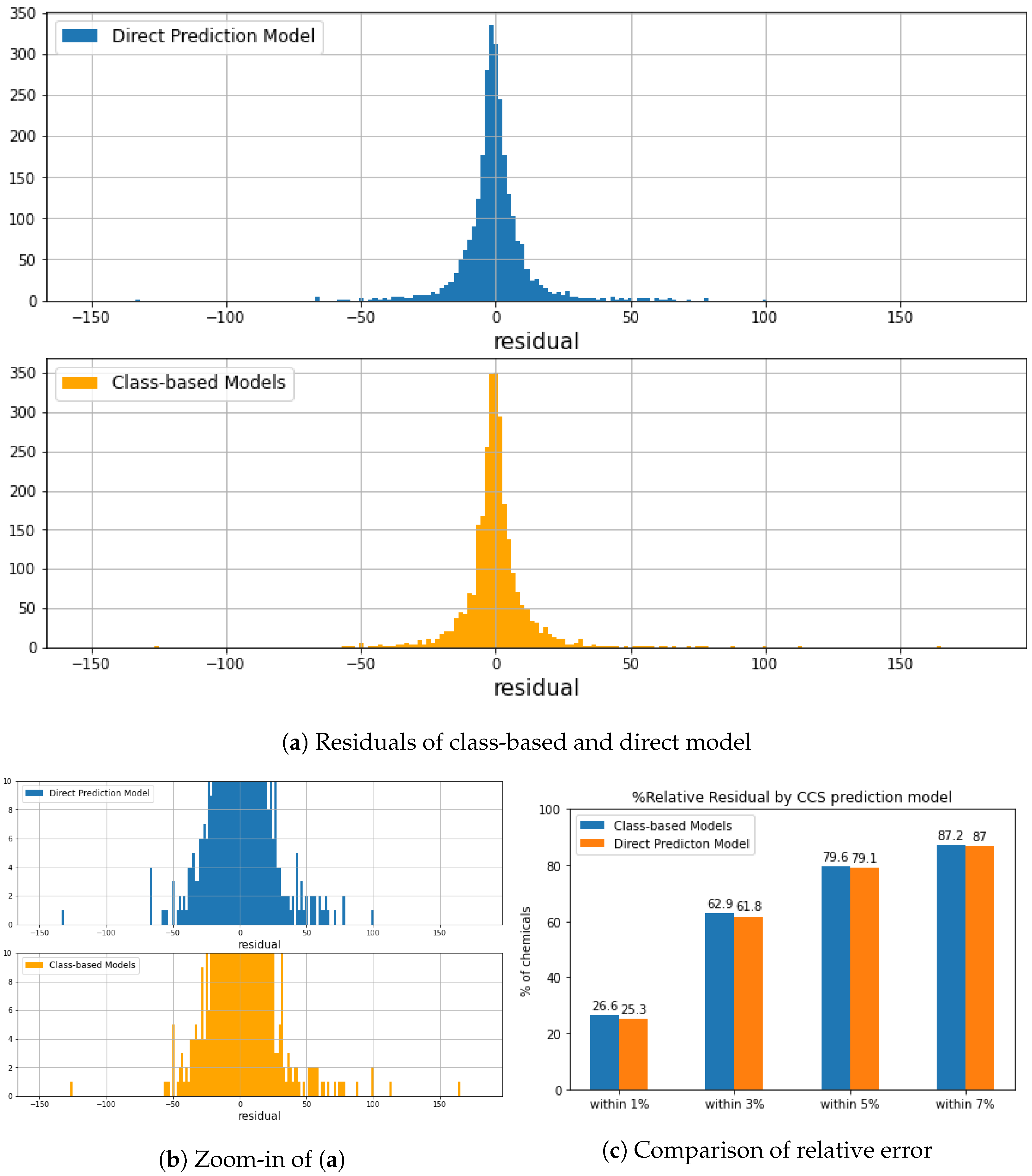
3.3. Evaluation of Regression Models
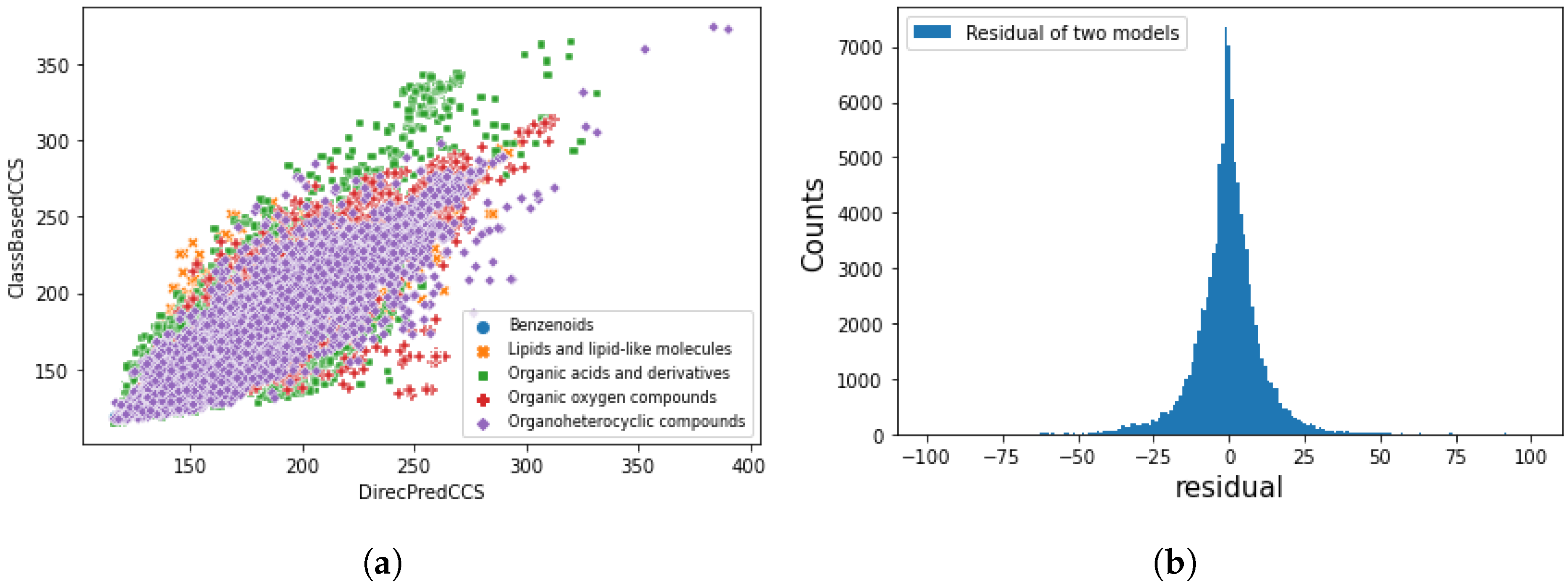
3.4. Application on SusDat
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Muir, D.C.; Howard, P.H. Are there other persistent organic pollutants? A challenge for environmental chemists. Environ. Sci. Technol. 2006, 40, 7157–7166. [Google Scholar] [CrossRef] [PubMed]
- Howard, P.H.; Muir, D.C. Identifying new persistent and bioaccumulative organics among chemicals in commerce II: Pharmaceuticals. Environ. Sci. Technol. 2011, 45, 6938–6946. [Google Scholar] [CrossRef] [PubMed]
- Escher, B.I.; Stapleton, H.M.; Schymanski, E.L. Tracking complex mixtures of chemicals in our changing environment. Science 2020, 367, 388–392. [Google Scholar] [CrossRef] [PubMed]
- Newton, S.R.; McMahen, R.L.; Sobus, J.R.; Mansouri, K.; Williams, A.J.; McEachran, A.D.; Strynar, M.J. Suspect screening and non-targeted analysis of drinking water using point-of-use filters. Environ. Pollut. 2018, 234, 297–306. [Google Scholar] [CrossRef] [PubMed]
- Shi, Q.; Xiong, Y.; Kaur, P.; Sy, N.D.; Gan, J. Contaminants of emerging concerns in recycled water: Fate and risks in agroecosystems. Sci. Total Environ. 2022, 814, 152527. [Google Scholar] [CrossRef] [PubMed]
- Rizzo, L.; Gernjak, W.; Krzeminski, P.; Malato, S.; McArdell, C.S.; Perez, J.A.S.; Schaar, H.; Fatta-Kassinos, D. Best available technologies and treatment trains to address current challenges in urban wastewater reuse for irrigation of crops in EU countries. Sci. Total Environ. 2020, 710, 136312. [Google Scholar] [CrossRef]
- Manaia, C.M. Assessing the risk of antibiotic resistance transmission from the environment to humans: Non-direct proportionality between abundance and risk. Trends Microbiol. 2017, 25, 173–181. [Google Scholar] [CrossRef]
- López-Pacheco, I.Y.; Silva-Núñez, A.; Salinas-Salazar, C.; Arévalo-Gallegos, A.; Lizarazo-Holguin, L.A.; Barceló, D.; Iqbal, H.M.; Parra-Saldívar, R. Anthropogenic contaminants of high concern: Existence in water resources and their adverse effects. Sci. Total Environ. 2019, 690, 1068–1088. [Google Scholar] [CrossRef]
- Ma, Y.; He, X.; Qi, K.; Wang, T.; Qi, Y.; Cui, L.; Wang, F.; Song, M. Effects of environmental contaminants on fertility and reproductive health. J. Environ. Sci. 2019, 77, 210–217. [Google Scholar] [CrossRef]
- Alygizakis, N.A.; Samanipour, S.; Hollender, J.; Ibáñez, M.; Kaserzon, S.; Kokkali, V.; Van Leerdam, J.A.; Mueller, J.F.; Pijnappels, M.; Reid, M.J.; et al. Exploring the potential of a global emerging contaminant early warning network through the use of retrospective suspect screening with high-resolution mass spectrometry. Environ. Sci. Technol. 2018, 52, 5135–5144. [Google Scholar] [CrossRef]
- Pedrazzani, R.; Bertanza, G.; Brnardić, I.; Cetecioglu, Z.; Dries, J.; Dvarionienė, J.; García-Fernández, A.J.; Langenhoff, A.; Libralato, G.; Lofrano, G.; et al. Opinion paper about organic trace pollutants in wastewater: Toxicity assessment in a European perspective. Sci. Total Environ. 2019, 651, 3202–3221. [Google Scholar] [CrossRef] [PubMed]
- Rueda-Ruzafa, L.; Cruz, F.; Roman, P.; Cardona, D. Gut microbiota and neurological effects of glyphosate. Neurotoxicology 2019, 75, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Lohmann, R.; Breivik, K.; Dachs, J.; Muir, D. Global fate of POPs: Current and future research directions. Environ. Pollut. 2007, 150, 150–165. [Google Scholar] [CrossRef] [PubMed]
- Samanipour, S.; Martin, J.W.; Lamoree, M.H.; Reid, M.J.; Thomas, K.V. Optimism for nontarget analysis in environmental chemistry. Environ. Sci. Technol. 2019, 53, 5529–5530. [Google Scholar] [CrossRef] [PubMed]
- Vermeulen, R.; Schymanski, E.L.; Barabási, A.L.; Miller, G.W. The exposome and health: Where chemistry meets biology. Science 2020, 367, 392–396. [Google Scholar] [CrossRef]
- Schymanski, E.L.; Jeon, J.; Gulde, R.; Fenner, K.; Ruff, M.; Singer, H.P.; Hollender, J. Identifying small molecules via high resolution mass spectrometry: Communicating confidence. Environ. Sci. Technol. 2014, 48, 2097–2098. [Google Scholar] [CrossRef]
- Schulze, B.; Jeon, Y.; Kaserzon, S.; Heffernan, A.L.; Dewapriya, P.; O’Brien, J.; Ramos, M.J.G.; Gorji, S.G.; Mueller, J.F.; Thomas, K.V.; et al. An assessment of quality assurance/quality control efforts in high resolution mass spectrometry non-target workflows for analysis of environmental samples. TrAC Trends Anal. Chem. 2020, 133, 116063. [Google Scholar] [CrossRef]
- Pérez-Lemus, N.; López-Serna, R.; Pérez-Elvira, S.I.; Barrado, E. Analytical methodologies for the determination of pharmaceuticals and personal care products (PPCPs) in sewage sludge: A critical review. Anal. Chim. Acta 2019, 1083, 19–40. [Google Scholar] [CrossRef]
- Hollender, J.; Schymanski, E.L.; Singer, H.P.; Ferguson, P.L. Nontarget screening with high resolution mass spectrometry in the environment: Ready to go? Environ. Sci. Technol. 2017, 51, 11505–11512. [Google Scholar] [CrossRef]
- Guo, Z.; Huang, S.; Wang, J.; Feng, Y.L. Recent advances in non-targeted screening analysis using liquid chromatography—High resolution mass spectrometry to explore new biomarkers for human exposure. Talanta 2020, 219, 121339. [Google Scholar] [CrossRef]
- Hollender, J.; Van Bavel, B.; Dulio, V.; Farmen, E.; Furtmann, K.; Koschorreck, J.; Kunkel, U.; Krauss, M.; Munthe, J.; Schlabach, M.; et al. High resolution mass spectrometry-based non-target screening can support regulatory environmental monitoring and chemicals management. Environ. Sci. Eur. 2019, 31, 42. [Google Scholar] [CrossRef]
- Knolhoff, A.M.; Callahan, J.H.; Croley, T.R. Mass accuracy and isotopic abundance measurements for HR-MS instrumentation: Capabilities for non-targeted analyses. J. Am. Soc. Mass Spectrom. 2014, 25, 1285–1294. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, F.; Sancho, J.V.; Ibáñez, M.; Abad, E.; Portolés, T.; Mattioli, L. Current use of high-resolution mass spectrometry in the environmental sciences. Anal. Bioanal. Chem. 2012, 403, 1251–1264. [Google Scholar] [CrossRef] [PubMed]
- Kaufmann, A. The current role of high-resolution mass spectrometry in food analysis. Anal. Bioanal. Chem. 2012, 403, 1233–1249. [Google Scholar] [CrossRef] [PubMed]
- Knolhoff, A.M.; Croley, T.R. Non-targeted screening approaches for contaminants and adulterants in food using liquid chromatography hyphenated to high resolution mass spectrometry. J. Chromatogr. A 2016, 1428, 86–96. [Google Scholar] [CrossRef] [PubMed]
- Kind, T.; Fiehn, O. Metabolomic database annotations via query of elemental compositions: Mass accuracy is insufficient even at less than 1 ppm. BMC Bioinform. 2006, 7, 234. [Google Scholar] [CrossRef]
- d’Atri, V.; Causon, T.; Hernandez-Alba, O.; Mutabazi, A.; Veuthey, J.L.; Cianferani, S.; Guillarme, D. Adding a new separation dimension to MS and LC–MS: What is the utility of ion mobility spectrometry? J. Sep. Sci. 2018, 41, 20–67. [Google Scholar] [CrossRef]
- Boelrijk, J.; van Herwerden, D.; Ensing, B.; Forré, P.; and Samanipour, S. Predicting RP-LC retention indices of structurally unknown chemicals from mass spectrometry data. ChemRxiv 2022. [Google Scholar] [CrossRef]
- Celma, A.; Ahrens, L.; Gago-Ferrero, P.; Hernández, F.; López, F.; Lundqvist, J.; Pitarch, E.; Sancho, J.V.; Wiberg, K.; Bijlsma, L. The relevant role of ion mobility separation in LC-HRMS based screening strategies for contaminants of emerging concern in the aquatic environment. Chemosphere 2021, 280, 130799. [Google Scholar] [CrossRef]
- Mairinger, T.; Causon, T.J.; Hann, S. The potential of ion mobility–mass spectrometry for non-targeted metabolomics. Curr. Opin. Chem. Biol. 2018, 42, 9–15. [Google Scholar] [CrossRef]
- Goscinny, S.; Joly, L.; De Pauw, E.; Hanot, V.; Eppe, G. Travelling-wave ion mobility time-of-flight mass spectrometry as an alternative strategy for screening of multi-class pesticides in fruits and vegetables. J. Chromatogr. A 2015, 1405, 85–93. [Google Scholar] [CrossRef] [PubMed]
- Hill, H.H., Jr.; Siems, W.F.; St. Louis, R.H. Ion mobility spectrometry. Anal. Chem. 1990, 62, 1201A–1209A. [Google Scholar] [CrossRef] [PubMed]
- Borsdorf, H.; Eiceman, G.A. Ion mobility spectrometry: Principles and applications. Appl. Spectrosc. Rev. 2006, 41, 323–375. [Google Scholar] [CrossRef]
- Eiceman, G.A.; Karpas, Z. Ion Mobility Spectrometry; CRC Press: Boca Raton, FL, USA, 2005. [Google Scholar]
- Hernández-Mesa, M.; D’atri, V.; Barknowitz, G.; Fanuel, M.; Pezzatti, J.; Dreolin, N.; Ropartz, D.; Monteau, F.; Vigneau, E.; Rudaz, S.; et al. Interlaboratory and interplatform study of steroids collision cross section by traveling wave ion mobility spectrometry. Anal. Chem. 2020, 92, 5013–5022. [Google Scholar] [CrossRef]
- Stow, S.M.; Causon, T.J.; Zheng, X.; Kurulugama, R.T.; Mairinger, T.; May, J.C.; Rennie, E.E.; Baker, E.S.; Smith, R.D.; McLean, J.A.; et al. An interlaboratory evaluation of drift tube ion mobility–mass spectrometry collision cross section measurements. Anal. Chem. 2017, 89, 9048–9055. [Google Scholar] [CrossRef] [PubMed]
- Hinnenkamp, V.; Klein, J.; Meckelmann, S.W.; Balsaa, P.; Schmidt, T.C.; Schmitz, O.J. Comparison of CCS values determined by traveling wave ion mobility mass spectrometry and drift tube ion mobility mass spectrometry. Anal. Chem. 2018, 90, 12042–12050. [Google Scholar] [CrossRef] [PubMed]
- Feuerstein, M.L.; Hernández-Mesa, M.; Kiehne, A.; Le Bizec, B.; Hann, S.; Dervilly, G.; Causon, T. Comparability of Steroid Collision Cross Sections Using Three Different IM-HRMS Technologies: An Interplatform Study. J. Am. Soc. Mass Spectrom. 2022. [Google Scholar] [CrossRef]
- Borsdorf, H.; Mayer, T.; Zarejousheghani, M.; Eiceman, G.A. Recent developments in ion mobility spectrometry. Appl. Spectrosc. Rev. 2011, 46, 472–521. [Google Scholar] [CrossRef]
- Celma, A.; Sancho, J.V.; Schymanski, E.L.; Fabregat-Safont, D.; Ibanez, M.; Goshawk, J.; Barknowitz, G.; Hernandez, F.; Bijlsma, L. Improving target and suspect screening high-resolution mass spectrometry workflows in environmental analysis by ion mobility separation. Environ. Sci. Technol. 2020, 54, 15120–15131. [Google Scholar] [CrossRef]
- Menger, F.; Celma, A.; Schymanski, E.L.; Lai, F.Y.; Bijlsma, L.; Wiberg, K.; Hernández, F.; Sancho, J.V.; Lutz, A. Enhancing Spectral Quality in Complex Environmental Matrices: Supporting Suspect and Non-Target Screening in Zebra Mussels with Ion Mobility. SSRN Electron. J. 2022. [Google Scholar] [CrossRef]
- Izquierdo-Sandoval, D.; Fabregat-Safont, D.; Lacalle-Bergeron, L.; Sancho, J.V.; Hernández, F.; Portoles, T. Benefits of Ion Mobility Separation in GC-APCI-HRMS Screening: From the Construction of a CCS Library to the Application to Real-World Samples. Anal. Chem. 2022, 94, 9040–9047. [Google Scholar] [CrossRef] [PubMed]
- Gabelica, V.; Marklund, E. Fundamentals of ion mobility spectrometry. Curr. Opin. Chem. Biol. 2018, 42, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Ross, D.H.; Xu, L. Determination of drugs and drug metabolites by ion mobility-mass spectrometry: A review. Anal. Chim. Acta 2021, 1154, 338270. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Shen, X.; Tu, J.; Zhu, Z.J. Large-Scale Prediction of Collision Cross-Section Values for Metabolites in Ion Mobility-Mass Spectrometry. Anal. Chem. 2016, 88, 11084–11091. [Google Scholar] [CrossRef] [PubMed]
- Plante, P.L.; Francovic-Fontaine, É.; May, J.C.; McLean, J.A.; Baker, E.S.; Laviolette, F.; Marchand, M.; Corbeil, J. Predicting Ion Mobility Collision Cross-Sections Using a Deep Neural Network: DeepCCS. Anal. Chem. 2019, 91, 5191–5199. [Google Scholar] [CrossRef]
- Zhou, Z.; Tu, J.; Zhu, Z.J. Advancing the large-scale CCS database for metabolomics and lipidomics at the machine-learning era. Curr. Opin. Chem. Biol. 2018, 42, 34–41. [Google Scholar] [CrossRef]
- Mollerup, C.B.; Mardal, M.; Dalsgaard, P.W.; Linnet, K.; Barron, L.P. Prediction of collision cross section and retention time for broad scope screening in gradient reversed-phase liquid chromatography-ion mobility-high resolution accurate mass spectrometry. J. Chromatogr. A 2018, 1542, 82–88. [Google Scholar] [CrossRef]
- Zhou, Z.; Luo, M.; Chen, X.; Yin, Y.; Xiong, X.; Wang, R.; Zhu, Z.J. Ion mobility collision cross-section atlas for known and unknown metabolite annotation in untargeted metabolomics. Nat. Commun. 2020, 11, 4334. [Google Scholar] [CrossRef]
- Ross, D.H.; Cho, J.H.; Xu, L. Breaking down structural diversity for comprehensive prediction of ion-neutral collision cross sections. Anal. Chem. 2020, 92, 4548–4557. [Google Scholar] [CrossRef]
- Bijlsma, L.; Bade, R.; Celma, A.; Mullin, L.; Cleland, G.; Stead, S.; Hernandez, F.; Sancho, J.V. Prediction of collision cross-section values for small molecules: Application to pesticide residue analysis. Anal. Chem. 2017, 89, 6583–6589. [Google Scholar] [CrossRef]
- Celma, A.; Bade, R.; Sancho, J.V.; Hernández, F.; Humpries, M.; Bijslma, L. Prediction of Retention Time and Collision Cross Section (CCSH+, CCSH-and CCSNa+) of Emerging Contaminants Using Multiple Adaptive Regression Splines. 2022. Available online: https://doi.org/10.21203/rs.3.rs-1249834/v1 (accessed on 13 January 2022).
- Cereto-Massagué, A.; Ojeda, M.J.; Valls, C.; Mulero, M.; Garcia-Vallvé, S.; Pujadas, G. Molecular fingerprint similarity search in virtual screening. Methods 2015, 71, 58–63. [Google Scholar] [CrossRef] [PubMed]
- Swain, M. PubChemPy: A Way to Interact with PubChem in Python. 2014. Available online: https://pubchempy.readthedocs.io/en/latest/ (accessed on 13 January 2022).
- Landrum, G. RDKit: Open-Source Cheminformatics. 2006. Available online: https://doi.org/10.5281/zenodo.3732262 (accessed on 13 January 2022).
- Weininger, D. SMILES, a chemical language and information system. 1. Introduction to methodology and encoding rules. J. Chem. Inf. Comput. Sci. 1988, 28, 31–36. [Google Scholar] [CrossRef]
- Weininger, D.; Weininger, A.; Weininger, J.L. SMILES. 2. Algorithm for generation of unique SMILES notation. J. Chem. Inf. Comput. Sci. 1989, 29, 97–101. [Google Scholar] [CrossRef]
- Nilakantan, R.; Bauman, N.; Dixon, J.S.; Venkataraghavan, R. Topological torsion: A new molecular descriptor for SAR applications. Comparison with other descriptors. J. Chem. Inf. Comput. Sci. 1987, 27, 82–85. [Google Scholar] [CrossRef]
- Capecchi, A.; Probst, D.; Reymond, J.L. One molecular fingerprint to rule them all: Drugs, biomolecules, and the metabolome. J. Cheminformatics 2020, 12, 1–15. [Google Scholar] [CrossRef]
- Picache, J.A.; Rose, B.S.; Balinski, A.; Leaptrot, K.L.; Sherrod, S.D.; May, J.C.; McLean, J.A. Collision cross section compendium to annotate and predict multi-omic compound identities. Chem. Sci. 2019, 10, 983–993. [Google Scholar] [CrossRef]
- Zheng, X.; Aly, A.N.; Zhou, Y.; Dupuis, K.T.; Bilbao, A.; Paurus, V.L.; Orton, D.J.; Wilson, R.; Payne, S.H.; Smith, R.D.; et al. A structural examination and collision cross section database for over 500 metabolites and xenobiotics using drift tube ion mobility spectrometry. Chem. Sci. 2017, 8, 7724–7736. [Google Scholar] [CrossRef]
- Zheng, X.; Dupuis, K.T.; Aly, N.A.; Zhou, Y.; Smith, F.B.; Tang, K.; Smith, R.D.; Baker, E.S. Utilizing ion mobility spectrometry and mass spectrometry for the analysis of polycyclic aromatic hydrocarbons, polychlorinated biphenyls, polybrominated diphenyl ethers and their metabolites. Anal. Chim. Acta 2018, 1037, 265–273. [Google Scholar] [CrossRef]
- Bajusz, D.; Rácz, A.; Héberger, K. Why is Tanimoto index an appropriate choice for fingerprint-based similarity calculations? J. Cheminform. 2015, 7, 20. [Google Scholar] [CrossRef]
- Hines, K.M.; Ross, D.H.; Davidson, K.L.; Bush, M.F.; Xu, L. Large-Scale Structural Characterization of Drug and Drug-Like Compounds by High-Throughput Ion Mobility-Mass Spectrometry. Anal. Chem. 2017, 89, 9023–9030. [Google Scholar] [CrossRef]
- Belova, L.; Caballero-Casero, N.; van Nuijs, A.L.N.; Covaci, A. Ion Mobility-High-Resolution Mass Spectrometry (IM-HRMS) for the Analysis of Contaminants of Emerging Concern (CECs): Database Compilation and Application to Urine Samples. Anal. Chem. 2021, 93, 6428–6436. [Google Scholar] [CrossRef] [PubMed]
- Schymanski, E.; Zhang, J.; Thiessen, P.; Bolton, E. Experimental CCS Values in Pubchem. Zenodo 2022. [Google Scholar] [CrossRef]
- Svetnik, V.; Liaw, A.; Tong, C.; Culberson, J.C.; Sheridan, R.P.; Feuston, B.P. Random forest: A classification and regression tool for compound classification and QSAR modeling. J. Chem. Inf. Comput. Sci. 2003, 43, 1947–1958. [Google Scholar] [CrossRef] [PubMed]
- Belova, L.; Celma, A.; Van Haesendonck, G.; Lemière, F.; Sancho, J.V.; Covaci, A.; van Nuijs, A.L.; Bijlsma, L. Revealing the differences in collision cross section values of small organic molecules acquired by different instrumental designs and prediction models. Anal. Chim. Acta 2022, 1229, 340361. [Google Scholar] [CrossRef] [PubMed]
- Dulio, V.; Koschorreck, J.; Van Bavel, B.; Van den Brink, P.; Hollender, J.; Munthe, J.; Schlabach, M.; Aalizadeh, R.; Agerstrand, M.; Ahrens, L.; et al. The NORMAN association and the European partnership for chemicals risk assessment (PARC): Let’s cooperate! Environ. Sci. Eur. 2020, 32, 100. [Google Scholar] [CrossRef]








| Super Class | Training | Test | F1 Score | Accuracy |
|---|---|---|---|---|
| Benzenoids | 181 | 46 | 0.905 | 0.935 |
| Lipids and lipid-like molecules | 189 | 47 | 0.909 | 0.889 |
| Organic acids and derivatives | 184 | 46 | 0.848 | 0.813 |
| Organic oxygen compounds | 142 | 36 | 0.861 | 0.861 |
| Organoheterocyclic compounds | 140 | 35 | 0.822 | 0.857 |
| Training | Test | ||||
|---|---|---|---|---|---|
| Dataset | Data | R | Data | R | MRE (%) |
| All | 10,659 | 0.972 | 2665 | 0.958 | 2.20 |
| Benzenoids | 1930 | 0.942 | 483 | 0.869 | 1.89 |
| Lipids and lipid-like molecules | 3675 | 0.940 | 919 | 0.932 | 2.33 |
| Organic acids and derivatives | 1392 | 0.950 | 348 | 0.901 | 2.21 |
| Organic oxygen compounds | 754 | 0.925 | 189 | 0.860 | 2.33 |
| Organoheterocyclic compounds | 2907 | 0.960 | 724 | 0.933 | 1.96 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, F.; van Herwerden, D.; Preud’homme, H.; Samanipour, S. Collision Cross Section Prediction with Molecular Fingerprint Using Machine Learning. Molecules 2022, 27, 6424. https://doi.org/10.3390/molecules27196424
Yang F, van Herwerden D, Preud’homme H, Samanipour S. Collision Cross Section Prediction with Molecular Fingerprint Using Machine Learning. Molecules. 2022; 27(19):6424. https://doi.org/10.3390/molecules27196424
Chicago/Turabian StyleYang, Fan, Denice van Herwerden, Hugues Preud’homme, and Saer Samanipour. 2022. "Collision Cross Section Prediction with Molecular Fingerprint Using Machine Learning" Molecules 27, no. 19: 6424. https://doi.org/10.3390/molecules27196424
APA StyleYang, F., van Herwerden, D., Preud’homme, H., & Samanipour, S. (2022). Collision Cross Section Prediction with Molecular Fingerprint Using Machine Learning. Molecules, 27(19), 6424. https://doi.org/10.3390/molecules27196424