
Homology Modeling, Molecular Docking, Molecular Dynamic Simulation, and Drug-Likeness of the Modified Alpha-Mangostin against the β-Tubulin Protein of Acanthamoeba Keratitis


Abstract
1. Introduction
2. Results
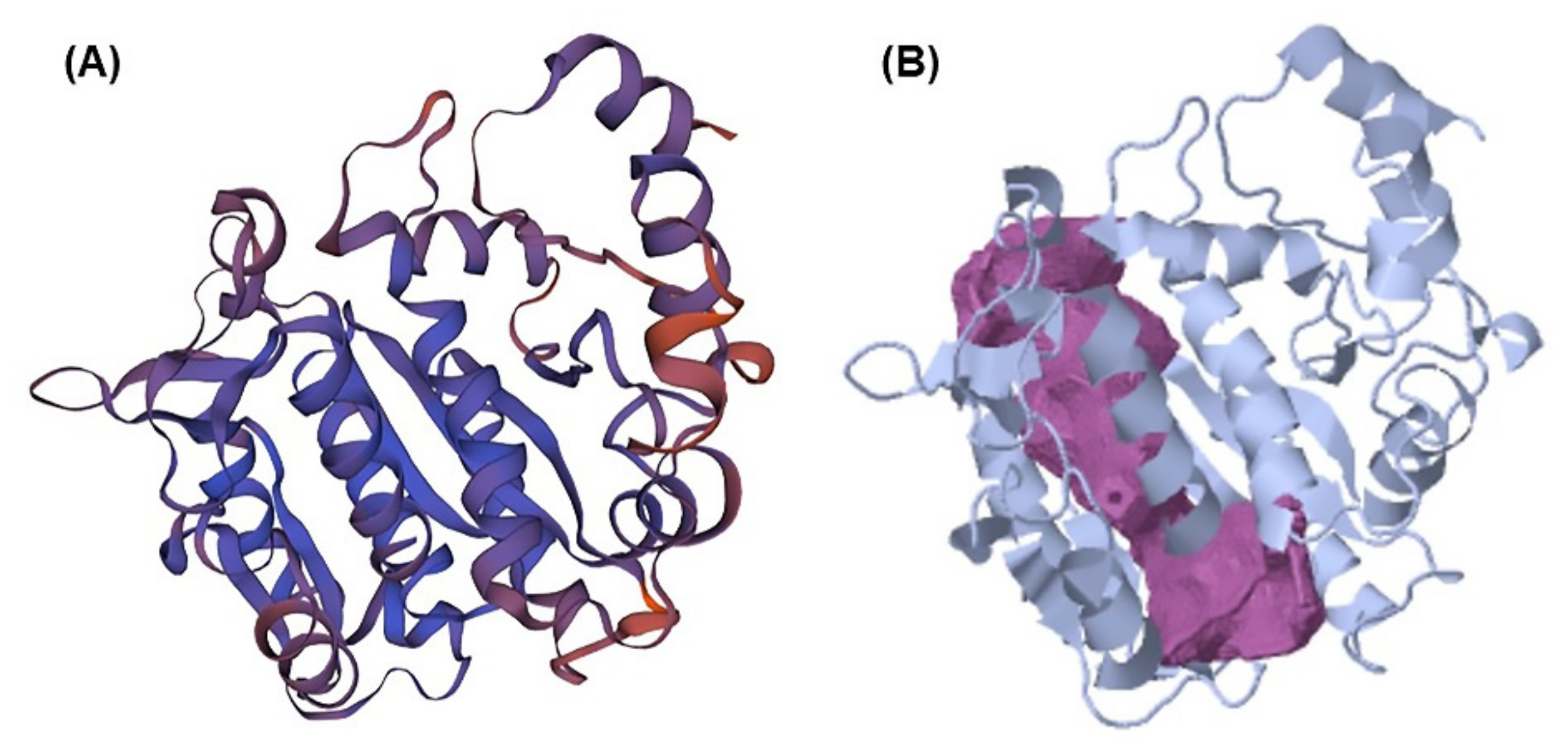
2.1. Beta-Tubulin (BT) Using Homology Modeling
2.2. Pocket Binding Analysis
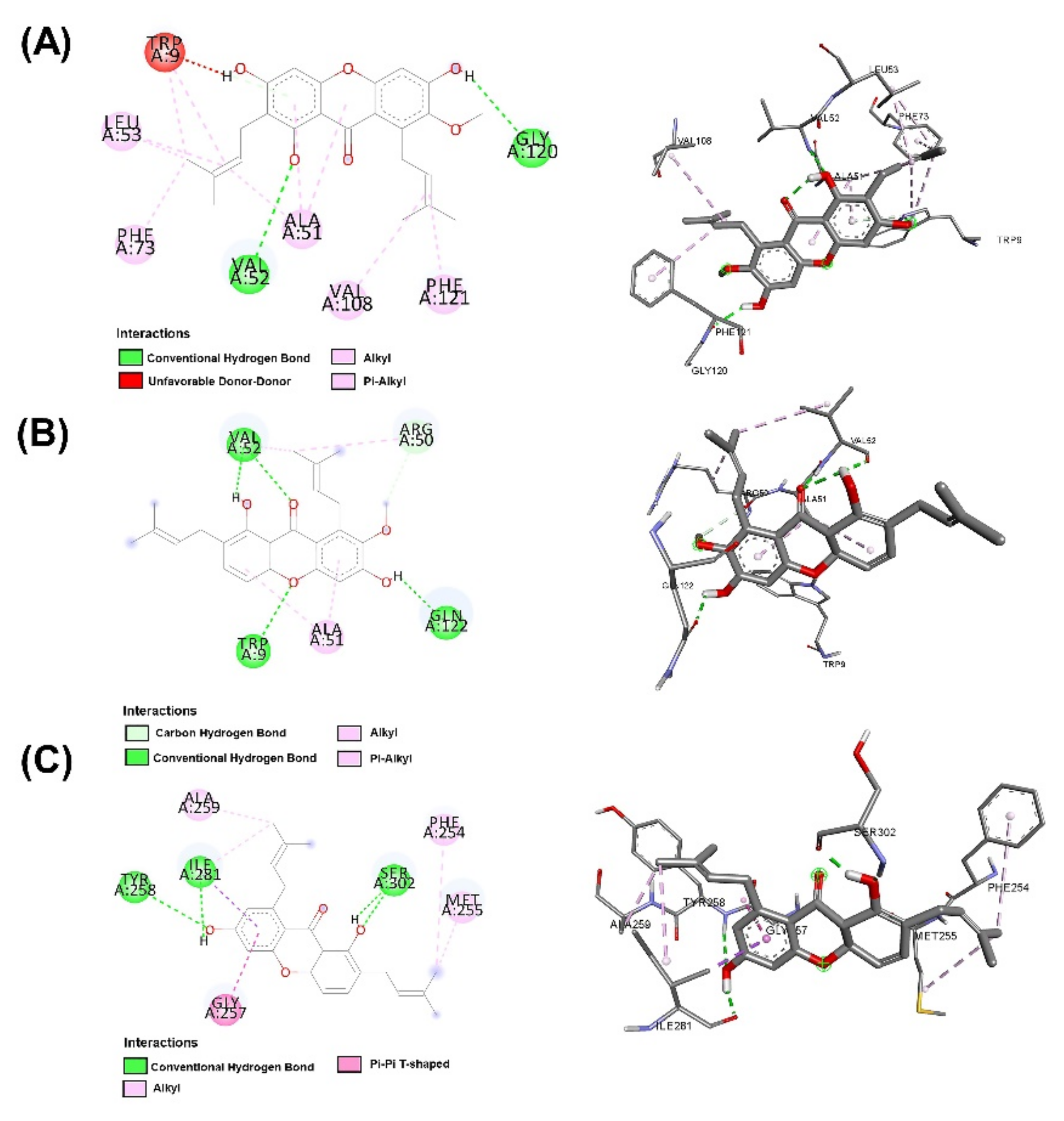
2.3. Molecular Docking
2.4. Quantum Chemical Calculations
2.5. Molecular Dynamics
2.6. Synthesis and Structural Analysis via NMR
2.7. Physicochemical and Pharmacokinetic Analysis of the Compounds
3. Discussion
4. Materials and Methods
4.1. Evaluation and Homology Modeling of the Beta-Tubulin Protein of Acanthamoeba Keratitis
4.2. Screening of Compounds Using Arguslab and Autodock
4.3. Quantum Chemical Calculations
4.4. Molecular Dynamics Modeling
4.5. Predicting Chemical Shifts in NMR Based on Knowledge of the Structure
4.6. Physicochemical and Pharmacokinetic Analysis of the Compounds Using the SwissADME Tool
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Padzik, M.; Baltaza, W.; Conn, D.B.; Szaflik, J.P.; Chomicz, L.J.A.O.A.; Medicine, E. Effect of povidone iodine, chlorhexidine digluconate and toyocamycin on amphizoic amoebic strains, infectious agents of Acanthamoeba keratitis–a growing threat to human health worldwide. Ann. Agric. Environ. Med. 2018, 25, 725–731. [Google Scholar] [CrossRef]
- Khan, N.A. Acanthamoeba: Biology and increasing importance in human health. FEMS Microbiol. Rev. 2006, 30, 564–595. [Google Scholar] [CrossRef] [PubMed]
- Niyyati, M.; Dodangeh, S.; Lorenzo-Morales, J. A Review of the Current Research Trends in the Application of Medicinal Plants as a Source for Novel Therapeutic Agents Against Acanthamoeba Infections. Iran. J. Pharm. Res. IJPR 2016, 15, 893–900. [Google Scholar]
- Tremblay, M.R.; Nevalainen, M.; Nair, S.J.; Porter, J.R.; Castro, A.C.; Behnke, M.L.; Yu, L.-C.; Hagel, M.; White, K.; Faia, K.; et al. Semisynthetic Cyclopamine Analogues as Potent and Orally Bioavailable Hedgehog Pathway Antagonists. J. Med. Chem. 2008, 51, 6646–6649. [Google Scholar] [CrossRef]
- Paddon, C.J.; Westfall, P.J.; Pitera, D.J.; Benjamin, K.; Fisher, K.; McPhee, D.; Leavell, M.D.; Tai, A.; Main, A.; Eng, D.; et al. High-level semi-synthetic production of the potent antimalarial artemisinin. Nature 2013, 496, 528–532. [Google Scholar] [CrossRef]
- Henriquez Fiona, L.; Ingram Paul, R.; Muench Stephen, P.; Rice David, W.; Roberts Craig, W. Molecular Basis for Resistance of Acanthamoeba Tubulins to All Major Classes of Antitubulin Compounds. Antimicrob. Agents Chemother. 2008, 52, 1133–1135. [Google Scholar] [CrossRef]
- Mungroo, M.R.; Khan, N.A.; Maciver, S.; Siddiqui, R. Opportunistic free-living amoebal pathogens. Pathog. Glob. Health 2022, 116, 70–84. [Google Scholar] [CrossRef]
- Dawson, P.J.; Gutteridge, W.E.; Gull, K. A comparison of the interaction of anthelmintic benzimidazoles with tubulin isolated from mammalian tissue and the parasitic nematode Ascaridia galli. Biochem. Pharmacol. 1984, 33, 1069–1074. [Google Scholar] [CrossRef]
- Ellis, G.C.; Phillips, J.B.; O’Rourke, S.; Lyczak, R.; Bowerman, B. Maternally expressed and partially redundant β-tubulins in Caenorhabditis elegans are autoregulated. J. Cell Sci. 2004, 117, 457–464. [Google Scholar] [CrossRef]
- Canta, A.; Chiorazzi, A.; Cavaletti, G. Tubulin: A target for antineoplastic drugs into the cancer cells but also in the peripheral nervous system. Curr. Med. Chem. 2009, 16, 1315–1324. [Google Scholar] [CrossRef]
- Fennell, B.J.; Naughton, J.A.; Dempsey, E.; Bell, A. Cellular and molecular actions of dinitroaniline and phosphorothioamidate herbicides on Plasmodium falciparum: Tubulin as a specific antimalarial target. Mol. Biochem. Parasitol. 2006, 145, 226–238. [Google Scholar] [CrossRef]
- Aleyasin, H.; Karuppagounder, S.S.; Kumar, A.; Sleiman, S.; Basso, M.; Ma, T.; Siddiq, A.; Chinta, S.J.; Brochier, C.; Langley, B.; et al. Antihelminthic Benzimidazoles Are Novel HIF Activators That Prevent Oxidative Neuronal Death via Binding to Tubulin. Antioxid. Redox Signal. 2014, 22, 121–134. [Google Scholar] [CrossRef] [PubMed]
- Chatterji, B.P.; Jindal, B.; Srivastava, S.; Panda, D. Microtubules as antifungal and antiparasitic drug targets. Expert Opin. Ther. Pat. 2011, 21, 167–186. [Google Scholar] [CrossRef]
- Katiyar, S.K.; Gordon, V.R.; McLaughlin, G.L.; Edlind, T.D. Antiprotozoal activities of benzimidazoles and correlations with beta-tubulin sequence. Antimicrob. Agents Chemother. 1994, 38, 2086–2090. [Google Scholar] [CrossRef] [PubMed]
- Ansori, A.N.M.; Fadholly, A.; Hayaza, S.; Susilo, R.J.K.; Inayatillah, B.; Winarni, D.; Husen, S.A. A review on medicinal properties of mangosteen (Garcinia mangostana L.). Res. J. Pharm. Technol. 2020, 13, 974–982. [Google Scholar] [CrossRef]
- Ketsa, S.; Paull, R.E. 1—Mangosteen (Garcinia mangostana L.). In Postharvest Biology and Technology of Tropical and Subtropical Fruits; Yahia, E.M., Ed.; Woodhead Publishing: Cambridge, UK, 2011; pp. 1–32e. [Google Scholar] [CrossRef]
- Pedraza-Chaverri, J.; Cárdenas-Rodríguez, N.; Orozco-Ibarra, M.; Pérez-Rojas, J.M. Medicinal properties of mangosteen (Garcinia mangostana). Food Chem. Toxicol. 2008, 46, 3227–3239. [Google Scholar] [CrossRef]
- Gutierrez-Orozco, F.; Thomas-Ahner, J.M.; Berman-Booty, L.D.; Galley, J.D.; Chitchumroonchokchai, C.; Mace, T.; Suksamrarn, S.; Bailey, M.T.; Clinton, S.K.; Lesinski, G.B.; et al. Dietary α-mangostin, a xanthone from mangosteen fruit, exacerbates experimental colitis and promotes dysbiosis in mice. Mol. Nutr. Food Res. 2014, 58, 1226–1238. [Google Scholar] [CrossRef]
- Cassileth, B. Mangosteen (Garcinia mangostana). Oncology 2011, 2011, 844. [Google Scholar]
- Jung, H.-A.; Su, B.-N.; Keller, W.J.; Mehta, R.G.; Kinghorn, A.D. Antioxidant Xanthones from the Pericarp of Garcinia mangostana (Mangosteen). J. Agric. Food Chem. 2006, 54, 2077–2082. [Google Scholar] [CrossRef]
- Gutierrez-Orozco, F.; Failla, M.L. Biological Activities and Bioavailability of Mangosteen Xanthones: A Critical Review of the Current Evidence. Nutrients 2013, 5, 3163–3183. [Google Scholar] [CrossRef]
- Manimekalai, I.; Sivakumari, K.; Ashok, K.; Rajesh, S.J. Antioxidant and anticancer potential of mangosteen fruit, Garcinia mangostana against hepatocellular carcinoma (HePG-2) cell line. World J. Pharm. Pharm. Sci. 2016, 5, 253–293. [Google Scholar]
- Obolskiy, D.; Pischel, I.; Siriwatanametanon, N.; Heinrich, M. Garcinia mangostana L.: A phytochemical and pharmacological review. Phytother. Res. 2009, 23, 1047–1065. [Google Scholar] [CrossRef] [PubMed]
- Hemshekhar, M.; Sunitha, K.; Santhosh, M.S.; Devaraja, S.; Kemparaju, K.; Vishwanath, B.S.; Niranjana, S.R.; Girish, K.S. An overview on genus garcinia: Phytochemical and therapeutical aspects. Phytochem. Rev. 2011, 10, 325–351. [Google Scholar] [CrossRef]
- Nakatani, K.; Atsumi, M.; Arakawa, T.; Oosawa, K.; Shimura, S.; Nakahata, N.; Ohizumi, Y. Inhibitions of Histamine Release and Prostaglandin E2 Synthesis by Mangosteen, a Thai Medicinal Plant. Biol. Pharm. Bull. 2002, 25, 1137–1141. [Google Scholar] [CrossRef] [PubMed]
- Kasai, K.; Ito, Y.; Nitta, A.; Ariyoshi, K.; Nakamura, T.; Miura, T. Metal coordination by L-amino acid oxidase derived from flounder Platichthys stellatus is structurally essential and regulates antibacterial activity. Appl. Microbiol. Biotechnol. 2020, 104, 9645–9654. [Google Scholar] [CrossRef]
- Ma, M.; Stoyanova, M.; Rademacher, G.; Dutcher, S.K.; Brown, A.; Zhang, R. Structure of the Decorated Ciliary Doublet Microtubule. Cell 2019, 179, 909–922.e912. [Google Scholar] [CrossRef]
- Studer, G.; Rempfer, C.; Waterhouse, A.M.; Gumienny, R.; Haas, J.; Schwede, T. QMEANDisCo—Distance constraints applied on model quality estimation. Bioinformatics 2020, 36, 1765–1771. [Google Scholar] [CrossRef]
- Hemmati, S.A. Identification of novel antagonists of the ecdysone receptor from the desert locust (Schistocerca gregaria) by in silico modelling. Plant Prot. (Sci. J. Agric.) 2021, 44, 135–146. [Google Scholar]
- Ongtanasup, T.; Prommee, N.; Jampa, O.; Limcharoen, T.; Wanmasae, S.; Nissapatorn, V.; Paul, A.K.; Pereira, M.d.L.; Wilairatana, P.; Nasongkla, N.; et al. The Cholesterol-Modulating Effect of the New Herbal Medicinal Recipe from Yellow Vine (Coscinium fenestratum (Goetgh.)), Ginger (Zingiber officinale Roscoe.), and Safflower (Carthamus tinctorius L.) on Suppressing PCSK9 Expression to Upregulate LDLR Expression in HepG2 Cells. Plants 2022, 11, 1835. [Google Scholar]
- Ongtanasup, T.; Wanmasae, S.; Srisang, S.; Manaspon, C.; Net-anong, S.; Eawsakul, K. In silico investigation of ACE2 and the main protease of SARS-CoV-2 with phytochemicals from Myristica fragrans (Houtt.) for the discovery of a novel COVID-19 drug. Saudi J. Biol. Sci. 2022, 29, 103389. [Google Scholar] [CrossRef]
- Eawsakul, K.; Panichayupakaranant, P.; Ongtanasup, T.; Warinhomhoun, S.; Noonong, K.; Bunluepuech, K.J.H. Computational study and in vitro alpha-glucosidase inhibitory effects of medicinal plants from a Thai folk remedy. Heliyon 2021, 7, e08078. [Google Scholar] [CrossRef] [PubMed]
- Pearson, R.J. Hard and Soft Acids and Bases, Dowden, Hutchinson and Ross. Inorg Chem. 1993, 26, 250–255. [Google Scholar]
- Lee, L.-H. Correlation between Lewis Acid−Base Surface Interaction Components and Linear Solvation Energy Relationship Solvatochromic α and β Parameters. Langmuir 1996, 12, 1681–1687. [Google Scholar] [CrossRef]
- Zlatopolskiy, B.D.; Radzom, M.; Zeeck, A.; de Meijere, A. Synthesis and Precursor-Directed Biosynthesis of New Hormaomycin Analogues. Eur. J. Org. Chem. 2006, 2006, 1525–1534. [Google Scholar] [CrossRef]
- Akhtar, R.; Zahoor, A.F.; Rasool, N.; Ahmad, M.; Ali, K.G. Recent trends in the chemistry of Sandmeyer reaction: A review. Mol. Divers. 2022, 26, 1837–1873. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.; Keshipeddy, S.; Zhang, Y.; Wei, X.; Savoie, J.; Patel, N.D.; Yee, N.K.; Senanayake, C.H. Efficient Monophosphorus Ligands for Palladium-Catalyzed Miyaura Borylation. Org. Lett. 2011, 13, 1366–1369. [Google Scholar] [CrossRef]
- Souto, J.A.; Stockman, R.A.; Ley, S.V.J.O.; Chemistry, B. Development of a flow method for the hydroboration/oxidation of olefins. Org. Biomol. Chem. 2015, 13, 3871–3877. [Google Scholar] [CrossRef] [PubMed]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef]
- Huang, S.-H.; Jong, A.Y. Cellular mechanisms of microbial proteins contributing to invasion of the blood–brain barrier. Cell. Microbiol. 2001, 3, 277–287. [Google Scholar] [CrossRef]
- Chancellor, M.B.; Staskin, D.R.; Kay, G.G.; Sandage, B.W.; Oefelein, M.G.; Tsao, J.W. Blood-Brain Barrier Permeation and Efflux Exclusion of Anticholinergics Used in the Treatment of Overactive Bladder. Drugs Aging 2012, 29, 259–273. [Google Scholar] [CrossRef]
- Kim, R.B. Drugs as P-glycoprotein substrates, inhibitors, and inducers. Drug Metab. Rev. 2002, 34, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Balimane, P.V.; Chong, S. A combined cell based approach to identify P-glycoprotein substrates and inhibitors in a single assay. Int. J. Pharm. 2005, 301, 80–88. [Google Scholar] [CrossRef] [PubMed]
- Testa, B.; Krämer, S.D. The Biochemistry of Drug Metabolism—An Introduction. Chem. Biodivers. 2006, 3, 1053–1101. [Google Scholar] [CrossRef] [PubMed]
- Hollenberg, P.F. Characteristics and common properties of inhibitors, inducers, and activators of CYP enzymes. Drug Metab. Rev. 2002, 34, 17–35. [Google Scholar] [CrossRef]
- Kirchmair, J.; Göller, A.H.; Lang, D.; Kunze, J.; Testa, B.; Wilson, I.D.; Glen, R.C.; Schneider, G. Predicting drug metabolism: Experiment and/or computation? Nat. Rev. Drug Discov. 2015, 14, 387–404. [Google Scholar] [CrossRef]
- Huang, S.-M.; Strong, J.M.; Zhang, L.; Reynolds, K.S.; Nallani, S.; Temple, R.; Abraham, S.; Habet, S.A.; Baweja, R.K.; Burckart, G.J.; et al. New Era in Drug Interaction Evaluation: US Food and Drug Administration Update on CYP Enzymes, Transporters, and the Guidance Process. J. Clin. Pharmacol. 2008, 48, 662–670. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 1997, 23, 3–25. [Google Scholar] [CrossRef]
- Veber, D.F.; Johnson, S.R.; Cheng, H.-Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular Properties That Influence the Oral Bioavailability of Drug Candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef]
- Egan, W.J.; Merz, K.M.; Baldwin, J.J. Prediction of Drug Absorption Using Multivariate Statistics. J. Med. Chem. 2000, 43, 3867–3877. [Google Scholar] [CrossRef]
- Siddiqui, R.; Aqeel, Y.; Khan, N.A. The Development of Drugs against Acanthamoeba Infections. Antimicrob. Agents Chemother. 2016, 60, 6441–6450. [Google Scholar] [CrossRef]
- Schwede, T.; Kopp, J.R.; Guex, N.; Peitsch, M.C. SWISS-MODEL: An automated protein homology-modeling server. Nucleic Acids Res. 2003, 31, 3381–3385. [Google Scholar] [CrossRef] [PubMed]
- Benkert, P.; Künzli, M.; Schwede, T. QMEAN server for protein model quality estimation. Nucleic Acids Res. 2009, 37, W510–W514. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, W.; Littlejohn, T.G. Swiss-PDB Viewer (Deep View). Brief. Bioinform. 2001, 2, 195–197. [Google Scholar] [CrossRef]
- Ramachandran, S.; Kota, P.; Ding, F.; Dokholyan, N.V. Automated minimization of steric clashes in protein structures. Proteins Struct. Funct. Bioinform. 2011, 79, 261–270. [Google Scholar] [CrossRef]
- Kota, P.; Ding, F.; Ramachandran, S.; Dokholyan, N.V. Gaia: Automated quality assessment of protein structure models. Bioinformatics 2011, 27, 2209–2215. [Google Scholar] [CrossRef]
- Amera, G.M.; Khan, R.J.; Pathak, A.; Kumar, A.; Singh, A.K. Structure based in-silico study on UDP-N-acetylmuramoyl-L-alanyl-D-glutamate-2,6-diaminopimelate ligase (MurE) from Acinetobacter baumannii as a drug target against nosocomial infections. Inform. Med. Unlocked 2019, 16, 100216. [Google Scholar] [CrossRef]
- Laskowski, R.; MacArthur, M.; Thornton, J. PROCHECK: Validation of protein-structure coordinates. Int. Tables Crystallogr. 2006, in press. [Google Scholar]
- Xu, Y.; Wang, S.; Hu, Q.; Gao, S.; Ma, X.; Zhang, W.; Shen, Y.; Chen, F.; Lai, L.; Pei, J. CavityPlus: A web server for protein cavity detection with pharmacophore modelling, allosteric site identification and covalent ligand binding ability prediction. Nucleic Acids Res. 2018, 46, W374–W379. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Chen, J.; Cheng, T.; Gindulyte, A.; He, J.; He, S.; Li, Q.; Shoemaker, B.A.; Thiessen, P.A.; Yu, B.; et al. PubChem in 2021: New data content and improved web interfaces. Nucleic Acids Res. 2021, 49, D1388–D1395. [Google Scholar] [CrossRef]
- Hanwell, M.D.; Curtis, D.E.; Lonie, D.C.; Vandermeersch, T.; Zurek, E.; Hutchison, G.R. Avogadro: An advanced semantic chemical editor, visualization, and analysis platform. J. Cheminform. 2012, 4, 17. [Google Scholar] [CrossRef]
- Oda, A.; Takahashi, O. Validation of ArgusLab Efficiencies for Binding Free Energy Calculations. Chem-Bio Inform. J. 2009, 9, 52–61. [Google Scholar] [CrossRef]
- Morris, G.M.; Goodsell, D.S.; Huey, R.; Hart, W.E.; Halliday, S.; Belew, R.; Olson, A.J. AutoDock. In Automated Docking of Flexible Ligands to Receptor-User Guide; The Scripps Research Institute, Molecular Graphics Laboratory, Department of Molecular Biology: La Jolla, CA, USA, 2001. [Google Scholar]
- Biovia, D.S. Discovery Studio Visualizer; BIOVIA: San Diego, CA, USA, 2017. [Google Scholar]
- Loschen, C.; Klamt, A. COSMOquick: A Novel Interface for Fast σ-Profile Composition and Its Application to COSMO-RS Solvent Screening Using Multiple Reference Solvents. Ind. Eng. Chem. Res. 2012, 51, 14303–14308. [Google Scholar] [CrossRef]
- Gautam, L.K.; Sharma, P.; Capalash, N. Structural insight into substrate binding of Acinetobacter baumannii polyphosphate-AMP phosphotransferase (PPK2), a novel drug target. Biochem. Biophys. Res. Commun. 2022, 626, 107–113. [Google Scholar] [CrossRef]
- Bekker, H.; Berendsen, H.; Dijkstra, E.; Achterop, S.; Vondrumen, R.; VANDERSPOEL, D.; Sijbers, A.; Keegstra, H.; Renardus, M. Gromacs-a parallel computer for molecular-dynamics simulations. In Proceedings of the 4th International Conference on Computational Physics (PC 92), Annecy, France, 21–25 September 1992; pp. 252–256. [Google Scholar]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef]
- Schüttelkopf, A.W.; Van Aalten, D.M.F. PRODRG: A tool for high-throughput crystallography of protein–ligand complexes. Acta Crystallogr. D Biol. Crystallogr. 2004, 60, 1355–1363. [Google Scholar] [CrossRef] [PubMed]
- Swamy, P.M.G.; Abbas, N.; Dhiwar, P.S.; Singh, E.; Ghara, A.; Das, A. Discovery of potential Aurora-A kinase inhibitors by 3D QSAR pharmacophore modeling, virtual screening, docking, and MD simulation studies. J. Biomol. Struct. Dyn. 2021, 1–22. [Google Scholar] [CrossRef]
- Bode, J.W. Reactor ChemAxon Ltd., Maramaros koz 2/a, Budapest, 1037 Hungary. www.chemaxon.com. Contact ChemAxon for pricing information. J. Am. Chem. Soc. 2004, 126, 15317. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | Residues | Druggability | Pred.Max pKd |
|---|---|---|---|
| 1 | GLY1, ASN2, GLN3, ILE4, GLY5, LYS6, LYS7, PHE8, TRP9, ASP33, ARG34, ILE35, ASN36, VAL37, TYR38, PHE39, THR40, GLU41, PRO49, ARG50, ALA51, VAL52, LEU53, VAL54, ASP55, LEU56, GLU57, PRO58, GLY59, THR60, MET61, ILE64, PHE73, PHE78, GLY84, ALA85, GLY86, ASN87, ASN88, VAL104, VAL107, VAL108, ARG109, LYS110, GLU111, ALA112, GLU113, ASN114, SER115, ASP116, LEU117, LEU118, GLN119, GLY120, PHE121, GLN122, VAL123, CYS124, HIS125, SER126, LEU127, GLY128, GLY129, GLY130, THR131, GLY132, SER133, GLY134, MET135, GLY136, THR137, LEU139, ILE140, ILE143, PHE147, ARG150, MET151, MET152, CYS153, PHE155, VAL157, MET158, PRO159, ASP165, THR166, GLU169, ASN172, ASN192, LEU195, TYR210, LEU213, ASN214, VAL217, MET221, VAL224, THR225, SER227, LEU228, ARG229, PHE230, SER236, ASP237, LEU238, ARG239 | Strong | 11.69 |
| 2 | GLU10, VAL11, ILE12, ASP14, GLU15, MET151, CYS153, PHE155, MET158, VAL181, GLN186, VAL187, MET188, CYS189, ILE190, HIS215, SER218, GLN219, VAL220, MET221, SER222, GLY223, VAL224, THR225, ALA226, ARG229, PHE230, PRO231, LEU234, SER236, ASP237, LEU238, ARG239, LYS240, LEU241, ALA242, VAL243, ASN244, LEU245, ILE246, PRO247, PHE248, ARG250, LEU251, HIS252, PHE253, PHE254, MET255, VAL256, GLY257, TYR258, ALA259, PRO260, LEU261, THR262, ARG270, ASN271, PHE272, ASN273, VAL274, ALA275, GLU276, ILE277, THR278, GLN279, GLN280, ILE281, PHE282, ASP283, ALA284, ASN286, ILE287, MET288, ALA289, ALA290, CYS291, ASP292, PRO293, ARG294, HIS295, GLY296, ARG297, TYR298, LEU299, THR300, ALA301, SER302, ALA303, VAL304, PHE305, ARG306, GLY307, LYS308, VAL309, GLU313, VAL314, ASP315, GLN316, GLN317, MET318, LEU319, ASN320 | Strong | 11.61 |
| 3 | ARG144, PRO148, ASP149, ARG150, MET151, GLN179, LEU180, VAL181, GLU182, ASN183, ALA184, ASP185, GLN186, LEU238, ARG239, LYS240, LEU241, ALA242, VAL243, ASN244, LEU245, ILE246, PRO247, PHE248, PRO249, ARG250, LEU251, HIS252 | Weak | 11.48 |
| 4 | ASP14, GLU15, HIS16, ASP27, ASP28, PRO29, LEU30, GLN31, LEU32, GLN219, GLY223, ALA226, SER227, LEU228, ARG229, PHE230, PRO231, GLY232, GLN233, LEU234, ASN235, SER236, ASP237, LYS240, LEU241, ASN244, LEU245, TYR258, ALA259, PRO260, PHE282, THR300, ALA301, SER302, ALA303, VAL304, PHE305, ARG306, GLY307, LYS308, VAL309, SER310, THR311, LYS312, VAL314, ASP315, GLN316, MET318 | Weak | 11.31 |
| 5 | ASN2, GLN3, ILE4, LYS6, LYS7, GLU10, GLY59, THR60, MET61, ASP62, ALA63, ILE64, ARG65, SER66, GLY67, VAL68, ASN209, TYR210, SER211, ASP212, ASN214, HIS215 | Weak | 8.39 |
| 6 | SER160, PRO161, LYS162, ASP191, ASN192, GLU193, ALA194, LEU195, TYR196, ASP197, ILE198, ARG201, ASP283, ALA284, LYS285, ASN286, ILE287, MET288, ALA289, ALA290, CYS291, ASP292, PRO293, ARG294, HIS295 | Weak | 7.32 |
| 7 | ASP55, LEU56, PRO58, VAL79, PHE80, GLY81, GLN82, SER83, GLY84, ALA85, LYS91, THR95, GLU96, GLY97, GLU99, LEU100, VAL101, SER103, MET135 | Weak | 7.29 |
| 8 | ASP197, PHE200, ARG201, THR202, LEU203, LYS204, PRO260, LEU261, THR262, ALA263, PRO264, ASN265, SER266, THR267, TYR269, ARG270, ASN271, GLU276, GLN279, GLN280, ILE281, PHE282, ASP283, ALA284, LYS285, ASN286 | Weak | 6.73 |
| 9 | ALA85, GLY86, ASN87, ASN88, TRP89, ALA90, LYS91, TYR94, THR166A, VAL167, VAL168, PRO170, TYR171, ASN172, THR174, LEU175 | Weak | 6.43 |
| Compounds | Smile | IUPAC Name | Binding Energy (kcal/mol) | Inhibition Constant, Ki | |
|---|---|---|---|---|---|
| Arguslab | Autodock | ||||
 | C1(=C(C(=CC3=C1C(C2C(C=C(C(=C2O)CC=C(C)C)O)O3)=O)O)OC)CC=C(C)C | 1,3,6-Trihydroxy-7-methoxy-2,8-bis(3-methylbut-2-en-1-yl)-9H-xanthen-9-one | −11.22 | −10.18 | 34.35 nM |
 | C1(=C(C(=CC3=C1C(C2C(C=CC(=C2O)CC=C(C)C)O3)=O)O)OC)CC=C(C)C | 1,6-dihydroxy-7-methoxy-2,8-bis(3-methylbut-2-en-1-yl)-9H-xanthen-9-one | −11.81 | −10.56 | 18.02 nM |
 | C1(=CC(=CC3=C1C(C2C(C=CC(=C2O)CC=C(C)C)O3)=O)O)CC=C(C)C | 1,6-dihydroxy-2,8-bis(3-methylbut-2-en-1-yl)-9H-xanthen-9-one | −12.17 | −10.52 | 19.43 nM |
| Quantum Chemistry Parameters | Alpha-Mangostin | 1,6-dihydroxy-7-methoxy-2,8-bis(3-methylbut-2-en-1-yl)-9H-xanthen-9-one | 1,6-dihydroxy-2,8-bis(3-methylbut-2-en-1-yl)-9H-xanthen-9-one |
|---|---|---|---|
| Electron affinity | 0.951 | 0.723 | 0.653 |
| Chemical potential | −4.874 | −4.807 | −4.739 |
| Hardness | 3.923 | 4.084 | 4.086 |
| Electrophilicity | 3.028 | 2.829 | 2.748 |
| HOMO | −8.797 | −8.890 | −8.825 |
| LUMO | −0.951 | −0.723 | −0.653 |
| Properties | Alpha-Mangostin | 1,6-dihydroxy-7-methoxy-2,8-bis(3-methylbut-2-en-1-yl)-9H-xanthen-9-one | 1,6-dihydroxy-2,8-bis(3-methylbut-2-en-1-yl)-9H-xanthen-9-one |
|---|---|---|---|
| Physicochemical Properties | |||
| Formula | C24H28O6 | C24H28O5 | C23H26O4 |
| Molecular weight | 412.48 g/mol | 396.48 g/mol | 366.45 g/mol |
| Num. H-bond acceptors | 6 | 5 | 4 |
| Num. H-bond donors | 3 | 2 | 2 |
| Molar Refractivity | 116.12 | 114.55 | 108.06 |
| Pharmacokinetics | |||
| GI absorption | High | High | High |
| BBB permeant | No | No | No |
| P-gp substrate | Yes | No | No |
| CYP1A2 inhibitor | No | No | No |
| CYP2C19 inhibitor | No | No | Yes |
| CYP2C9 inhibitor | Yes | Yes | Yes |
| CYP2D6 inhibitor | No | No | No |
| CYP3A4 inhibitor | Yes | Yes | Yes |
| Compounds | Lipinski | Veber | Egan |
|---|---|---|---|
| Alpha-mangostin | Yes; 0 violation | Yes | Yes |
| 1,6-dihydroxy-7-methoxy-2,8-bis(3-methylbut-2-en-1-yl)-9H-xanthen-9-one | Yes; 0 violation | Yes | Yes |
| 1,6-dihydroxy-2,8-bis(3-methylbut-2-en-1-yl)-9H-xanthen-9-one | Yes; 0 violation | Yes | Yes |
| Targeted Protein | Protein Sequences |
|---|---|
| Beta-Tubulin (Acanthamoeba keratitis) | GNQIGKKFWEVIADEHGIDGTGKYIGDDPLQLDRINVYFTEASGGNYVPRAVLVDLEPGTMDAIRSGVHGKLFRPDNFVFGQSGAGNNWAKGHYTEGAELVDSVLDVVRKEAENSDLLQGFQVCHSLGGGTGSGMGTLLISKIREEFPDRMMCTFSVMPSPKVSDTVVEPYN ATLSVHQLVENADQVMCIDNEALYDICFRTLKLSNPNYSDLNHLVSQVMSGVTASLR FPGQLNSDLRKLAVNLIPFPRLHFFMVGYAPLTAPNSTAYRNFNVAEITQQIFDAKNIM AACDPRHGRYLTASAVFRGKVSTKEVDQQMLN |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ongtanasup, T.; Mazumder, A.; Dwivedi, A.; Eawsakul, K. Homology Modeling, Molecular Docking, Molecular Dynamic Simulation, and Drug-Likeness of the Modified Alpha-Mangostin against the β-Tubulin Protein of Acanthamoeba Keratitis. Molecules 2022, 27, 6338. https://doi.org/10.3390/molecules27196338
Ongtanasup T, Mazumder A, Dwivedi A, Eawsakul K. Homology Modeling, Molecular Docking, Molecular Dynamic Simulation, and Drug-Likeness of the Modified Alpha-Mangostin against the β-Tubulin Protein of Acanthamoeba Keratitis. Molecules. 2022; 27(19):6338. https://doi.org/10.3390/molecules27196338
Chicago/Turabian StyleOngtanasup, Tassanee, Anisha Mazumder, Anupma Dwivedi, and Komgrit Eawsakul. 2022. "Homology Modeling, Molecular Docking, Molecular Dynamic Simulation, and Drug-Likeness of the Modified Alpha-Mangostin against the β-Tubulin Protein of Acanthamoeba Keratitis" Molecules 27, no. 19: 6338. https://doi.org/10.3390/molecules27196338
APA StyleOngtanasup, T., Mazumder, A., Dwivedi, A., & Eawsakul, K. (2022). Homology Modeling, Molecular Docking, Molecular Dynamic Simulation, and Drug-Likeness of the Modified Alpha-Mangostin against the β-Tubulin Protein of Acanthamoeba Keratitis. Molecules, 27(19), 6338. https://doi.org/10.3390/molecules27196338