

Lewis Acid-Induced Dinitrogen Cleavage in an Anionic Side-on End-on Bound Dinitrogen Diniobium Hydride Complex

Abstract
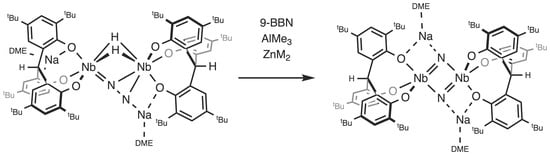
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
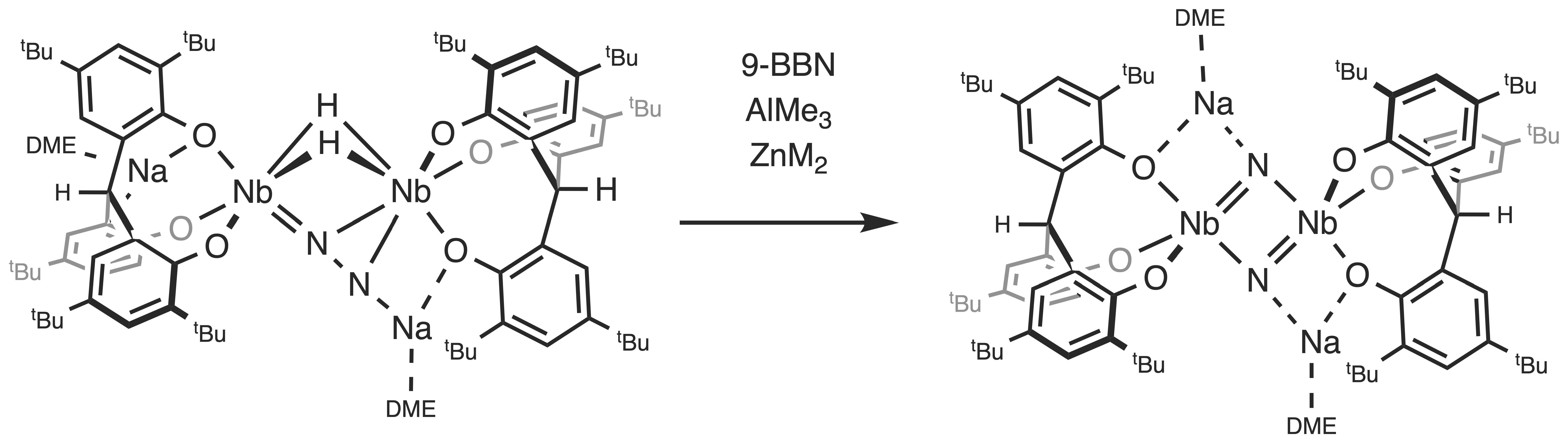
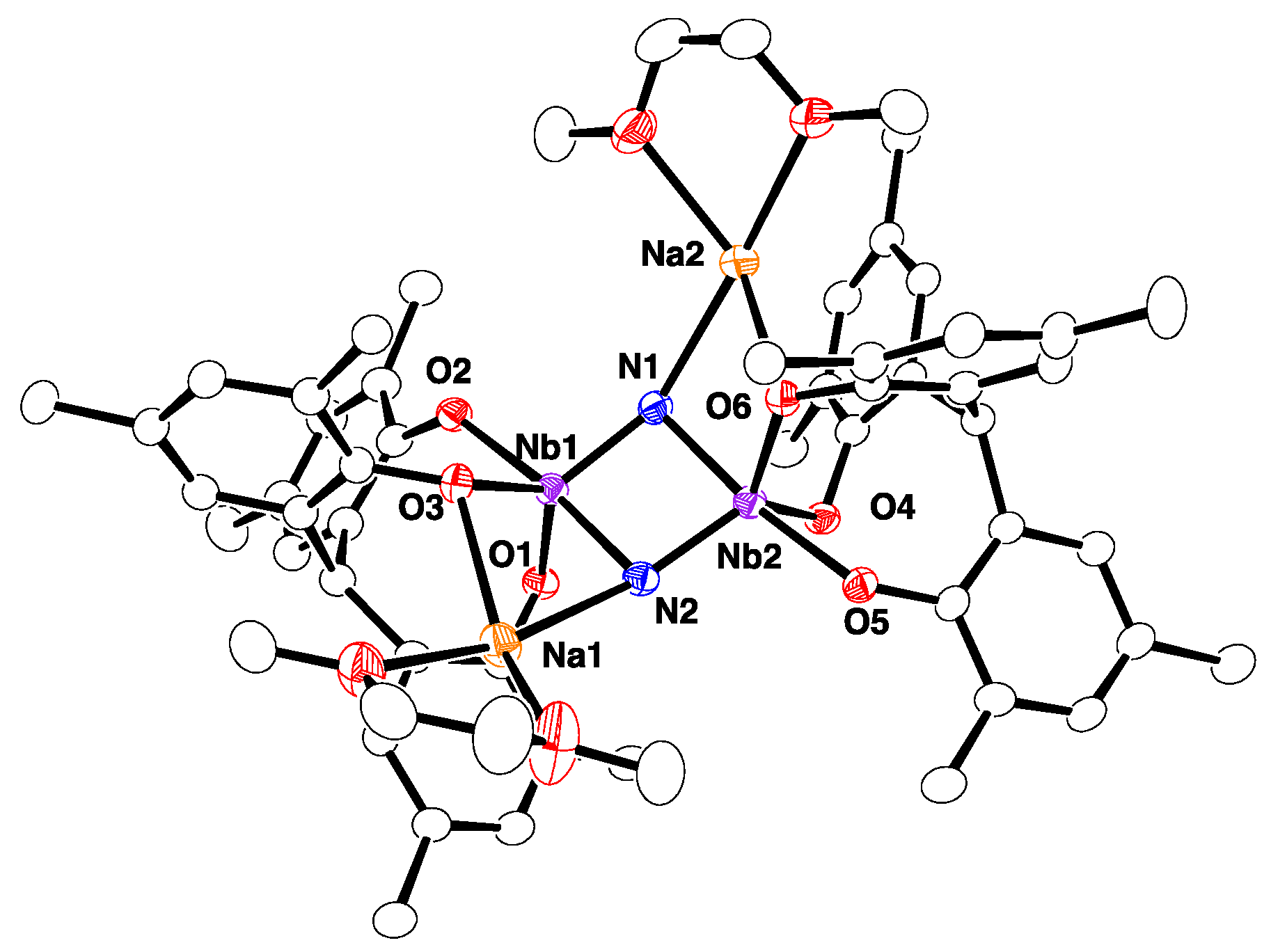
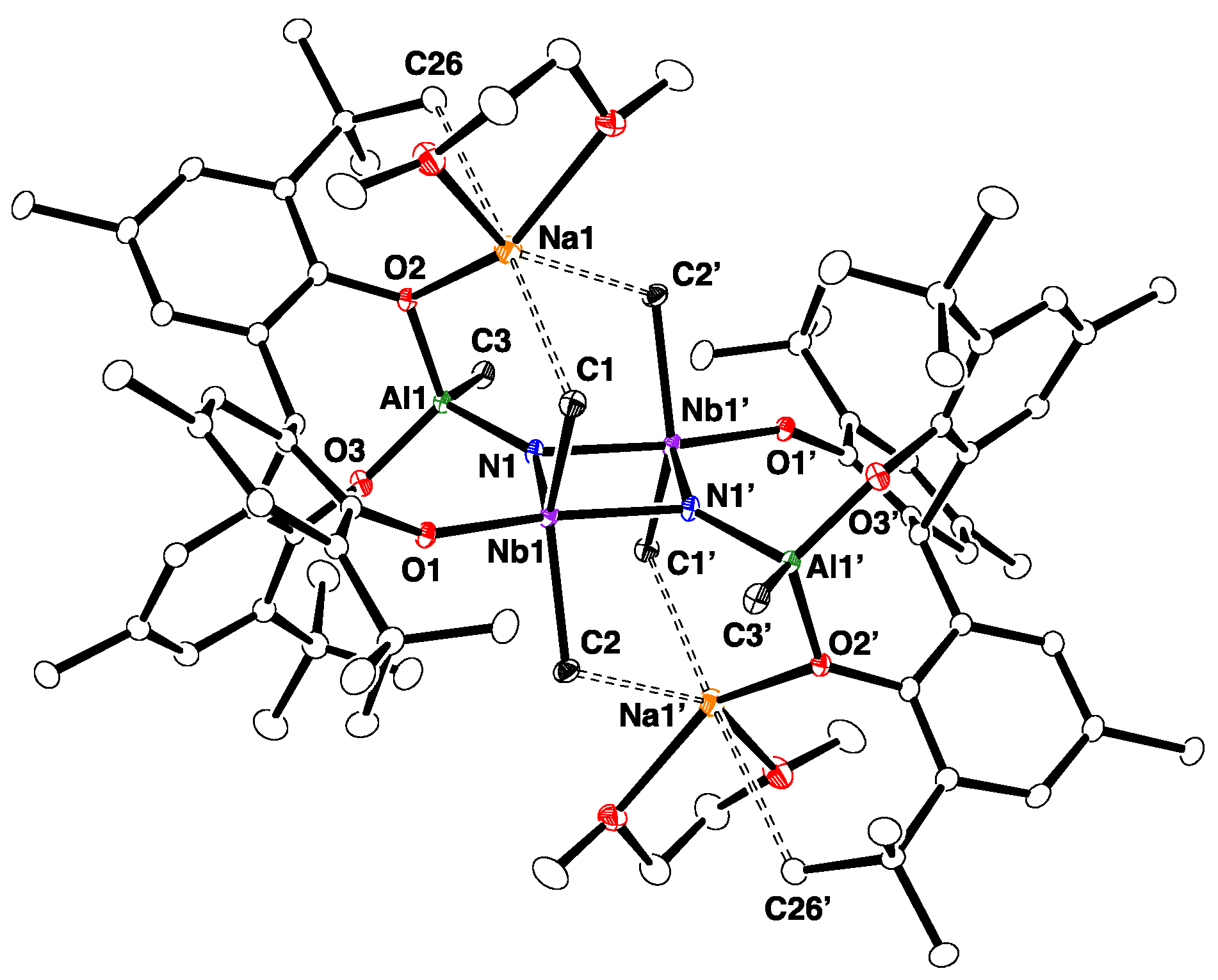
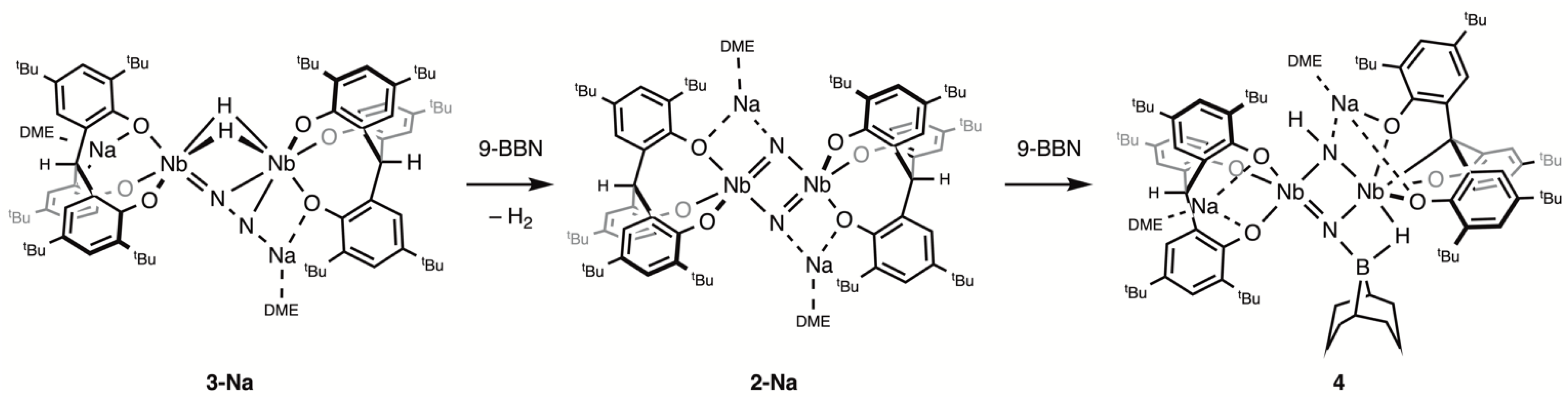
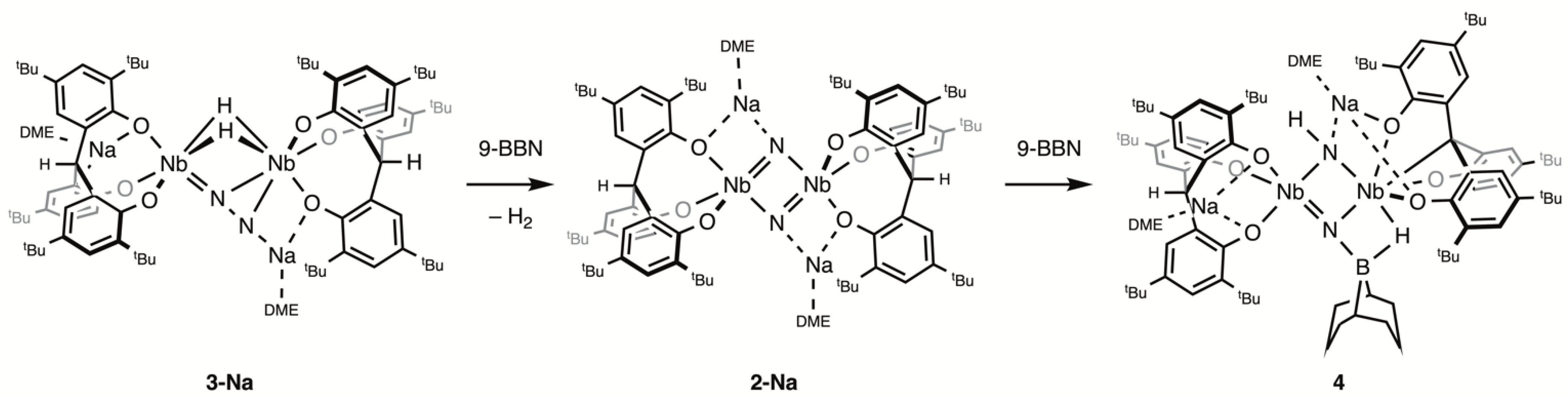
2.1. Reaction with 9-BBN
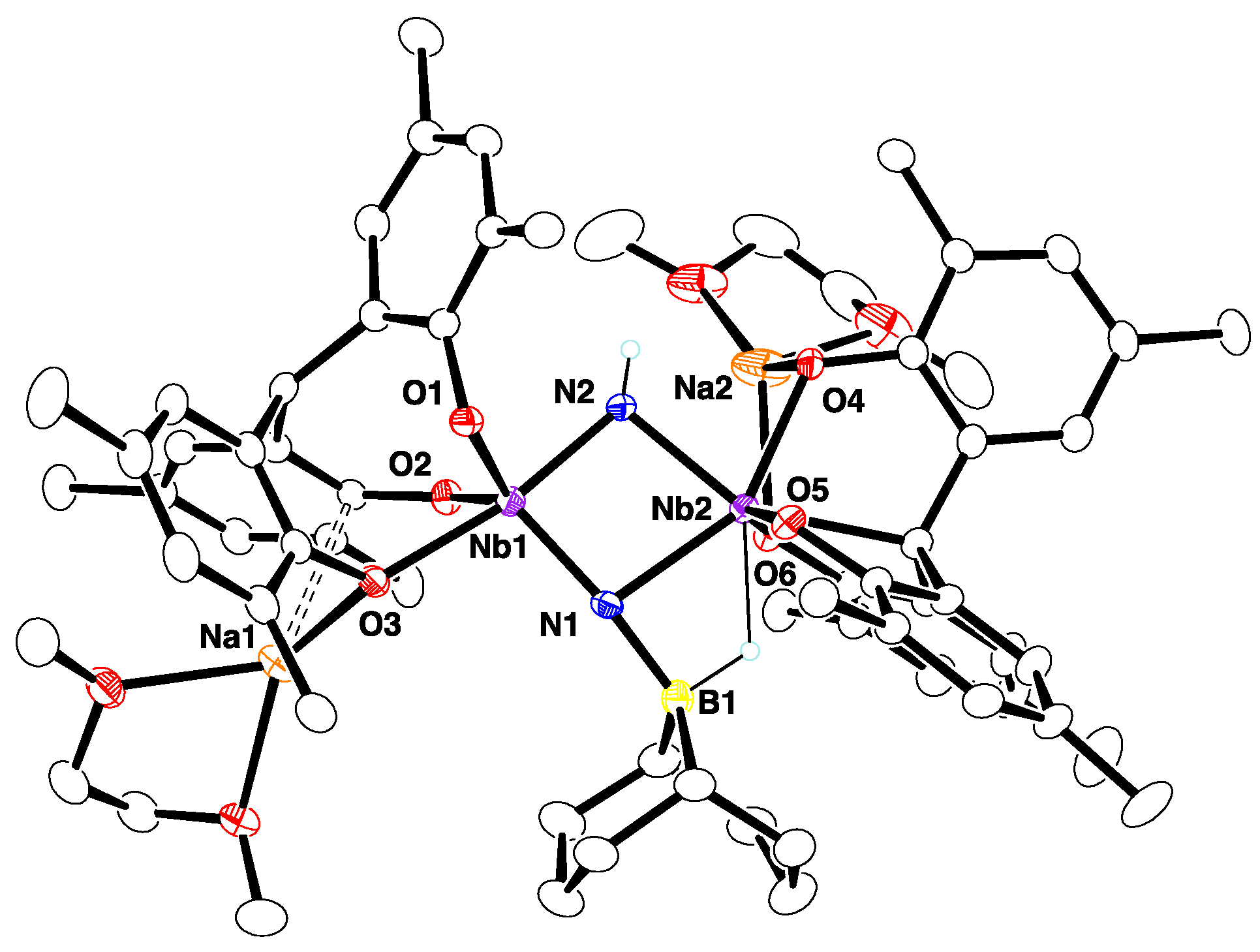
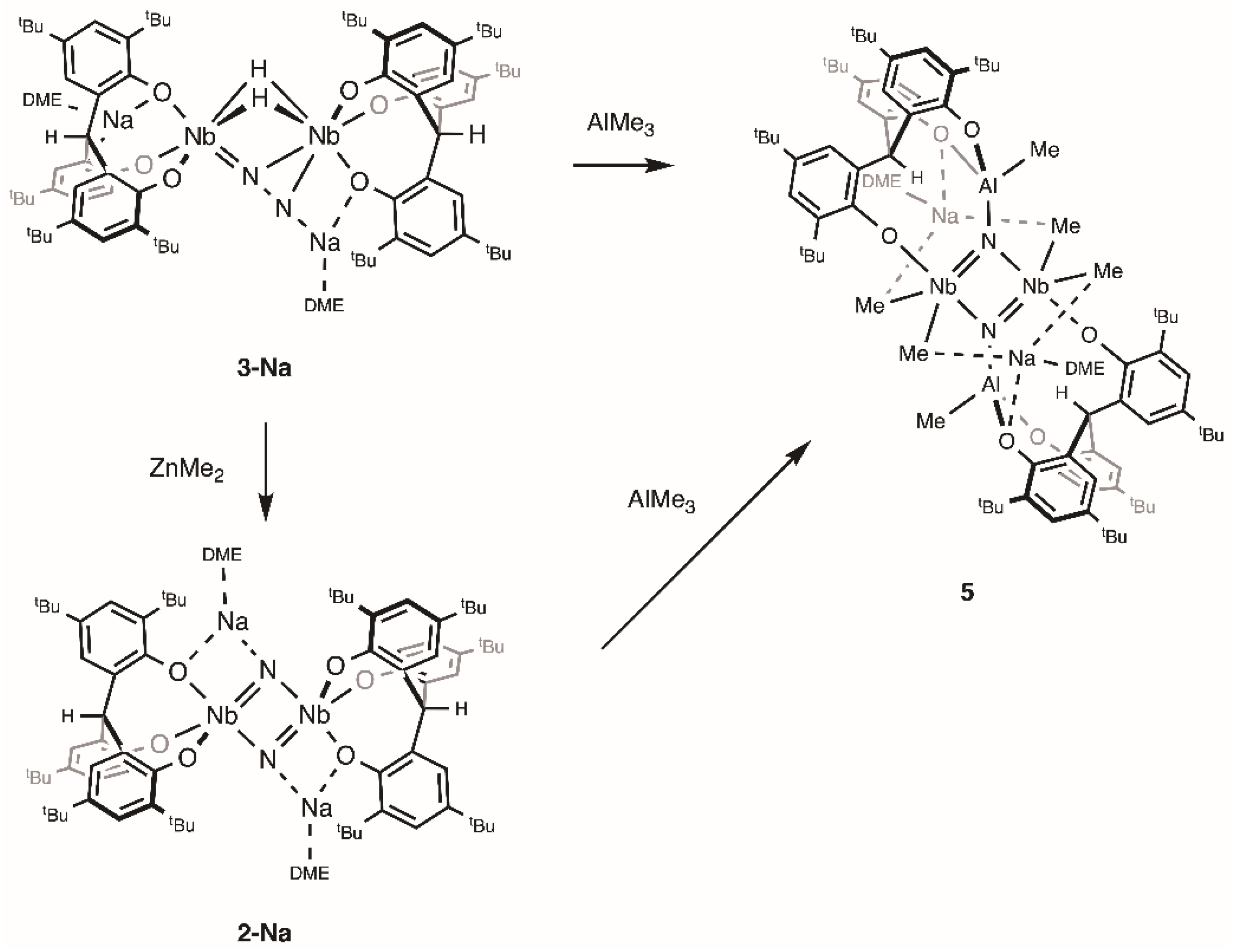
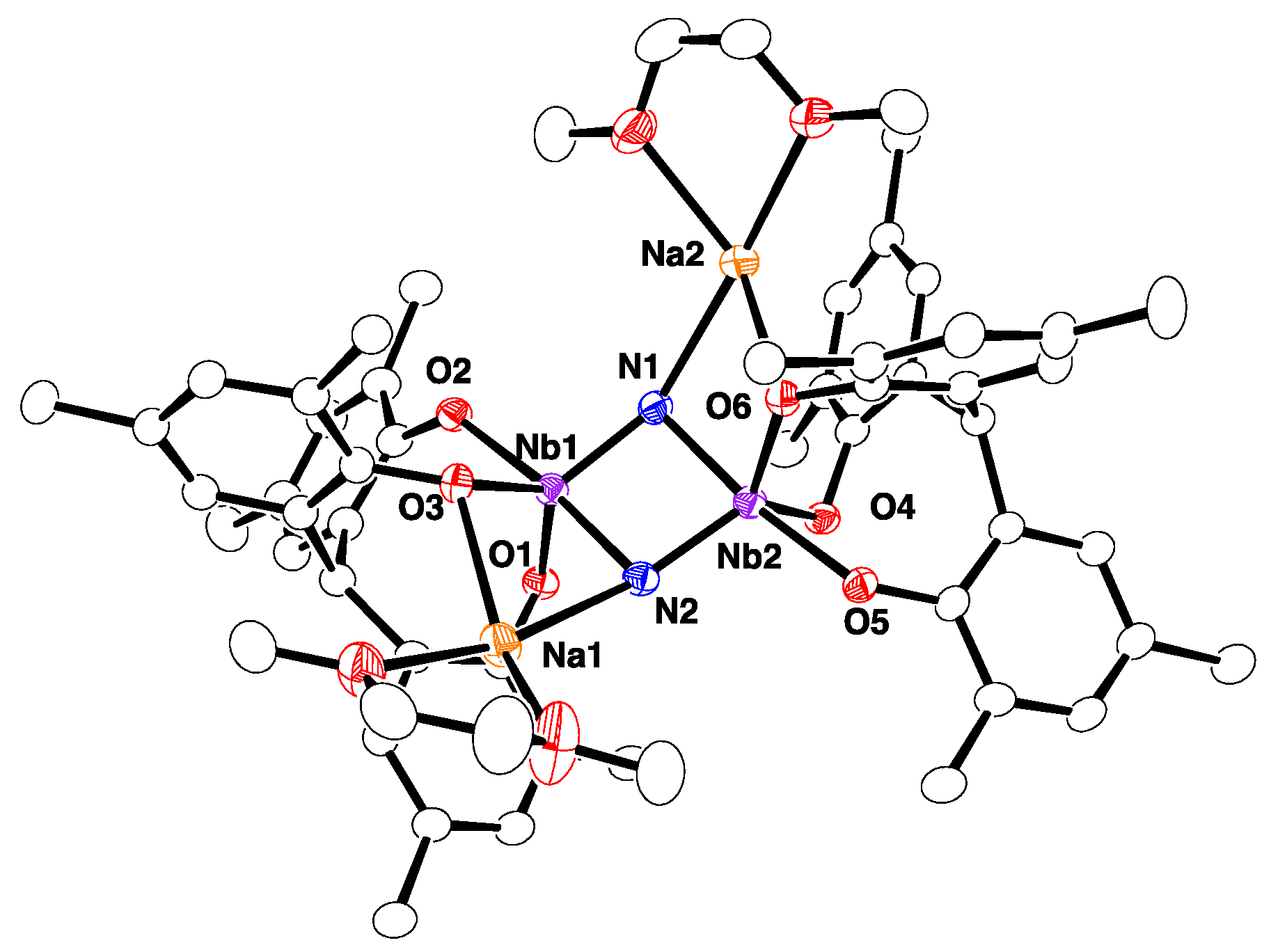
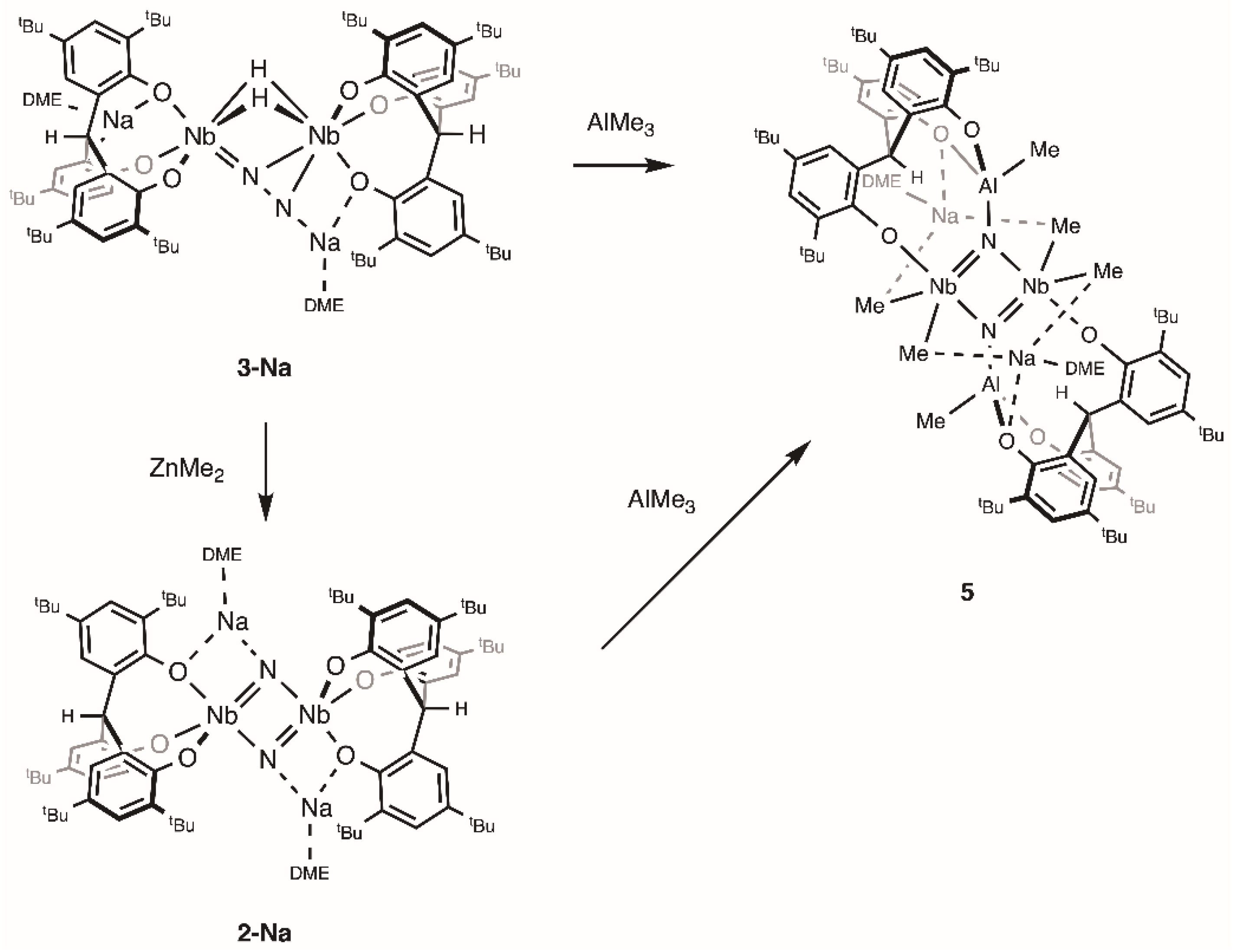
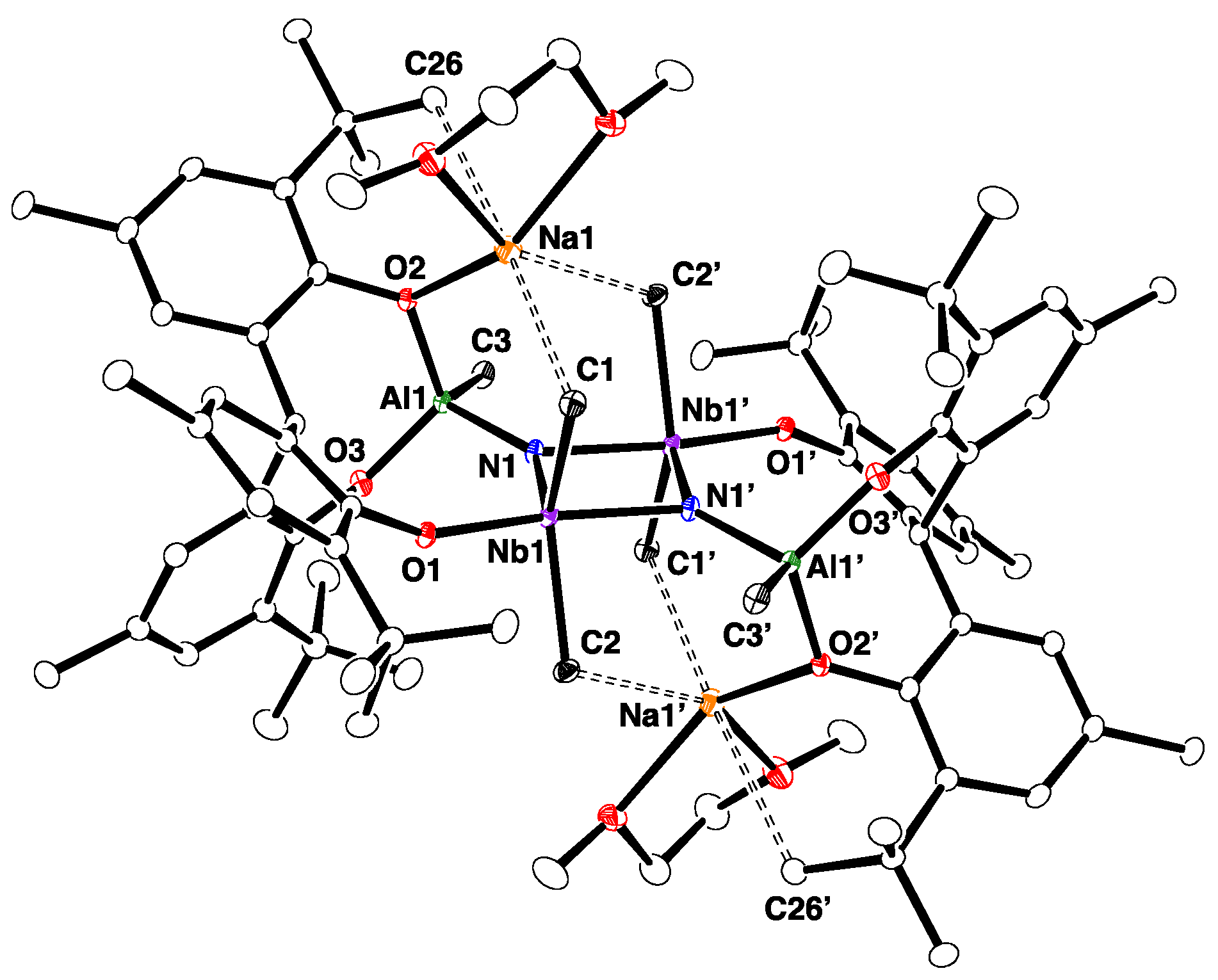
2.2. Reactions with ZnMe2 and AlMe3
3. Conclusions
4. Materials and Methods
4.1. General Information
4.2. Synthesis of 2-Na
4.3. Synthesis of 2-Na-15N
4.4. Synthesis of 4
4.5. Reaction of 2-Na with 9-BBN
4.6. Reaction of 2-K-15N with 9-BBN
4.7. Reaction of 3-Na with ZnMe2
4.8. Synthesis of 5
4.9. Reaction of 2-Na with AlMe3
4.10. Reaction of 2-K-15N with AlMe3
4.11. X-ray Crystallography
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Allen, A.D.; Senoff, C.V. Nitrogenpentammineruthenium(II) complexes. Chem. Commun. 1965, 621–622. [Google Scholar] [CrossRef]
- Hidai, M.; Mizobe, Y. Recent Advances in the Chemistry of Dinitrogen Complexes. Chem. Rev. 1995, 95, 1115–1133. [Google Scholar] [CrossRef]
- Fryzuk, M.D.; Johnson, S.A. The Continuing Story of Dinitrogen Activation. Coord. Chem. Rev. 2000, 200, 379–409. [Google Scholar]
- Walter, M.D. Recent Advances in Transition Metal-Catalyzed Dinitrogen Activation. Adv. Organomet. Chem. 2016, 65, 261–377. [Google Scholar]
- Burford, R.J.; Fryzuk, M.D. Examining the Relationship between Coordination Mode and Reactivity of Dinitrogen. Nat. Chem. Rev. 2017, 1, 1–13. [Google Scholar]
- Kim, S.; Loose, F.; Chirik, P.J. Beyond Ammonia: Nitrogen–Element Bond Forming Reactions with Coordinated Dinitrogen. Chem Rev. 2020, 120, 5637–5681. [Google Scholar]
- Singh, D.; Buratto, W.R.; Torres, J.F.; Murray, L.J. Activation of Dinitrogen by Polynuclear Metal Complexes. Chem. Rev. 2020, 120, 5517–5581. [Google Scholar]
- Masero, F.; Perrin, M.A.; Dey, S.; Mougel, V. Dinitrogen Fixation: Rationalizing Strategies Utilizing Molecular Complexes. Chem. Eur. J. 2021, 27, 3892–3928. [Google Scholar]
- Tanabe, Y.; Nishibayashi, Y. Comprehensive Insights into Synthetic Nitrogen Fixation Assisted by Molecular Catalysts under Ambient or Mild Conditions. Chem. Soc. Rev. 2021, 50, 5201–5242. [Google Scholar]
- Lv, Z.-J.; Wei, J.; Zhang, W.-X.; Chen, P.; Deng, D.; Shi, Z.-J.; Xi, Z. Direct Transformation of Dinitrogen: Synthesis of N-Containing Organic Compounds via N–C bond Formation. Natl. Sci. Rev. 2020, 7, 1564–1583. [Google Scholar]
- Fryzuk, M.D.; Johnson, S.A.; Retting, S.J. New Mode of Coordination for the Dinitrogen Ligand: A Dinuclear Tantalum Complex with a Bridging N2 Unit That Is Both Side-On and End-On. J. Am. Chem. Soc. 1998, 120, 11024–11025. [Google Scholar] [CrossRef]
- Fryzuk, M.D.; Johnson, S.A.; Patrick, B.O.; Albinati, A.; Mason, S.A.; Koetzle, T.F. New Mode of Coordination for the Dinitrogen Ligand: Formation, Bonding, and Reactivity of a Tantalum Complex with a Bridging N2 Unit That Is Both Side-On and End-On. J. Am. Chem. Soc. 2001, 123, 3960–3973. [Google Scholar] [CrossRef] [PubMed]
- Studt, F.; MacKay, B.A.; Fryzuk, M.D.; Tuczek, F. Spectroscopic Properties and Quantum Chemistry-Based Normal Coordinate Analysis (QCB-NCA) of a Dinuclear Tantalum Complex Exhibiting the Novel Side-On End-On Bridging Geometry of N2: Correlations to Electronic Structure and Reactivity. J. Am. Chem. Soc. 2004, 126, 280–290. [Google Scholar] [CrossRef] [PubMed]
- Burford, R.J.; Yeo, A.; Fryzuk, M.D. Dinitrogen Activation by Group 4 and Group 5 metal Complexes Supported by Phosphine-Amido Containing Ligand Manifolds. Coord. Chem. Rev. 2017, 334, 84–99. [Google Scholar] [CrossRef]
- Wang, B.; Luo, G.; Nishiura, M.; Hu, S.; Shima, T.; Luo, Y.; Hou, Z. Dinitrogen Activation by Dihydrogen and a PNP-Ligated Titanium Complex. J. Am. Chem. Soc. 2017, 139, 1818–1821. [Google Scholar] [CrossRef]
- Mo, Z.; Shima, T.; Hou, Z. Synthesis and Diverse Transformation of a Dinitrogen Dititanium Hydride Complex Bearing Rigid Acridane-Based PNP-Pincer Ligands. Angew. Chem. Int. Ed. 2020, 59, 8635–8644. [Google Scholar] [CrossRef]
- Pun, D.; Lobkovsky, E.; Chirik, P.J. Indenyl Zirconium Dinitrogen Chemistry: N2 Coordination to an Isolated Zirconium Sandwich and Synthesis of Side-on, End-on Dinitrogen Compounds. J. Am. Chem. Soc. 2008, 130, 6047–6054. [Google Scholar] [CrossRef]
- Geri, J.B.; Shanahan, J.P.; Szymczak, N.K. Testing the Push-Pull Hypothesis: Lewis Acid Augmented N2 Activation at Iron. J. Am. Chem. Soc. 2017, 139, 5952–5956. [Google Scholar] [CrossRef] [Green Version]
- Apps, S.L.; White, A.J.P.; Miller, P.W.; Long, N.J. Synthesis and Reactivity of an N-Triphos Mo(0) Dinitrogen Complex. Dalton Trans. 2018, 47, 11386–11395. [Google Scholar] [CrossRef]
- Takahashi, T.; Kodama, T.; Watakabe, A.; Uchida, Y.; Hidai, M. Preparation and Characterization of Novel MU3-N2 Mixed Metal Complexes. J. Am. Chem. Soc. 1983, 105, 1680–1682. [Google Scholar] [CrossRef]
- Klein, H.F.; Ellrich, K.; Ackermann, K. Hetero-Bimetallic Dinitrogen Activation: X-ray Structure of the (Me3P)3CoN2AlMe2 Dimer. J. Chem. Soc. Chem. Commun. 1983, 16, 888–889. [Google Scholar] [CrossRef]
- Chatt, J.; Crabtree, R.H.; Jeffery, E.A.; Richards, R.L. The Basic Strengths of some Dinitrogen Complexes of Molybdenum(0), Tungsten(0), Rhenium(0), and Osmium(II). J. Chem. Soc. Dalton Trans. 1973, 11, 1167–1172. [Google Scholar] [CrossRef]
- Studt, F.; MacKay, B.A.; Johnson, S.A.; Patrick, B.O.; Fryzuk, M.D.; Tuczek, F. Lewis Adducts of the Side-On End-On Dinitrogen-Bridged Complex [{(NPN)Ta}2(μ-H)2(μ-η1:η2-N2)] with AlMe3, GaMe3, and B(C6F5)3: Synthesis, Structure, and Spectroscopic Properties. Chem. Eur. J. 2005, 11, 604–618. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, S.; Ishida, Y.; Kameo, H.; Sakaki, S.; Kawaguchi, H. Couterion Dependence of Dinitrogen Activation and Functionalization by a Diniobium Hydride Anion. Angew. Chem. Int. Ed. 2020, 59, 13444–13450. [Google Scholar] [CrossRef] [PubMed]
- Akagi, F.; Matsuo, T.; Kawaguchi, H. Dinitrogen Cleavage by a Diniobium Tetrahydride Complex: Formation of a Nitride and Its Conversion into Imide Species. Angew. Chem. Int. Ed. 2007, 46, 8778–8781. [Google Scholar] [CrossRef] [PubMed]
- Caselli, A.; Solari, E.; Scopelliti, R.; Floriani, C.; Re, N.; Rizzoli, C.; Chiesi-Villa, A. Dinitrogen Rearranging over a Metal–Oxo Surface and Cleaving to Nitride: From the End-On to the Side-On Bonding Mode, to the Stepwise Cleavage of the N≡N Bonds Assisted by NbIII-calix[4]arene. J. Am. Chem. Soc. 2000, 122, 3652–3670. [Google Scholar] [CrossRef]
- Coffinet, A.; Specklin, D.; Vendier, L.; Etienne, M.; Simonneau, A. Frustrated Lewis Pair Chemistry Enables N2 Borylation by Formal 1,3-Addition of a B−H Bond in the Coordination Sphere of Tungsten. Chem. Eur. J. 2019, 25, 14300–14303. [Google Scholar] [CrossRef]
- Coffinet, A.; Zhang, D.; Vendier, L.; Bontemps, S.; Simonneau, A. Borane-Catalysed Dinitrogen Borylation by 1,3-B–H Bond Addition. Dalton Trans. 2021, 50, 5582–5589. [Google Scholar] [CrossRef]
- Semproni, S.; Chirik, P.J. Dinitrogen Borylation with Group 4 Metallocene Complexes. Eur. J. Inorg. Chem. 2013, 2013, 3907–3915. [Google Scholar] [CrossRef]
- Ishino, H.; Ishii, Y.; Hidai, M. Synthsis of Boryldiazenido Complexes from Tungsten Dinitorgen Complexes. Chem. Lett. 1998, 27, 677–678. [Google Scholar] [CrossRef]
- Fryzuk, M.D.; MacKay, B.A.; Johnson, S.A.; Patrick, B.O. Hydroboration of Coordinated Dinitrogen: A New Reaction for the N2 ligand that Results in Its Functionalization and Cleavage. Angew. Chem. Int. Ed. 2002, 41, 3709–3712. [Google Scholar] [CrossRef]
- MacKay, B.A.; Johnson, S.A.; Patrick, B.O.; Fryzuk, M.D. Functionalization and Cleavage of Coordinated Dinitrogen via Hydroboration Using Primary and Secondary Boranes. Can. J. Chem. 2005, 83, 315–323. [Google Scholar] [CrossRef]
- Espada, M.F.; Bennaamane, S.; Liao, Q.; Saffon-Merceron, N.; Massou, S.; Clot, E.; Nebra, N.; Fustier-Boutignon, M.; Mézailles, N. Room-Temperature Functionalization of N2 to Borylamine at a Molybdenum Complex. Angew. Chem. Int. Ind. 2018, 57, 12865–12868. [Google Scholar] [CrossRef] [PubMed]
- Hughes, E.W. The Crystal Structure of Ammonia-Borane, H3NBH3. J. Am. Chem. Soc. 1956, 78, 502–503. [Google Scholar] [CrossRef]
- Li, J.; Gao, D.; Hu, H.; Cui, C. Reaction of a Bulky Amine Borane with Lanthanide Trialkyls. Formation of Alkyl Lanthanide Imide Complexes. New J. Chem. 2015, 39, 7567–7570. [Google Scholar] [CrossRef]
- Gardiner, M.G.; Raston, C.L.; Skelton, B.W.; White, A.H. Anionic and Neutral Aluminum Bis(N,N′-di-tert-butylethylenediamide) Complexes: [Al{[N(t-Bu)CH2]2}2]− and [Al{[N(t-Bu)CH2]2}2]•. Inorg. Chem. 1997, 36, 12795–12803. [Google Scholar] [CrossRef]
- Schulz, S.; Thomas, F.; Priesmann, W.M.; Neiger, M. Syntheses and X-ray Structures of Base-Stabilized Iminoalanes. Organometallics 2006, 25, 1392–1398. [Google Scholar] [CrossRef]
- Dagorne, S.; Lavanant, L.; Welter, R.; Chassenieux, C.; Haquette, P.; Jaouen, G. Synthesis and Structural Characterization of Neutral and Cationic Alkylaluminum Complexes Based on Bidentate Aminophenolate Ligands. Organometallics 2003, 22, 3732–3741. [Google Scholar] [CrossRef]
- Rutherford, D.; Atwood, D.A. Unusual Alkylaluminum Amides, Adducts, and Aluminates Containing Lithium. J. Am. Chem. Soc. 1996, 118, 11535–11540. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXT—Integrated Space-Group and Crystal-Structure Determination. Acta Cryst. 2015, A71, 3–8. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal Structure Refinement with SHELXL. Acta Cryst. 2015, C71, 3–8. [Google Scholar]
- Spek, A.L. Structure Validation in Chemical Crystallography. Acta Cryst. 2009, 65, 148–155. [Google Scholar] [CrossRef] [PubMed]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A Complete Structure Solution, Refinement and Analysis Program. J. Appl. Cryst. 2009, 42, 339–341. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Suzuki, N.; Ishida, Y.; Kawaguchi, H. Lewis Acid-Induced Dinitrogen Cleavage in an Anionic Side-on End-on Bound Dinitrogen Diniobium Hydride Complex. Molecules 2022, 27, 5553. https://doi.org/10.3390/molecules27175553
Suzuki N, Ishida Y, Kawaguchi H. Lewis Acid-Induced Dinitrogen Cleavage in an Anionic Side-on End-on Bound Dinitrogen Diniobium Hydride Complex. Molecules. 2022; 27(17):5553. https://doi.org/10.3390/molecules27175553
Chicago/Turabian StyleSuzuki, Naofumi, Yutaka Ishida, and Hiroyuki Kawaguchi. 2022. "Lewis Acid-Induced Dinitrogen Cleavage in an Anionic Side-on End-on Bound Dinitrogen Diniobium Hydride Complex" Molecules 27, no. 17: 5553. https://doi.org/10.3390/molecules27175553
APA StyleSuzuki, N., Ishida, Y., & Kawaguchi, H. (2022). Lewis Acid-Induced Dinitrogen Cleavage in an Anionic Side-on End-on Bound Dinitrogen Diniobium Hydride Complex. Molecules, 27(17), 5553. https://doi.org/10.3390/molecules27175553