

Designing Dual Inhibitors of Autotaxin-LPAR GPCR Axis



Abstract
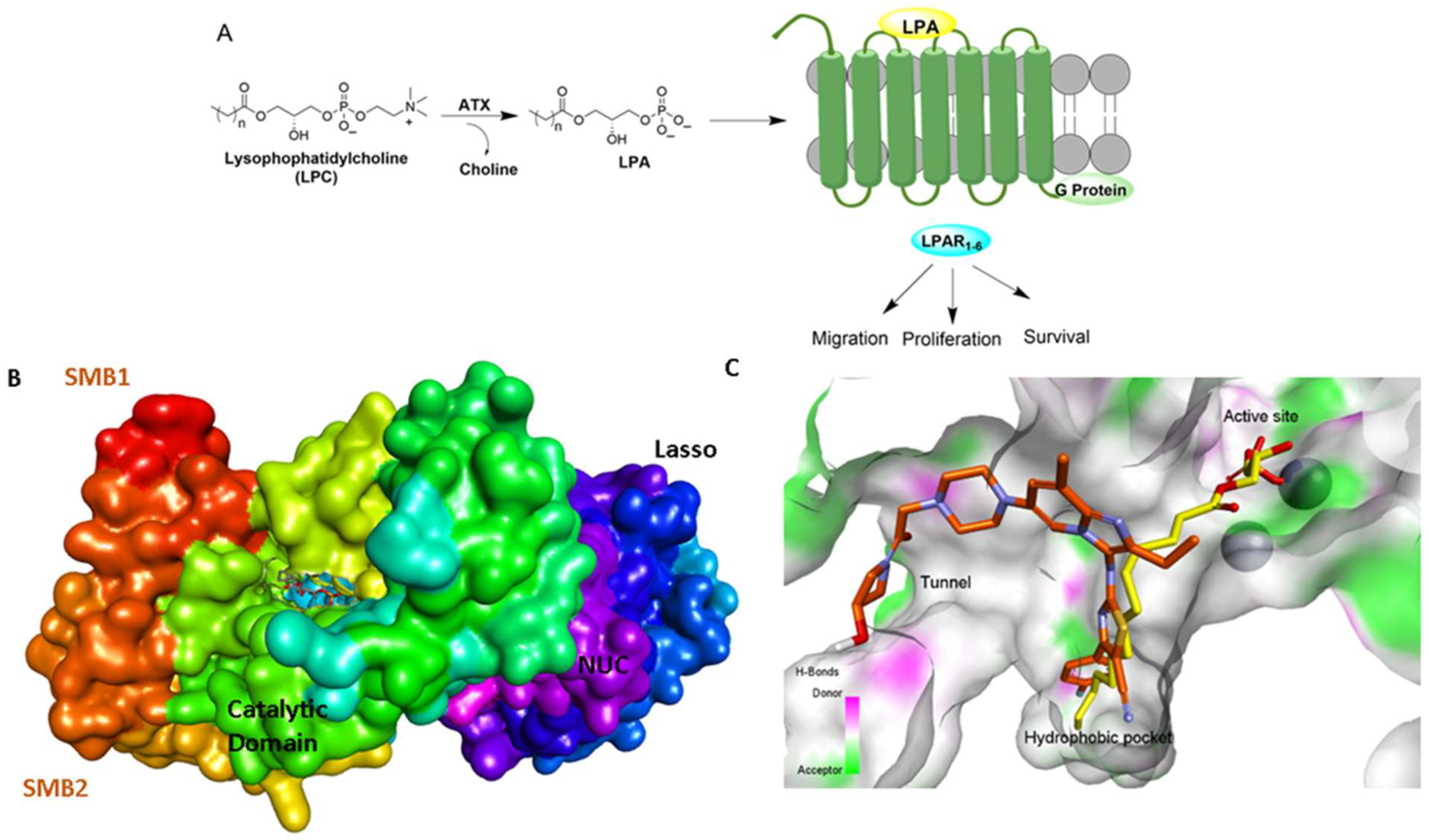
1. Introduction
2. Structural Aspects of ATX Inhibitors
3. ATX-LPAR1 GPCR Axis in Idiopathic Pulmonary Fibrosis and Cancer
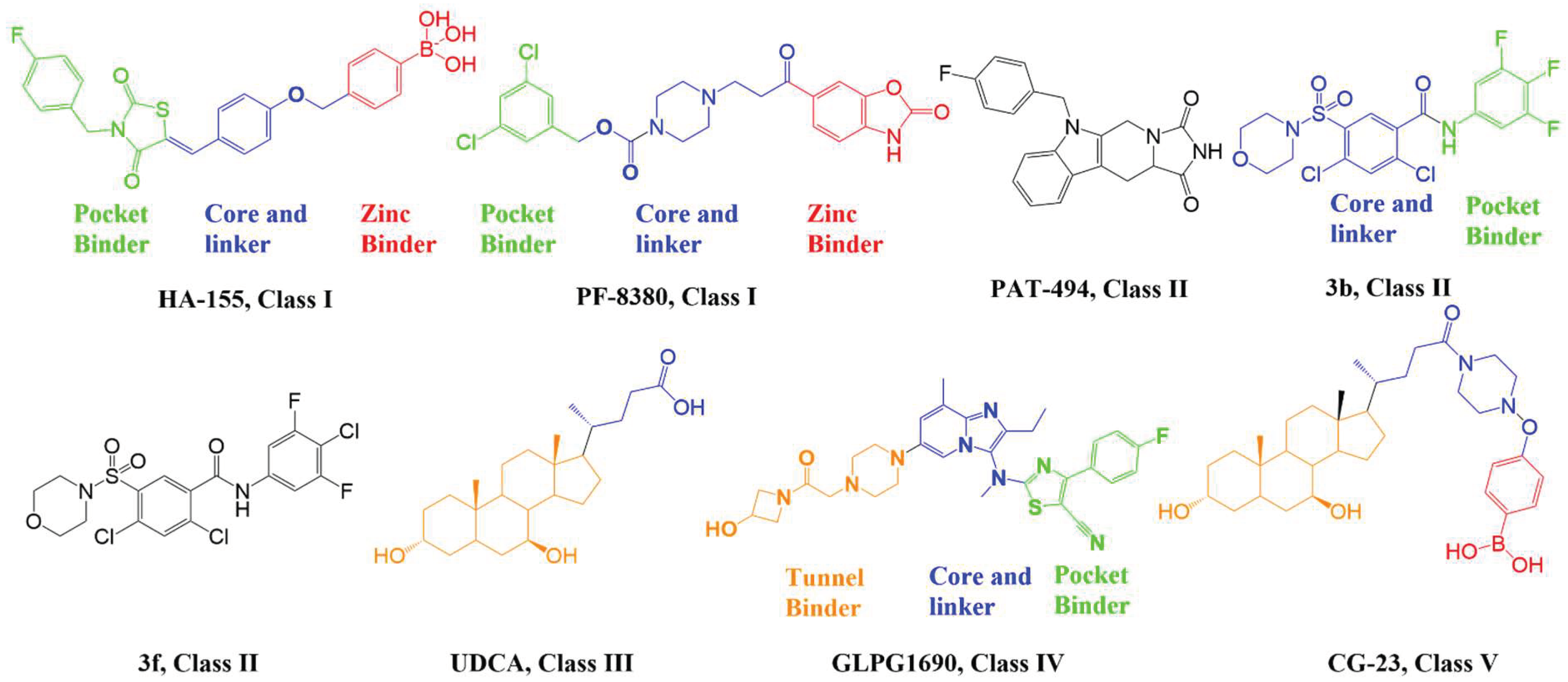
4. Progress with Different Classes of ATX Inhibitors
4.1. Orthosteric Inhibitors (Class I)
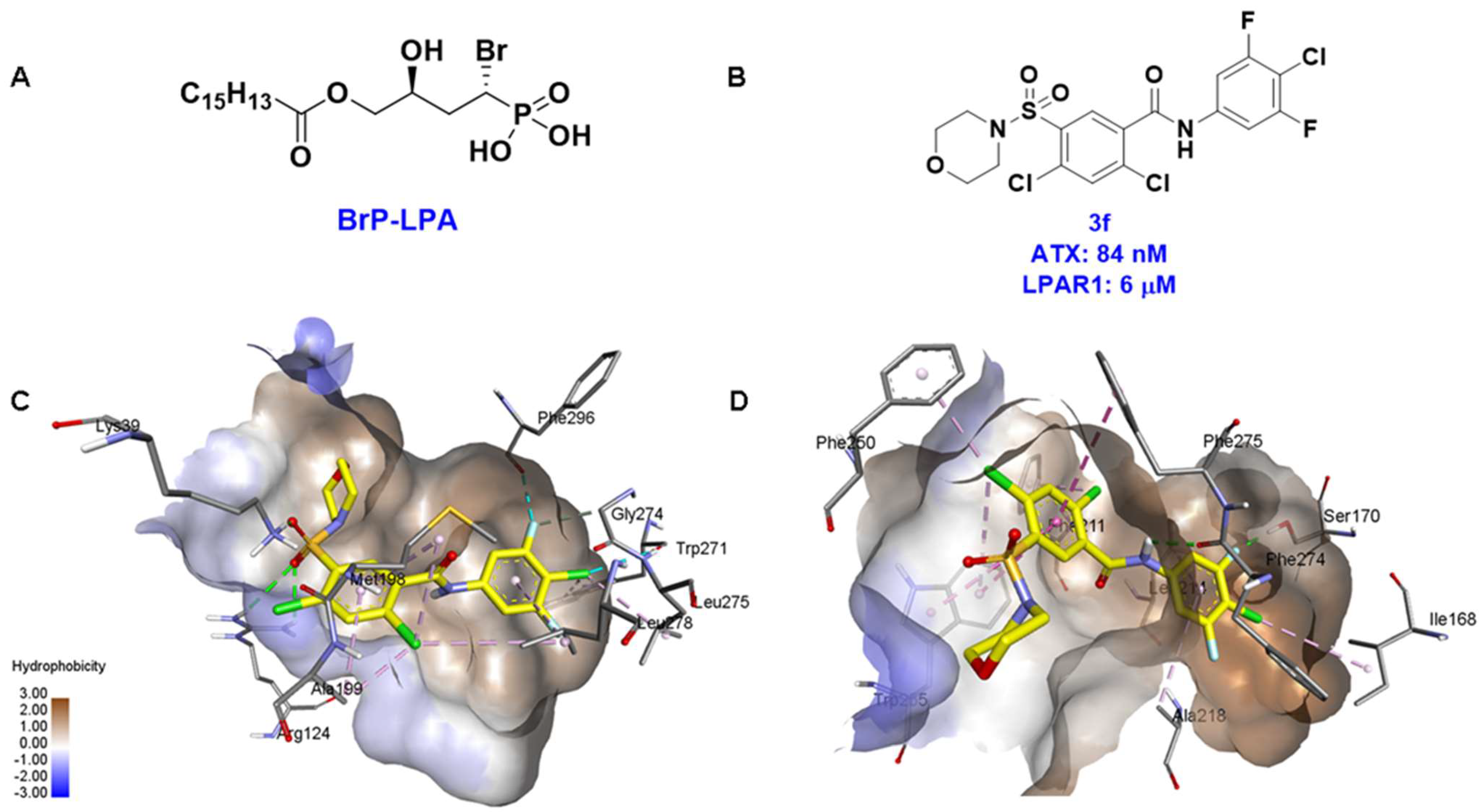
4.2. Hydrophobic Pocket Inhibitors (Class II)
4.3. Allosteric Tunnel Inhibitors (Class III)
4.4. Pocket-Tunnel Hybrids (Class IV)
4.5. Tunnel-Pocket Hybrid Inhibitors (Class V)
5. Biological Prospective of ATX-LPAR1 Dual Inhibition
6. Strategies for Designing Dual Targeting Inhibitors

7. The Ins and Outs of ATX Activity Measurement
8. Conclusions and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Banerjee, S.; Norman, D.D.; Lee, S.C.; Parrill, A.L.; Pham, T.C.T.; Baker, D.L.; Tigyi, G.J.; Miller, D.D. Highly Potent Non-Carboxylic Acid Autotaxin Inhibitors Reduce Melanoma Metastasis and Chemotherapeutic Resistance of Breast Cancer Stem Cells. J. Med. Chem. 2017, 60, 1309–1324. [Google Scholar] [CrossRef] [PubMed]
- Lei, H.; Wang, X.; Zhao, G.; Li, T.; Cui, Y.; Wu, H.; Yang, J.; Jiang, N.; Zhai, X. Design, synthesis and promising anti-tumor efficacy of novel imidazo[1,2-a]pyridine derivatives as potent autotaxin allosteric inhibitors. Eur. J. Med. Chem. 2022, 236, 114307. [Google Scholar] [CrossRef] [PubMed]
- Lei, H.; Guo, M.; Li, X.; Jia, F.; Li, C.; Yang, Y.; Cao, M.; Jiang, N.; Ma, E.; Zhai, X. Discovery of Novel Indole-Based Allosteric Highly Potent ATX Inhibitors with Great In Vivo Efficacy in a Mouse Lung Fibrosis Model. J. Med. Chem. 2020, 63, 7326–7346. [Google Scholar] [CrossRef]
- Jia, Y.; Li, Y.; Xu, X.-D.; Tian, Y.; Shang, H. Design and Development of Autotaxin Inhibitors. Pharmaceuticals 2021, 14, 1203. [Google Scholar] [CrossRef]
- Tigyi, G.; Dacheux, M.A.; Lin, K.-H.; Yue, J.; Norman, D.; Benyó, Z.; Lee, S.C. Anti-cancer strategies targeting the autotaxin-lysophosphatidic acid receptor axis: Is there a path forward? Cancer Metastasis Rev. 2021, 40, 3–5. [Google Scholar] [CrossRef] [PubMed]
- Valdés-Rives, S.A.; González-Arenas, A. Autotaxin-Lysophosphatidic Acid: From Inflammation to Cancer Development. Mediat. Inflamm. 2017, 2017, 9173090. [Google Scholar] [CrossRef]
- Tigyi, G.; Lin, K.H.; Jang, I.H.; Lee, S.C. Revisiting the role of lysophosphatidic acid in stem cell biology. Exp. Biol. Med. 2021, 246, 1802–1809. [Google Scholar] [CrossRef]
- Seo, E.J.; Kwon, Y.W.; Jang, I.H.; Kim, D.K.; Lee, S.I.; Choi, E.J.; Kim, K.H.; Suh, D.S.; Lee, J.H.; Choi, K.U.; et al. Autotaxin Regulates Maintenance of Ovarian Cancer Stem Cells through Lysophosphatidic Acid-Mediated Autocrine Mechanism. Stem Cells 2016, 34, 551–564. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Oda, S.K.; Shotts, K.; Donovan, E.E.; Strauch, P.; Pujanauski, L.M.; Victorino, F.; Al-Shami, A.; Fujiwara, Y.; Tigyi, G.; et al. Lysophosphatidic acid receptor 5 inhibits B cell antigen receptor signaling and antibody response. J. Immunol. 2014, 193, 85–95. [Google Scholar] [CrossRef]
- Lee, S.C.; Dacheux, M.A.; Norman, D.D.; Balazs, L.; Torres, R.M.; Augelli-Szafran, C.E.; Tigyi, G.J. Regulation of Tumor Immunity by Lysophosphatidic Acid. Cancers 2020, 12, 1202. [Google Scholar] [CrossRef]
- Mathew, D.; Kremer, K.N.; Strauch, P.; Tigyi, G.; Pelanda, R.; Torres, R.M. LPA5 Is an Inhibitory Receptor That Suppresses CD8 T-Cell Cytotoxic Function via Disruption of Early TCR Signaling. Front. Immunol. 2019, 10, 1159. [Google Scholar] [CrossRef] [PubMed]
- Oda, S.K.; Strauch, P.; Fujiwara, Y.; Al-Shami, A.; Oravecz, T.; Tigyi, G.; Pelanda, R.; Torres, R.M. Lysophosphatidic Acid Inhibits CD8 T-cell Activation and Control of Tumor Progression. Cancer Immunol. Res. 2013, 1, 245–255. [Google Scholar] [CrossRef] [PubMed]
- Yung, Y.C.; Stoddard, N.C.; Mirendil, H.; Chun, J. Lysophosphatidic Acid signaling in the nervous system. Neuron 2015, 85, 669–682. [Google Scholar] [CrossRef] [PubMed]
- Rivera, R.R.; Lin, M.E.; Bornhop, E.C.; Chun, J. Conditional Lpar1 gene targeting identifies cell types mediating neuropathic pain. Faseb J. 2020, 34, 8833–8842. [Google Scholar] [CrossRef]
- Ueda, H.; Matsunaga, H.; Olaposi, O.I.; Nagai, J. Lysophosphatidic acid: Chemical signature of neuropathic pain. Biochim. Biophys. Acta 2013, 1831, 61–73. [Google Scholar] [CrossRef]
- Ninou, I.; Magkrioti, C.; Aidinis, V. Autotaxin in Pathophysiology and Pulmonary Fibrosis. Front. Med. 2018, 5, 180. [Google Scholar] [CrossRef]
- Ninou, I.; Kaffe, E.; Müller, S.; Budd, D.C.; Stevenson, C.S.; Ullmer, C.; Aidinis, V. Pharmacologic targeting of the ATX/LPA axis attenuates bleomycin-induced pulmonary fibrosis. Pulm. Pharmacol. Ther. 2018, 52, 32–40. [Google Scholar] [CrossRef]
- Murphy, B.; Sum, C.-S.; Wang, T.; Heiry, R.; Kalinowski, S.; Hung, C.-P.; Chu, C.-H.; Azzara, A.; Milinda, Z.; Burns, L. LPA1 antagonist BMS-986278 for idiopathic pulmonary fibrosis: Preclinical pharmacological in vitro and in vivo evaluation. Eur. Respir. J. 2019, 54 (Suppl. 63), PA5383. [Google Scholar]
- Corte, T.J.; Lancaster, L.; Swigris, J.J.; Maher, T.M.; Goldin, J.G.; Palmer, S.M.; Suda, T.; Ogura, T.; Minnich, A.; Zhan, X. Phase 2 trial design of BMS-986278, a lysophosphatidic acid receptor 1 (LPA1) antagonist, in patients with idiopathic pulmonary fibrosis (IPF) or progressive fibrotic interstitial lung disease (PF-ILD). BMJ Open Respir. Res. 2021, 8, e001026. [Google Scholar] [CrossRef]
- Swaney, J.; Chapman, C.; Correa, L.; Stebbins, K.; Bundey, R.; Prodanovich, P.; Fagan, P.; Baccei, C.; Santini, A.; Hutchinson, J. A novel, orally active LPA1 receptor antagonist inhibits lung fibrosis in the mouse bleomycin model. Br. J. Pharmacol. 2010, 160, 1699–1713. [Google Scholar] [CrossRef]
- Montesi, S.B.; Mathai, S.K.; Brenner, L.N.; Gorshkova, I.A.; Berdyshev, E.V.; Tager, A.M.; Shea, B.S. Docosatetraenoyl LPA is elevated in exhaled breath condensate in idiopathic pulmonary fibrosis. BMC Pulm. Med. 2014, 14, 5. [Google Scholar] [CrossRef] [PubMed]
- Ma, B.; Zhang, L.; Sun, L.; Xin, Z.; Kumaravel, G.; Marcotte, D.; Chodaparambil, J.V.; Wang, Q.; Wehr, A.; Jing, J.; et al. Discovery of Potent Selective Nonzinc Binding Autotaxin Inhibitor BIO-32546. ACS Med. Chem. Lett. 2021, 12, 1124–1129. [Google Scholar] [CrossRef] [PubMed]
- Bhattarai, S.; Sharma, S.; Ara, H.; Subedi, U.; Sun, G.; Li, C.; Bhuiyan, M.S.; Kevil, C.; Armstrong, W.P.; Minvielle, M.T.; et al. Disrupted Blood-Brain Barrier and Mitochondrial Impairment by Autotaxin-Lysophosphatidic Acid Axis in Postischemic Stroke. J. Am. Heart Assoc. 2021, 10, e021511. [Google Scholar] [CrossRef] [PubMed]
- Flammier, S.; Peyruchaud, O.; Bourguillault, F.; Duboeuf, F.; Davignon, J.-L.; Norman, D.D.; Isaac, S.; Marotte, H.; Tigyi, G.; Machuca-Gayet, I.; et al. Osteoclast-Derived Autotaxin, a Distinguishing Factor for Inflammatory Bone Loss. Arthritis Rheumatol. 2019, 71, 1801–1811. [Google Scholar] [CrossRef]
- Erenel, H.; Yilmaz, N.; Cift, T.; Bulut, B.; Sozen, I.; Aslan Cetin, B.; Gezer, A.; Ekmekci, H.; Kaya, B.; Tuten, A. Maternal serum autotaxin levels in early- and late-onset preeclampsia. Hypertens. Pregnancy 2017, 36, 310–314. [Google Scholar] [CrossRef]
- Matas-Rico, E.; Frijlink, E.; van der Haar Àvila, I.; Menegakis, A.; van Zon, M.; Morris, A.J.; Koster, J.; Salgado-Polo, F.; de Kivit, S.; Lança, T.; et al. Autotaxin impedes anti-tumor immunity by suppressing chemotaxis and tumor infiltration of CD8(+) T cells. Cell Rep. 2021, 37, 110013. [Google Scholar] [CrossRef]
- Banerjee, S.; Norman, D.D.; Deng, S.; Fakayode, S.O.; Lee, S.C.; Parrill, A.L.; Li, W.; Miller, D.D.; Tigyi, G.J. Molecular modelling guided design, synthesis and QSAR analysis of new small molecule non-lipid autotaxin inhibitors. Bioorg. Chem. 2020, 103, 104188. [Google Scholar] [CrossRef]
- Katsifa, A.; Kaffe, E.; Nikolaidou-Katsaridou, N.; Economides, A.N.; Newbigging, S.; McKerlie, C.; Aidinis, V. The Bulk of Autotaxin Activity Is Dispensable for Adult Mouse Life. PLoS ONE 2015, 10, e0143083. [Google Scholar] [CrossRef]
- Lin, S.; Haque, A.; Raeman, R.; Guo, L.; He, P.; Denning, T.L.; El-Rayes, B.; Moolenaar, W.H.; Yun, C.C. Autotaxin determines colitis severity in mice and is secreted by B cells in the colon. Faseb J. 2019, 33, 3623–3635. [Google Scholar] [CrossRef]
- Hausmann, J.; Kamtekar, S.; Christodoulou, E.; Day, J.E.; Wu, T.; Fulkerson, Z.; Albers, H.M.; van Meeteren, L.A.; Houben, A.J.; van Zeijl, L.; et al. Moolenaar, W.H.; Perrakis, A. Structural basis of substrate discrimination and integrin binding by autotaxin. Nat. Struct. Mol. Biol. 2011, 18, 198–204. [Google Scholar] [CrossRef]
- Nishimasu, H.; Okudaira, S.; Hama, K.; Mihara, E.; Dohmae, N.; Inoue, A.; Ishitani, R.; Takagi, J.; Aoki, J.; Nureki, O. Crystal structure of autotaxin and insight into GPCR activation by lipid mediators. Nat. Struct. Mol. Biol. 2011, 18, 205–212. [Google Scholar] [CrossRef]
- Clark, J.M.; Salgado-Polo, F.; Macdonald, S.J.F.; Barrett, T.N.; Perrakis, A.; Jamieson, C. Structure-Based Design of a Novel Class of Autotaxin Inhibitors Based on Endogenous Allosteric Modulators. J. Med. Chem. 2022, 65, 6338–6351. [Google Scholar] [CrossRef] [PubMed]
- Fells, J.I.; Lee, S.C.; Norman, D.D.; Tsukahara, R.; Kirby, J.R.; Nelson, S.; Seibel, W.; Papoian, R.; Patil, R.; Miller, D.D.; et al. Targeting the hydrophobic pocket of autotaxin with virtual screening of inhibitors identifies a common aromatic sulfonamide structural motif. FEBS J. 2014, 281, 1017–1028. [Google Scholar] [CrossRef]
- Cuozzo, J.W.; Clark, M.A.; Keefe, A.D.; Kohlmann, A.; Mulvihill, M.; Ni, H.; Renzetti, L.M.; Resnicow, D.I.; Ruebsam, F.; Sigel, E.A.; et al. Novel Autotaxin Inhibitor for the Treatment of Idiopathic Pulmonary Fibrosis: A Clinical Candidate Discovered Using DNA-Encoded Chemistry. J. Med. Chem. 2020, 63, 7840–7856. [Google Scholar] [CrossRef] [PubMed]
- Drugmaker Galapagos Keeps ‘Appetite’ for Risk after Clinical Setbacks. Reuters. Reported by SarahMorland in Gdansk and Edited by Larry King. Available online: https://www.reuters.com/article/us-galapagos-outlook-idUKKBN2AJ218 (accessed on 20 July 2022).
- Bano, S.; Al-Rashida, M.; Alharthy, R.D.; Khan, I.A.; Iqbal, J. Nucleotide pyrophosphatase/phosphodiesterases (NPPs) including NPP1 and NPP2/ ATX as important drug targets: A patent review (2015–2020). Exp. Opin. Ther. Pat. 2022, 32, 743–751. [Google Scholar] [CrossRef]
- Figueiredo, M. With Positive Early Findings, Phase 2 Cudetaxestat Trial Expected Soon. Pulmonary Fibrosis News, 24 May 2022. [Google Scholar]
- Blade Therapeutics Announces FDA Activation of IND Application to Investigate Cudetaxestat, a Non-Competitive Autotaxin Inhibitor, in Idiopathic Pulmonary Fibrosis (IPF). Available online: https://www.businesswire.com/news/home/20210825005020/en/Blade-Therapeutics-Announces-FDA-Activation-of-IND-Application-to-Investigate-Cudetaxestat-a-Non-Competitive-Autotaxin-Inhibitor-in-Idiopathic-Pulmonary-Fibrosis-IPF (accessed on 20 July 2022).
- Abdel-Magid, A.F. Lysophosphatidic Acid Receptor 1 Antagonists for the Treatment of Fibrosis. ACS Med. Chem. Lett. 2019, 10, 1378–1379. [Google Scholar] [CrossRef] [PubMed]
- Palmer, S.M.; Snyder, L.; Todd, J.L.; Soule, B.; Christian, R.; Anstrom, K.; Luo, Y.; Gagnon, R.; Rosen, G. Randomized, Double-Blind, Placebo-Controlled, Phase 2 Trial of BMS-986020, a Lysophosphatidic Acid Receptor Antagonist for the Treatment of Idiopathic Pulmonary Fibrosis. CHEST 2018, 154, 1061–1069. [Google Scholar] [CrossRef]
- Decato, B.E.; Leeming, D.J.; Sand, J.M.B.; Fischer, A.; Du, S.; Palmer, S.M.; Karsdal, M.; Luo, Y.; Minnich, A. LPA1 antagonist BMS-986020 changes collagen dynamics and exerts antifibrotic effects in vitro and in patients with idiopathic pulmonary fibrosis. Respir. Res. 2022, 23, 61. [Google Scholar] [CrossRef]
- Allanore, Y.; Distler, O.; Jagerschmidt, A.; Illiano, S.; Ledein, L.; Boitier, E.; Agueusop, I.; Denton, C.P.; Khanna, D. Lysophosphatidic Acid Receptor 1 Antagonist SAR100842 for Patients With Diffuse Cutaneous Systemic Sclerosis: A Double-Blind, Randomized, Eight-Week Placebo-Controlled Study Followed by a Sixteen-Week Open-Label Extension Study. Arthritis Rheumatol. 2018, 70, 1634–1643. [Google Scholar] [CrossRef]
- She, S.; Zhang, Q.; Shi, J.; Yang, F.; Dai, K. Roles of Autotaxin/Autotaxin-Lysophosphatidic Acid Axis in the Initiation and Progression of Liver Cancer. Front. Oncol. 2022, 12, 922945. [Google Scholar] [CrossRef]
- Tigyi, G.J.; Yue, J.; Norman, D.D.; Szabo, E.; Balogh, A.; Balazs, L.; Zhao, G.; Lee, S.C. Regulation of tumor cell—Microenvironment interaction by the autotaxin-lysophosphatidic acid receptor axis. Adv. Biol. Regul. 2019, 71, 183–193. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Gonzalo, F.R.; Izpisua Belmonte, J.C. Albumin-associated lipids regulate human embryonic stem cell self-renewal. PLoS ONE 2008, 3, e1384. [Google Scholar] [CrossRef] [PubMed]
- Stavish, D.; Boiers, C.; Price, C.; Frith, T.J.R.; Halliwell, J.; Saldana-Guerrero, I.; Wray, J.; Brown, J.; Carr, J.; James, C.; et al. Generation and trapping of a mesoderm biased state of human pluripotency. Nat. Commun. 2020, 11, 4989. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.; Suh, D.-S.; Lee, S.C.; Tigyi, G.J.; Kim, J.H. Role of autotaxin in cancer stem cells. Cancer Metastasis Rev. 2018, 37, 509–518. [Google Scholar] [CrossRef]
- Liu, S.; Umezu-Goto, M.; Murph, M.; Lu, Y.; Liu, W.; Zhang, F.; Yu, S.; Stephens, L.C.; Cui, X.; Murrow, G.; et al. Expression of autotaxin and lysophosphatidic acid receptors increases mammary tumorigenesis, invasion, and metastases. Cancer Cell 2009, 15, 539–550. [Google Scholar] [CrossRef]
- Chrencik, J.E.; Roth, C.B.; Terakado, M.; Kurata, H.; Omi, R.; Kihara, Y.; Warshaviak, D.; Nakade, S.; Asmar-Rovira, G.; Mileni, M.; et al. Crystal Structure of Antagonist Bound Human Lysophosphatidic Acid Receptor 1. Cell 2015, 161, 1633–1643. [Google Scholar] [CrossRef]
- Parrill, A.L.; Echols, U.; Nguyen, T.; Pham, T.C.; Hoeglund, A.; Baker, D.L. Virtual screening approaches for the identification of non-lipid autotaxin inhibitors. Bioorg. Med. Chem. 2008, 16, 1784–1795. [Google Scholar] [CrossRef] [PubMed]
- Hoeglund, A.B.; Bostic, H.E.; Howard, A.L.; Wanjala, I.W.; Best, M.D.; Baker, D.L.; Parrill, A.L. Optimization of a Pipemidic Acid Autotaxin Inhibitor. J. Med. Chem. 2010, 53, 1056–1066. [Google Scholar] [CrossRef]
- Albers, H.M.H.G.; van Meeteren, L.A.; Egan, D.A.; van Tilburg, E.W.; Moolenaar, W.H.; Ovaa, H. Discovery and Optimization of Boronic Acid Based Inhibitors of Autotaxin. J. Med. Chem. 2010, 53, 4958–4967. [Google Scholar] [CrossRef]
- Albers, H.M.H.G.; Hendrickx, L.J.D.; van Tol, R.J.P.; Hausmann, J.; Perrakis, A.; Ovaa, H. Structure-Based Design of Novel Boronic Acid-Based Inhibitors of Autotaxin. J. Med. Chem. 2011, 54, 4619–4626. [Google Scholar] [CrossRef]
- Kawaguchi, M.; Okabe, T.; Okudaira, S.; Nishimasu, H.; Ishitani, R.; Kojima, H.; Nureki, O.; Aoki, J.; Nagano, T. Screening and X-ray Crystal Structure-based Optimization of Autotaxin (ENPP2) Inhibitors, Using a Newly Developed Fluorescence Probe. ACS Chem. Biol. 2013, 8, 1713–1721. [Google Scholar] [CrossRef] [PubMed]
- Pantsar, T.; Singha, P.; Nevalainen, T.J.; Koshevoy, I.; Leppänen, J.; Poso, A.; Niskanen, J.M.A.; Pasonen-Seppänen, S.; Savinainen, J.R.; Laitinen, T.; et al. Design, synthesis, and biological evaluation of 2,4-dihydropyrano[2,3-c]pyrazole derivatives as autotaxin inhibitors. Eur. J. Pharm. Sci. 2017, 107, 97–111. [Google Scholar] [CrossRef] [PubMed]
- Jing, T.; Miao, X.; Jiang, F.; Guo, M.; Xing, L.; Zhang, J.; Zuo, D.; Lei, H.; Zhai, X. Discovery and optimization of tetrahydropyrido[4,3-d]pyrimidine derivatives as novel ATX and EGFR dual inhibitors. Bioorg. Med. Chem. 2018, 26, 1784–1796. [Google Scholar] [CrossRef] [PubMed]
- Jiang, N.; Zhou, Y.; Zhu, M.; Zhang, J.; Cao, M.; Lei, H.; Guo, M.; Gong, P.; Su, G.; Zhai, X. Optimization and evaluation of novel tetrahydropyrido[4,3-d]pyrimidine derivatives as ATX inhibitors for cardiac and hepatic fibrosis. Eur. J. Med. Chem. 2020, 187, 111904. [Google Scholar] [CrossRef] [PubMed]
- Miller, L.M.; Keune, W.-J.; Castagna, D.; Young, L.C.; Duffy, E.L.; Potjewyd, F.; Salgado-Polo, F.; Engel García, P.; Semaan, D.; Pritchard, J.M.; et al. Structure–Activity Relationships of Small Molecule Autotaxin Inhibitors with a Discrete Binding Mode. J. Med. Chem. 2017, 60, 722–748. [Google Scholar] [CrossRef]
- Shah, P.; Cheasty, A.; Foxton, C.; Raynham, T.; Farooq, M.; Gutierrez, I.F.; Lejeune, A.; Pritchard, M.; Turnbull, A.; Pang, L.; et al. Discovery of potent inhibitors of the lysophospholipase autotaxin. Bioorg. Med. Chem. Lett. 2016, 26, 5403–5410. [Google Scholar] [CrossRef]
- Balijepalli, P.; Sitton, C.C.; Meier, K.E. Lysophosphatidic Acid Signaling in Cancer Cells: What Makes LPA So Special? Cells 2021, 10, 2059. [Google Scholar] [CrossRef]
- Gupta, P.B.; Onder, T.T.; Jiang, G.; Tao, K.; Kuperwasser, C.; Weinberg, R.A.; Lander, E.S. Identification of selective inhibitors of cancer stem cells by high-throughput screening. Cell 2009, 138, 645–659. [Google Scholar] [CrossRef]
- Lee, S.-C.; Fujiwara, Y.; Liu, J.; Yue, J.; Shimizu, Y.; Norman, D.D.; Wang, Y.; Tsukahara, R.; Szabo, E.; Patil, R.; et al. Autotaxin and LPA1 and LPA5 Receptors Exert Disparate Functions in Tumor Cells versus the Host Tissue Microenvironment in Melanoma Invasion and Metastasis. Mol. Cancer Res. 2015, 13, 174–185. [Google Scholar] [CrossRef]
- Zhang, H.; Xu, X.; Gajewiak, J.; Tsukahara, R.; Fujiwara, Y.; Liu, J.; Fells, J.I.; Perygin, D.; Parrill, A.L.; Tigyi, G.; et al. Dual Activity Lysophosphatidic Acid Receptor Pan-Antagonist/Autotaxin Inhibitor Reduces Breast Cancer Cell Migration In vitro and Causes Tumor Regression In vivo. Cancer Res. 2009, 69, 5441–5449. [Google Scholar] [CrossRef]
- Leblanc, R.; Lee, S.C.; David, M.; Bordet, J.C.; Norman, D.D.; Patil, R.; Miller, D.; Sahay, D.; Ribeiro, J.; Clezardin, P.; et al. Interaction of platelet-derived autotaxin with tumor integrin alphaVbeta3 controls metastasis of breast cancer cells to bone. Blood 2014, 124, 3141–3150. [Google Scholar] [CrossRef] [PubMed]
- Leblanc, R.; Peyruchaud, O. The role of platelets and megakaryocytes in bone metastasis. J. Bone Oncol. 2016, 5, 109–111. [Google Scholar] [CrossRef] [PubMed]
- Sahay, D.; Leblanc, R.; Grunewald, T.G.; Ambatipudi, S.; Ribeiro, J.; Clézardin, P.; Peyruchaud, O. The LPA1/ZEB1/miR-21-activation pathway regulates metastasis in basal breast cancer. Oncotarget 2015, 6, 20604–20620. [Google Scholar] [CrossRef]
- Iwaki, Y.; Ohhata, A.; Nakatani, S.; Hisaichi, K.; Okabe, Y.; Hiramatsu, A.; Watanabe, T.; Yamamoto, S.; Nishiyama, T.; Kobayashi, J.; et al. ONO-8430506: A Novel Autotaxin Inhibitor That Enhances the Antitumor Effect of Paclitaxel in a Breast Cancer Model. ACS Med. Chem. Lett. 2020, 11, 1335–1341. [Google Scholar] [CrossRef] [PubMed]
- Venkatraman, G.; Benesch, M.G.; Tang, X.; Dewald, J.; McMullen, T.P.; Brindley, D.N. Lysophosphatidate signaling stabilizes Nrf2 and increases the expression of genes involved in drug resistance and oxidative stress responses: Implications for cancer treatment. Faseb J. 2015, 29, 772–785. [Google Scholar] [CrossRef]
- Erstad, D.J.; Tager, A.M.; Hoshida, Y.; Fuchs, B.C. The autotaxin-lysophosphatidic acid pathway emerges as a therapeutic target to prevent liver cancer. Mol. Cell Oncol. 2017, 4, e1311827. [Google Scholar] [CrossRef]
- Nikitopoulou, I.; Oikonomou, N.; Karouzakis, E.; Sevastou, I.; Nikolaidou-Katsaridou, N.; Zhao, Z.; Mersinias, V.; Armaka, M.; Xu, Y.; Masu, M.; et al. Autotaxin expression from synovial fibroblasts is essential for the pathogenesis of modeled arthritis. J. Exp. Med. 2012, 209, 925–933. [Google Scholar] [CrossRef]
- Miyabe, Y.; Miyabe, C.; Iwai, Y.; Takayasu, A.; Fukuda, S.; Yokoyama, W.; Nagai, J.; Jona, M.; Tokuhara, Y.; Ohkawa, R.; et al. Necessity of Lysophosphatidic Acid Receptor 1 for Development of Arthritis. Arthritis Rheum. 2013, 65, 2037–2047. [Google Scholar] [CrossRef]
- Orosa, B.; García, S.; Martínez, P.; González, A.; Gómez-Reino, J.J.; Conde, C. Lysophosphatidic acid receptor inhibition as a new multipronged treatment for rheumatoid arthritis. Ann. Rheum. Dis. 2014, 73, 298–305. [Google Scholar] [CrossRef]
- Nikitopoulou, I.; Kaffe, E.; Sevastou, I.; Sirioti, I.; Samiotaki, M.; Madan, D.; Prestwich, G.D.; Aidinis, V. A metabolically-stabilized phosphonate analog of lysophosphatidic acid attenuates collagen-induced arthritis. PLoS ONE 2013, 8, e70941. [Google Scholar] [CrossRef]
- Proschak, E.; Stark, H.; Merk, D. Polypharmacology by Design: A Medicinal Chemist’s Perspective on Multitargeting Compounds. J. Med. Chem. 2019, 62, 420–444. [Google Scholar] [CrossRef] [PubMed]
- Benek, O.; Korabecny, J.; Soukup, O. A Perspective on Multi-target Drugs for Alzheimer’s Disease. Trends Pharmacol. Sci. 2020, 41, 434–445. [Google Scholar] [CrossRef] [PubMed]
- Shuai, W.; Wang, G.; Zhang, Y.; Bu, F.; Zhang, S.; Miller, D.D.; Li, W.; Ouyang, L.; Wang, Y. Recent Progress on Tubulin Inhibitors with Dual Targeting Capabilities for Cancer Therapy. J. Med. Chem. 2021, 64, 7963–7990. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Zhao, Y.; Liu, Y.; Sun, D.; Zhen, Y.; Liu, J.; Fu, L.; Zhang, L.; Ouyang, L. Discovery of a Novel Dual-Target Inhibitor of ERK1 and ERK5 That Induces Regulated Cell Death to Overcome Compensatory Mechanism in Specific Tumor Types. J. Med. Chem. 2020, 63, 3976–3995. [Google Scholar] [CrossRef]
- Smith, A.D.; Roda, D.; Yap, T.A. Strategies for modern biomarker and drug development in oncology. J. Hematol. Oncol. 2014, 7, 70. [Google Scholar] [CrossRef]
- Sun, D.; Zhao, Y.; Zhang, S.; Zhang, L.; Liu, B.; Ouyang, L. Dual-target kinase drug design: Current strategies and future directions in cancer therapy. Eur. J. Med. Chem. 2020, 188, 112025. [Google Scholar] [CrossRef]
- Banerjee, S.; Yadav, S.; Banerjee, S.; Fakayode, S.O.; Parvathareddy, J.; Reichard, W.; Surendranathan, S.; Mahmud, F.; Whatcott, R.; Thammathong, J.; et al. Drug Repurposing to Identify Nilotinib as a Potential SARS-CoV-2 Main Protease Inhibitor: Insights from a Computational and In Vitro Study. J. Chem. Inform. Model. 2021, 61, 5469–5483. [Google Scholar] [CrossRef]
- Arnst, K.E.; Banerjee, S.; Chen, H.; Deng, S.; Hwang, D.-J.; Li, W.; Miller, D.D. Current advances of tubulin inhibitors as dual acting small molecules for cancer therapy. Med. Res. Rev. 2019, 39, 1398–1426. [Google Scholar] [CrossRef]
- Morphy, R.; Rankovic, Z. Designed Multiple Ligands. An Emerging Drug Discovery Paradigm. J. Med. Chem. 2005, 48, 6523–6543. [Google Scholar] [CrossRef]
- Cavalli, A.; Bolognesi, M.L.; Minarini, A.; Rosini, M.; Tumiatti, V.; Recanatini, M.; Melchiorre, C. Multi-target-Directed Ligands To Combat Neurodegenerative Diseases. J. Med. Chem. 2008, 51, 347–372. [Google Scholar] [CrossRef]
- Zhang, W.; Pei, J.; Lai, L. Computational Multitarget Drug Design. J. Chem. Inf. Model. 2017, 57, 403–412. [Google Scholar] [CrossRef] [PubMed]
- Sonawane, V.; Mohd Siddique, M.U.; Jadav, S.S.; Sinha, B.N.; Jayaprakash, V.; Chaudhuri, B. Cink4T, a quinazolinone-based dual inhibitor of Cdk4 and tubulin polymerization, identified via ligand-based virtual screening, for efficient anticancer therapy. Eur. J. Med. Chem. 2019, 165, 115–132. [Google Scholar] [CrossRef] [PubMed]
- Jesionowska, A.; Cecerska, E.; Dolegowska, B. Methods for quantifying lysophosphatidic acid in body fluids: A review. Anal. Biochem. 2014, 453, 38–43. [Google Scholar] [CrossRef] [PubMed]
- Ferry, G.; Tellier, E.; Try, A.; Grés, S.; Naime, I.; Simon, M.F.; Rodriguez, M.; Boucher, J.; Tack, I.; Gesta, S.; et al. Autotaxin is released from adipocytes, catalyzes lysophosphatidic acid synthesis, and activates preadipocyte proliferation. Up-regulated expression with adipocyte differentiation and obesity. J. Biol. Chem. 2003, 278, 18162–18169. [Google Scholar] [CrossRef] [PubMed]
- Baker, D.L.; Fujiwara, Y.; Pigg, K.R.; Tsukahara, R.; Kobayashi, S.; Murofushi, H.; Uchiyama, A.; Murakami-Murofushi, K.; Koh, E.; Bandle, R.W.; et al. Carba analogs of cyclic phosphatidic acid are selective inhibitors of autotaxin and cancer cell invasion and metastasis. J. Biol. Chem. 2006, 281, 22786–22793. [Google Scholar] [CrossRef]
- Chen, J.H.; Zou, F.; Wang, N.D.; Xie, S.W.; Zhang, X. Production and application of LPA polyclonal antibody. Bioorg. Med. Chem. Lett. 2000, 10, 1691–1693. [Google Scholar] [CrossRef]
- Baker, D.L.; Umstot, E.S.; Desiderio, D.M.; Tigyi, G.J. Quantitative analysis of lysophosphatidic acid in human blood fractions. Ann. N. Y. Acad. Sci. 2000, 905, 267–269. [Google Scholar] [CrossRef]
- Shan, L.; Jaffe, K.; Li, S.; Davis, L. Quantitative determination of lysophosphatidic acid by LC/ESI/MS/MS employing a reversed phase HPLC column. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2008, 864, 22–28. [Google Scholar] [CrossRef]
- Bollinger, J.G.; Ii, H.; Sadilek, M.; Gelb, M.H. Improved method for the quantification of lysophospholipids including enol ether species by liquid chromatography-tandem mass spectrometry. J. Lipid Res. 2010, 51, 440–447. [Google Scholar] [CrossRef]
- Umezu-Goto, M.; Kishi, Y.; Taira, A.; Hama, K.; Dohmae, N.; Takio, K.; Yamori, T.; Mills, G.B.; Inoue, K.; Aoki, J.; et al. Autotaxin has lysophospholipase D activity leading to tumor cell growth and motility by lysophosphatidic acid production. J. Cell Biol. 2002, 158, 227–233. [Google Scholar] [CrossRef]
- Ferguson, C.G.; Bigman, C.S.; Richardson, R.D.; van Meeteren, L.A.; Moolenaar, W.H.; Prestwich, G.D. Fluorogenic Phospholipid Substrate to Detect Lysophospholipase D/Autotaxin Activity. Org. Lett. 2006, 8, 2023–2026. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Zhao, J.; He, D.; Berdyshev, E.; Zhong, M.; Salgia, R.; Morris, A.J.; Smyth, S.S.; Natarajan, V.; Zhao, Y. Autotaxin induces lung epithelial cell migration through lysoPLD activity-dependent and -independent pathways. Biochem. J. 2011, 439, 45–55. [Google Scholar] [CrossRef] [PubMed]

 .
.

{kind=link}
{kind=link}
{kind=link}
| ID | Structure | Biological Efficacy (IC50: nM) | Mode of Binding | Cocrystal Structure (PDB) |
|---|---|---|---|---|
| 1 |  | LPC Assay: 43.6 (ATX) | Pocket binder (Class II) | N.D. |
| 2 |  | FS-3 Assay: 9 (ATX) 14450 (LPAR1) | Pocket binder (Class II) | N.D. |
| 3 |  | FS-3 Assay: 84 (ATX) 6100 (LPAR1) | Pocket binder (Class II) | N.D. |
| 4 |  | FS-3 Assay: 219 (ATX) | Pocket binder (Class II) | N.D. |
| 5 |  | FS-3 Assay: 218 (ATX) | Pocket binder (Class II) | N.D. |
| 6 |  | LPC Assay: 87 (ATX) | Pocket binder (class II) | N.D. |
| 7 |  | LPC Assay: 24.2 (ATX) | Pocket Binder (Class II) | N.D. |
| 8 |  | LPC Assay: 15.3 (ATX) | Pocket Binder (Class II) | N.D. |
| 9 |  | LPC Assay: >300 | Tunnel Binder (Class III) | 5LQQ |
| 10 |  | LPC Assay: 81 (ATX) | Tunnel Binder (Class III) | 5LQQ |
| 11 |  | LPC Assay: 1 (ATX) | Pocket-Tunnel Hybrid (Class IV) | 5LIA |
| 12 |  | LPC Assay: 1.01 (ATX) | Pocket-Tunnel Hybrid (Class IV) | N.D. |
| 13 |  | FS-3 Assay: 3.4 (ATX) | Pocket-Tunnel Hybrid (Class IV) | N.D. |
| 14 |  | Plasma hlPA: 53 (ATX) ATX FRET: 1 (ATX) | Pocket-Tunnel Hybrid (Class IV) | 7MFH |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Banerjee, S.; Lee, S.; Norman, D.D.; Tigyi, G.J. Designing Dual Inhibitors of Autotaxin-LPAR GPCR Axis. Molecules 2022, 27, 5487. https://doi.org/10.3390/molecules27175487
Banerjee S, Lee S, Norman DD, Tigyi GJ. Designing Dual Inhibitors of Autotaxin-LPAR GPCR Axis. Molecules. 2022; 27(17):5487. https://doi.org/10.3390/molecules27175487
Chicago/Turabian StyleBanerjee, Souvik, Suechin Lee, Derek D. Norman, and Gabor J. Tigyi. 2022. "Designing Dual Inhibitors of Autotaxin-LPAR GPCR Axis" Molecules 27, no. 17: 5487. https://doi.org/10.3390/molecules27175487
APA StyleBanerjee, S., Lee, S., Norman, D. D., & Tigyi, G. J. (2022). Designing Dual Inhibitors of Autotaxin-LPAR GPCR Axis. Molecules, 27(17), 5487. https://doi.org/10.3390/molecules27175487