
Synthesis and In Silico Docking Study towards M-Pro of Novel Heterocyclic Compounds Derived from Pyrazolopyrimidinone as Putative SARS-CoV-2 Inhibitors


 ,
,  , ,
, ,  and
and
Abstract
:
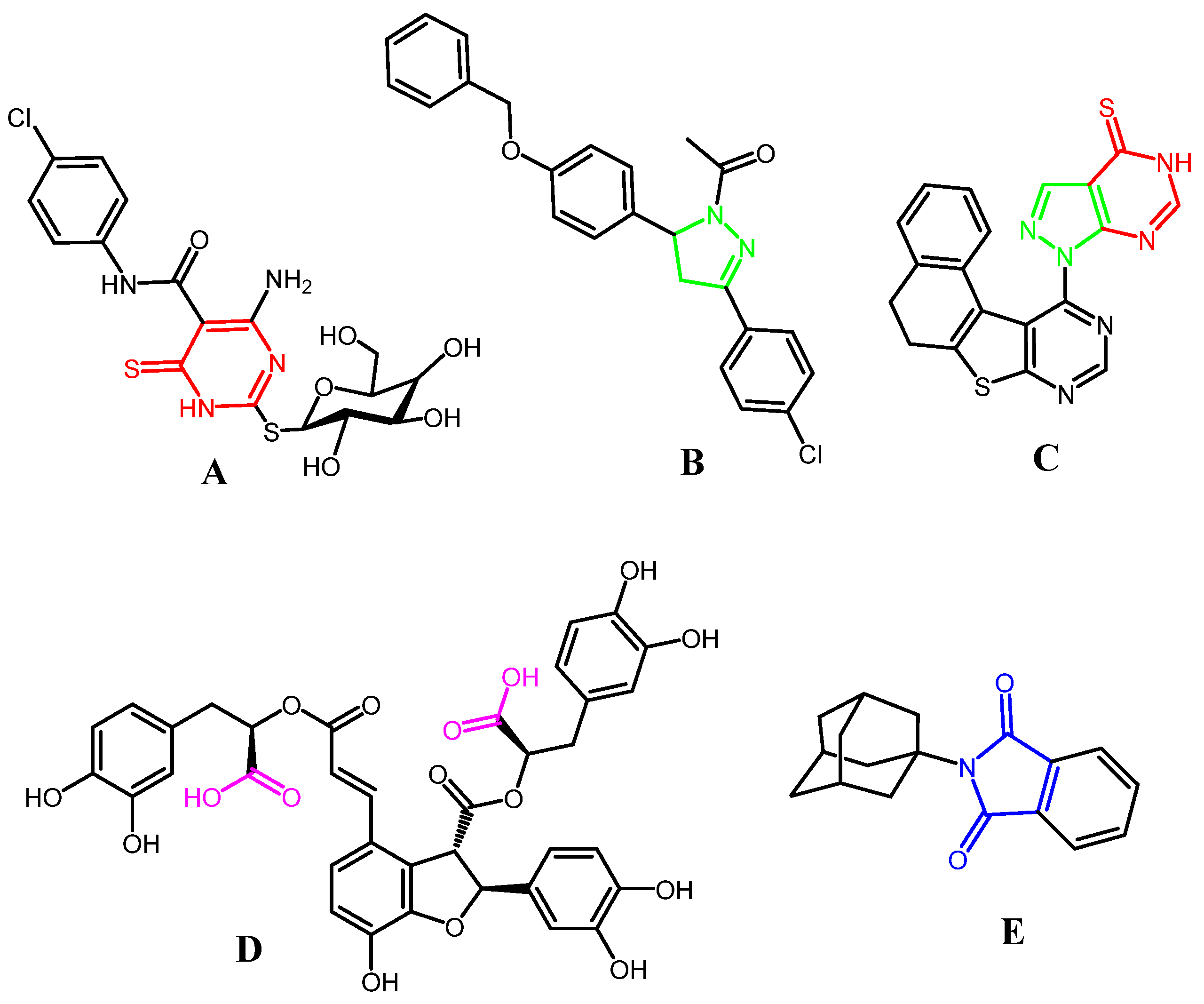
1. Introduction
2. Results and Discussion
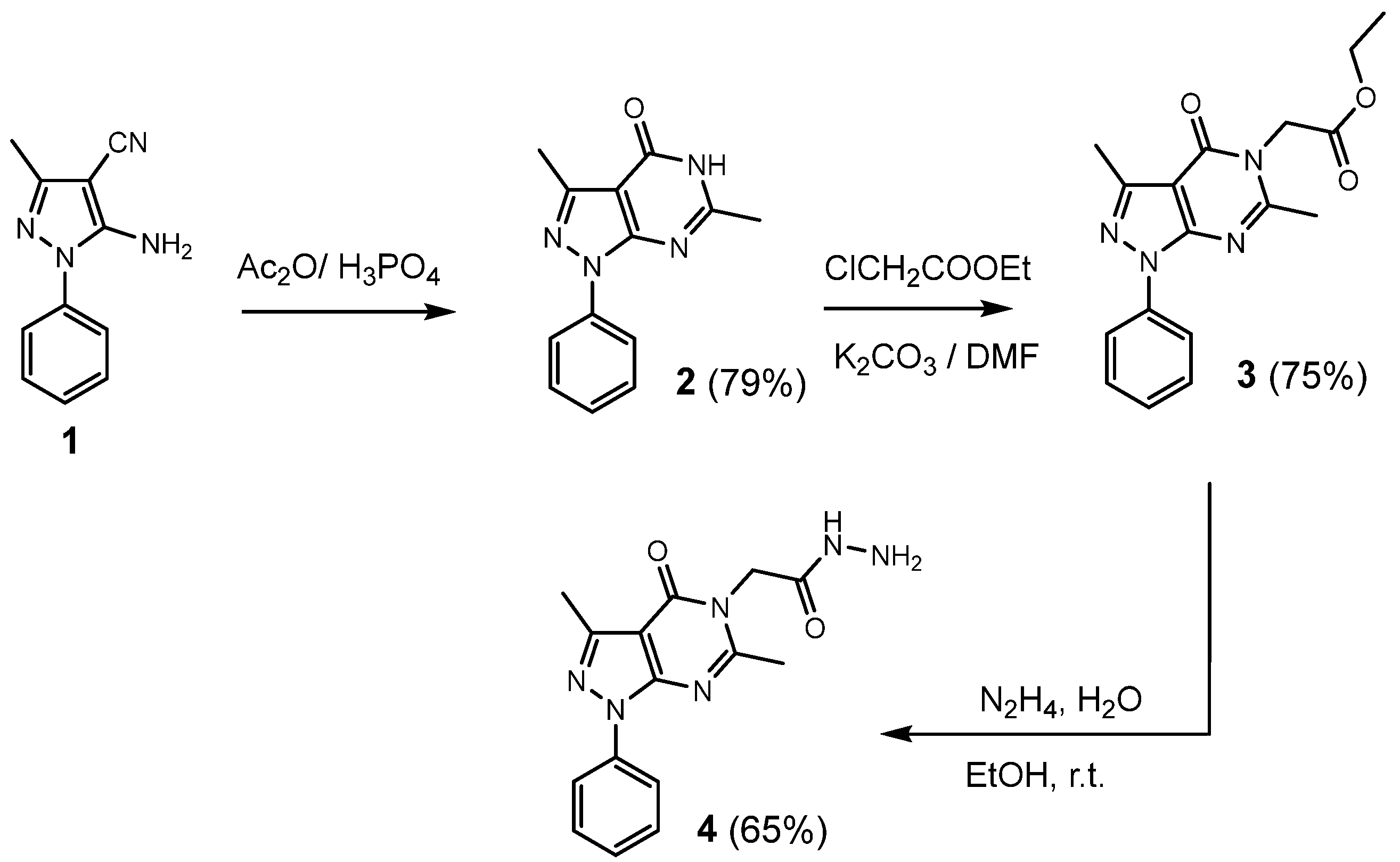
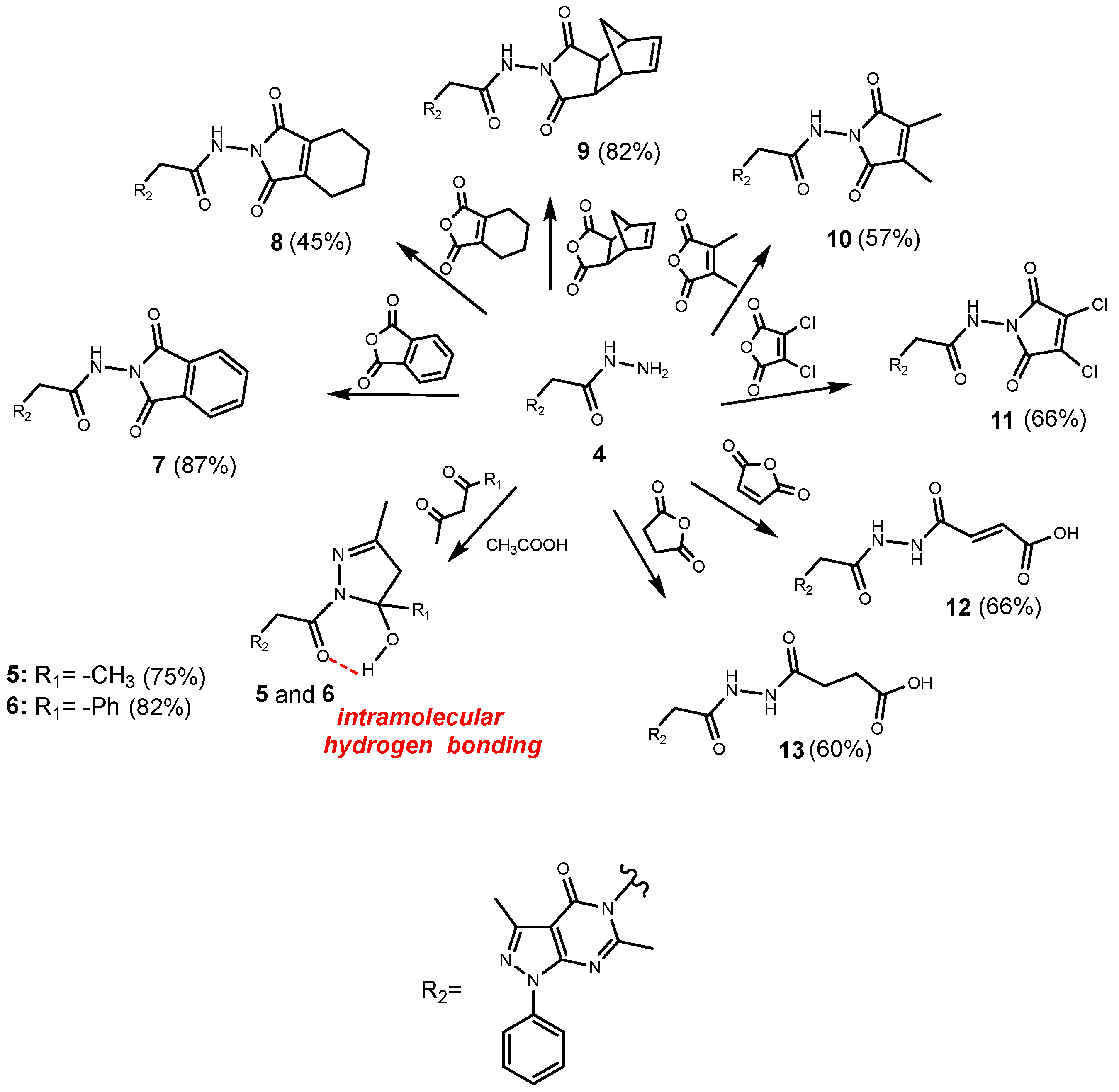
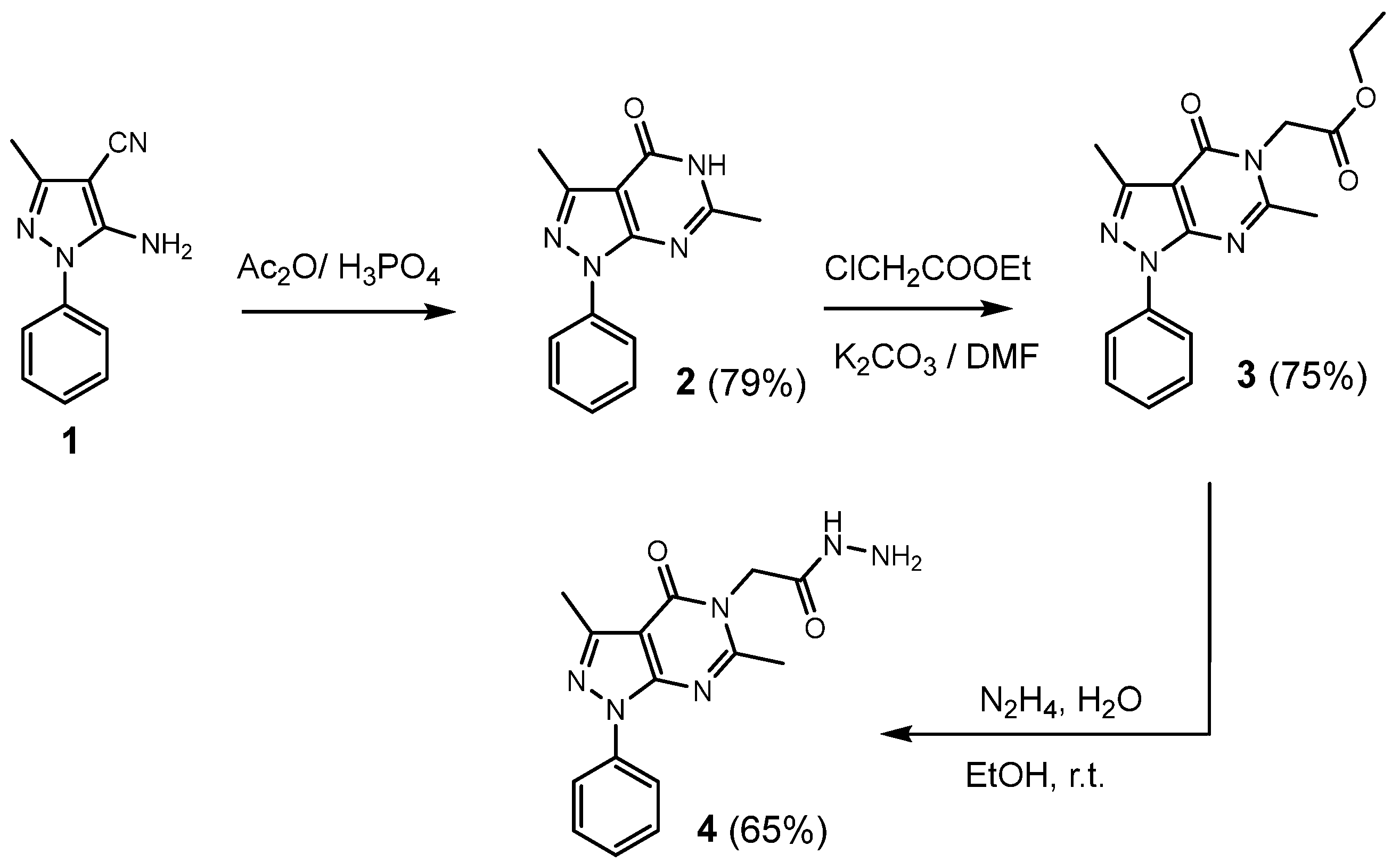
2.1. Chemistry
2.2. Molecular Docking Studies
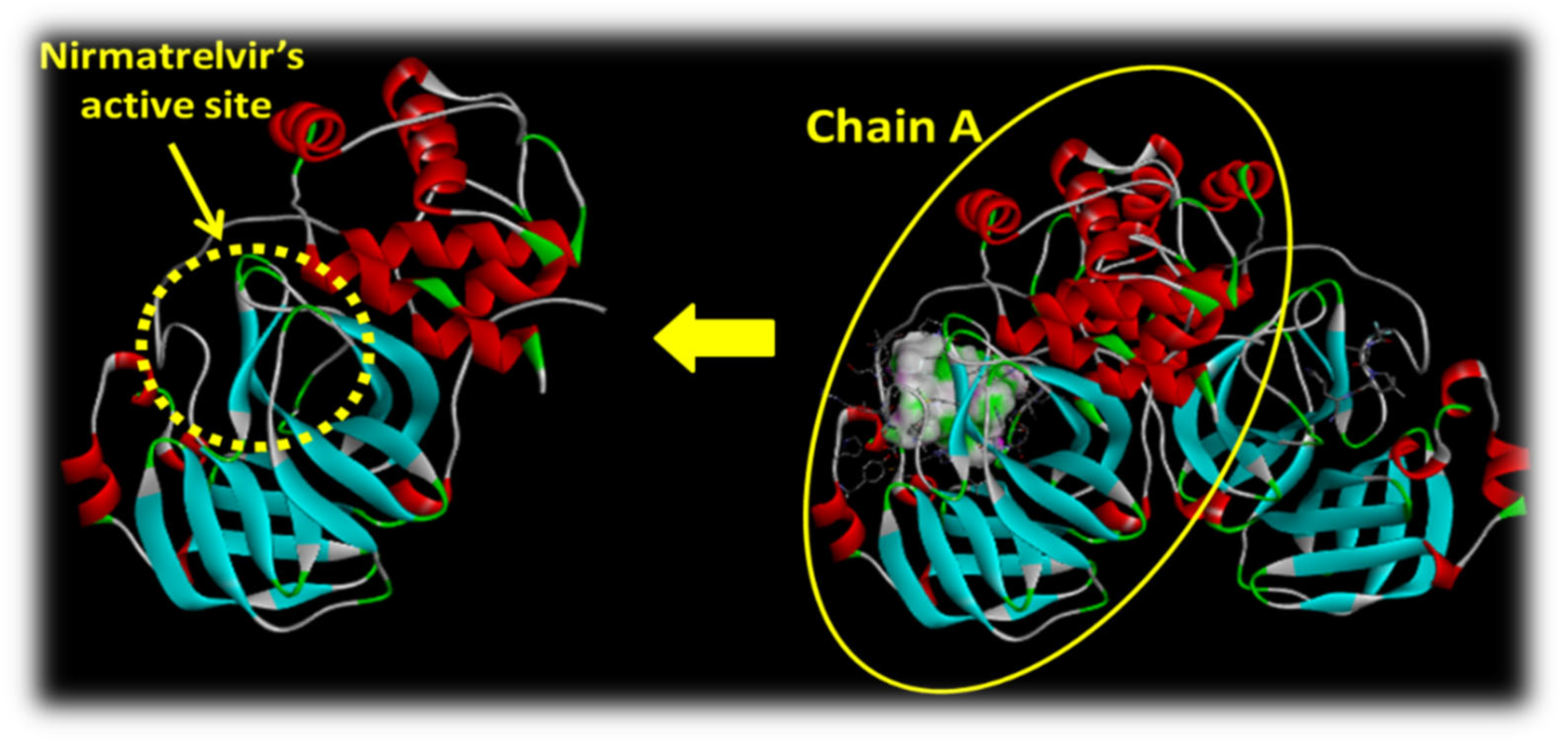
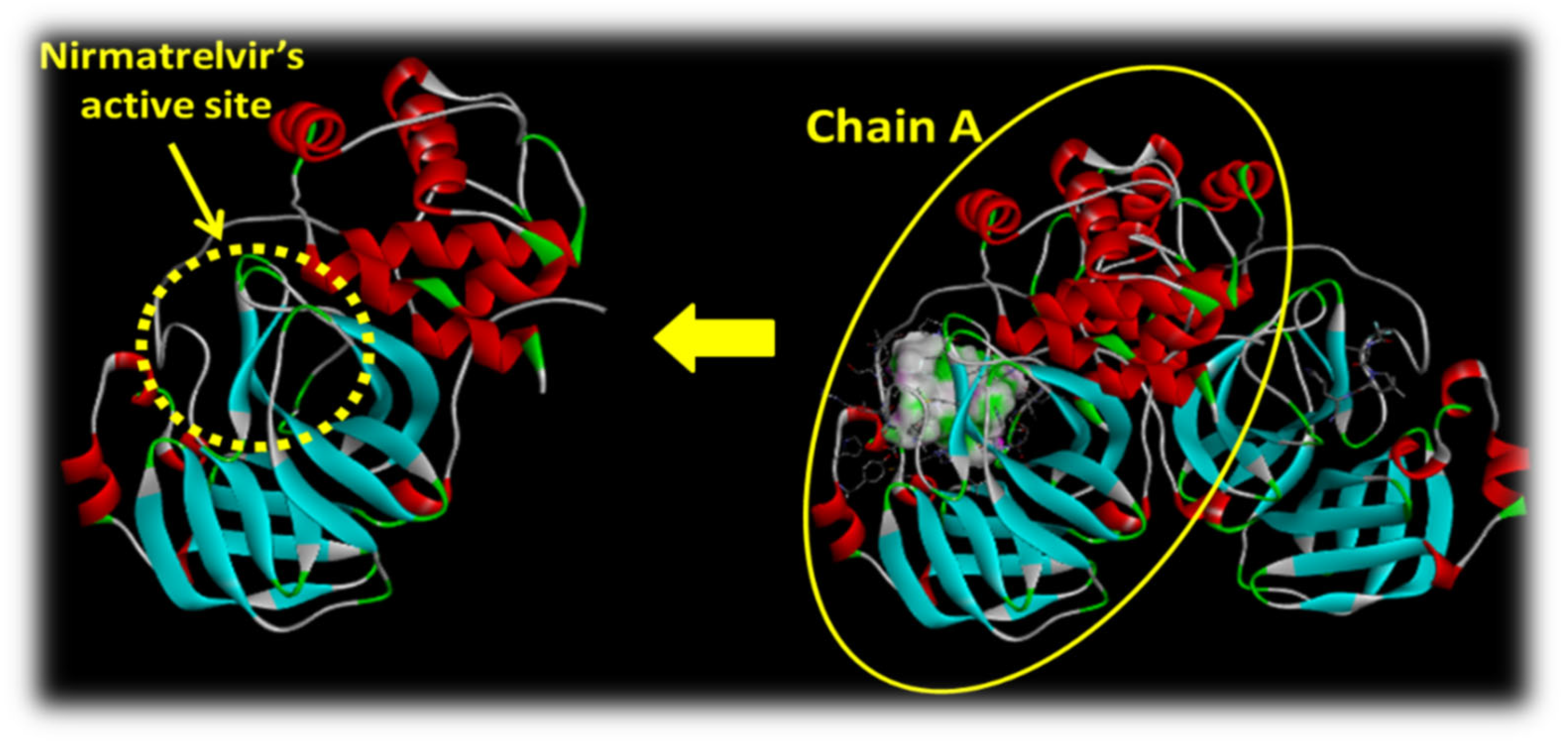
2.2.1. Insight into the Protein Used
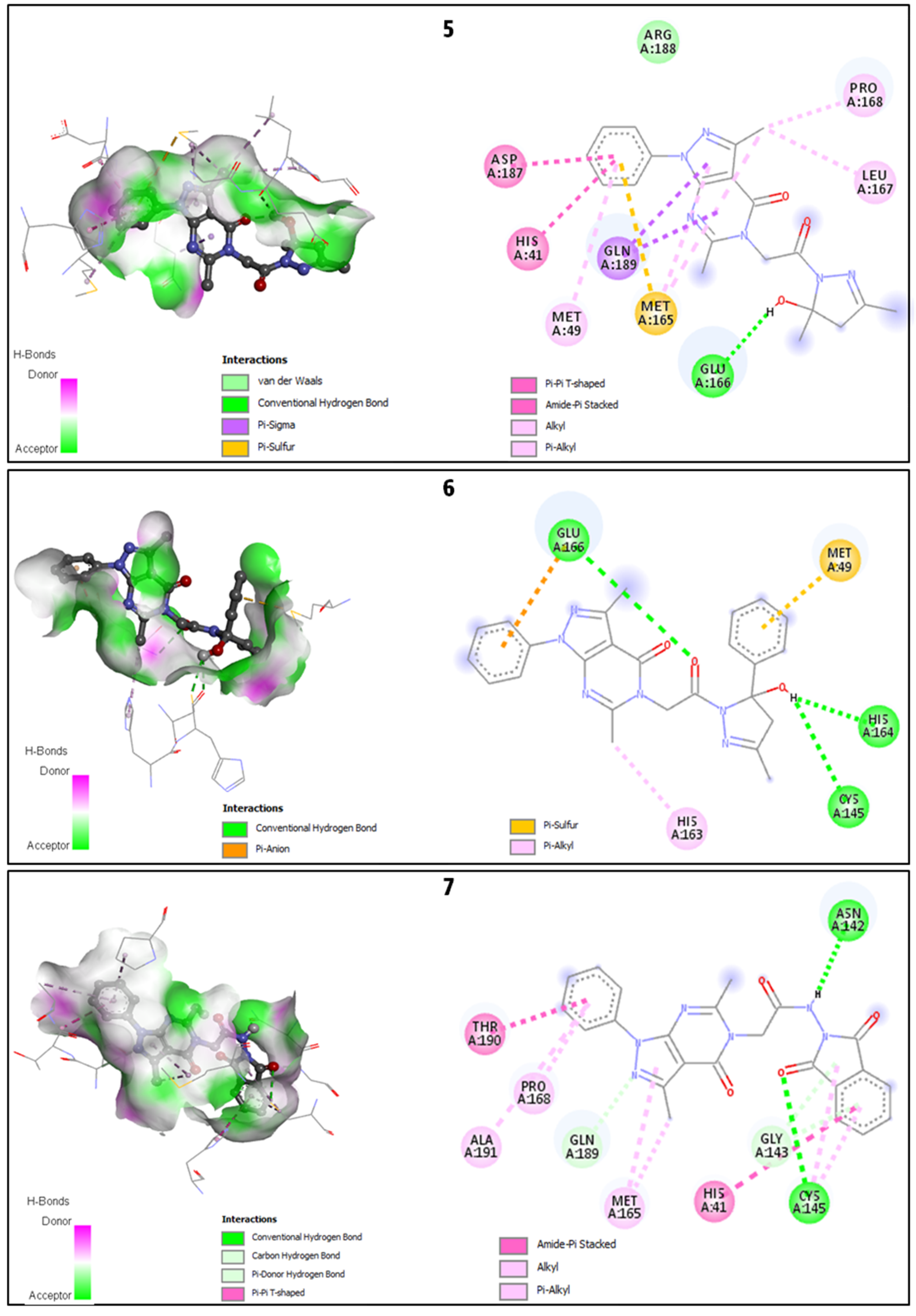
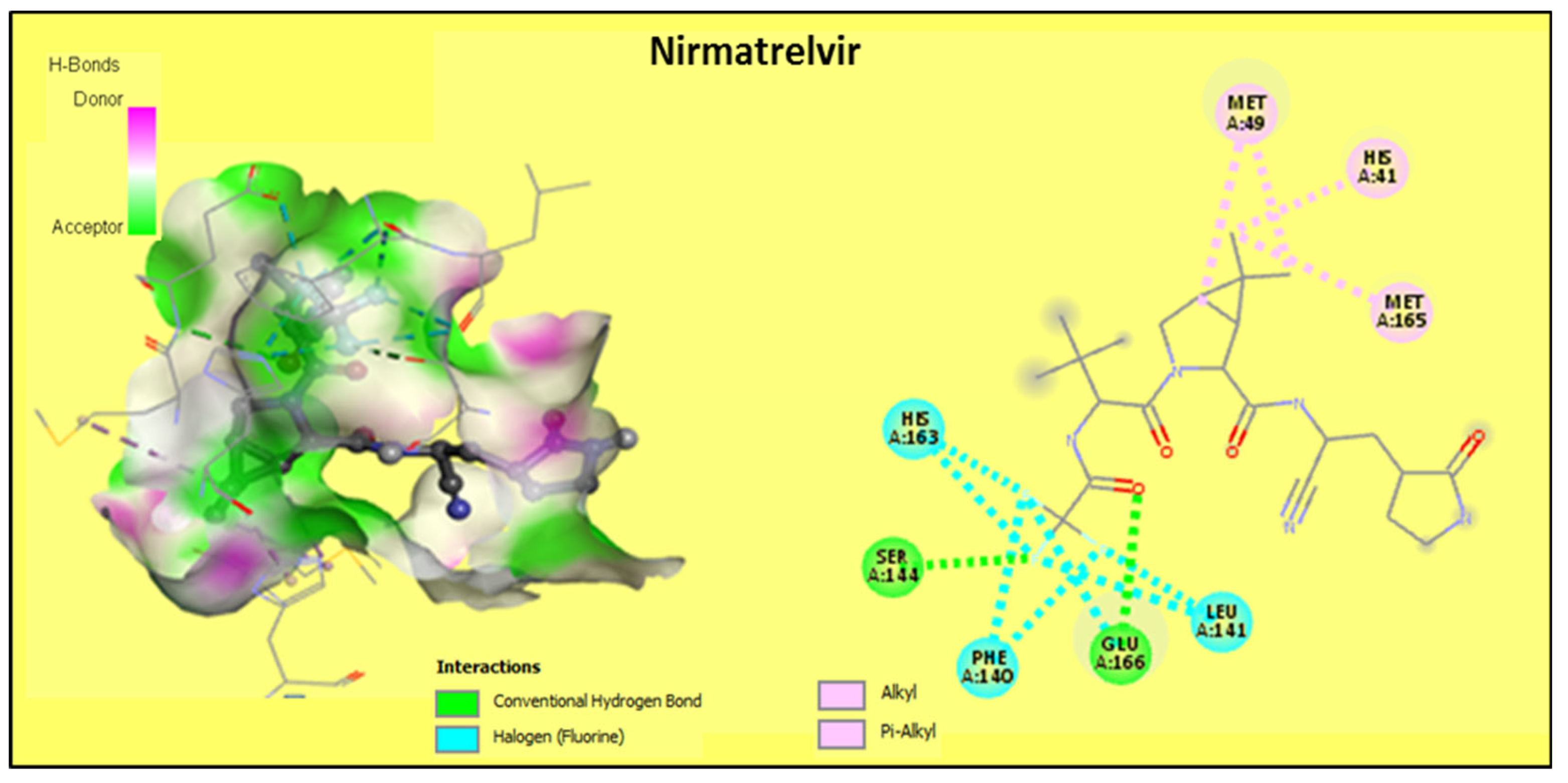
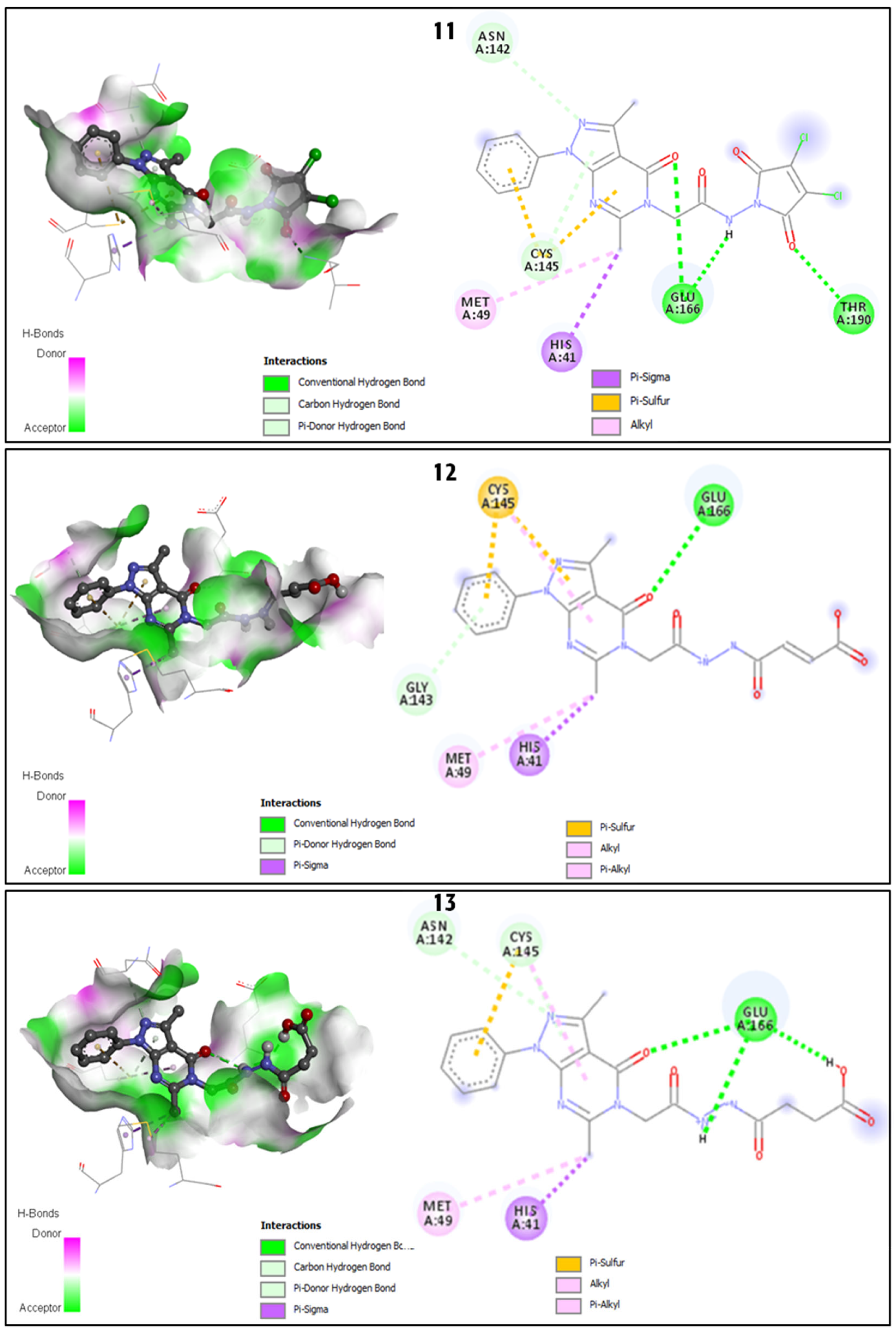
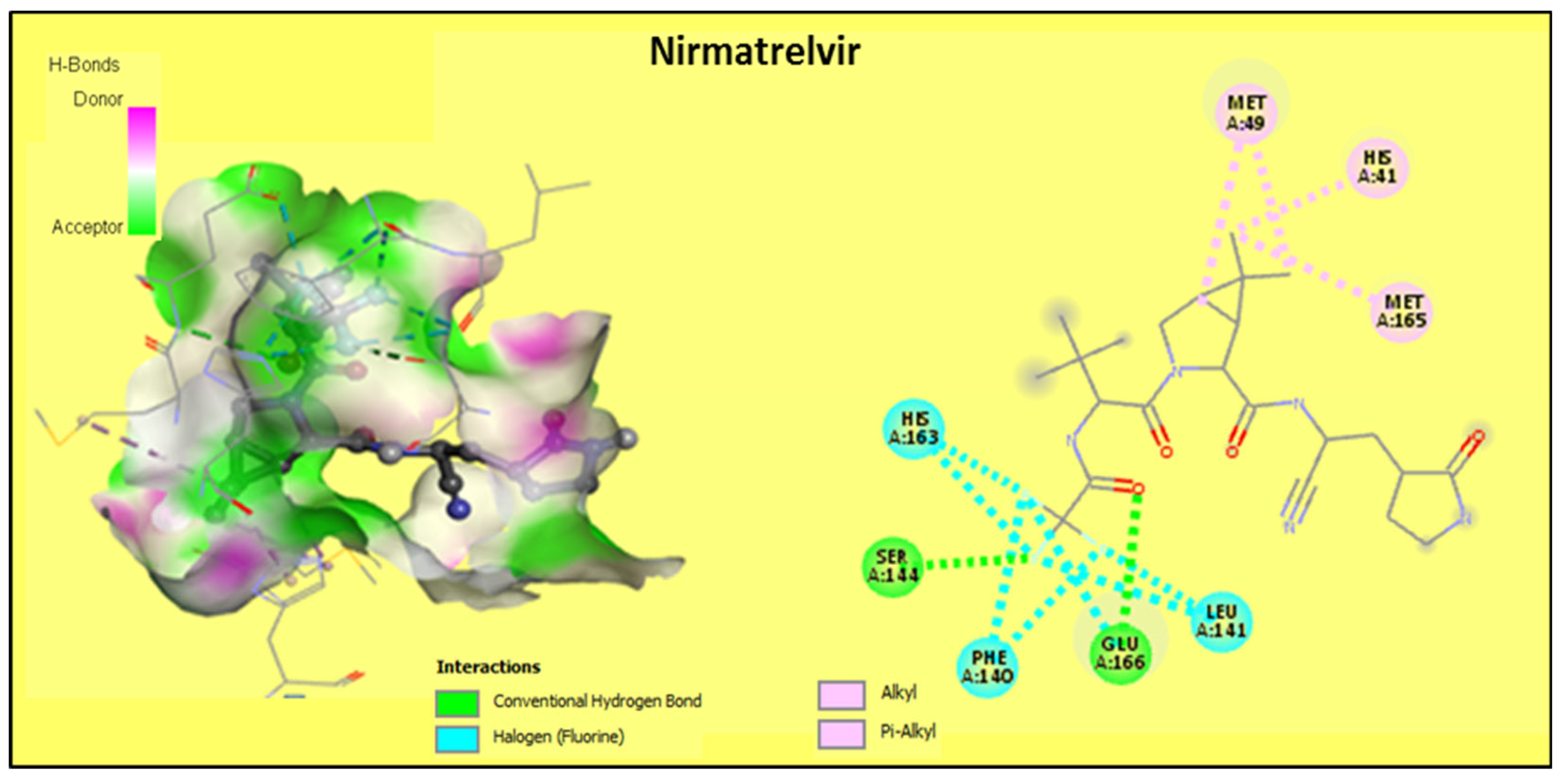
2.2.2. Discussion of the Different Interactions Formed
2.2.3. Binding Energies Analysis
2.3. Prediction of Drug-Likeness Descriptors and Toxicity
2.3.1. Predicted Molecular Properties and Drug-Likeness
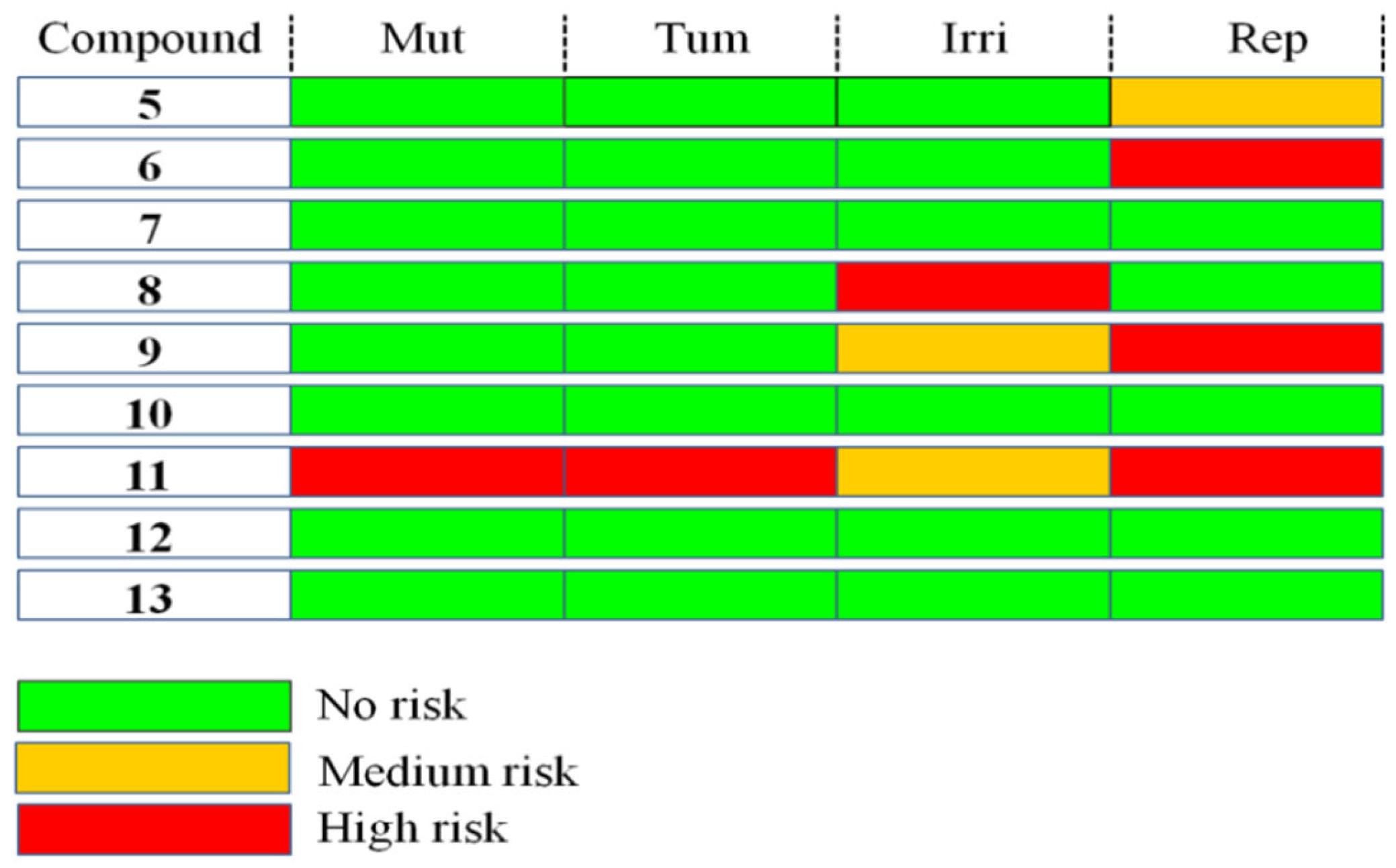
2.3.2. Toxicity Analysis
3. Materials and Methods
3.1. Chemistry
3.1.1. General Procedure for the Synthesis of 3,6-dimethyl-1-phenyl-1,5-dihydro-4H-pyrazolo [3,4-d]pyrimidin-4-one (2)
3.1.2. General Procedure for the Synthesis of Ethyl 2-(3,6-dimethyl-4-oxo-1-phenyl-1,4-dihydro-5H-pyrazolo[3,4-d]pyrimidin-5-yl)acetate (3)
3.1.3. General Procedure for the Synthesis of 2-(3,6-dimethyl-4-oxo-1-phenyl-1,4-dihydro-5H-pyrazolo[3,4-d]pyrimidin-5-yl)acetohydrazide (4)
3.1.4. General Procedure for the Synthesis of the Compounds 5 and 6
3.1.5. General Procedure for the Synthesis of the Compounds 7–13
3.2. Molecular Docking Procedure
3.3. Chemoinformatics
3.3.1. ADMET Properties
3.3.2. Toxicity Prediction
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Hu, B.; Guo, H.; Zhou, P.; Shi, Z.L. Characteristics of SARS-CoV-2 and COVID-19. Nat. Rev. Microbiol. 2021, 19, 141–154. [Google Scholar] [CrossRef]
- World Health Organization (WHO). 14.9 Million Excess Deaths Associated with the COVID-19 Pandemic in 2020 and 2021. Available online: https://www.who.int/news/item/05-05-2022-14.9-million-excess-deaths-were-associated-with-the-covid-19-pandemic-in-2020-and-2021 (accessed on 7 July 2022).
- Bheenaveni, R.S. India’s Indigenous Idea of Herd Immunity: The Solution for COVID-19? Tradit. Med. Res. 2020, 5, 182. [Google Scholar]
- Al-Shamsi, H.O.; Alhazzani, W.; Alhuraiji, A.; Coomes, E.A.; Chemaly, R.F.; Almuhanna, M.; Wolff, R.A.; Ibrahim, N.K.; Chua, M.L.K.; Hotte, S.J.; et al. A Practical Approach to the Management of Cancer Patients During the Novel Coronavirus Disease 2019 (COVID-19) Pandemic: An International Collaborative Group. Oncologist 2020, 25, e936–e945. [Google Scholar] [CrossRef] [PubMed]
- Corey, L.; Beyrer, C.; Cohen, M.S.; Michael, N.L.; Bedford, T.; Rolland, M. SARS-CoV-2 Variants in Patients with Immunosuppression. N. Engl. J. Med. 2021, 385, 562–566. [Google Scholar] [CrossRef]
- Ibrahim, M.; Abdelrahman, A.; Hussien, T.; Badr, E.; Mohamed, T.; El-Seedi, H.; Pare, P.; Efferth, T.; Hegazy, M.E. In silico drug discovery of major metabolites from spices as SARS-CoV-2 main protease inhibitors. Comput. Biol. Med. 2020, 126, 104046. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Lin, D.; Sun, X.; Curth, U.; Drosten, C.; Sauerhering, L.; Becker, S.; Rox, K.; Hilgenfeld, R. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved a-ketoamide inhibitors. Science 2020, 368, 409–412. [Google Scholar] [CrossRef]
- Bhardwaj, V.; Singh, R.; Sharma, J.; Rajendran, V.; Purohit, R.; Kumar, S. Identification of bioactive molecules from Tea plant as SARS-CoV-2 main protease inhibitors. J. Biomol. Struct. Dyn. 2020, 10, 3449–3458. [Google Scholar] [CrossRef]
- Kumar, S.; Thambiraja, T.S.; Karuppanan, K.; Subramaniam, G. Omicron and Delta Variant of SARS-CoV-2: A Comparative Computational Study of Spike Protein. J. Med. Virol. 2022, 94, 1641–1649. [Google Scholar] [CrossRef]
- Chen, J.; Wang, R.; Gilby, N.B.; Wei, G.-W. Omicron Variant (B.1.1.529): Infectivity, Vaccine Breakthrough, and Antibody Resistance. J. Chem. Inf. Model. 2022, 62, 412–422. [Google Scholar] [CrossRef]
- Pulliam, J.R.C.; van Schalkwyk, C.; Govender, N.; von Gottberg, A.; Cohen, C.; Groome, M.J.; Dushoff, J.; Mlisana, K.; Moultrie, H. Increased Risk of SARS-CoV-2 Reinfection Associated with Emergence of the Omicron Variant in South Africa. Science 2022, 376, eabn4947. [Google Scholar] [CrossRef]
- Graham, F. Daily Briefing: Omicron Coronavirus Variant Puts Scientists on Alert. Nature 2021. Available online: https://www.nature.com/articles/d41586-021-03564-6 (accessed on 7 July 2022). [CrossRef] [PubMed]
- Karim, S.S.A.; Karim, Q.A. Omicron SARS-CoV-2 Variant: A New Chapter in the COVID-19 Pandemic. Lancet 2021, 398, 2126–2128. [Google Scholar] [CrossRef]
- Commissioner O. of the Coronavirus (COVID-19) Update: FDA Issues Emergency Use Authorization for Potential COVID-19 Treatment. Available online: https://www.fda.gov/news-events/press-announcements/coronavirus-covid-19-update-fda-issues-emergency-use-authorization-potential-covid-19-treatment (accessed on 6 June 2022).
- EMA Treatments and Vaccines for COVID-19. Available online: https://www.ema.europa.eu/en/human-regulatory/overview/public-health-threats/coronavirus-disease-covid-19/treatments-vaccines-covid-19 (accessed on 6 June 2022).
- Wang, Z.; Chen, X.; Lu, Y.; Chen, F.; Zhang, W. Clinical Characteristics and Therapeutic Procedure for Four Cases with 2019 Novel Coronavirus Pneumonia Receiving Combined Chinese and Western Medicine Treatment. Biosci. Trends 2020, advpub. [Google Scholar] [CrossRef] [PubMed]
- Eastin, C.; Eastin, T. Epidemiological Characteristics of 2143 Pediatric Patients with 2019 Coronavirus Disease in China. J. Emerg. Med. 2020, 58, 712–713. [Google Scholar] [CrossRef]
- Wang, C.; Horby, P.W.; Hayden, F.G.; Gao, G.F. A Novel Coronavirus Outbreak of Global Health Concern. Lancet 2020, 395, 470–473. [Google Scholar] [CrossRef]
- UniProt Consortium UniProt: A Worldwide Hub of Protein Knowledge. Nucleic Acids Res. 2019, 47, D506–D515. [CrossRef]
- Pickett, B.E.; Greer, D.S.; Zhang, Y.; Stewart, L.; Zhou, L.; Sun, G.; Gu, Z.; Kumar, S.; Zaremba, S.; Larsen, C.N.; et al. Virus Pathogen Database and Analysis Resource (ViPR): A Comprehensive Bioinformatics Database and Analysis Resource for the Coronavirus Research Community. Viruses 2012, 4, 3209–3226. [Google Scholar] [CrossRef]
- Shu, Y.; McCauley, J. GISAID: Global Initiative on Sharing All Influenza Data—From Vision to Reality. Eurosurveilliance 2017, 22, 30494. [Google Scholar] [CrossRef]
- Gasteiger, E.; Gattiker, A.; Hoogland, C.; Ivanyi, I.; Appel, R.D.; Bairoch, A. ExPASy: The Proteomics Server for in-Depth Protein Knowledge and Analysis. Nucleic Acids Res. 2003, 31, 3784–3788. [Google Scholar] [CrossRef]
- Kumar, S.; Mathavan, S.; Jin, W.J.; Azman, N.A.B.; Subramanaiam, D.; Zainalabidin, N.A.B.; Lingadaran, D.; Sattar, Z.B.A.; Manickam, D.L.; Anbananthan, P.S.; et al. COVID-19 Vaccine Candidates by Identification of B and T Cell Multi-Epitopes Against SARS-COV-2. Preprints 2020, 2020080092. [Google Scholar] [CrossRef]
- Graham, B.S. Rapid COVID-19 Vaccine Development. Science 2020, 368, 945–946. [Google Scholar] [CrossRef] [PubMed]
- Heaton, P.M. The Covid-19 Vaccine-Development Multiverse. N. Engl. J. Med. 2020, 383, 1986–1988. [Google Scholar] [CrossRef] [PubMed]
- Tan, J.-N.; Li, H.; Gu, Y. Water Mediated Trapping of Active Methylene Intermediates Generated by IBX-Induced Oxidation of Baylis–Hillman Adducts with Nucleophiles. Green Chem. 2010, 12, 1772–1782. [Google Scholar] [CrossRef]
- Thomas, N.; Zachariah, S.M. Pharmacological activities of chromene derivatives: An overview. Asian J. Pharm. Clin. Res. 2013, 6, 11–15. [Google Scholar]
- Horchani, M.; Heise, N.V.; Hoenke, S.; Csuk, R.; Harrath, A.H.; Jannet, H.B.; Romdhane, A. Synthesis and In Silico Docking of New Pyrazolo[4,3-e]Pyrido[1,2-a]Pyrimidine-Based Cytotoxic Agents. Int. J. Mol. Sci. 2021, 22, 10258. [Google Scholar] [CrossRef]
- Horchani, M.; Hajlaoui, A.; Harrath, A.H.; Mansour, L.; Ben Jannet, H.; Romdhane, A. New Pyrazolo-Triazolo-Pyrimidine Derivatives as Antibacterial Agents: Design and Synthesis, Molecular Docking and DFT Studies. J. Mol. Struct. 2020, 1199, 127007. [Google Scholar] [CrossRef]
- Chortani, S.; Nimbarte, V.D.; Horchani, M.; Ben Jannet, H.; Romdhane, A. Synthesis, Biological Evaluation and Molecular Docking Analysis of Novel Benzopyrimidinone Derivatives as Potential Anti-Tyrosinase Agents. Bioorg. Chem. 2019, 92, 103270. [Google Scholar] [CrossRef]
- Cherif, M.; Horchani, M.; Al-Ghamdi, Y.O.; Almalki, S.G.; Alqurashi, Y.E.; Ben Jannet, H.; Romdhane, A. New Pyrano-1,2,3-Triazolopyrimidinone Derivatives as Anticholinesterase and Antibacterial Agents: Design, Microwave-Assisted Synthesis and Molecular Docking Study. J. Mol. Struct. 2020, 1220, 128685. [Google Scholar] [CrossRef]
- Mamdouh, A.A.; Galal, H.E.; Nashwa, M.M. Anti-Covid-19 Drug Analogues: Synthesis of Novel Pyrimidine Thioglycosides as Antiviral Agents Against SARS-COV-2 and Avian Influenza H5N1 Viruses. ACS Omega 2021, 6, 16890–16904. [Google Scholar] [CrossRef]
- El-Sabbagh, O.I.; Baraka, M.M.; Ibrahim, S.M.; Pannecouque, C.; Andrei, G.; Snoeck, R.; Balzarini, J.; Rashad, A.A. Synthesis and Antiviral Activity of New Pyrazole and Thiazole Derivatives. Eur. J. Med. Chem. 2009, 44, 3746–3753. [Google Scholar] [CrossRef]
- Rashad, A.E.; Hegab, M.I.; Abdel-Megeid, R.E.; Micky, J.A.; Abdel-Megeid, F.M.E. Synthesis and Antiviral Evaluation of Some New Pyrazole and Fused Pyrazolopyrimidine Derivatives. Bioorg. Med. Chem. 2008, 16, 7102–7106. [Google Scholar] [CrossRef] [PubMed]
- Elebeedy, D.; Elkhatib, W.F.; Kandeil, A.; Ghanem, A.; Kutkat, O.; Alnajjar, R.; Saleh, M.A.; Maksoud, A.I.A.E.; Badawy, I.; Al-Karmalawy, A.A. Anti-SARS-CoV-2 Activities of Tanshinone IIA, Carnosic Acid, Rosmarinic Acid, Salvianolic Acid, Baicalein, and Glycyrrhetinic Acid between Computational and in Vitro Insights. RSC Adv. 2021, 11, 29267–29286. [Google Scholar] [CrossRef] [PubMed]
- Mandić, L.; Benčić, P.; Mlinarić-Majerski, K.; Liekens, S.; Snoeck, R.; Andrei, G.; Kralj, M.; Basarić, N. Substituted Adamantylphthalimides: Synthesis, Antiviral and Antiproliferative Activity. Arch. Pharm. 2020, 353, 2000024. [Google Scholar] [CrossRef]
- Gan, X.; Hu, D.; Chen, Z.; Wang, Y.; Song, B. Synthesis and Antiviral Evaluation of Novel 1,3,4-Oxadiazole/Thiadiazole-Chalcone Conjugates. Bioorg. Med. Chem. Lett. 2017, 27, 4298–4301. [Google Scholar] [CrossRef]
- Sławiński, J.; Szafrański, K.; Pogorzelska, A.; Żołnowska, B.; Kawiak, A.; Macur, K.; Belka, M.; Bączek, T. Novel 2-Benzylthio-5-(1,3,4-Oxadiazol-2-Yl)Benzenesulfonamides with Anticancer Activity: Synthesis, QSAR Study, and Metabolic Stability. Eur. J. Med. Chem. 2017, 132, 236–248. [Google Scholar] [CrossRef] [PubMed]
- Naresh Kumar, R.; Poornachandra, Y.; Nagender, P.; Santhosh Kumar, G.; Krishna Swaroop, D.; Ganesh Kumar, C.; Narsaiah, B. Synthesis of Novel Nicotinohydrazide and (1,3,4-Oxadiazol-2-Yl)-6-(Trifluoromethyl)Pyridine Derivatives as Potential Anticancer Agents. Bioorg. Med. Chem. Lett. 2016, 26, 4829–4831. [Google Scholar] [CrossRef]
- Ghosh, R.; Chakraborty, A.; Biswas, A.; Chowdhuri, S. Identification of polyphenols from Broussonetiapapyrifera as SARS CoV-2 main protease inhibitors using in silico docking and molecular dynamics simulation approaches. J. Biomol. Struct. Dyn. 2021, 39, 6747–6760. [Google Scholar] [CrossRef]
- Ye, Z.; Yang, Y.; Li, X.; Cao, D.; Ouyang, D. An Integrated Transfer Learning and Multitask Learning Approach for Pharmacokinetic Parameter Prediction. Mol. Pharm. 2019, 16, 533–541. [Google Scholar] [CrossRef]
- Saraswat, J.; Singh, P.; Patel, R. A Computational Approach for the Screening of Potential Antiviral Compounds against SARS-CoV-2 Protease: Ionic Liquid vs Herbal and Natural Compounds. J. Mol. Liq. 2021, 326, 115298. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the Speed and Accuracy of Docking with a New Scoring Function, Efficient Optimization and Multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- Greasley, S.E.; Noell, S.; Plotnikova, O.; Ferre, R.A.; Liu, W.; Bolanos, B.; Fennell, K.; Nicki, J.; Craig, T.; Zhu, Y.; et al. Structural Basis for Nirmatrelvir in Vitro Efficacy against SARS-CoV-2 Variants. J. Biol. Chem. 2022, 298, 101972. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ligand | Binding Affinity (kcal/mol) | Intermolecular Interactions | |
|---|---|---|---|
| Conventional Hydrogen Bonds | Interacting Amino Acid Residues | ||
| 5 | −7.5 | 1 | HIS41, MET49, MET165, GLU166 *, LEU167, PRO168, ASP187, GLN189 |
| 6 | −7.7 | 3 | MET49, CYS145 *, HIS163, HIS164 *, GLU166 * |
| 7 | −8.0 | 2 | HIS41, ASN142 *, GLY143, CYS145 *, MET165, PRO168, GLN189, THR190, ALA191 |
| 8 | −8.2 | 3 | THR26 *, HIS41, MET49, GLY143 *, CYS145, HIS163, MET165, GLU166 * |
| 9 | −7.4 | 2 | HIS41, MET49, ASN142, CYS145, MET165, GLU166 **, PRO168 |
| 10 | −7.7 | 3 | HIS41, MET49, ASN142, CYS145, GLU166 **, THR190 * |
| 11 | −7.6 | 3 | HIS41, MET49, GLY143, CYS145, GLU166 **, THR190 * |
| 12 | −7.0 | 1 | HIS41, MET49, GLY143, CYS145, GLU166 * |
| 13 | −7.8 | 3 | HIS41, MET49, ASN142, CYS145, GLU166 *** |
| Nirmatrelvir R | −7.7 | 2 | HIS41, MET49, PHE140, LEU141, SER144 *, HIS163, MET165, GLU166 * |
| Compd. No. | MW a | nHA b | nHD c | logP(o/w) d | Nrotbo e | TPSA f | MV g | %Abs h |
|---|---|---|---|---|---|---|---|---|
| Acceptable Value | <500 | <10 | <5 | <5 | ≤10 | <140 | 500 | 100% |
| 5 | 394.180 | 9 | 1 | 0.654 | 4 | 105.610 | 388.873 | 72.56 |
| 6 | 456.190 | 9 | 1 | 1.515 | 5 | 105.610 | 458.888 | 72.56 |
| 7 | 442.140 | 10 | 1 | 1.101 | 5 | 119.190 | 430.449 | 67.87 |
| 8 | 446.170 | 10 | 1 | 1.327 | 5 | 127.200 | 435.722 | 65.11 |
| 9 | 458.170 | 10 | 1 | 1.014 | 5 | 127.200 | 444.462 | 65.11 |
| 10 | 420.150 | 10 | 1 | 0.902 | 5 | 119.190 | 409.687 | 67.87 |
| 11 | 460.050 | 10 | 1 | 1.263 | 5 | 119.190 | 405.517 | 67.87 |
| 12 | 410.130 | 11 | 3 | −0.505 | 8 | 148.870 | 392.441 | 57.63 |
| 13 | 412.150 | 11 | 3 | −0.562 | 9 | 148.210 | 395.078 | 57.86 |
| Compd. No. | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 |
|---|---|---|---|---|---|---|---|---|---|
| Pfizer Rule | + | + | + | + | + | + | + | + | + |
| Lipinski Rule | + | + | + | + | + | + | + | + | + |
| i Nviolation Lipinski (Acceptable Value ≤ 1) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Horchani, M.; Heise, N.V.; Csuk, R.; Ben Jannet, H.; Harrath, A.H.; Romdhane, A. Synthesis and In Silico Docking Study towards M-Pro of Novel Heterocyclic Compounds Derived from Pyrazolopyrimidinone as Putative SARS-CoV-2 Inhibitors. Molecules 2022, 27, 5303. https://doi.org/10.3390/molecules27165303
Horchani M, Heise NV, Csuk R, Ben Jannet H, Harrath AH, Romdhane A. Synthesis and In Silico Docking Study towards M-Pro of Novel Heterocyclic Compounds Derived from Pyrazolopyrimidinone as Putative SARS-CoV-2 Inhibitors. Molecules. 2022; 27(16):5303. https://doi.org/10.3390/molecules27165303
Chicago/Turabian StyleHorchani, Mabrouk, Niels V. Heise, René Csuk, Hichem Ben Jannet, Abdel Halim Harrath, and Anis Romdhane. 2022. "Synthesis and In Silico Docking Study towards M-Pro of Novel Heterocyclic Compounds Derived from Pyrazolopyrimidinone as Putative SARS-CoV-2 Inhibitors" Molecules 27, no. 16: 5303. https://doi.org/10.3390/molecules27165303
APA StyleHorchani, M., Heise, N. V., Csuk, R., Ben Jannet, H., Harrath, A. H., & Romdhane, A. (2022). Synthesis and In Silico Docking Study towards M-Pro of Novel Heterocyclic Compounds Derived from Pyrazolopyrimidinone as Putative SARS-CoV-2 Inhibitors. Molecules, 27(16), 5303. https://doi.org/10.3390/molecules27165303