
Epidermal Growth Factor Receptor Kinase Inhibitor Ameliorates β-Amyloid Oligomer-Induced Alzheimer Disease in Swiss Albino Mice


Abstract
:1. Introduction
2. Results
2.1. Effect of GE on AD-Induced Behavioral Changes
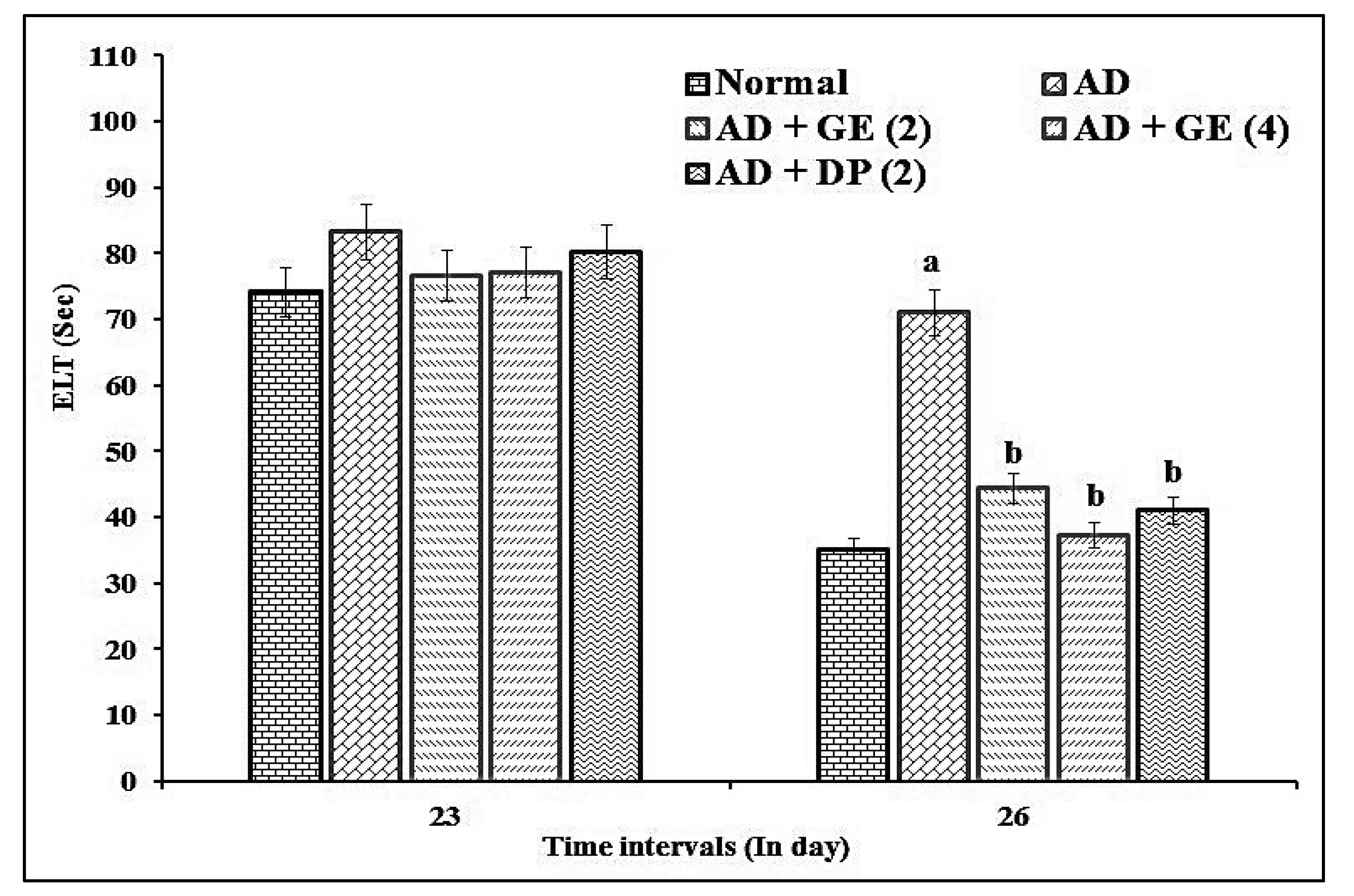
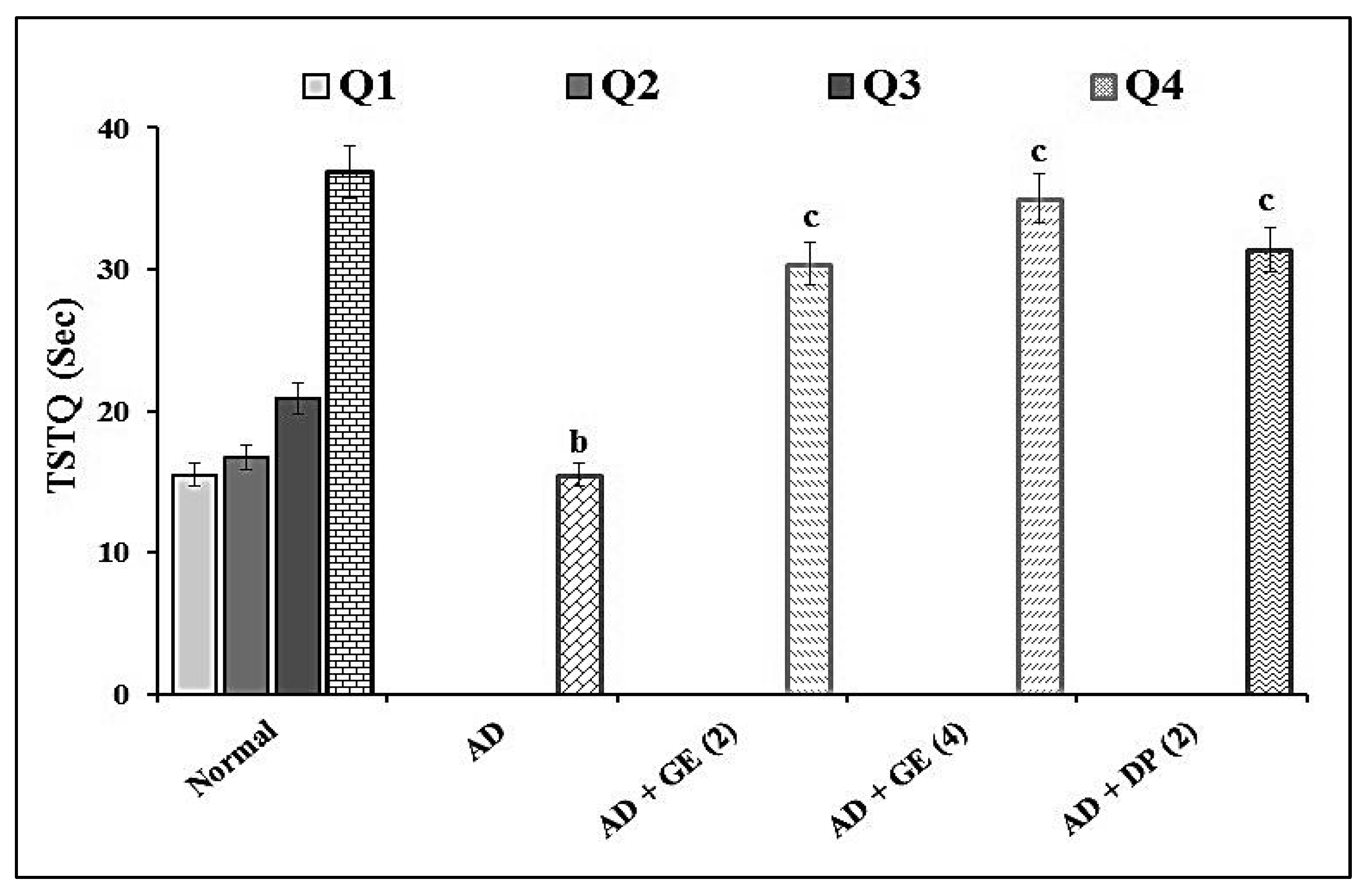
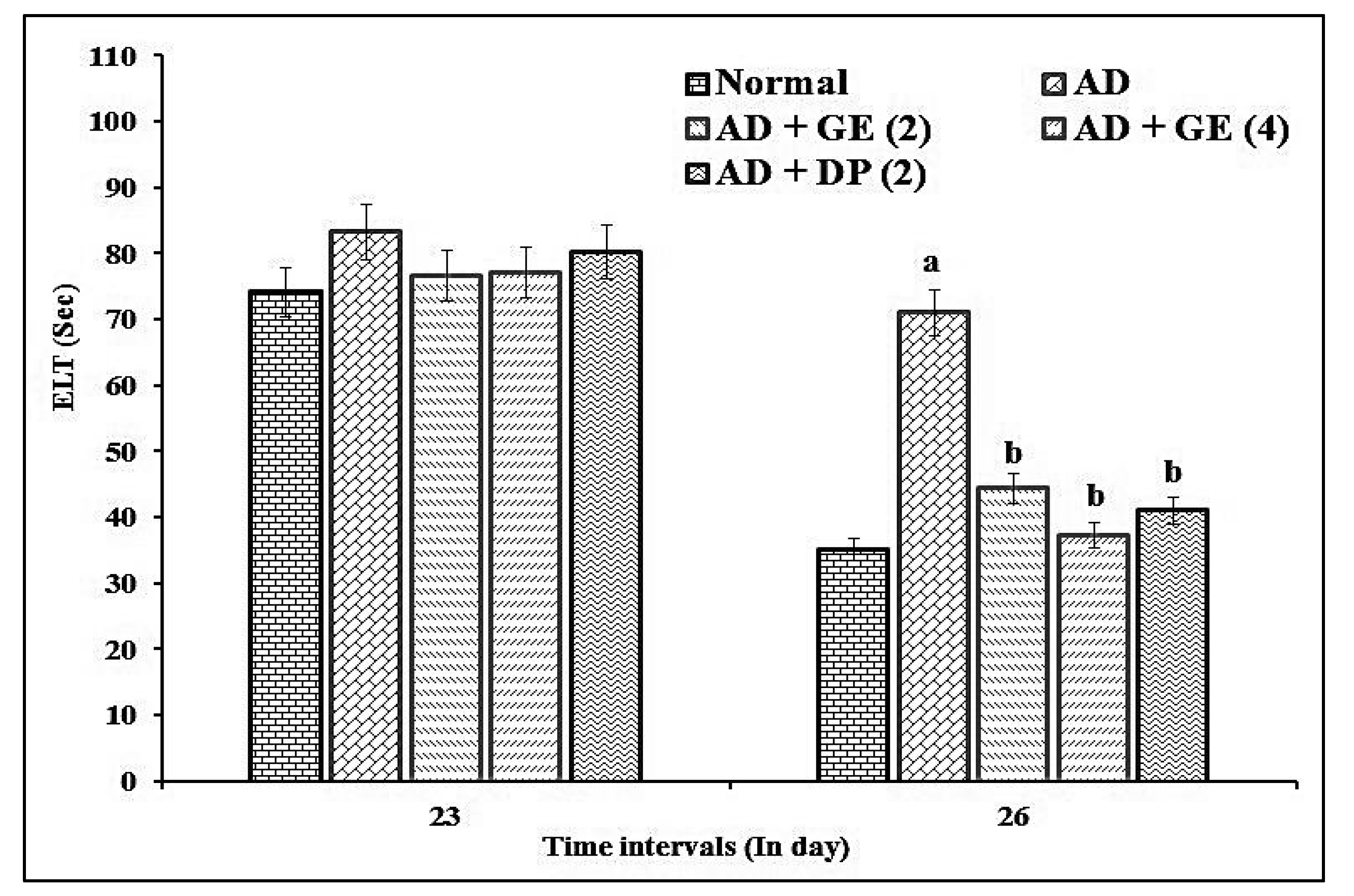
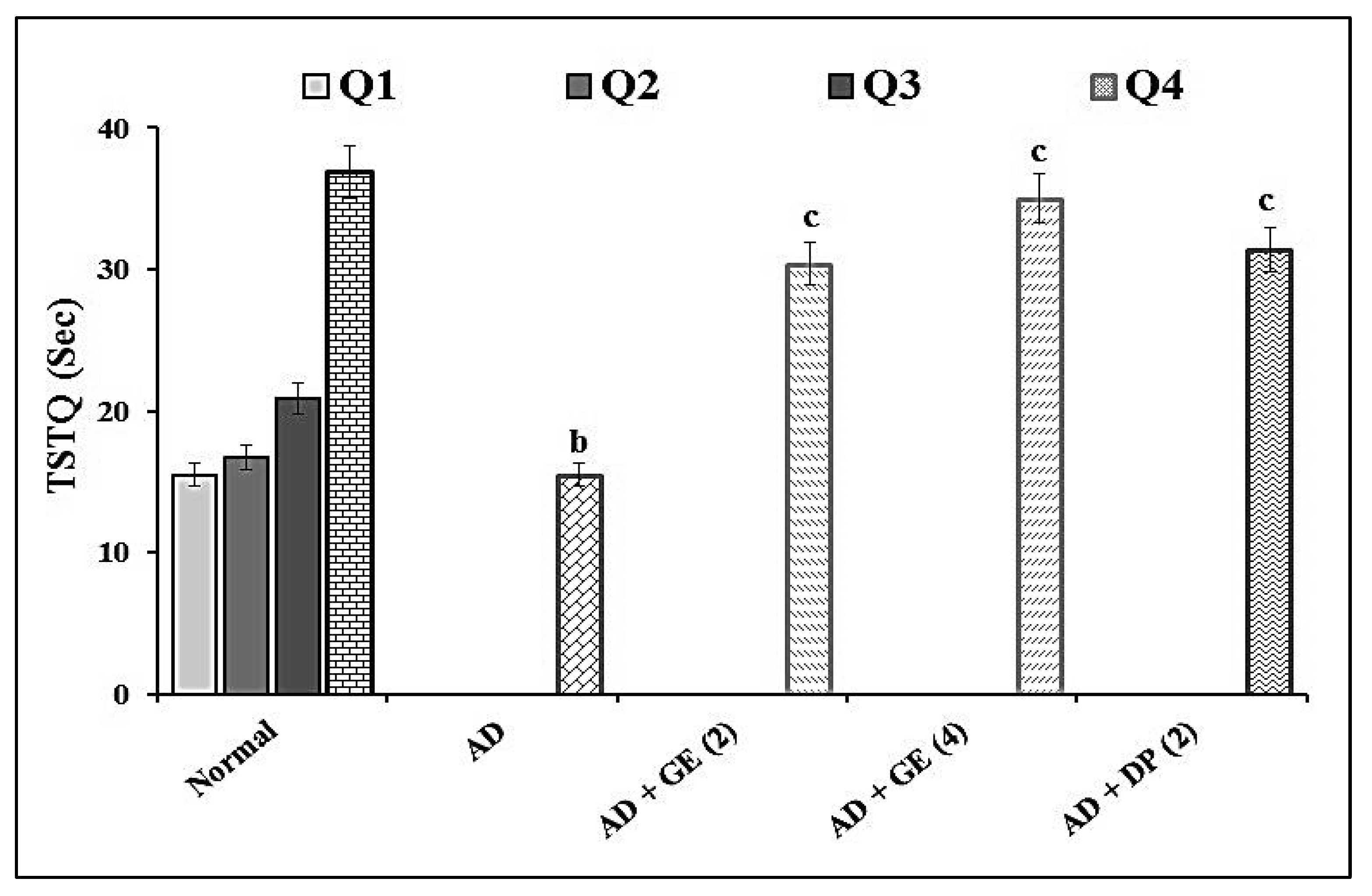
2.2. Effect of GE on Escape Latency Time (ELT) and Time Spent in the Target Quadrant (TSTQ) in MWM Test
2.3. Biomarker Estimations
Effect of GE on Aβ Oligomer-Induced AD Tissue Biomarker Changes
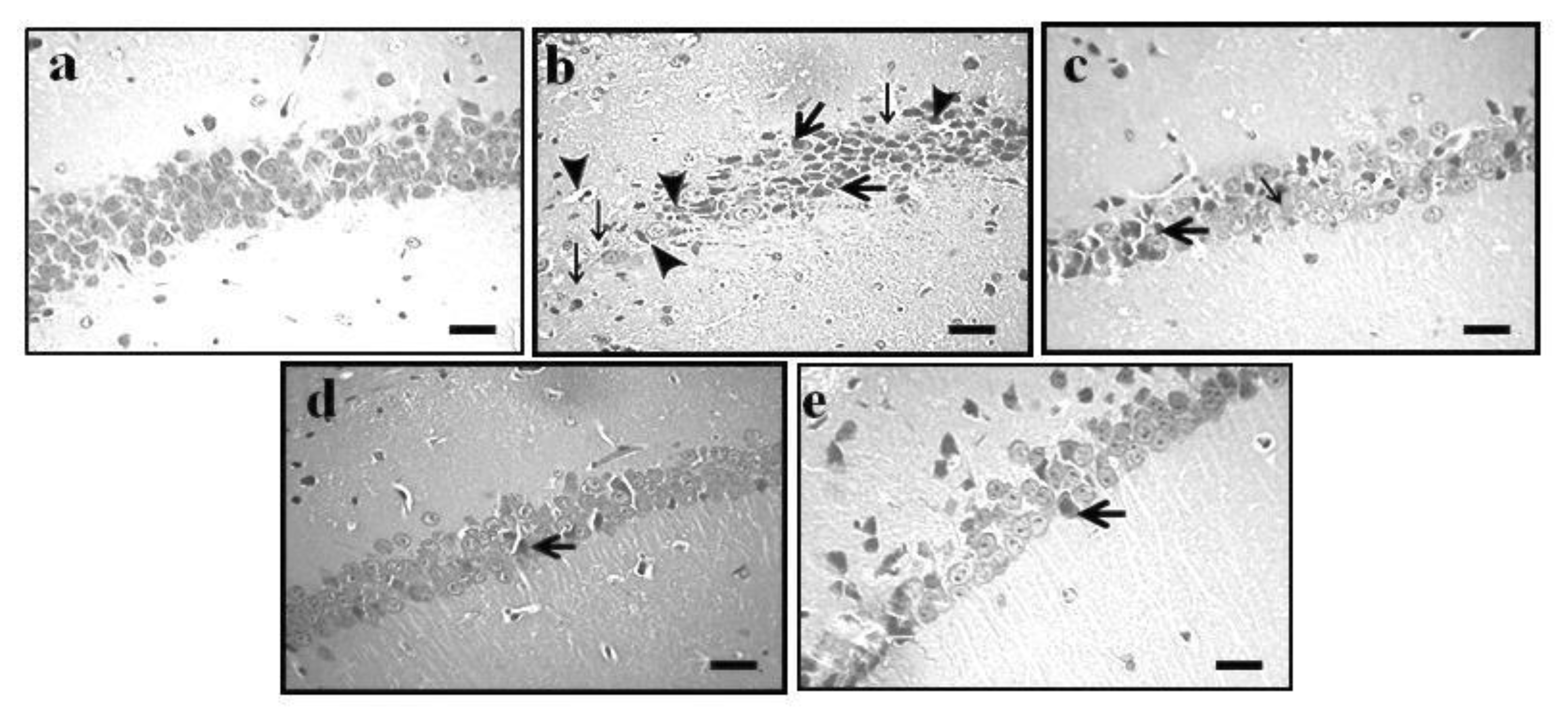
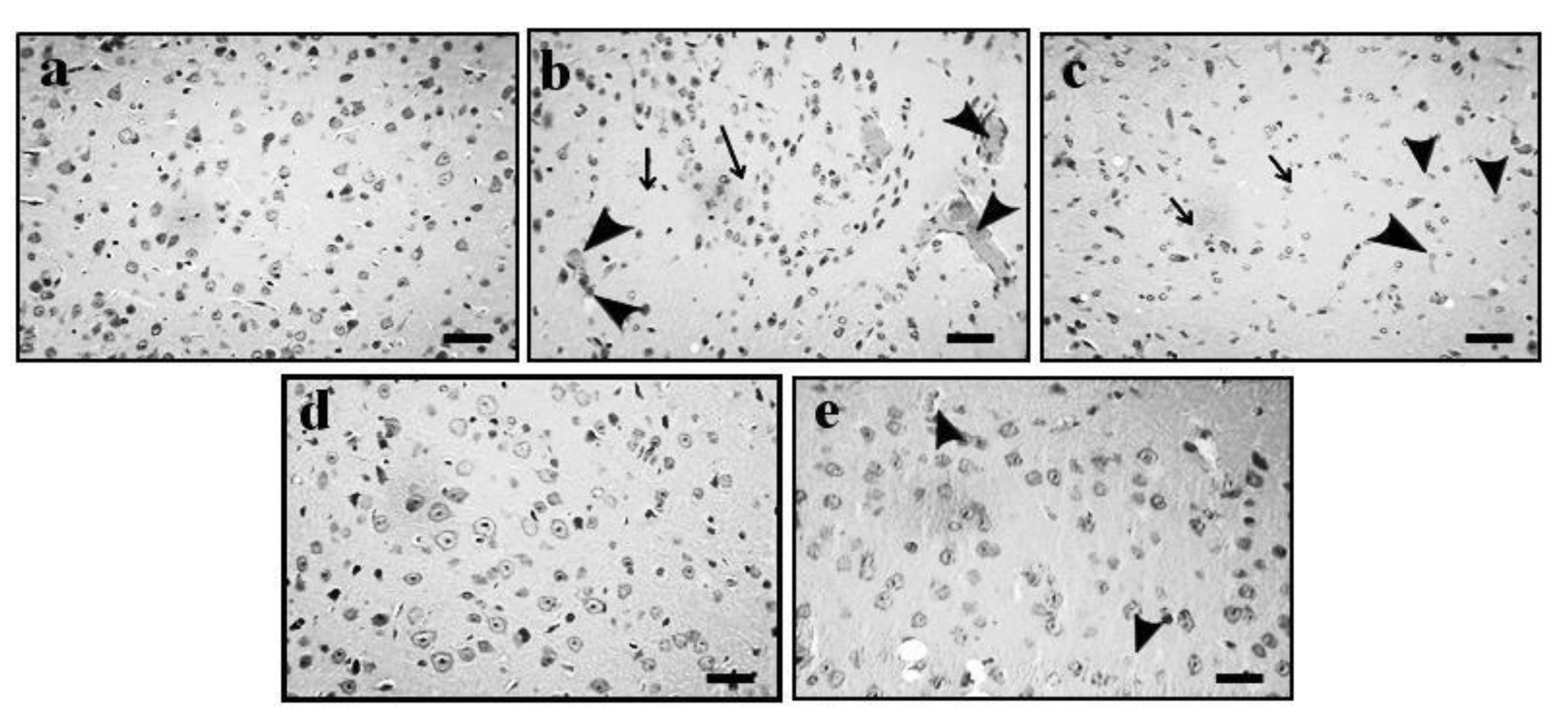
2.4. Effect of GE on AD-Induced Histopathological Changes
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Chemicals
4.3. Preparation of Aβ1–42 Oligomer
4.4. Induction of AD Mouse Model
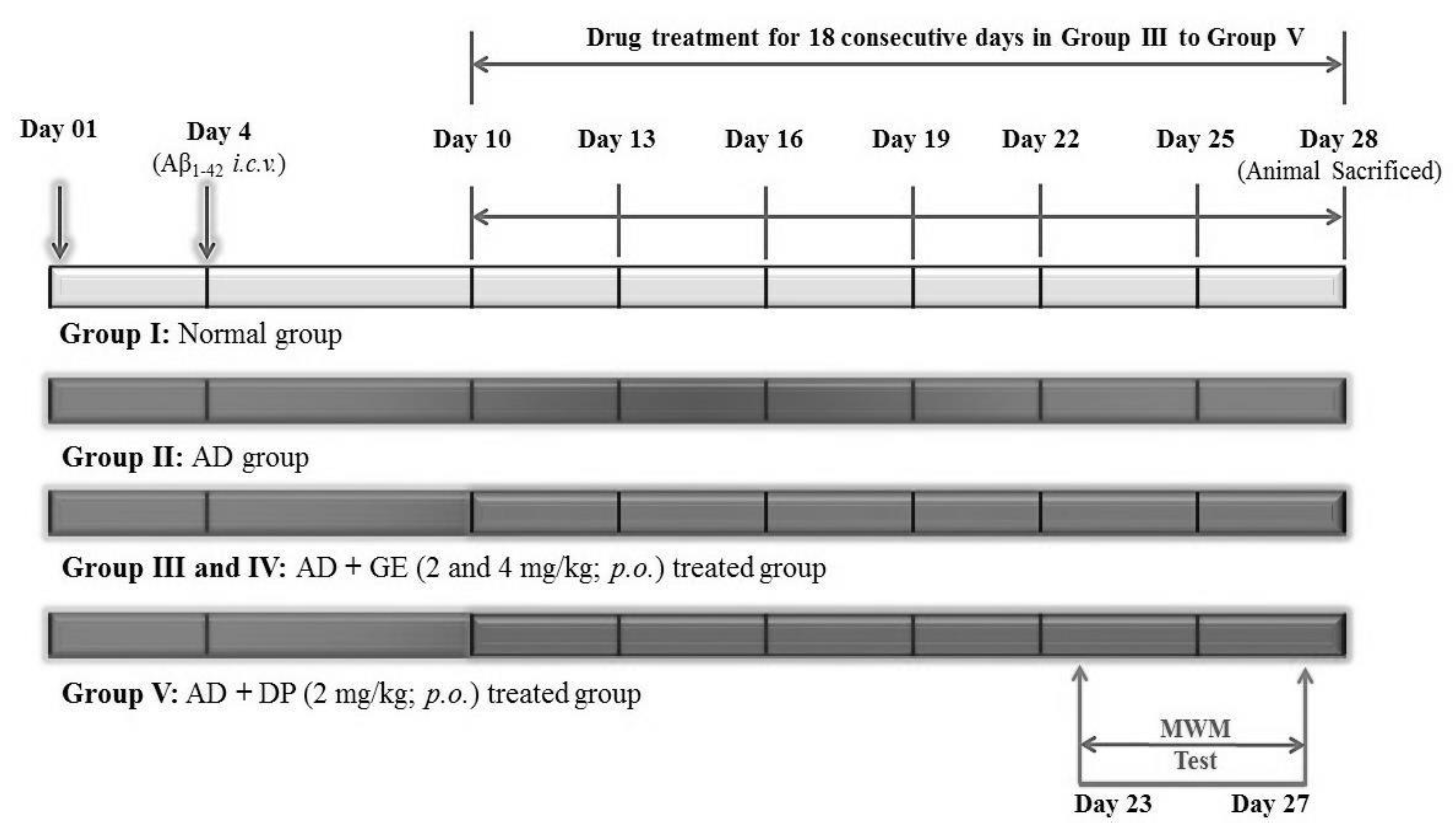
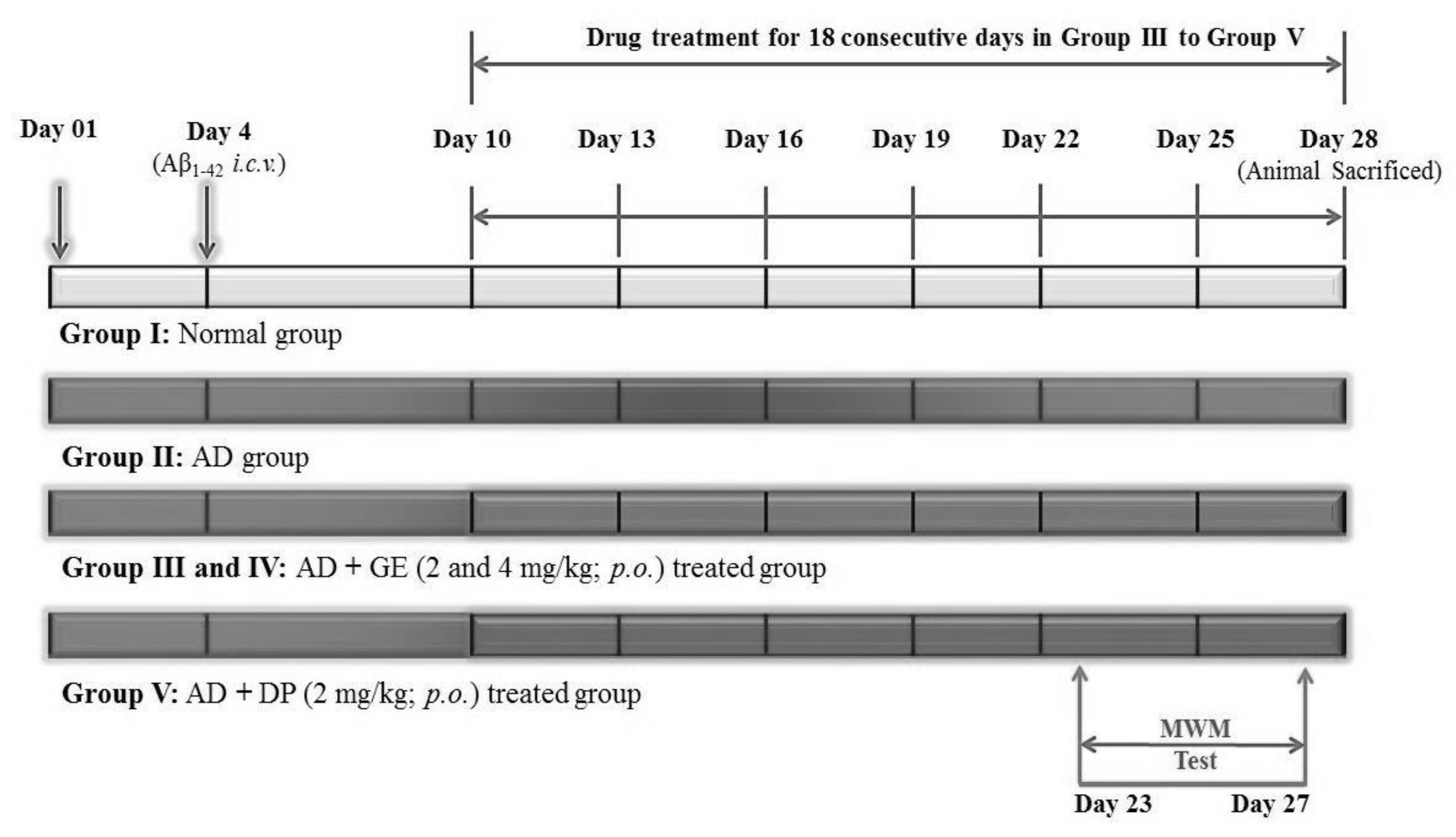
4.5. Experimental Protocol
4.6. Screening of the Effect of GE in AD-Associated Neurobehavioral Changes
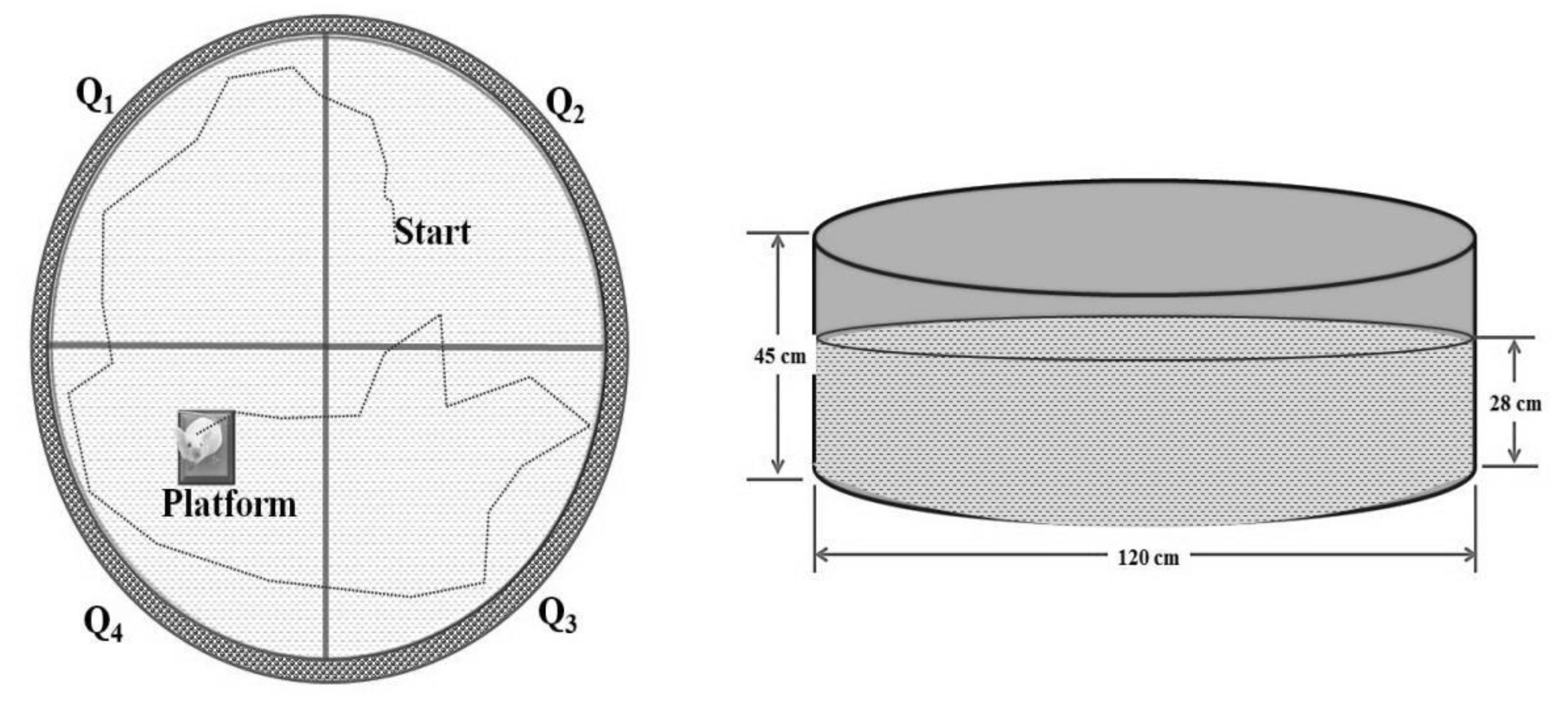
Assessment of Learning and Memory Using the Morris Water Maze (MWM) Test
4.7. Estimations of Tissue Biomarker Changes
4.7.1. Estimation of AChE as an Indication of Cholinergic Neurochemical Alteration in CNS
4.7.2. Estimation of TBARS as an Indication of Lipid Peroxidation
4.7.3. Estimation of GSH as an Indication of Oxidative Stress
4.8. Analysis of Mouse Brain Histopathological Changes
4.9. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Association, A. 2019 Alzheimer’s Disease Facts and Figures. Alzheimer’s Dement. 2019, 15, 321–387. [Google Scholar] [CrossRef]
- Burns, A. Diagnosis and Management of Alzheimer’s Disease. Dialogues Clin. Neurosci. 2000, 2, 129–138. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, S.; Ahmad, R.; Khare, S.K. Alzheimer’s Disease and Its Treatment by Different Approaches: A Review. Eur. J. Med. Chem. 2021, 216, 113320. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.T.; Nguyen, T.D.; Nguyen, T.K.O.; Vo, T.K. Advances in Developing Therapeutic Strategies for Alzheimer’s Disease. Biomed. Pharmacother. 2021, 139, 111623. [Google Scholar] [CrossRef] [PubMed]
- Cummings, J. Drug Development for Psychotropic, Cognitive-Enhancing, and Disease-Modifying Treatments for Alzheimer’s Disease. J. Neuropsychiatry Clin. Neurosci. 2021, 33, 3–13. [Google Scholar] [CrossRef]
- Olajide, O.J.; Gbadamosi, I.T.; Yawson, E.O.; Arogundade, T.; Lewu, F.S.; Ogunrinola, K.Y.; Adigun, O.O.; Bamisi, O.; Lambe, E.; Arietarhire, L.O.; et al. Hippocampal Degeneration and Behavioral Impairment During Alzheimer-Like Pathogenesis Involves Glutamate Excitotoxicity. J. Mol. Neurosci. 2021, 71, 1205–1220. [Google Scholar] [CrossRef]
- Mangalmurti, A.; Lukens, J.R. How Neurons Die in Alzheimer’s Disease: Implications for Neuroinflammation. Curr. Opin. Neurobiol. 2022, 75, 102575. [Google Scholar] [CrossRef]
- Sarkar, S.; Chegu Krishnamurthi, M. Nutritional, Dietary, and Lifestyle Approaches for Prevention and Management of Alzheimer’s Disease. In Role of Nutrients in Neurological Disorders; Springer: Berlin/Heidelberg, Germany, 2022; pp. 61–84. [Google Scholar]
- Müller, L.; Kirschstein, T.; Köhling, R.; Kuhla, A.; Teipel, S. Neuronal Hyperexcitability in APP SWE/PS1dE9 Mouse Models of Alzheimer’s Disease. J. Alzheimer’s Dis. 2021, 81, 855–869. [Google Scholar] [CrossRef]
- Hampel, H.; Hardy, J.; Blennow, K.; Chen, C.; Perry, G.; Kim, S.H.; Villemagne, V.L.; Aisen, P.; Vendruscolo, M.; Iwatsubo, T. The Amyloid-β Pathway in Alzheimer’s Disease. Mol. Psychiatry 2021, 26, 5481–5503. [Google Scholar] [CrossRef]
- Wagner, B.; Natarajan, A.; Grünaug, S.; Kroismayr, R.; Wagner, E.F.; Sibilia, M. Neuronal Survival Depends on EGFR Signaling in Cortical but Not Midbrain Astrocytes. EMBO J. 2006, 25, 752–762. [Google Scholar] [CrossRef]
- Neto, E.; Leitão, L.; Mateus, J.; Sousa, D.M.; Conceição, F.; Alves, C.J.; Oreffo, R.O.C.; West, J.; Aguiar, P.; Lamghari, M. Sensory Neurons Sprouting Is Dependent on Osteoclast-Derived Extracellular Vesicles Involving the Activation of Epidermal Growth Factor Receptors. BioRxiv 2020. [Google Scholar] [CrossRef]
- Wang, L.; Chiang, H.-C.; Wu, W.; Liang, B.; Xie, Z.; Yao, X.; Ma, W.; Du, S.; Zhong, Y. Epidermal Growth Factor Receptor Is a Preferred Target for Treating Amyloid-Β–induced Memory Loss. Proc. Natl. Acad. Sci. USA 2012, 109, 16743–16748. [Google Scholar] [CrossRef] [PubMed]
- Mellinghoff, I.K.; Wang, M.Y.; Vivanco, I.; Haas-Kogan, D.A.; Zhu, S.; Dia, E.Q.; Lu, K.V.; Yoshimoto, K.; Huang, J.H.Y.; Chute, D.J.; et al. Molecular Determinants of the Response of Glioblastomas to EGFR Kinase Inhibitors. N. Engl. J. Med. 2005, 353, 2012–2024. [Google Scholar] [CrossRef] [PubMed]
- Wakeling, A.E.; Barker, A.J.; Davies, D.H.; Brown, D.S.; Green, L.R.; Cartlidge, S.A.; Woodburn, J.R. Specific Inhibition of Epidermal Growth Factor Receptor Tyrosine Kinase by 4-Anilinoquinazolines. Breast Cancer Res. Treat. 1996, 38, 67–73. [Google Scholar] [CrossRef]
- Mansour, H.M.; Fawzy, H.M.; El-Khatib, A.S.; Khattab, M.M. Potential Repositioning of Anti-Cancer EGFR Inhibitors in Alzheimer’s Disease: Current Perspectives and Challenging Prospects. Neuroscience 2021, 469, 191–196. [Google Scholar] [CrossRef] [PubMed]
- Pao, W.; Miller, V.; Zakowski, M.; Doherty, J.; Politi, K.; Sarkaria, I.; Singh, B.; Heelan, R.; Rusch, V.; Fulton, L.; et al. EGF Receptor Gene Mutations Are Common in Lung Cancers from “never Smokers” and Are Associated with Sensitivity of Tumors to Gefitinib and Erlotinib. Proc. Natl. Acad. Sci. USA 2004, 101, 13306–13311. [Google Scholar] [CrossRef] [PubMed]
- Woodburn, J.R. The Epidermal Growth Factor Receptor and Its Inhibition in Cancer Therapy. Pharmacol. Ther. 1999, 82, 241–250. [Google Scholar] [CrossRef]
- Srinivasalu, V.K.; Subramaniam, N.; Vidhyadharan, S.; Murthy, S.P. 29 Management of Recurrent and Metastatic Head and Neck Cancer. Compr. Manag. Head Neck Cancer 2021, 396. [Google Scholar] [CrossRef]
- Mansour, H.M.; Fawzy, H.M.; El-Khatib, A.S.; Khattab, M.M. Repurposed Anti-Cancer Epidermal Growth Factor Receptor Inhibitors: Mechanisms of Neuroprotective Effects in Alzheimer’s Disease. Neural Regen. Res. 2022, 17, 1913–1918. [Google Scholar] [CrossRef]
- Dai, T.; Jiang, W.; Wang, M.; Guo, Z.; Dai, R. Influence of Two-Period Cross-over Design on the Bioequivalence Study of Gefitinib Tablets in Beagle Dogs. Eur. J. Pharm. Sci. 2021, 165, 105933. [Google Scholar] [CrossRef]
- Beeg, M.; Battocchio, E.; De Luigi, A.; Colombo, L.; Natale, C.; Cagnotto, A.; Corbelli, A.; Fiordaliso, F.; Diomede, L.; Salmona, M. Nonphosphorylated Tau Slows down Aβ1–42 Aggregation, Binds to Aβ1–42 Oligomers, and Reduces Aβ1–42 Toxicity. J. Biol. Chem. 2021, 296, 100664. [Google Scholar] [CrossRef] [PubMed]
- Zheng, M.; Liu, J.; Ruan, Z.; Tian, S.; Ma, Y.; Zhu, J.; Li, G. Intrahippocampal Injection of Aβ1-42 Inhibits Neurogenesis and down-Regulates IFN-γ and NF-κB Expression in Hippocampus of Adult Mouse Brain. Amyloid 2013, 20, 13–20. [Google Scholar] [CrossRef] [PubMed]
- Picone, P.; Nuzzo, D.; Giacomazza, D.; Di Carlo, M. β-Amyloid Peptide: The Cell Compartment Multi-Faceted Interaction in Alzheimer’s Disease. Neurotox. Res. 2020, 37, 250–263. [Google Scholar] [CrossRef]
- Du Yan, S.; Chen, X.; Fu, J.; Chen, M.; Zhu, H.; Roher, A.; Slattery, T.; Zhao, L.; Nagashima, M.; Morser, J.; et al. RAGE and Amyloid-β Peptide Neurotoxicity in Alzheimer’s Disease. Nature 1996, 382, 685–691. [Google Scholar] [CrossRef]
- Park, J.-S.; Kam, T.-I.; Lee, S.; Park, H.; Oh, Y.; Kwon, S.-H.; Song, J.-J.; Kim, D.; Kim, H.; Jhaldiyal, A.; et al. Blocking Microglial Activation of Reactive Astrocytes Is Neuroprotective in Models of Alzheimer’s Disease. Acta Neuropathol. Commun. 2021, 9, 78. [Google Scholar] [CrossRef] [PubMed]
- P, S.H.K.; Babu, T.D.; Pareeth, C.M.; Joshy, G.; Mathew, D.; Thayyil, M.S. Antioxidant Activity of Erlotinib and Gefitinib: Theoretical and Experimental Insights. Free. Radic. Res. 2022, 56, 196–208. [Google Scholar]
- Alaaeldin, R.; Hassan, H.A.; Abdel-Rahman, I.M.; Mohyeldin, R.H.; Youssef, N.; Allam, A.E.; Abdelwahab, S.F.; Zhao, Q.-L.; Fathy, M. A New EGFR Inhibitor from Ficus Benghalensis Exerted Potential Anti-Inflammatory Activity via Akt/PI3K Pathway Inhibition. Curr. Issues Mol. Biol. 2022, 44, 2967–2981. [Google Scholar] [CrossRef]
- Cheng, K.-C.; Chen, Y.-H.; Wu, C.-L.; Lee, W.-P.; Cheung, C.H.A.; Chiang, H.-C. Rac1 and Akt Exhibit Distinct Roles in Mediating Aβ-Induced Memory Damage and Learning Impairment. Mol. Neurobiol. 2021, 58, 5224–5238. [Google Scholar] [CrossRef]
- Zhang, Q.; Li, R.; Chen, X.; Lee, S.B.; Pan, J.; Xiong, D.; Hu, J.; Miller, M.S.; Szabo, E.; Lubet, R.A.; et al. Effect of Weekly or Daily Dosing Regimen of Gefitinib in Mouse Models of Lung Cancer. Oncotarget 2017, 8, 72447. [Google Scholar] [CrossRef]
- Rehman, I.U.; Ahmad, R.; Khan, I.; Lee, H.J.; Park, J.; Ullah, R.; Choi, M.J.; Kang, H.Y.; Kim, M.O. Nicotinamide Ameliorates Amyloid Beta-Induced Oxidative Stress-Mediated Neuroinflammation and Neurodegeneration in Adult Mouse Brain. Biomedicines 2021, 9, 408. [Google Scholar] [CrossRef]
- Thamban Chandrika, N.; Fosso, M.Y.; Tsodikov, O.V.; LeVine, H.; Garneau-Tsodikova, S. Combining Chalcones with Donepezil to Inhibit Both Cholinesterases and Aβ Fibril Assembly. Molecules 2020, 25, 77. [Google Scholar] [CrossRef] [PubMed]
- Crawford, S. Anti-Inflammatory/antioxidant Use in Long-Term Maintenance Cancer Therapy: A New Therapeutic Approach to Disease Progression and Recurrence. Ther. Adv. Med. Oncol. 2014, 6, 52–68. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.-H.; Chen, M.-C.; Chiu, T.-H.; Li, Y.-H.; Yu, W.-C.; Liao, W.-L.; Oner, M.; Yu, C.-T.R.; Wu, C.-C.; Yang, T.-Y.; et al. Arecoline Promotes Migration of A549 Lung Cancer Cells through Activating the EGFR/Src/FAK Pathway. Toxins 2019, 11, 185. [Google Scholar] [CrossRef] [PubMed]
- Pahal, S.; Gupta, A.; Choudhary, P.; Chaudhary, A.; Singh, S. Network Pharmacological Evaluation of Withania Somnifera Bioactive Phytochemicals for Identifying Novel Potential Inhibitors against Neurodegenerative Disorder. J. Biomol. Struct. Dyn. 2021. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Cheuk, I.W.Y.; Shin, V.Y.; Kwong, A. Acetylcholine Receptors: Key Players in Cancer Development. Surg. Oncol. 2019, 31, 46–53. [Google Scholar] [CrossRef]
- Li, J.; Wang, C.; Zhang, J.H.; Cai, J.-M.; Cao, Y.-P.; Sun, X.-J. Hydrogen-Rich Saline Improves Memory Function in a Rat Model of Amyloid-Beta-Induced Alzheimer’s Disease by Reduction of Oxidative Stress. Brain Res. 2010, 1328, 152–161. [Google Scholar] [CrossRef]
- Paxinos, G.; Franklin, K.B.J. The Mouse Brain in Stereotaxic Coordinates; Gulf Professional Publishing: Houston, TX, USA, 2004. [Google Scholar]
- Park, S.J.; Jung, J.M.; Lee, H.E.; Lee, Y.W.; Kim, D.H.; Kim, J.M.; Hong, J.G.; Lee, C.H.; Jung, I.H.; Cho, Y.-B.; et al. The Memory Ameliorating Effects of INM-176, an Ethanolic Extract of Angelica Gigas, against Scopolamine-or Aβ₁–₄₂-Induced Cognitive Dysfunction in Mice. J. Ethnopharmacol. 2012, 143, 611–620. [Google Scholar] [CrossRef]
- Morris, R. Developments of a Water-Maze Procedure for Studying Spatial Learning in the Rat. J. Neurosci. Methods 1984, 11, 47–60. [Google Scholar] [CrossRef]
- Parle, M. Animal Models for Testing Memory. Asia Pac. J. Pharm. 2004, 16, 101–120. [Google Scholar]
- Ellman, G.L.; Courtney, K.D.; Andres, V., Jr.; Featherstone, R.M. A New and Rapid Colorimetric Determination of Acetylcholinesterase Activity. Biochem. Pharmacol. 1961, 7, 88–95. [Google Scholar] [CrossRef]
- Ohkawa, H.; Ohishi, N.; Yagi, K. Assay for Lipid Peroxides in Animal Tissues by Thiobarbituric Acid Reaction. Anal. Biochem. 1979, 95, 351–358. [Google Scholar] [CrossRef]
- Ellman, G.L. Tissue Sulfhydryl Groups. Arch. Biochem. Biophys. 1959, 82, 70–77. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Groups | Hippocampus | Cortex | Cerebellum |
|---|---|---|---|
| Normal | 21.5 ± 1.5 | 20.7 ± 2.3 | 20.2 ± 1.3 |
| AD | 41.1 ± 1.3 a | 42.5 ± 1.2 a | 36.4 ± 1.5 a |
| AD + GE (2) | 28.9 ± 0.7 b | 29.2 ± 1.7 b | 30.3 ± 1.7 b |
| AD + GE (4) | 23.5 ± 1.1 b | 21.4 ± 1.6 b | 20.5 ± 1.6 b |
| AD + DP (1) | 22.3 ± 1.3 b | 22.9 ± 1.4 b | 21.3 ± 1.4 b |
| Groups | Hippocampus | Cortex | Cerebellum |
|---|---|---|---|
| Normal | 4.2 ± 0.11 | 4.2 ± 0.13 | 3.5 ± 0.13 |
| AD | 7.5 ± 0.09 a | 7.9 ± 0.23 a | 5.9 ± 0.08 a |
| AD + GE (2) | 4.9 ± 0.05 b | 4.7 ± 0.14 b | 4.3 ± 0.11 b |
| AD + GE (4) | 4.3 ± 0.02 b | 4.3 ± 0.15 b | 3.7 ± 0.09 b |
| AD + DP (1) | 4.2 ± 0.03 b | 4.2 ± 0.08 b | 3.6 ± 0.14 b |
| Groups | Hippocampus | Cortex | Cerebellum |
|---|---|---|---|
| Normal | 81.6 ± 1.4 | 88.1 ± 1.3 | 74.6 ± 2.1 |
| AD | 52.9 ± 0.5 a | 58.5 ± 1.6 a | 54.2 ± 0.8 a |
| AD + GE (2) | 66.9 ± 1.6 b | 70.2 ± 2.1 b | 66.9 ± 1.7 b |
| AD + GE (4) | 77.6 ± 1.2 b | 83.7 ± 2.2 b | 71.7 ± 2.2 b |
| AD + DP (1) | 79.7 ± 1.5 b | 86.4 ± 1.3 b | 73.1 ± 1.2 b |
| Days | Quadrants | |||
|---|---|---|---|---|
| Day 1 | Q1 | Q2 | Q3 | Q4 |
| Day 2 | Q2 | Q3 | Q4 | Q1 |
| Day 3 | Q3 | Q4 | Q1 | Q2 |
| Day 4 | Q4 | Q1 | Q2 | Q3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dhamodharan, J.; Sekhar, G.; Muthuraman, A. Epidermal Growth Factor Receptor Kinase Inhibitor Ameliorates β-Amyloid Oligomer-Induced Alzheimer Disease in Swiss Albino Mice. Molecules 2022, 27, 5182. https://doi.org/10.3390/molecules27165182
Dhamodharan J, Sekhar G, Muthuraman A. Epidermal Growth Factor Receptor Kinase Inhibitor Ameliorates β-Amyloid Oligomer-Induced Alzheimer Disease in Swiss Albino Mice. Molecules. 2022; 27(16):5182. https://doi.org/10.3390/molecules27165182
Chicago/Turabian StyleDhamodharan, Jagadeesh, Ganthimathy Sekhar, and Arunachalam Muthuraman. 2022. "Epidermal Growth Factor Receptor Kinase Inhibitor Ameliorates β-Amyloid Oligomer-Induced Alzheimer Disease in Swiss Albino Mice" Molecules 27, no. 16: 5182. https://doi.org/10.3390/molecules27165182
APA StyleDhamodharan, J., Sekhar, G., & Muthuraman, A. (2022). Epidermal Growth Factor Receptor Kinase Inhibitor Ameliorates β-Amyloid Oligomer-Induced Alzheimer Disease in Swiss Albino Mice. Molecules, 27(16), 5182. https://doi.org/10.3390/molecules27165182