
Structural Insights into the Role of β3 nAChR Subunit in the Activation of Nicotinic Receptors

 ,
,  , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
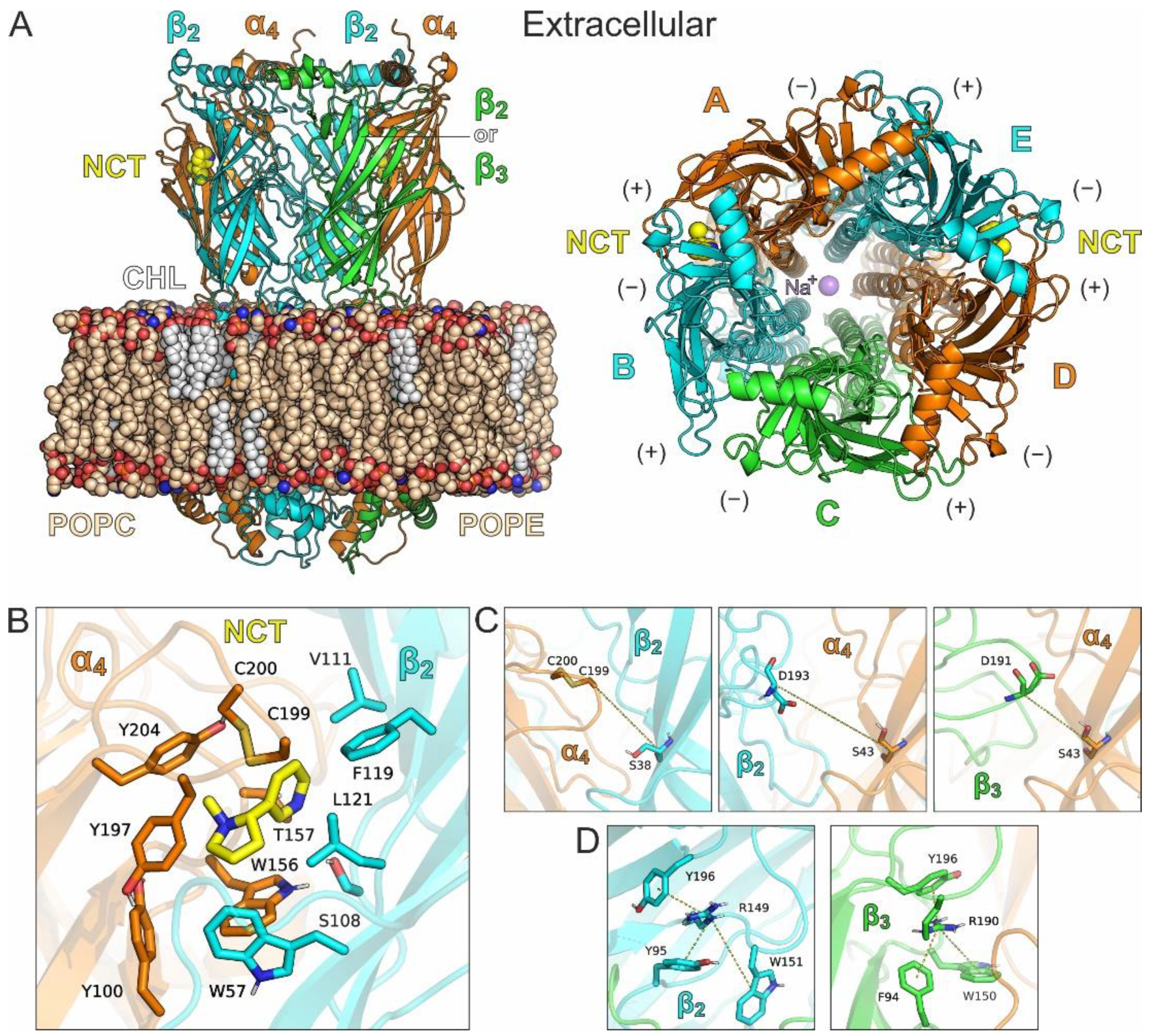
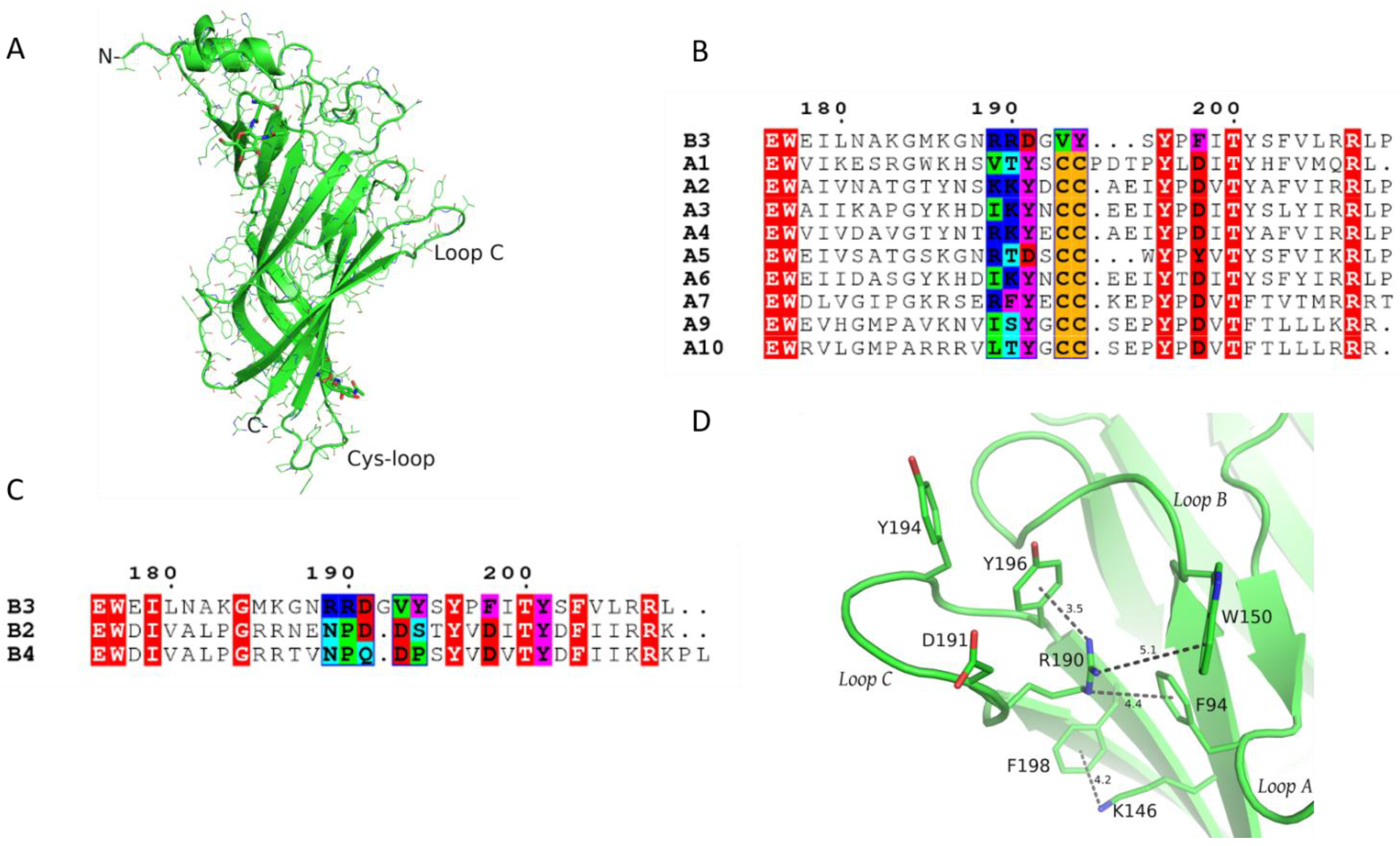
1.1. Crystal Structure of β3 ECD and Description of Its Unique Features
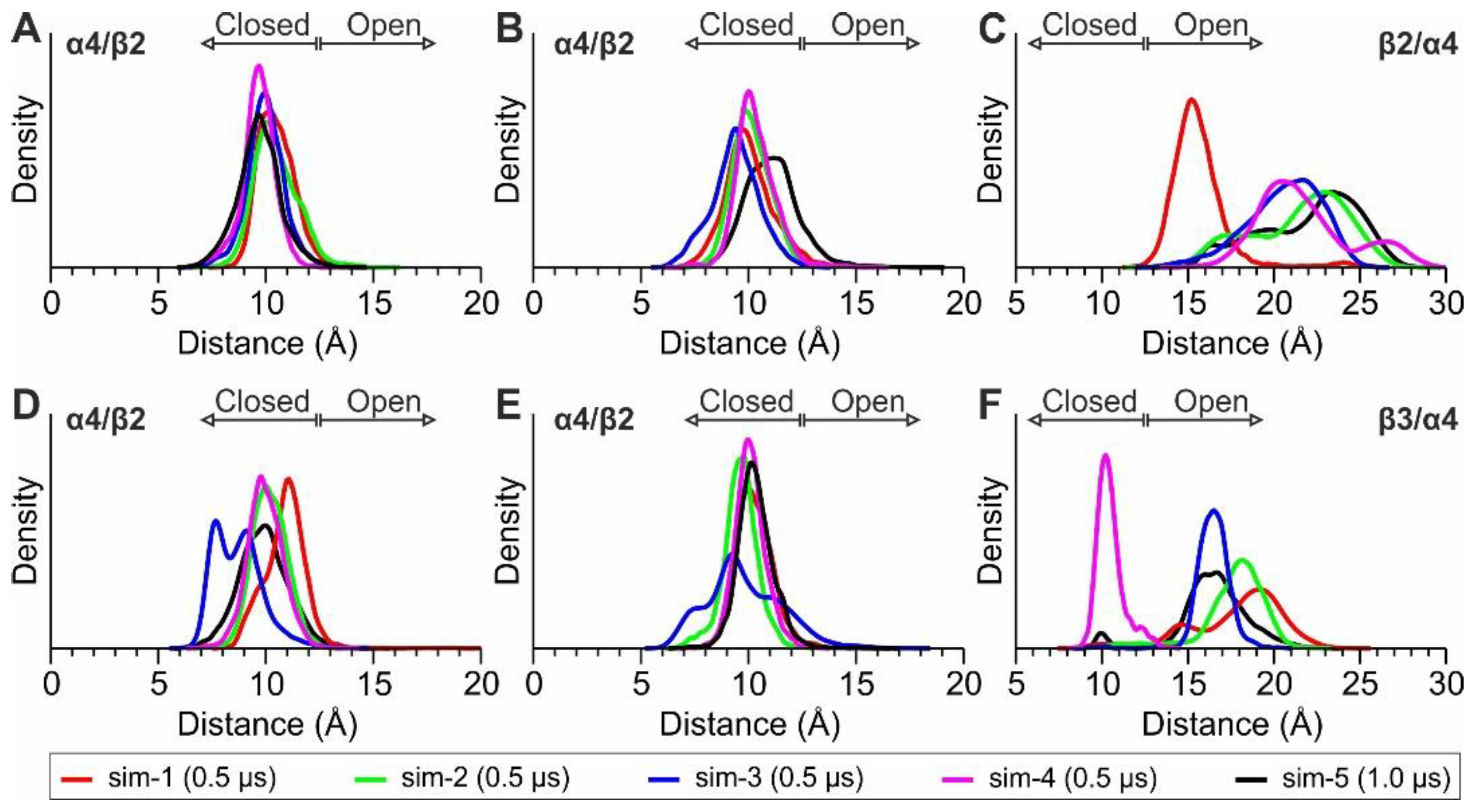
1.2. MD Simulations of α4β2 and α4β2β3 nAChRs
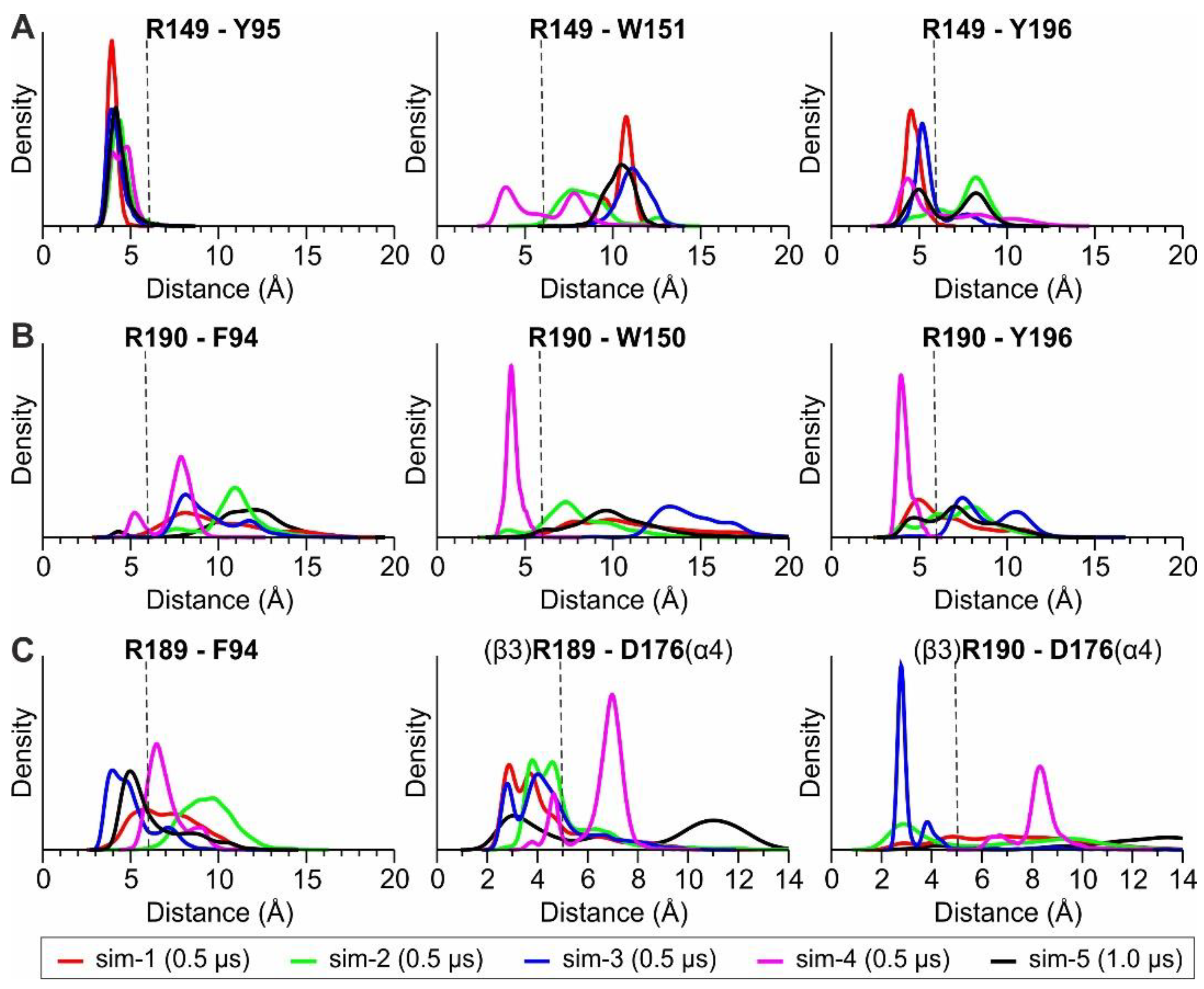
1.2.1. MD Simulations with Nicotine Bound Explicitly to α4(+)/β2(−) Interfaces
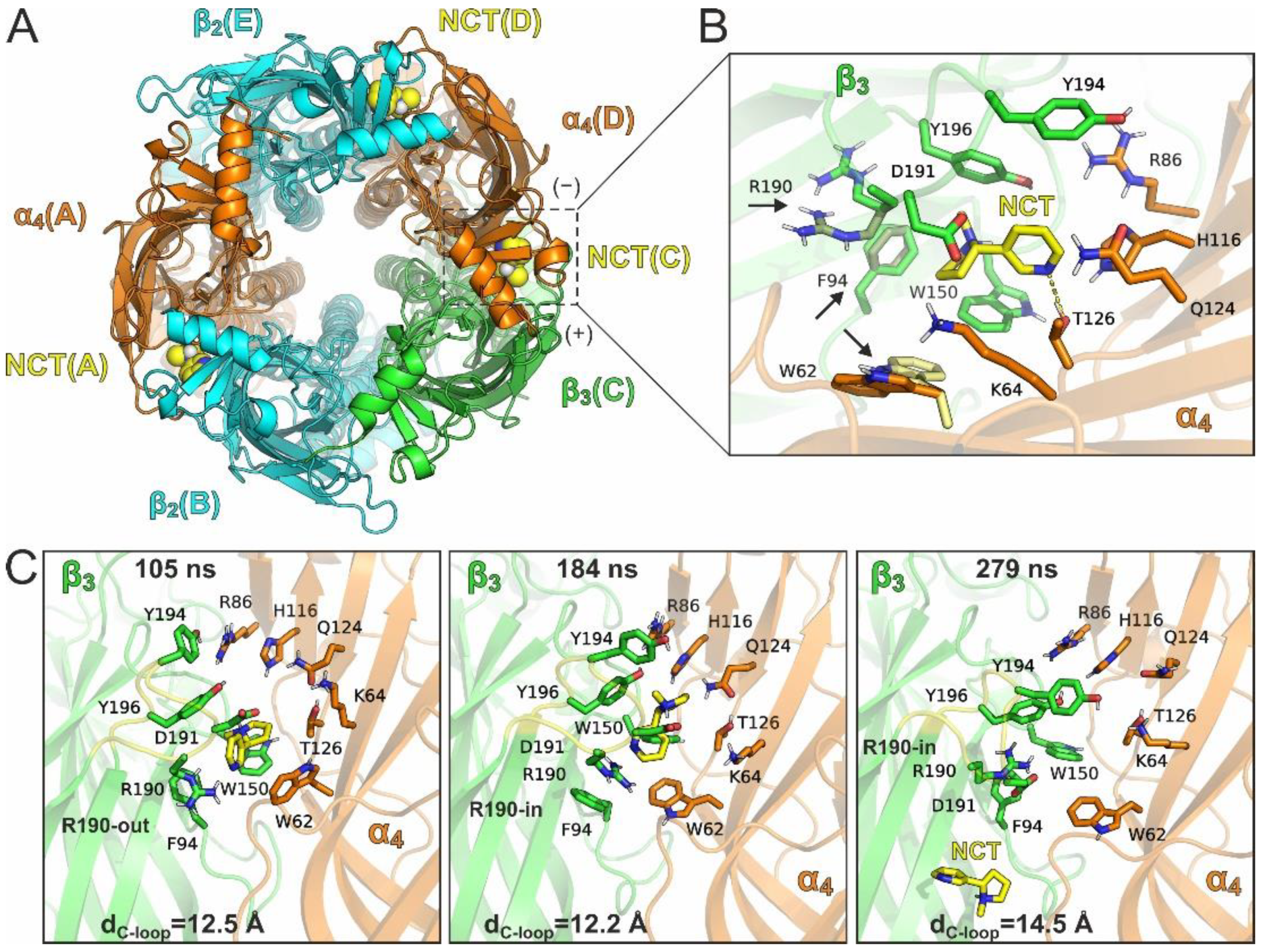
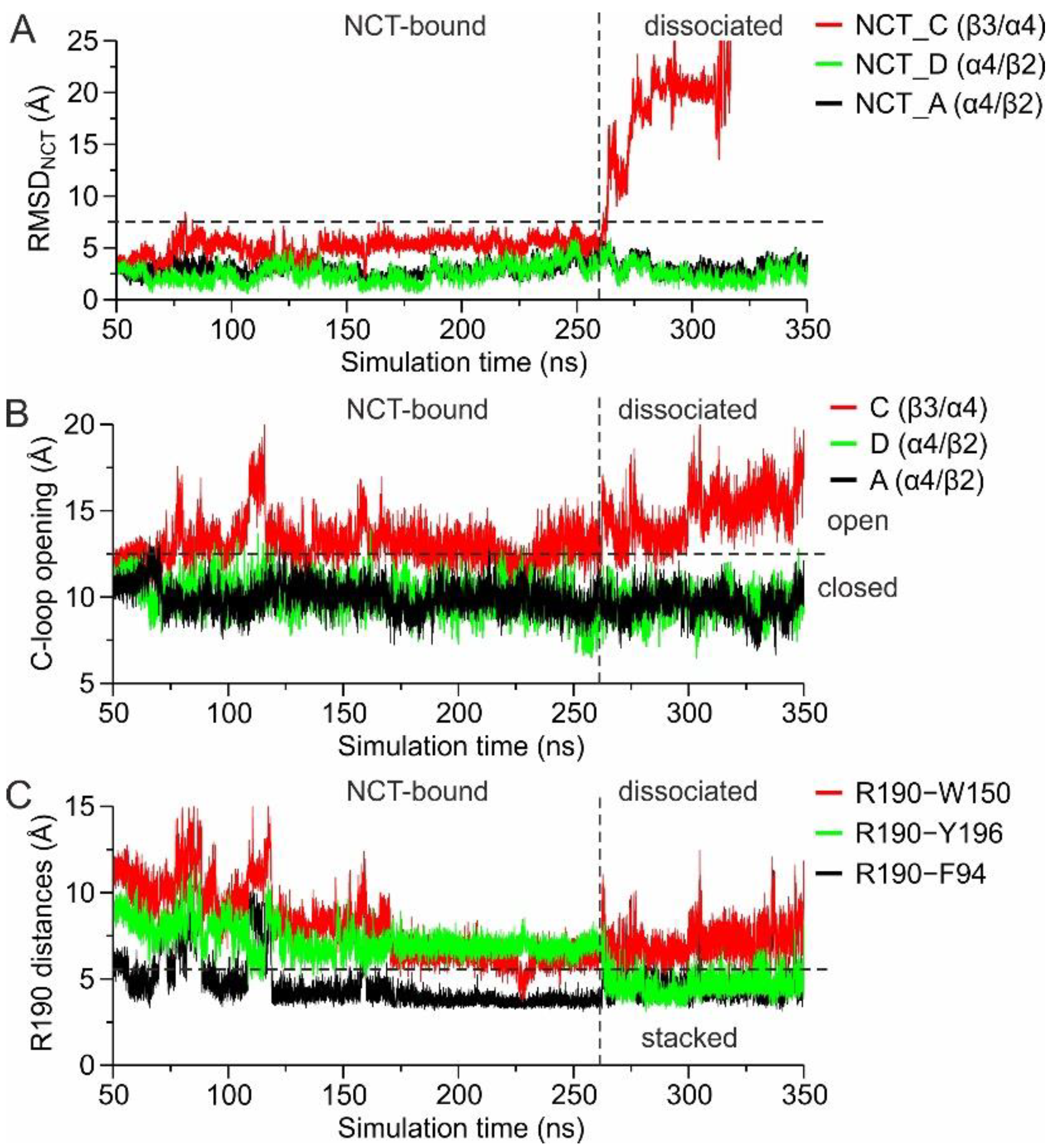
1.2.2. MD Simulations with an Additional Nicotine Bound to the β3(+)/α4(−) Interface
2. Discussion
3. Conclusions
4. Materials and Methods
4.1. Construct Design
4.2. Protein Expression and Purification
4.3. Crystallization and Data Collection
4.4. Structure Determination and Refinement
4.5. Computational Methods
4.5.1. Preparation of the Systems
4.5.2. Molecular Dynamics (MD) Simulations
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Albuquerque, E.X.; Pereira, E.F.R.; Alkondon, M.; Rogers, S.W. Mammalian Nicotinic Acetylcholine Receptors: From Structure to Function. Physiol. Rev. 2009, 89, 73–120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lester, H.A.; Dibas, M.I.; Dahan, D.S.; Leite, J.F.; Dougherty, D.A. Cys-loop receptors: New twists and turns. Trends Neurosci. 2004, 27, 329–336. [Google Scholar] [CrossRef] [PubMed]
- Sine, S.M.; Engel, A.G. Recent advances in Cys-loop receptor structure and function. Nature 2006, 440, 448–455. [Google Scholar] [CrossRef]
- Nemecz, A.; Prevost, M.S.; Menny, A.; Corringer, P.J. Emerging Molecular Mechanisms of Signal Transduction in Pentameric Ligand-Gated Ion Channels. Neuron 2016, 90, 452–470. [Google Scholar] [CrossRef] [PubMed]
- Millar, N.S.; Gotti, C. Diversity of vertebrate nicotinic acetylcholine receptors. Neuropharmacology 2009, 56, 237–246. [Google Scholar] [CrossRef] [Green Version]
- Unwin, N. Refined structure of the nicotinic acetylcholine receptor at 4A resolution. J. Mol. Biol. 2005, 346, 967–989. [Google Scholar] [CrossRef]
- Brejc, K.; van Dijk, W.J.; Klaassen, R.V.; Schuurmans, M.; van Der Oost, J.; Smit, A.B.; Sixma, T.K. Crystal structure of an ACh-binding protein reveals the ligand-binding domain of nicotinic receptors. Nature 2001, 411, 269–276. [Google Scholar] [CrossRef]
- Gotti, C.; Moretti, M.; Gaimarri, A.; Zanardi, A.; Clementi, F.; Zoli, M. Heterogeneity and complexity of native brain nicotinic receptors. Biochem. Pharmacol. 2007, 74, 1102–1111. [Google Scholar] [CrossRef]
- Corringer, P.-J.; Poitevin, F.; Prevost, M.S.; Sauguet, L.; Delarue, M.; Changeux, J.-P. Structure and Pharmacology of Pentameric Receptor Channels: From Bacteria to Brain. Structure 2012, 20, 941–956. [Google Scholar] [CrossRef] [Green Version]
- Kuryatov, A.; Onksen, J.; Lindstrom, J. Roles of accessory subunits in alpha4beta2(*) nicotinic receptors. Mol. Pharmacol. 2008, 74, 132–143. [Google Scholar] [CrossRef]
- Groot-Kormelink, P.J.; Boorman, J.P.; Sivilotti, L.G. Formation of functional α3β4α5 human neuronal nicotinic receptors in Xenopus oocytes: A reporter mutation approach. Br. J. Pharmacol. 2001, 134, 789–796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tumkosit, P.; Kuryatov, A.; Luo, J.; Lindstrom, J. β3 Subunits Promote Expression and Nicotine-Induced Up-Regulation of Human Nicotinic α6* Nicotinic Acetylcholine Receptors Expressed in Transfected Cell Lines. Mol. Pharmacol. 2006, 70, 1358–1368. [Google Scholar] [CrossRef] [Green Version]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alföldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020, 581, 434–443. [Google Scholar] [CrossRef] [PubMed]
- Hoft, N.R.; Corley, R.P.; McQueen, M.B.; Schlaepfer, I.R.; Huizinga, D.; Ehringer, M.A. Genetic association of the CHRNA6 and CHRNB3 genes with tobacco dependence in a nationally representative sample. Neuropsychopharmacology 2009, 34, 698–706. [Google Scholar] [CrossRef] [Green Version]
- Jain, A.; Kuryatov, A.; Wang, J.; Kamenecka, T.M.; Lindstrom, J. Unorthodox Acetylcholine Binding Sites Formed by alpha5 and beta3 Accessory Subunits in alpha4beta2* Nicotinic Acetylcholine Receptors. J. Biol. Chem. 2016, 291, 23452–23463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prevost, M.S.; Bouchenaki, H.; Barilone, N.; Gielen, M.; Corringer, P.J. Concatemers to re-investigate the role of α5 in α4β2 nicotinic receptors. Cell. Mol. Life Sci. 2021, 78, 1051–1064. [Google Scholar] [CrossRef] [PubMed]
- Rucktooa, P.; Smit, A.B.; Sixma, T.K. Insight in nAChR subtype selectivity from AChBP crystal structures. Biochem. Pharmacol. 2009, 78, 777–787. [Google Scholar] [CrossRef] [Green Version]
- Zouridakis, M.; Giastas, P.; Zarkadas, E.; Chroni-Tzartou, D.; Bregestovski, P.; Tzartos, S.J. Crystal structures of free and antagonist-bound states of human ± 9 nicotinic receptor extracellular domain. Nat. Struct. Mol. Biol. 2014, 21, 976–980. [Google Scholar] [CrossRef]
- Unwin, N. Acetylcholine receptor channel imaged in the open state. Nature 1995, 373, 37–43. [Google Scholar] [CrossRef]
- Dellisanti, C.D.; Yao, Y.; Stroud, J.C.; Wang, Z.Z.; Chen, L. Crystal structure of the extracellular domain of nAChR alpha1 bound to alpha-bungarotoxin at 1.94 A resolution. Nat. Neurosci. 2007, 10, 953–962. [Google Scholar] [CrossRef]
- Zouridakis, M.; Papakyriakou, A.; Ivanov, I.A.; Kasheverov, I.E.; Tsetlin, V.; Tzartos, S.; Giastas, P. Crystal structure of the monomeric extracellular domain of α9 nicotinic receptor subunit in complex with α-conotoxin RgIA: Molecular dynamics insights into RgIA binding to α9α10 nicotinic receptors. Front. Pharmacol. 2019, 10, 474. [Google Scholar] [CrossRef] [PubMed]
- Kouvatsos, N.; Giastas, P.; Chroni-Tzartou, D.; Poulopoulou, C.; Tzartos, S.J. Crystal structure of a human neuronal nAChR extracellular domain in pentameric assembly: Ligand-bound α2 homopentamer. Proc. Natl. Acad. Sci. USA 2016, 113, 9635–9640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, S.-X.; Huang, S.; Bren, N.; Noridomi, K.; Dellisanti, C.D.; Sine, S.M.; Chen, L. Ligand-binding domain of an α7-nicotinic receptor chimera and its complex with agonist. Nat. Neurosci. 2011, 14, 1253–1259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morales-Perez, C.L.; Noviello, C.M.; Hibbs, R.E. X-ray structure of the human alpha4beta2 nicotinic receptor. Nature 2016, 538, 411–415. [Google Scholar] [CrossRef] [Green Version]
- Walsh, R.M., Jr.; Roh, S.H.; Gharpure, A.; Morales-Perez, C.L.; Teng, J.; Hibbs, R.E. Structural principles of distinct assemblies of the human alpha4beta2 nicotinic receptor. Nature 2018, 557, 261–265. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Liu, S.; Zhou, Y.; Zhang, M.; Chen, H.; Xu, H.E.; Sun, D.; Liu, L.; Tian, C. Structural basis of human α7 nicotinic acetylcholine receptor activation. Cell Res. 2021, 31, 713–716. [Google Scholar] [CrossRef]
- Noviello, C.M.; Gharpure, A.; Mukhtasimova, N.; Cabuco, R.; Baxter, L.; Borek, D.; Sine, S.M.; Hibbs, R.E. Structure and gating mechanism of the α7 nicotinic acetylcholine receptor. Cell 2021, 184, 2121–2134.e13. [Google Scholar] [CrossRef]
- Mukhtasimova, N.; Free, C.; Sine, S.M. Initial Coupling of Binding to Gating Mediated by Conserved Residues in the Muscle Nicotinic Receptor. J. Gen. Physiol. 2005, 126, 23–39. [Google Scholar] [CrossRef] [Green Version]
- Yu, R.; Tae, H.S.; Xu, Q.; Craik, D.J.; Adams, D.J.; Jiang, T.; Kaas, Q. Molecular dynamics simulations of dihydro-β-erythroidine bound to the human α4β2 nicotinic acetylcholine receptor. Br. J. Pharmacol. 2019, 176, 2750–2763. [Google Scholar] [CrossRef]
- Dash, B.; Lukas, R.J.; Li, M.D. A signal peptide missense mutation associated with nicotine dependence alters α2*-nicotinic acetylcholine receptor function. Neuropharmacology 2014, 79, 715–725. [Google Scholar] [CrossRef] [Green Version]
- Ahring, P.K.; Peters, D.; Christensen, J.K.; Jensen, M.L.; Harpsøe, K.; Balle, T. The (α4)3(β2)2 nAChR has a benzodiazepine-like modulatory binding site in the αα-subunit interface as revealed by studies with NS9283. Biochem. Pharmacol. 2011, 8, 1029–1030. [Google Scholar] [CrossRef]
- Mazzaferro, S.; Benallegue, N.; Carbone, A.; Gasparri, F.; Vijayan, R.; Biggin, P.C.; Moroni, M.; Bermudez, I. Additional Acetylcholine (ACh) binding site at α4/α4 interface of (α4β2) 2α4 nicotinic receptor influences agonist sensitivity. J. Biol. Chem. 2011, 286, 31043–31054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grupe, M.; Jensen, A.A.; Ahring, P.K.; Christensen, J.K.; Grunnet, M. Unravelling the mechanism of action of NS9283, a positive allosteric modulator of (α4)3(β2)2 nicotinic ACh receptors. Br. J. Pharmacol. 2013, 168, 2000–2010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahring, P.K.; Liao, V.W.Y.; Balle, T. Concatenated nicotinic acetylcholine receptors: A gift or a curse? J. Gen. Physiol. 2018, 150, 453–473. [Google Scholar] [CrossRef] [Green Version]
- Tapia, L.; Kuryatov, A.; Lindstrom, J. Ca 2+ Permeability of the (α4) 3 (β2) 2 Stoichiometry Greatly Exceeds That of (α4) 2 (β2) 3 Human Acetylcholine Receptors. Mol. Pharmacol. 2007, 71, 769–776. [Google Scholar] [CrossRef]
- Mesoy, S.M.; Bridgland-Taylor, M.; Lummis, S.C.R. Mutations of the nACh Receptor M4 Helix Reveal Different Phenotypes in Different Expression Systems: Could Lipids be Responsible? Front. Physiol. 2022, 13, 850782. [Google Scholar] [CrossRef]
- Nelson, M.E.; Kuryatov, A.; Choi, C.H.; Zhou, Y.; Lindstrom, J. Alternate stoichiometries of α4β2 nicotinic acetylcholine receptors. Mol. Pharmacol. 2003, 63, 332–341. [Google Scholar] [CrossRef] [Green Version]
- Giastas, P.; Zouridakis, M.; Tzartos, S.J. Understanding structure-function relationships of the human neuronal acetylcholine receptor: Insights from the first crystal structures of neuronal subunits. Br. J. Pharmacol. 2018, 175, 1880–1891. [Google Scholar] [CrossRef] [Green Version]
- Kamens, H.M.; Miyamoto, J.; Powers, M.S.; Ro, K.; Soto, M.; Cox, R.; Stitzel, J.A.; Ehringer, M.A. The β3 subunit of the nicotinic acetylcholine receptor: Modulation of gene expression and nicotine consumption. Neuropharmacology 2015, 99, 639–649. [Google Scholar] [CrossRef] [Green Version]
- Benallegue, N.; Mazzaferro, S.; Alcaino, C.; Bermudez, I. The additional ACh binding site at the alpha4(+)/alpha4(−) interface of the (alpha4beta2)2alpha4 nicotinic ACh receptor contributes to desensitization. Br. J. Pharm. 2013, 170, 304–316. [Google Scholar] [CrossRef] [Green Version]
- Stothard, P. The sequence manipulation suite: JavaScript programs for analyzing and formatting protein and DNA sequences. Biotechniques 2000, 28, 1102–1104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lazaridis, K.; Zisimopoulou, P.; Giastas, P.; Bitzopoulou, K.; Evangelakou, P.; Sideri, A.; Tzartos, S.J. Expression of human AChR extracellular domain mutants with improved characteristics. Int. J. Biol. Macromol. 2014, 63, 210–217. [Google Scholar] [CrossRef] [PubMed]
- Kabsch, W. XDS. Acta Cryst. D Biol. Cryst. 2010, 66, 125–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evans, P.R. An introduction to data reduction: Space-group determination, scaling and intensity statistics. Acta Crystallogr. Sect. D Biol. Cryst. 2011, 67, 282–292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCoy, A.J.; Grosse-Kunstleve, R.W.; Adams, P.D.; Winn, M.D.; Storoni, L.C.; Read, R.J. Phaser crystallographic software. J. Appl. Cryst. 2007, 40, 658–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Afonine, P.V.; Grosse-Kunstleve, R.W.; Echols, N.; Headd, J.J.; Moriarty, N.W.; Mustyakimov, M.; Terwilliger, T.C.; Urzhumtsev, A.; Zwart, P.H.; Adams, P.D. Towards automated crystallographic structure refinement with phenix.refine. Acta Cryst. Sect. D Biol. Cryst. 2012, 68, 352–367. [Google Scholar] [CrossRef] [Green Version]
- Emsley, P.; Lohkamp, B.; Scott, W.G.; Cowtan, K. Features and development of Coot. Acta Cryst. D Biol. Cryst. 2010, 66, 486–501. [Google Scholar] [CrossRef] [Green Version]
- Karplus, P.A.; Diederichs, K. Linking crystallographic model and data quality. Science 2012, 336, 1030–1033. [Google Scholar] [CrossRef] [Green Version]
- Fiser, A.; Šali, A. Modeller: Generation and Refinement of Homology-Based Protein Structure Models. Methods Enzymol. 2003, 374, 461–491. [Google Scholar]
- Jo, S.; Kim, T.; Iyer, V.G.; Im, W. CHARMM-GUI: A web-based graphical user interface for CHARMM. J. Comput. Chem. 2008, 29, 1859–1865. [Google Scholar] [CrossRef] [PubMed]
- Case, D.A.; Cheatham, T.E., III; Darden, T.; Gohlke, H.; Luo, R.; Merz, K.M., Jr.; Onufriev, A.; Simmerling, C.; Wang, B.; Woods, R.J. The Amber biomolecular simulation programs. J. Comput. Chem. 2005, 26, 1668–1688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the accuracy of protein side chain and backbone parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dickson, C.J.; Madej, B.D.; Skjevik, Å.A.; Betz, R.M.; Teigen, K.; Gould, I.R.; Walker, R.C. Lipid14: The Amber Lipid Force Field. J. Chem. Theory Comput. 2014, 10, 865–879. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef]
- Götz, A.W.; Williamson, M.J.; Xu, D.; Poole, D.; Le Grand, S.; Walker, R.C. Routine Microsecond Molecular Dynamics Simulations with AMBER on GPUs. 1. Generalized Born. J. Chem. Theory Comput. 2012, 8, 1542–1555. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N⋅log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef] [Green Version]
- Roe, D.R.; Cheatham, T.E. PTRAJ and CPPTRAJ: Software for Processing and Analysis of Molecular Dynamics Trajectory Data. J. Chem. Theory Comput. 2013, 9, 3084–3095. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Giastas, P.; Papakyriakou, A.; Tsafaras, G.; Tzartos, S.J.; Zouridakis, M. Structural Insights into the Role of β3 nAChR Subunit in the Activation of Nicotinic Receptors. Molecules 2022, 27, 4642. https://doi.org/10.3390/molecules27144642
Giastas P, Papakyriakou A, Tsafaras G, Tzartos SJ, Zouridakis M. Structural Insights into the Role of β3 nAChR Subunit in the Activation of Nicotinic Receptors. Molecules. 2022; 27(14):4642. https://doi.org/10.3390/molecules27144642
Chicago/Turabian StyleGiastas, Petros, Athanasios Papakyriakou, George Tsafaras, Socrates J. Tzartos, and Marios Zouridakis. 2022. "Structural Insights into the Role of β3 nAChR Subunit in the Activation of Nicotinic Receptors" Molecules 27, no. 14: 4642. https://doi.org/10.3390/molecules27144642
APA StyleGiastas, P., Papakyriakou, A., Tsafaras, G., Tzartos, S. J., & Zouridakis, M. (2022). Structural Insights into the Role of β3 nAChR Subunit in the Activation of Nicotinic Receptors. Molecules, 27(14), 4642. https://doi.org/10.3390/molecules27144642