
Photocrosslinking Probes Proximity of Thymine Modifiers Tethering Excitonically Coupled Dye Aggregates to DNA Holliday Junction

 , ,
, ,
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
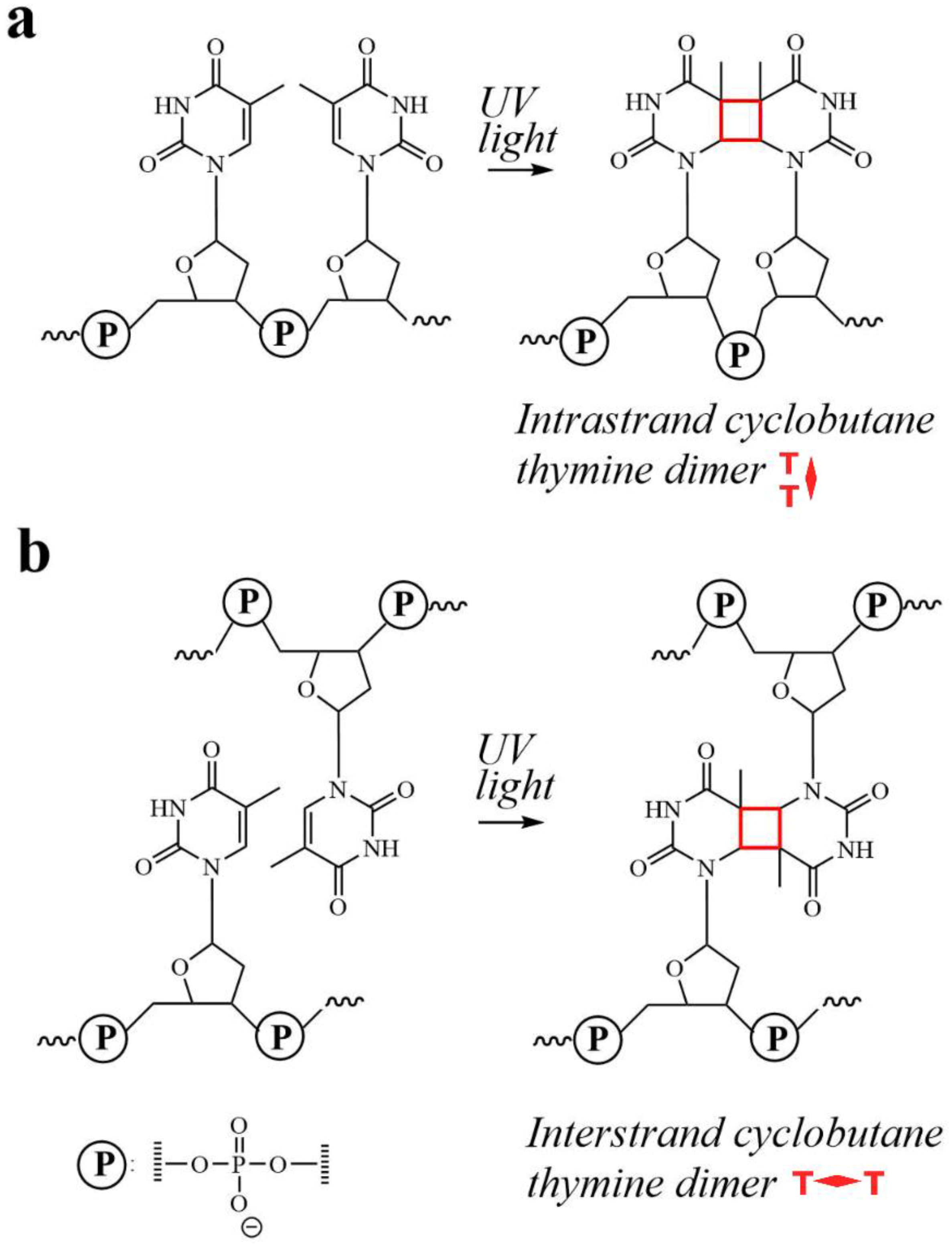
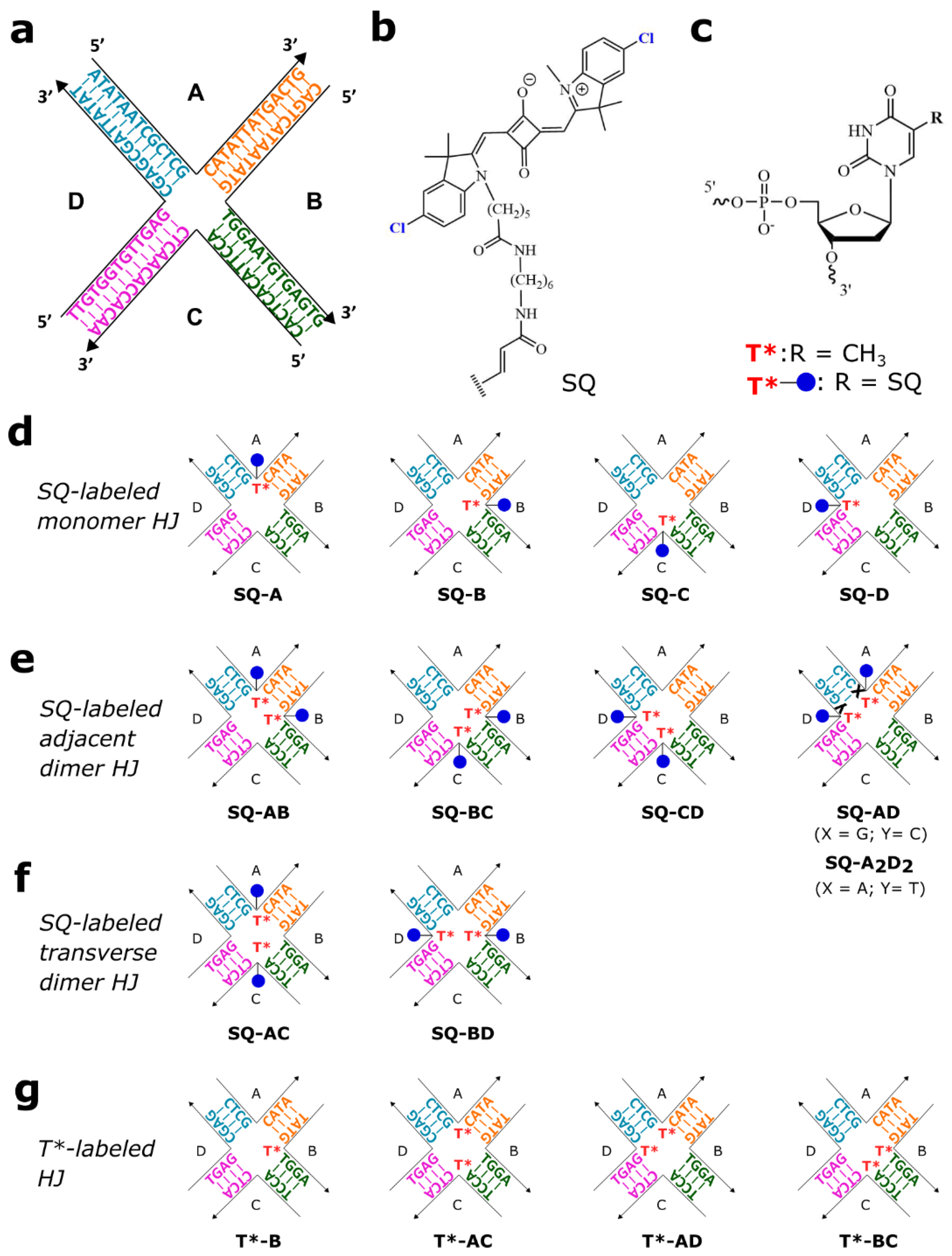
2.1. Molecular Design
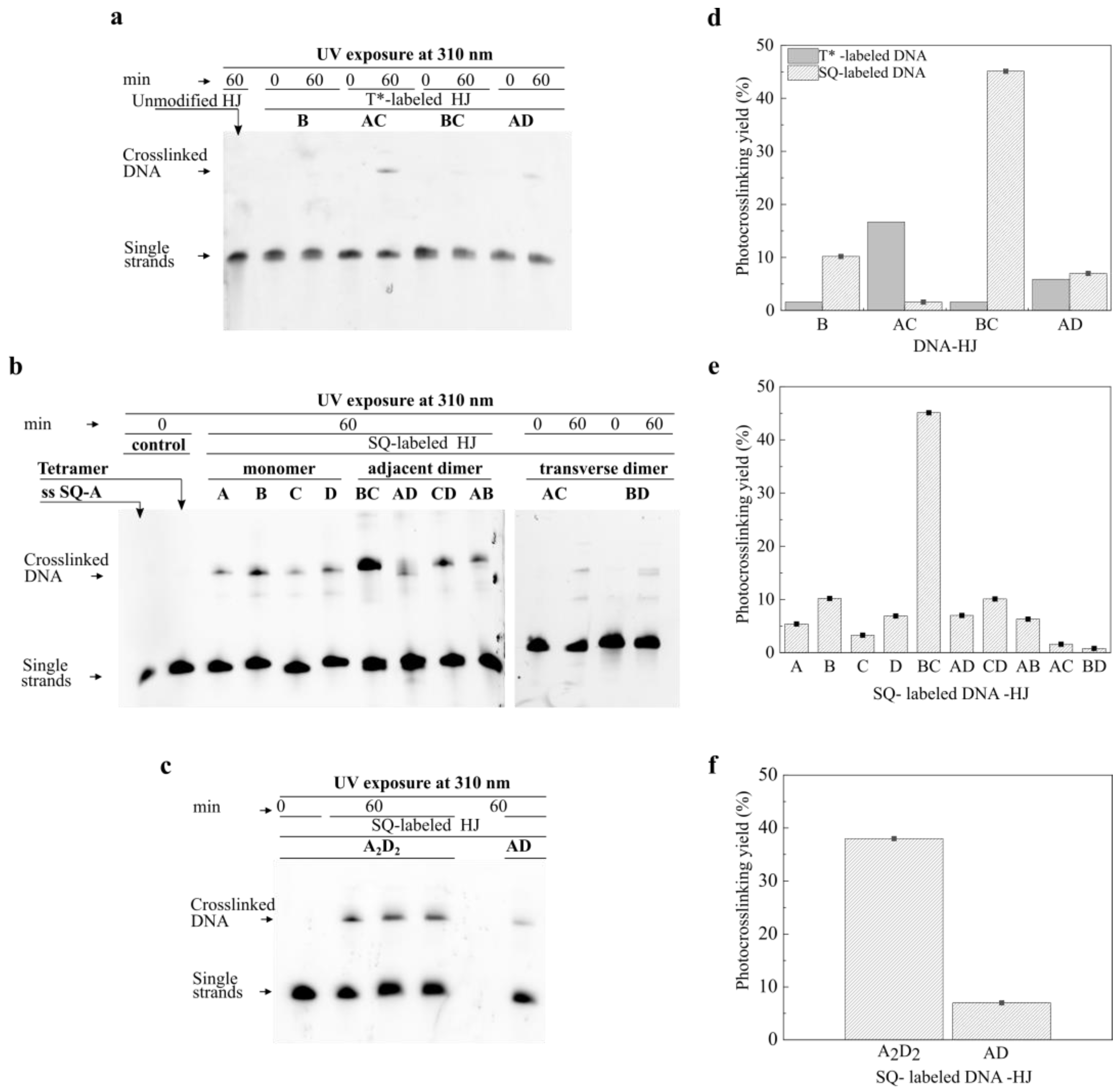
2.2. Photocrosslinking in HJ Constructs
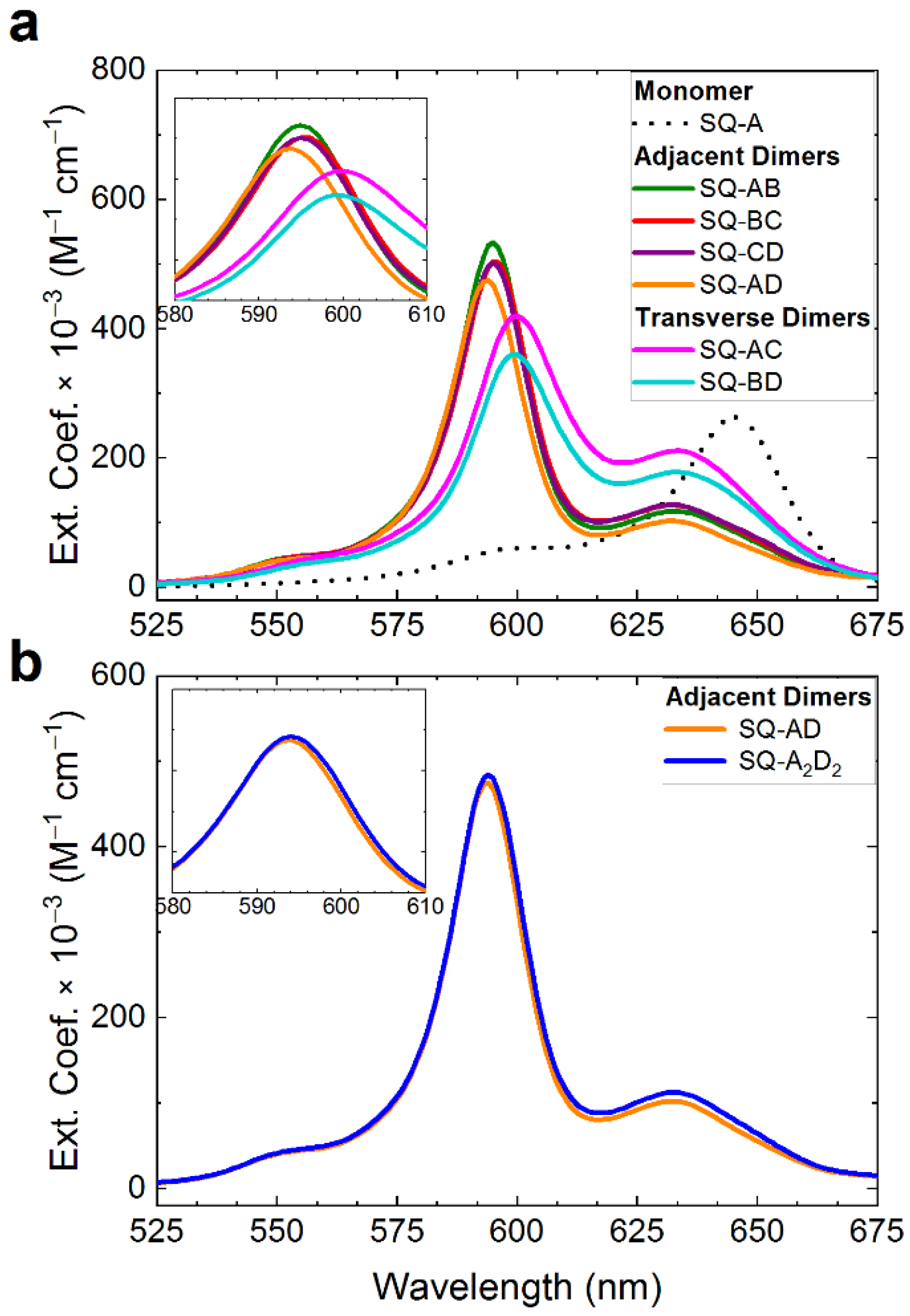
2.3. Squaraine Tetramer Templated by Doubly Crosslinked HJ
3. Materials and Methods
3.1. Sample Preparation
3.2. Photocrosslinking Reaction
3.3. Analytical Denaturing Gel Electrophoresis
3.4. Synthesis of SQ-Tetramer Templated by Doubly Crosslinked HJ
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Asanuma, H.; Fujii, T.; Kato, T.; Kashida, H. Coherent interactions of dyes assembled on DNA. J. Photochem. Photobiol. C Photochem. Rev. 2012, 13, 124–135. [Google Scholar] [CrossRef]
- Asanuma, H.; Shirasuka, K.; Takarada, T.; Kashida, H.; Komiyama, M. DNA-dye conjugates for controllable H aggregation(1). J. Am. Chem. Soc. 2003, 125, 2217–2223. [Google Scholar] [CrossRef]
- Cunningham, P.D.; Khachatrian, A.; Buckhout-White, S.; Deschamps, J.R.; Goldman, E.R.; Medintz, I.L.; Melinger, J.S. Resonance Energy Transfer in DNA Duplexes Labeled with Localized Dyes. J. Phys. Chem. B 2014, 118, 14555–14565. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, P.D.; Kim, Y.C.; Díaz, S.A.; Buckhout-White, S.; Mathur, D.; Medintz, I.L.; Melinger, J.S. Optical Properties of Vibronically Coupled Cy3 Dimers on DNA Scaffolds. J. Phys. Chem. B 2018, 122, 5020–5029. [Google Scholar] [CrossRef] [PubMed]
- Fujii, T.; Kashida, H.; Asanuma, H. Analysis of Coherent Heteroclustering of Different Dyes by Use of Threoninol Nucleotides for Comparison with the Molecular Exciton Theory. Chem.—A Eur. J. 2009, 15, 10092–10102. [Google Scholar] [CrossRef] [PubMed]
- Garo, F.; Haner, R. A DNA-based light-harvesting antenna. Angew. Chem. Int. Ed. 2012, 51, 916–919. [Google Scholar] [CrossRef]
- Häner, R.; Samain, F.; Malinovskii, V.L. DNA-Assisted Self-Assembly of Pyrene Foldamers. Chem.—A Eur. J. 2009, 15, 5701–5708. [Google Scholar] [CrossRef]
- Hart, S.M.; Chen, W.J.; Banal, J.L.; Bricker, W.P.; Dodin, A.; Markova, L.; Vyborna, Y.; Willard, A.P.; Häner, R.; Bathe, M.; et al. Engineering couplings for exciton transport using synthetic DNA scaffolds. Chem 2021, 7, 752–773. [Google Scholar] [CrossRef]
- Heussman, D.; Kittell, J.; Kringle, L.; Tamimi, A.; von Hippel, P.H.; Marcus, A.H. Measuring local conformations and conformational disorder of (Cy3)(2) dimer labeled DNA fork junctions using absorbance, circular dichroism and two-dimensional fluorescence spectroscopy. Faraday Discuss. 2019, 216, 211–235. [Google Scholar] [CrossRef]
- Ikeda, S.; Okamoto, A. Hybridization-Sensitive On–Off DNA Probe: Application of the Exciton Coupling Effect to Effective Fluorescence Quenching. Chem.-Asian J. 2008, 3, 958–968. [Google Scholar] [CrossRef]
- Kashida, H.; Asanuma, H.; Komiyama, M. Alternating Hetero H Aggregation of Different Dyes by Interstrand Stacking from Two DNA-Dye Conjugates. Angew. Chem. Int. Ed. 2004, 43, 6522–6525. [Google Scholar] [CrossRef] [PubMed]
- Kashida, H.; Tanaka, M.; Baba, S.; Sakamoto, T.; Kawai, G.; Asanuma, H.; Komiyama, M. Covalent Incorporation of Methyl Red Dyes into Double-Stranded DNA for Their Ordered Clustering. Chem.—A Eur. J. 2006, 12, 777–784. [Google Scholar] [CrossRef] [PubMed]
- Kringle, L.; Sawaya, N.P.D.; Widom, J.; Adams, C.; Raymer, M.G.; Aspuru-Guzik, A.; Marcus, A.H. Temperature-dependent conformations of exciton-coupled Cy3 dimers in double-stranded DNA. J. Chem. Phys. 2018, 148, 085101. [Google Scholar] [CrossRef]
- Li, S.; Langenegger, S.M.; Häner, R. Control of aggregation-induced emission by DNA hybridization. Chem. Commun. 2013, 49, 5835–5837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malinovskii, V.L.; Wenger, D.; Häner, R. Nucleic acid-guided assembly of aromatic chromophores. Chem. Soc. Rev. 2009, 39, 410–422. [Google Scholar] [CrossRef] [PubMed]
- Markova, L.I.; Malinovskii, V.L.; Patsenker, L.D.; Häner, R. Synthesis and properties of squaraine-modified DNA. Org. Biomol. Chem. 2012, 10, 8944–8947. [Google Scholar] [CrossRef]
- Markova, L.I.; Malinovskii, V.L.; Patsenker, L.D.; Häner, R. J- vs. H-type assembly: Pentamethine cyanine (Cy5) as a near-IR chiroptical reporter. Chem. Commun. 2013, 49, 5298–5300. [Google Scholar] [CrossRef] [Green Version]
- Mazuski, R.J.; Díaz, S.A.; Wood, R.E.; Lloyd, L.T.; Klein, W.P.; Mathur, D.; Melinger, J.S.; Engel, G.S.; Medintz, I.L. Ultrafast Excitation Transfer in Cy5 DNA Photonic Wires Displays Dye Conjugation and Excitation Energy Dependency. J. Phys. Chem. Lett. 2020, 11, 4163–4172. [Google Scholar] [CrossRef]
- Nicoli, F.; Roos, M.K.; Hemmig, E.A.; Di Antonio, M.; de Vivie-Riedle, R.; Liedl, T. Proximity-Induced H-Aggregation of Cyanine Dyes on DNA-Duplexes. J. Phys. Chem. A 2016, 120, 9941–9947. [Google Scholar] [CrossRef]
- Probst, M.; Wenger, D.; Biner, S.M.; Häner, R. The DNA three-way junction as a mould for tripartite chromophore assembly. Org. Biomol. Chem. 2011, 10, 755–759. [Google Scholar] [CrossRef]
- Sohail, S.H.; Otto, J.P.; Cunningham, P.D.; Kim, Y.C.; Wood, R.E.; Allodi, M.A.; Higgins, J.S.; Melinger, J.S.; Engel, G.S. DNA scaffold supports long-lived vibronic coherence in an indodicarbocyanine (Cy5) dimer. Chem. Sci. 2020, 11, 8546–8557. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Wu, Z.; Xu, X.; Yang, S. A Supramolecular Counter Circuit Based on Cyanine Dye Assembly. Chem.—A Eur. J. 2020, 26, 13235–13240. [Google Scholar] [CrossRef] [PubMed]
- Dietzsch, J.; Bialas, D.; Bandorf, J.; Würthner, F.; Höbartner, C. Tuning Exciton Coupling of Merocyanine Nucleoside Dimers by RNA, DNA and GNA Double Helix Conformations. Angew. Chem. Int. Ed. 2022, 61, e202116783. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Sha, R.; Knowlton, W.B.; Seeman, N.C.; Canary, J.W.; Yurke, B. Exciton Delocalization in a DNA-Templated Organic Semiconductor Dimer Assembly. ACS Nano 2022, 16, 1301–1307. [Google Scholar] [CrossRef]
- Chiriboga, M.; Diaz, S.A.; Mathur, D.; Hastman, D.A.; Melinger, J.S.; Veneziano, R.; Medintz, I.L. Understanding Self-Assembled Pseudoisocyanine Dye Aggregates in DNA Nanostructures and Their Exciton Relay Transfer Capabilities. J. Phys. Chem. B 2021, 126, 110–122. [Google Scholar] [CrossRef]
- Kasha, M. Energy Transfer Mechanisms and the Molecular Exciton Model for Molecular Aggregates. Radiat. Res. 1963, 20, 55. [Google Scholar] [CrossRef]
- Cannon, B.L.; Kellis, D.L.; Davis, P.H.; Lee, J.; Kuang, W.; Hughes, W.L.; Graugnard, E.; Yurke, B.; Knowlton, W.B. Excitonic AND Logic Gates on DNA Brick Nanobreadboards. ACS Photonics 2015, 2, 398–404. [Google Scholar] [CrossRef] [Green Version]
- Graugnard, E.; Kellis, D.L.; Bui, H.; Barnes, S.; Kuang, W.; Lee, J.; Hughes, W.L.; Knowlton, W.B.; Yurke, B. DNA-Controlled Excitonic Switches. Nano Lett. 2012, 12, 2117–2122. [Google Scholar] [CrossRef]
- Kellis, D.L.; Rehn, S.M.; Cannon, B.L.; Davis, P.H.; Graugnard, E.; Lee, J.; Yurke, B.; Knowlton, W.B. DNA-mediated excitonic upconversion FRET switching. New J. Phys. 2015, 17, 115007. [Google Scholar] [CrossRef]
- Ostroverkhova, O. Organic Optoelectronic Materials: Mechanisms and Applications. Chem. Rev. 2016, 116, 13279–13412. [Google Scholar] [CrossRef]
- Sawaya, N.P.D.; Rappoport, D.; Tabor, D.P.; Aspuru-Guzik, A. Excitonics: A Set of Gates for Molecular Exciton Processing and Signaling. ACS Nano 2018, 12, 6410–6420. [Google Scholar] [CrossRef] [PubMed]
- Scholes, G.D.; Rumbles, G. Excitons in nanoscale systems. Nat. Mater. 2006, 5, 683–696. [Google Scholar] [CrossRef] [PubMed]
- Wasielewski, M.R. Self-Assembly Strategies for Integrating Light Harvesting and Charge Separation in Artificial Photosynthetic Systems. Acc. Chem. Res. 2009, 42, 1910–1921. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Lebeck, A.R.; Dwyer, C. Nanoscale Resonance Energy Transfer-Based Devices for Probabilistic Computing. IEEE Micro 2015, 35, 72–84. [Google Scholar] [CrossRef]
- Wasielewski, M.R.; Forbes, M.D.E.; Frank, N.L.; Kowalski, K.; Scholes, G.D.; Yuen-Zhou, J.; Baldo, M.A.; Freedman, D.E.; Goldsmith, R.H.; Goodson, T.; et al. Exploiting chemistry and molecular systems for quantum information science. Nat. Rev. Chem. 2020, 4, 490–504. [Google Scholar] [CrossRef]
- Wang, M.; Silva, G.L.; Armitage, B.A. DNA-Templated Formation of a Helical Cyanine Dye J-Aggregate. J. Am. Chem. Soc. 2000, 122, 9977–9986. [Google Scholar] [CrossRef]
- Hart, S.M.; Wang, X.; Guo, J.; Bathe, M.; Schlau-Cohen, G.S. Tuning Optical Absorption and Emission Using Strongly Coupled Dimers in Programmable DNA Scaffolds. J. Phys. Chem. Lett. 2022, 13, 1863–1871. [Google Scholar] [CrossRef]
- Cannon, B.L.; Patten, L.K.; Kellis, D.L.; Davis, P.H.; Lee, J.; Graugnard, E.; Yurke, B.; Knowlton, W.B. Large Davydov Splitting and Strong Fluorescence Suppression: An Investigation of Exciton Delocalization in DNA-Templated Holliday Junction Dye Aggregates. J. Phys. Chem. A 2018, 122, 2086–2095. [Google Scholar] [CrossRef]
- Cannon, B.L.; Kellis, D.L.; Patten, L.K.; Davis, P.H.; Lee, J.; Graugnard, E.; Yurke, B.; Knowlton, W.B. Coherent Exciton Delocalization in a Two-State DNA-Templated Dye Aggregate System. J. Phys. Chem. A 2017, 121, 6905–6916. [Google Scholar] [CrossRef]
- Huff, J.S.; Davis, P.H.; Christy, A.; Kellis, D.L.; Kandadai, N.; Toa, Z.S.D.; Scholes, G.D.; Yurke, B.; Knowlton, W.B.; Pensack, R.D. DNA-Templated Aggregates of Strongly Coupled Cyanine Dyes: Nonradiative Decay Governs Exciton Lifetimes. J. Phys. Chem. Lett. 2019, 10, 2386–2392. [Google Scholar] [CrossRef]
- Huff, J.S.; Turner, D.B.; Mass, O.A.; Patten, L.K.; Wilson, C.K.; Roy, S.K.; Barclay, M.S.; Yurke, B.; Knowlton, W.B.; Davis, P.H.; et al. Excited-State Lifetimes of DNA-Templated Cyanine Dimer, Trimer, and Tetramer Aggregates: The Role of Exciton Delocalization, Dye Separation, and DNA Heterogeneity. J. Phys. Chem. B 2021, 125, 10240–10259. [Google Scholar] [CrossRef]
- Roy, S.K.; Mass, O.A.; Kellis, D.L.; Wilson, C.K.; Hall, J.A.; Yurke, B.; Knowlton, W.B. Exciton Delocalization and Scaffold Stability in Bridged Nucleotide-Substituted, DNA Duplex-Templated Cyanine Aggregates. J. Phys. Chem. B 2021, 125, 13670–13684. [Google Scholar] [CrossRef] [PubMed]
- Meares, A.; Susumu, K.; Mathur, D.; Lee, S.H.; Mass, O.A.; Lee, J.; Pensack, R.D.; Yurke, B.; Knowlton, W.B.; Melinger, J.S.; et al. Synthesis of Substituted Cy5 Phosphoramidite Derivatives and Their Incorporation into Oligonucleotides Using Automated DNA Synthesis. ACS Omega 2022, 7, 11002–11016. [Google Scholar] [CrossRef] [PubMed]
- Seeman, N.C. DNA in a material world. Nature 2003, 421, 427–431. [Google Scholar] [CrossRef] [PubMed]
- Seeman, N.C. Structural DNA nanotechnology: An overview. Breast Cancer 2005, 303, 143–166. [Google Scholar] [CrossRef] [Green Version]
- Barclay, M.S.; Roy, S.K.; Huff, J.S.; Mass, O.A.; Turner, D.B.; Wilson, C.K.; Kellis, D.L.; Terpetschnig, E.A.; Lee, J.; Davis, P.H.; et al. Rotaxane rings promote oblique packing and extended lifetimes in DNA-templated molecular dye aggregates. Commun. Chem. 2021, 4, 19. [Google Scholar] [CrossRef]
- Mass, O.A.; Wilson, C.K.; Barcenas, G.; Terpetschnig, E.A.; Obukhova, O.M.; Kolosova, O.S.; Tatarets, A.L.; Li, L.; Yurke, B.; Knowlton, W.B.; et al. Influence of Hydrophobicity on Excitonic Coupling in DNA-Templated Indolenine Squaraine Dye Aggregates. J. Phys. Chem. C 2022, 126, 3475–3488. [Google Scholar] [CrossRef]
- Mass, O.A.; Wilson, C.K.; Roy, S.K.; Barclay, M.S.; Patten, L.K.; Terpetschnig, E.A.; Lee, J.; Pensack, R.D.; Yurke, B.; Knowlton, W.B. Exciton Delocalization in Indolenine Squaraine Aggregates Templated by DNA Holliday Junction Scaffolds. J. Phys. Chem. B 2020, 124, 9636–9647. [Google Scholar] [CrossRef]
- Rappoport, Z.; Liebman, J.F. The Chemistry of Cyclobutanes 1; Wiley: Chichester, UK, 2005. [Google Scholar]
- Rauer, C.; Nogueira, J.J.; Marquetand, P.; González, L. Cyclobutane Thymine Photodimerization Mechanism Revealed by Nonadiabatic Molecular Dynamics. J. Am. Chem. Soc. 2016, 138, 15911–15916. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, J.; Nishiguchi, K.; Manabe, K.; Masutani, C.; Hanaoka, F.; Iwai, S. Photosensitized [2 + 2] cycloaddition of N -acetylated cytosine affords stereoselective formation of cyclobutane pyrimidine dimer. Nucleic Acids Res. 2010, 39, 1165–1175. [Google Scholar] [CrossRef] [Green Version]
- Su, D.G.T.; Fang, H.; Gross, M.L.; Taylor, J.-S.A. Photocrosslinking of human telomeric G-quadruplex loops by anti cyclobutane thymine dimer formation. Proc. Natl. Acad. Sci. USA 2009, 106, 12861–12866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerling, T.; Kube, M.; Kick, B.; Dietz, H. Sequence-programmable covalent bonding of designed DNA assemblies. Sci. Adv. 2018, 4, eaau1157. [Google Scholar] [CrossRef] [Green Version]
- Su, D.G.T.; Kao, J.L.-F.; Gross, M.L.; Taylor, J.-S.A. Structure Determination of an Interstrand-Type Cis-Anti Cyclobutane Thymine Dimer Produced in High Yield by UVB Light in an Oligodeoxynucleotide at Acidic pH. J. Am. Chem. Soc. 2008, 130, 11328–11337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spano, F.C. The Spectral Signatures of Frenkel Polarons in H- and J-Aggregates. Acc. Chem. Res. 2009, 43, 429–439. [Google Scholar] [CrossRef] [PubMed]
- Kühn, O.; Renger, T.; May, V. Theory of exciton-vibrational dynamics in molecular dimers. Chem. Phys. 1996, 204, 99–114. [Google Scholar] [CrossRef]
- von Hippel, P.H.; Johnson, N.P.; Marcus, A.H. Fifty years of DNA “breathing”: Reflections on old and new approaches. Biopolymers 2013, 99, 923–954. [Google Scholar]
- Hyeon, C.; Lee, J.; Yoon, J.; Hohng, S.; Thirumalai, D. Hidden complexity in the isomerization dynamics of Holliday junctions. Nat. Chem. 2012, 4, 907–914. [Google Scholar] [CrossRef]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 Years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Basu, S.; Cervantes-Salguero, K.; Yurke, B.; Knowlton, W.B.; Lee, J.; Mass, O.A. Photocrosslinking Probes Proximity of Thymine Modifiers Tethering Excitonically Coupled Dye Aggregates to DNA Holliday Junction. Molecules 2022, 27, 4006. https://doi.org/10.3390/molecules27134006
Basu S, Cervantes-Salguero K, Yurke B, Knowlton WB, Lee J, Mass OA. Photocrosslinking Probes Proximity of Thymine Modifiers Tethering Excitonically Coupled Dye Aggregates to DNA Holliday Junction. Molecules. 2022; 27(13):4006. https://doi.org/10.3390/molecules27134006
Chicago/Turabian StyleBasu, Shibani, Keitel Cervantes-Salguero, Bernard Yurke, William B. Knowlton, Jeunghoon Lee, and Olga A. Mass. 2022. "Photocrosslinking Probes Proximity of Thymine Modifiers Tethering Excitonically Coupled Dye Aggregates to DNA Holliday Junction" Molecules 27, no. 13: 4006. https://doi.org/10.3390/molecules27134006
APA StyleBasu, S., Cervantes-Salguero, K., Yurke, B., Knowlton, W. B., Lee, J., & Mass, O. A. (2022). Photocrosslinking Probes Proximity of Thymine Modifiers Tethering Excitonically Coupled Dye Aggregates to DNA Holliday Junction. Molecules, 27(13), 4006. https://doi.org/10.3390/molecules27134006