
Acid–Base Equilibrium and Self-Association in Relation to High Antitumor Activity of Selected Unsymmetrical Bisacridines Established by Extensive Chemometric Analysis

 , ,
, ,  , ,
, , 
Abstract
:1. Introduction
2. Results
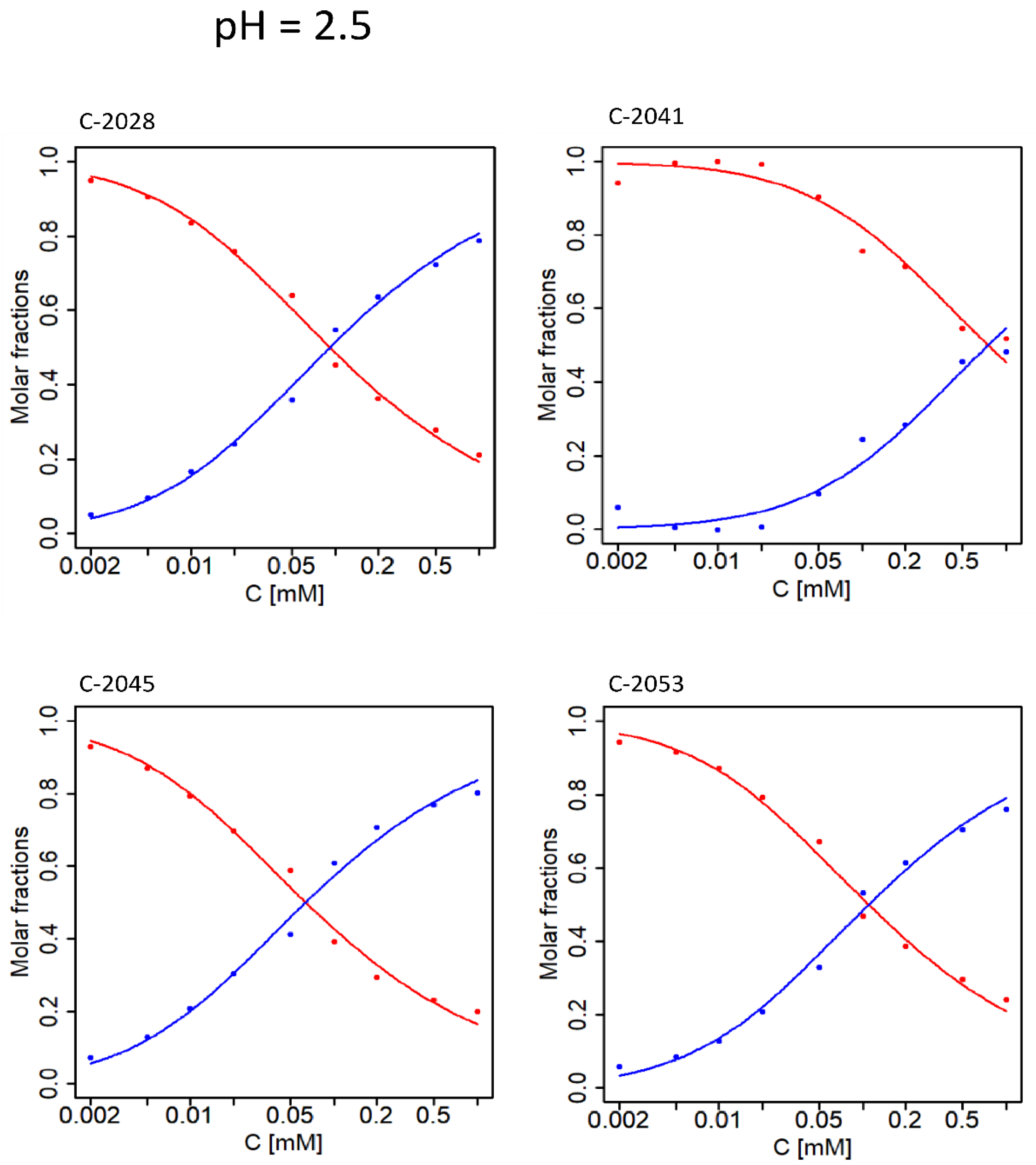
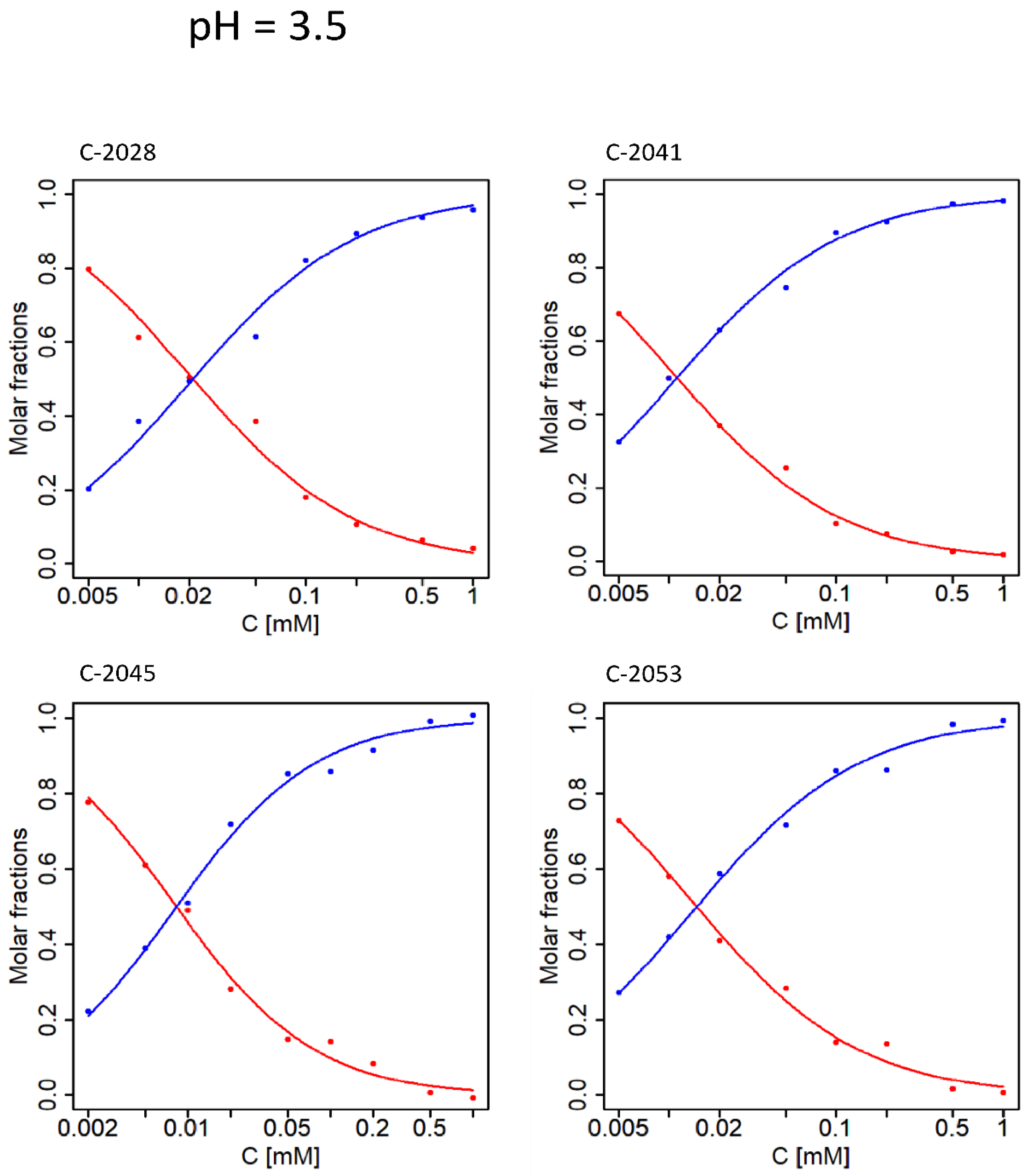
2.1. Determination of pKa Values for Studied Compounds
2.2. UA Structure Determination by NMR Spectroscopy
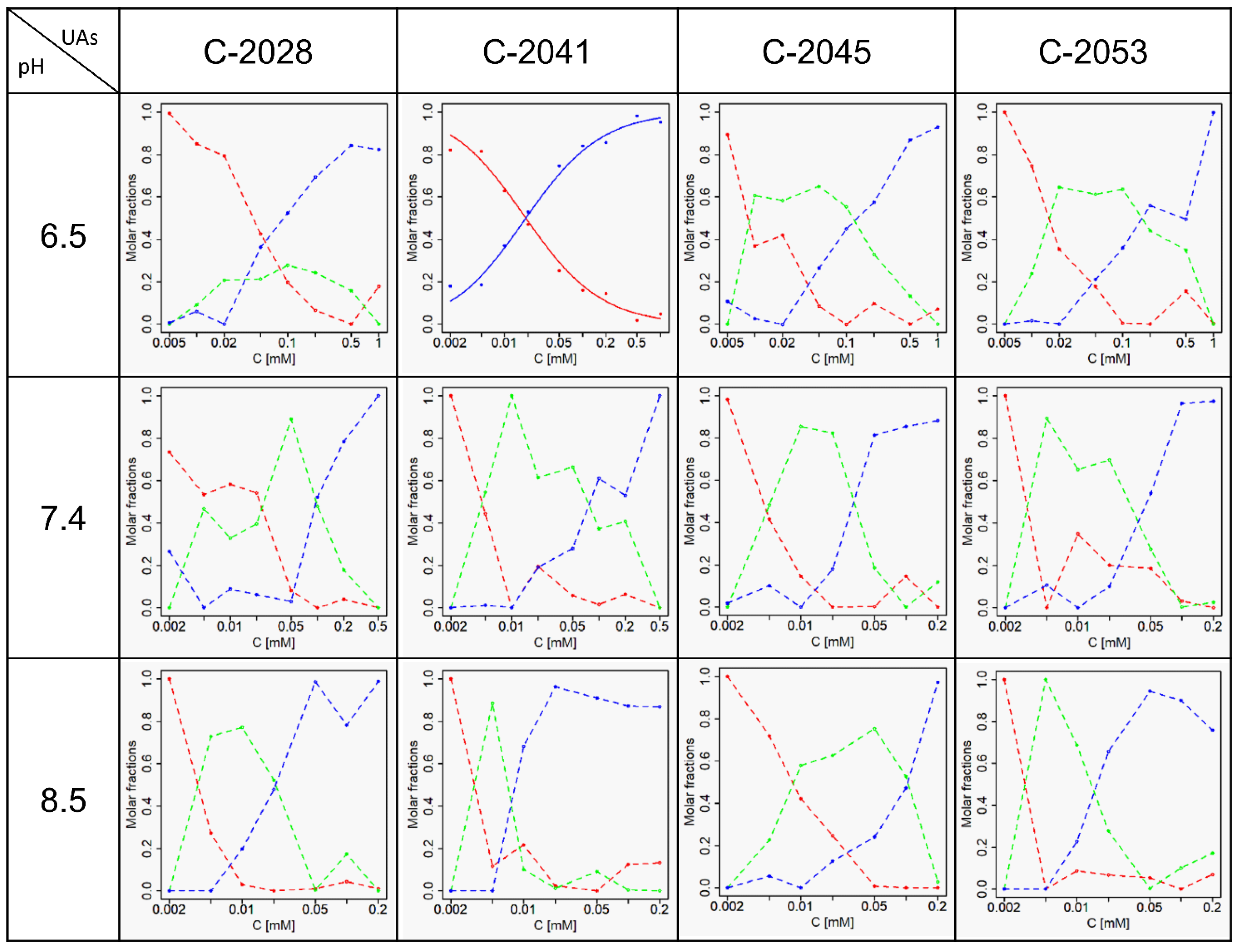
2.3. Self-Association of UAs
3. Discussion
4. Materials and Methods
4.1. Chemicals
4.2. General Procedures
4.3. pKa Determination
4.4. Aggregation Study
4.5. Chemometric Analysis: General Procedures
4.5.1. Chemometric Analysis for pKa Determination
4.5.2. Chemometric Analysis for Self-Association Study
4.6. NMR Studies of UA Protonation Forms
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Konopa, J.; Horowska, B.; Paluszkiewicz, E.; Borowa-Mazgaj, B.; Augustin, E.; Skwarska, A.; Mazerska, Z. Unsymmetrical Bisacridines with Antitumor Activity and Use Thereof. European Patent EP 3070078 A1, 10 October 2017. [Google Scholar]
- Konopa, J.; Horowska, B.; Paluszkiewicz, E.; Borowa-Mazgaj, B.; Augustin, E.; Skwarska, A.; Mazerska, Z. Asymmetric Bis-Acridines with Antitumour Activity and Their Uses. United States Patent US10202349B2, 12 February 2019. [Google Scholar]
- Paluszkiewicz, E.; Horowska, B.; Borowa-Mazgaj, B.; Peszyńska-Sularz, G.; Paradziej-Łukowicz, J.; Augustin, E.; Konopa, J.; Mazerska, Z. Design, synthesis and high antitumor potential of new unsymmetrical bisacridine derivatives towards human solid tumors, specifically pancreatic cancers and their unique ability to stabilize DNA G-quadruplexes. Eur. J. Med. Chem. 2020, 204, 112599. [Google Scholar] [CrossRef] [PubMed]
- Kulesza, J.; Pawłowska, M.; Augustin, E. The Influence of Antitumor Unsymmetrical Bisacridines on 3D Cancer Spheroids Growth and Viability. Molecules 2021, 26, 6262. [Google Scholar] [CrossRef] [PubMed]
- Pilch, J.; Matysiak-Brynda, E.; Kowalczyk, A.; Bujak, P.; Mazerska, Z.; Nowicka, A.M.; Augustin, E. New Unsymmetrical Bisacridine Derivatives Noncovalently Attached to Quaternary Quantum Dots Improve Cancer Therapy by Enhancing Cytotoxicity toward Cancer Cells and Protecting Normal Cells. ACS Appl. Mater. Interfaces 2020, 12, 17276–17289. [Google Scholar] [CrossRef] [PubMed]
- Pilch, J.; Kowalik, P.; Bujak, P.; Nowicka, A.M.; Augustin, E. Quantum Dots as a Good Carriers of Unsymmetrical Bisacridines for Modulating Cellular Uptake and the Biological Response in Lung and Colon Cancer Cells. Nanomaterials 2021, 11, 462. [Google Scholar] [CrossRef] [PubMed]
- Pilch, J.; Kowalik, P.; Kowalczyk, A.; Bujak, P.; Kasprzak, A.; Paluszkiewicz, E.; Augustin, E.; Nowicka, A.M. Foliate-Targeting Quantum Dots-β-Cyclodextrin Nanocarrier for Efficient Delivery of Unsymmetrical Bisacridines to Lung and Prostate Cancer Cells. Int. J. Mol. Sci. 2021, 23, 1261. [Google Scholar] [CrossRef] [PubMed]
- Mieszkowska, A.; Nowicka, A.; Kowalczyk, A.; Potęga, A.; Pawłowska, M.; Kosno, M.; Augustin, E.; Mazerska, Z. Metabolic Profiles of New Unsymmetrical Bisacridine Antitumor Agents in Electrochemical and Enzymatic Noncellular Systems and in Tumor Cells. Pharmaceuticals 2021, 14, 317. [Google Scholar] [CrossRef]
- Potęga, A.; Kosno, M.; Mazerska, Z. Novel insights into conjugation of antitumor-active unsymmetrical bisacridine C-2028 with glutathione: Characteristics of non-enzymatic and glutathione S-transferase-mediated reactions. J. Pharm. Anal. 2021, 11, 791–798. [Google Scholar] [CrossRef]
- WHO Chronicle. International Non-proprietary Names for Pharmaceutical Substances. Suppl. WHO Chron. 1976, 30, 1–18. [Google Scholar]
- Wiśniewska, A.; Chrapkowska, A.; Kot-Wasik, A.; Konopa, J.; Mazerska, Z. Metabolic transformations of antitumor imidazoacridinone, C-1311, with microsomal fractions of rat and human liver. Acta Biochim. Pol. 2007, 54, 831–838. [Google Scholar] [CrossRef] [Green Version]
- Gniazdowski, M.; Szmigiero, L. Nitracrine and its congeners-An overview. Gen. Pharmacol. 1995, 26, 473–481. [Google Scholar] [CrossRef]
- Laskowski, T.; Borzyszkowska, J.; Grynda, J.; Mazerski, J. C-1311 (Symadex), a potential anti-cancer drug, intercalates into DNA between A and G moieties. NMR-derived and MD-refined stereostructure of the d(GAGGCCTC) 2:C-1311 complex. J. Mol. Struct. 2017, 1141, 357–367. [Google Scholar] [CrossRef]
- Laskowski, T.; Czub, J.; Sowiński, P.; Mazerski, J. Intercalation complex of imidazoacridinone C-1311, a potential anticancer drug, with DNA helix d(CGATCG)2: Stereostructural studies by 2D NMR spectroscopy. J. Biomol. Struct. Dyn. 2016, 34, 653–663. [Google Scholar] [CrossRef] [PubMed]
- Bochman, M.L.; Paeschke, K.; Zakian, V.A. DNA secondary structures: Stability and function of G-quadruplex structures. Nat. Rev. Genet. 2012, 13, 770–780. [Google Scholar] [CrossRef] [Green Version]
- Kolesnikova, S.; Curtis, E.A. Structure and Function of Multimeric G-Quadruplexes. Molecules 2019, 24, 3074. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Yang, D. Sequence, Stability, Structure of G-Quadruplexes and Their Drug Interactions. Curr. Protoc. Nucleic Acid Chem. 2012, 50, 17.5.1–17.5.17. [Google Scholar] [CrossRef]
- Phan, A.T.; Luu, K.N.; Patel, D.J. Different loop arrangements of intramolecular human telomeric (3+1) G-quadruplexes in K+ solution. Nucleic Acids Res. 2006, 34, 5715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- White, K.A.; Grillo-Hill, B.K.; Barber, D.L. Cancer cell behaviors mediated by dysregulated pH dynamics at a glance. J. Cell Sci. 2017, 130, 663–669. [Google Scholar] [CrossRef] [Green Version]
- Galenkamp, K.M.O.; Sosicka, P.; Jung, M.; Recouvreux, M.V.; Zhang, Y.; Moldenhauer, M.R.; Brandi, G.; Freeze, H.H.; Commisso, C. Golgi acidification by NHE7 regulates cytosolic pH homeostasis in pancreatic cancer cells. Cancer Discov. 2020, 10, 823–835. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Li, Y.; Li, Z.; Shao, W.; Song, J.; Wei, J. Acidic tumor microenvironment promotes pancreatic cancer through miR-451a/MEF2D axis. J. Oncol. 2022, 12, ID3966386. [Google Scholar] [CrossRef]
- Petukh, M.; Stefl, S.; Alexov, E. The Role of Protonation States in Ligand-Receptor Recognition and Binding. Curr. Pharm. 2013, 19, 4182. [Google Scholar] [CrossRef] [Green Version]
- Onufriev, A.V.; Alexov, E. Protonation and pK changes in protein-ligand binding. Q. Rev. Biophys. 2013, 46, 181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Longo, G.S.; Pérez-Chávez, N.A.; Szleifer, I. How protonation modulates the interaction between proteins and pH-responsive hydrogel films. Curr. Opin. Colloid Interface Sci. 2019, 41, 27–39. [Google Scholar] [CrossRef]
- Nagesh, N.; Krishnaiah, A.; Dhople, V.M.; Sundaram, C.S.; Jagannadham, M.V. Noncovalent interaction of G-quadruplex DNA with acridine at low concentration monitored by MALDI-TOF mass spectrometry. Nucleosides Nucleotides Nucleic Acids 2007, 26, 303–315. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, R.; Artali, R.; Benoit, A.; Gargallo, R.; Eritja, R.; Ferguson, D.M.; Sham, Y.Y.; Mazzini, S. Structure and Stability of Human Telomeric G-Quadruplex with Preclinical 9-Amino Acridines. PLoS ONE 2013, 8, e57701. [Google Scholar] [CrossRef]
- Gowan, S.M.; Heald, R.; Stevens, M.F.; Kelland, L.R. Potent inhibition of telomerase by small-molecule pentacyclic acridines capable of interacting with G-quadruplexes. Mol. Pharmacol. 2001, 60, 981–988. [Google Scholar] [CrossRef]
- Nelder, J.A.; Mead, R. A Simplex Method for Function Minimization. Comput. J. 1965, 7, 308–313. [Google Scholar] [CrossRef]
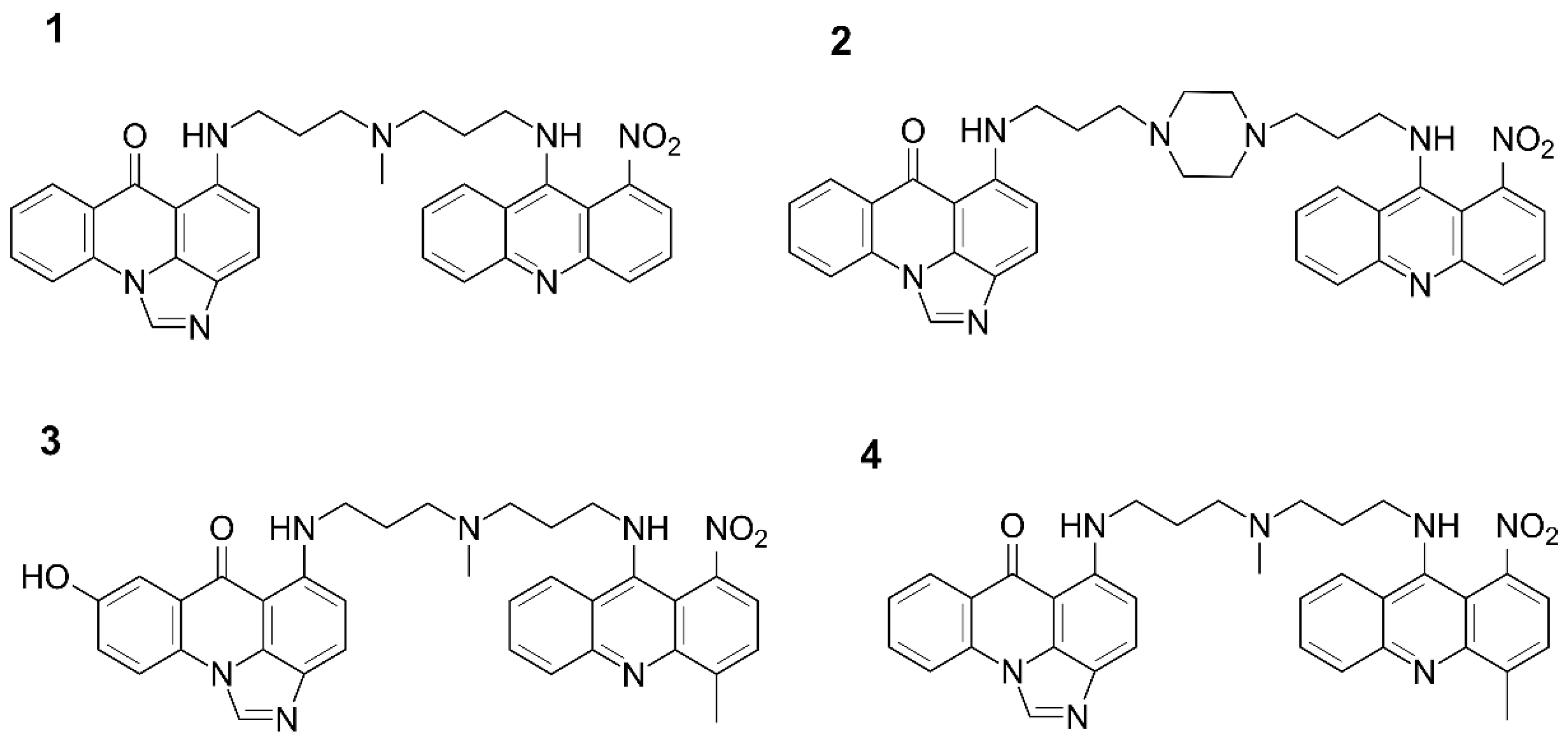
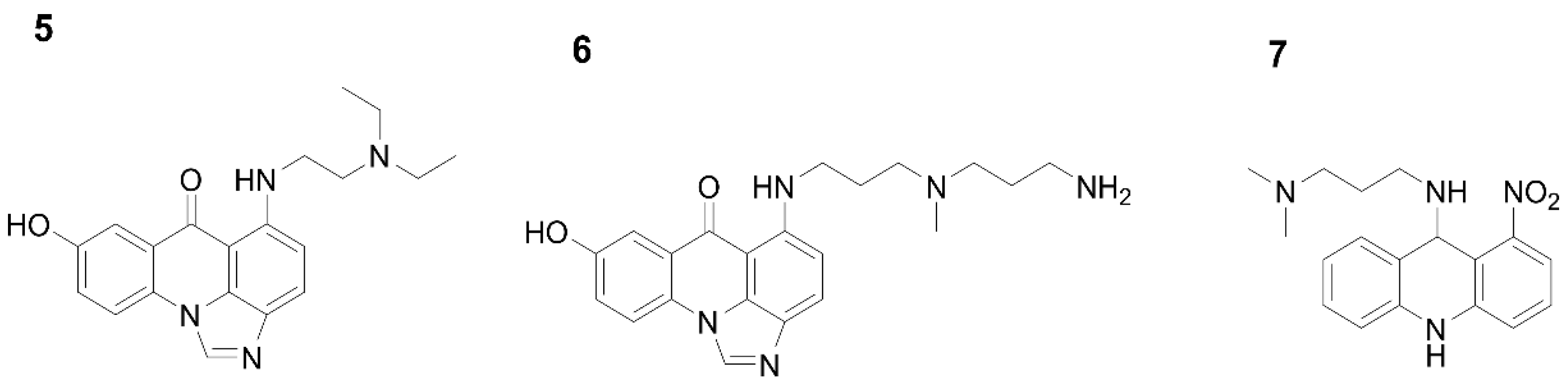

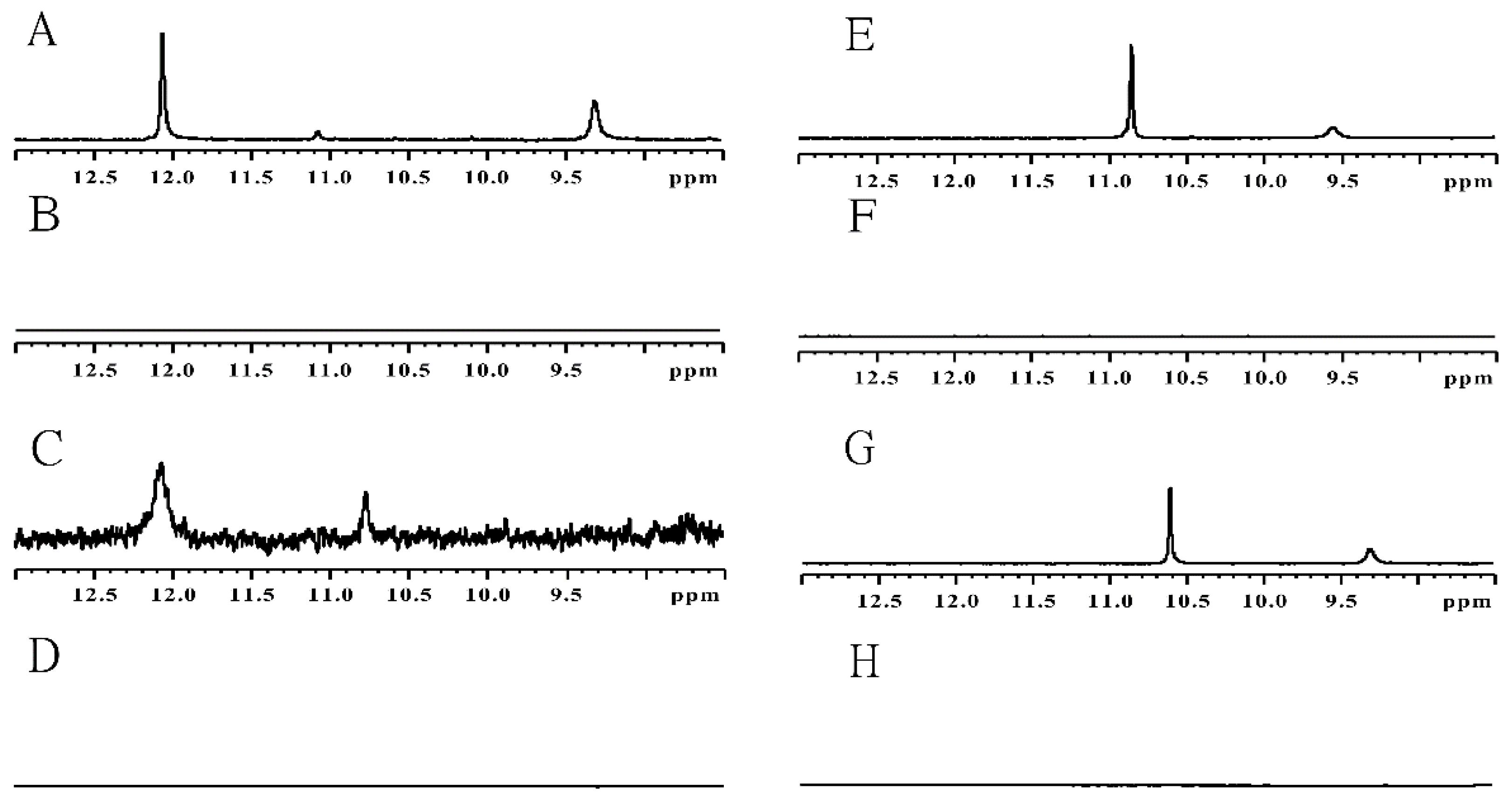
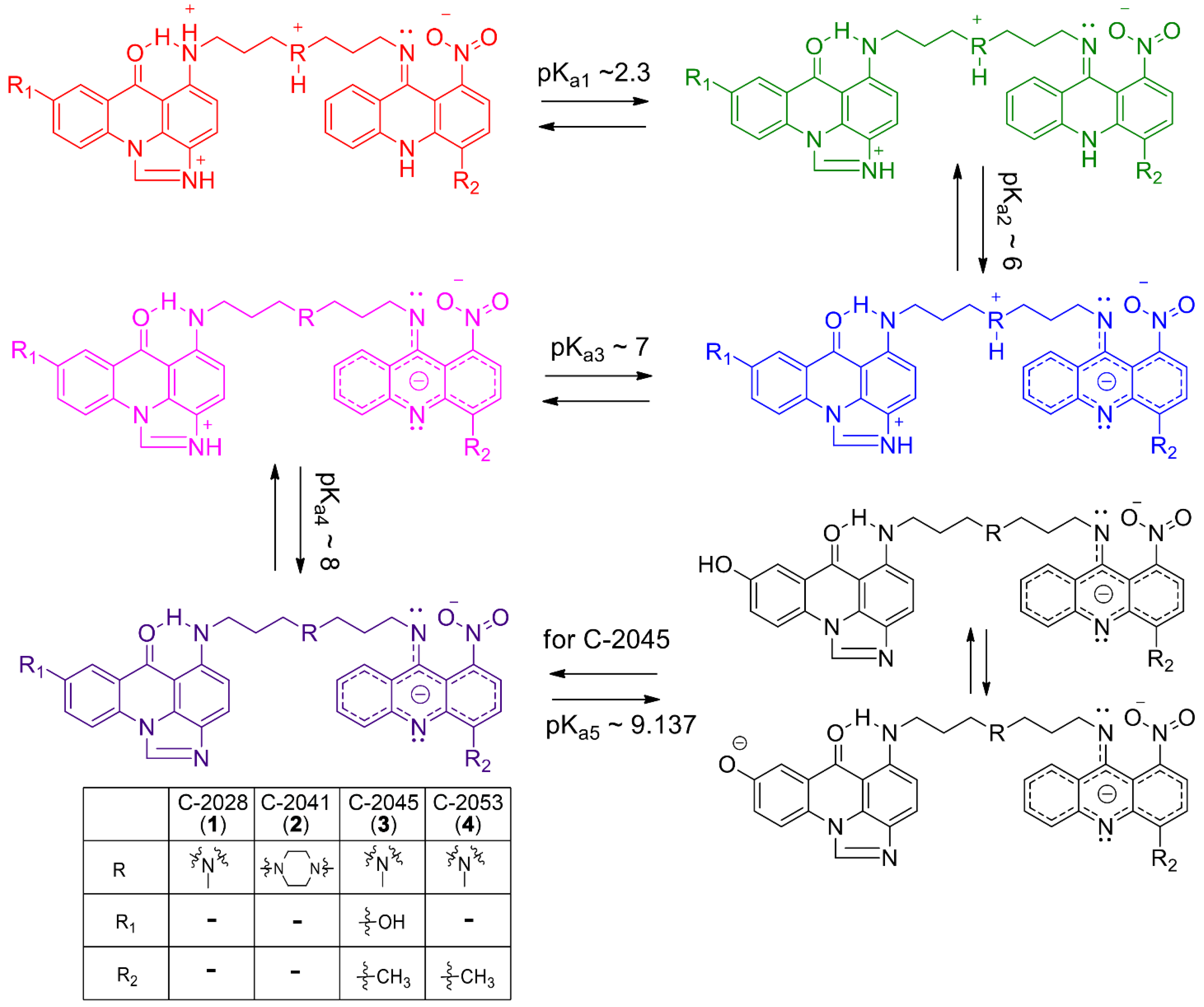

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | UAs | pKa1 | SD | pKa2 | SD | pKa3 | SD | pKa4 | SD | pKa5 | SD |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | C-2028 | 2.194 | 0.005 | 6.139 | 0.001 | 7.397 | 0.005 | 8.546 | 0.02 | - | - |
| 2 | C-2041 | 2.435 | 0.004 | 5.843 | 0.005 | 7.158 | 0.004 | - | - | - | - |
| 3 | C-2045 | 2.301 | 0.003 | 6.120 | 0.002 | 7.327 | 0.006 | - | - | 9.137 | 0.04 |
| 4 | C-2053 | 2.285 | 0.005 | 5.997 | 0.002 | 7.522 | 0.003 | 8.235 | 0.004 | - | - |
| No. | pKa1 | SD | pKa2 | SD | pKa3 | SD |
|---|---|---|---|---|---|---|
| 5 | 2.768 | 0.004 | 7.597 | 0.007 | 9.675 | 0.02 |
| 6 | 2.662 | 0.007 | 7.686 | 0.009 | - | - |
| 7 | - | - | 6.227 | 0.0007 | - | - |
| No. | pH | 2.5 | 3.5 | 6.5 | 7.4 | 8.5 | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| UAs | K | SD | K | SD | K | SD | K | SD | K | SD | |
| 1 | C-2028 | 10.89 * | 0.02 | 26.94 * | 0.033 | ~20 | ~40, ~10 | ~220, ~50 | |||
| 2 | C-2041 | 1.33 | 0.04 | 50.77 * | 0.02 | 33.25 * | 0.037 | ~200, ~13 | ~250, ~143 | ||
| 3 | C-2045 | 15.72 * | 0.026 | ~63 | ~125, ~40 | ~200, ~40 | ~125, ~10 | ||||
| 4 | C-2053 | 9.14 * | 0.026 | ~50 | ~60, ~5 | ~250, ~20 | ~400, ~60 | ||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kosno, M.; Laskowski, T.; Frackowiak, J.E.; Potęga, A.; Kurdyn, A.; Andrałojć, W.; Borzyszkowska-Bukowska, J.; Szwarc-Karabyka, K.; Mazerska, Z. Acid–Base Equilibrium and Self-Association in Relation to High Antitumor Activity of Selected Unsymmetrical Bisacridines Established by Extensive Chemometric Analysis. Molecules 2022, 27, 3995. https://doi.org/10.3390/molecules27133995
Kosno M, Laskowski T, Frackowiak JE, Potęga A, Kurdyn A, Andrałojć W, Borzyszkowska-Bukowska J, Szwarc-Karabyka K, Mazerska Z. Acid–Base Equilibrium and Self-Association in Relation to High Antitumor Activity of Selected Unsymmetrical Bisacridines Established by Extensive Chemometric Analysis. Molecules. 2022; 27(13):3995. https://doi.org/10.3390/molecules27133995
Chicago/Turabian StyleKosno, Michał, Tomasz Laskowski, Joanna E. Frackowiak, Agnieszka Potęga, Agnieszka Kurdyn, Witold Andrałojć, Julia Borzyszkowska-Bukowska, Katarzyna Szwarc-Karabyka, and Zofia Mazerska. 2022. "Acid–Base Equilibrium and Self-Association in Relation to High Antitumor Activity of Selected Unsymmetrical Bisacridines Established by Extensive Chemometric Analysis" Molecules 27, no. 13: 3995. https://doi.org/10.3390/molecules27133995
APA StyleKosno, M., Laskowski, T., Frackowiak, J. E., Potęga, A., Kurdyn, A., Andrałojć, W., Borzyszkowska-Bukowska, J., Szwarc-Karabyka, K., & Mazerska, Z. (2022). Acid–Base Equilibrium and Self-Association in Relation to High Antitumor Activity of Selected Unsymmetrical Bisacridines Established by Extensive Chemometric Analysis. Molecules, 27(13), 3995. https://doi.org/10.3390/molecules27133995