
Hydrogen Sulfide Biology and Its Role in Cancer

 , ,
, ,  ,
,
Abstract
1. Introduction
2. Physiological and Pathological Roles of H2S
3. Endogenous Production of H2S
4. Upregulation of Different H2S-Producing Enzymes in Cancer
4.1. CBS Expression in Cancer
4.2. CBS Function in Cancer
4.3. CSE Expression in Cancer
4.4. CSE Function in Cancer
4.5. Expression of 3-MST in Cancer
4.6. The Function of 3-MST in Cancer
5. Dual Role of H2S in Cancer
5.1. The Cancer-Promoting Effect of H2S
5.2. Anti-Cancer Effect of H2S
6. H2S Production and Programmed Cell Death
7. H2S as a Signaling Molecule and Role in Signaling Pathways
8. Protein Sulfhydration and Cancer
9. H2S-Mediated Persulfidation of NF-kB
10. H2S and DNA Repair
11. H2S and Immunomodulation in Cancer
12. H2S and Ferroptosis
13. H2S-Donating Compounds
14. The Potential of H2S in Cancer Therapy in Comparison to Other Complex Compounds
15. Tumor Markers Associated with H2S in Bodily Gas and Fluids
16. Conclusions and Future Directions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Zaorska, E.; Tomasova, L.; Koszelewski, D.; Ostaszewski, R.; Ufnal, M. Hydrogen Sulfide in Pharmacotherapy, Beyond the Hydrogen Sulfide-Donors. Biomolecules 2020, 10, 323. [Google Scholar] [CrossRef] [PubMed]
- PPowell, C.R.; Dillon, K.; Matson, J.B. A review of hydrogen sulfide (H2S) donors: Chemistry and potential therapeutic applications. Biochem. Pharmacol. 2018, 149, 110–123. [Google Scholar] [CrossRef] [PubMed]
- Askari, H.; Seifi, B.; Kadkhodaee, M.; Sanadgol, N.; Elshiekh, M.; Ranjbaran, M.; Ahghari, P. Protective effects of hydrogen sulfide on chronic kidney disease by reducing oxidative stress, inflammation and apoptosis. EXCLI J. 2018, 17, 14–23. [Google Scholar] [CrossRef] [PubMed]
- John, A.M.S.P.; Kundu, S.; Pushpakumar, S.; Fordham, M.; Weber, G.; Mukhopadhyay, M.; Sen, U. GYY4137, a Hydrogen Sulfide Donor Modulates miR194-Dependent Collagen Realignment in Diabetic Kidney. Sci. Rep. 2017, 7, 10924. [Google Scholar] [CrossRef] [PubMed]
- Dong, Q.; Yang, B.; Han, J.-G.; Zhang, M.-M.; Liu, W.; Zhang, X.; Yu, H.-L.; Liu, Z.-G.; Zhang, S.-H.; Li, T.; et al. A novel hydrogen sulfide-releasing donor, HA-ADT, suppresses the growth of human breast cancer cells through inhibiting the PI3K/AKT/mTOR and Ras/Raf/MEK/ERK signaling pathways. Cancer Lett. 2019, 455, 60–72. [Google Scholar] [CrossRef] [PubMed]
- Zhen, Y.; Wu, Q.; Ding, Y.; Zhang, W.; Zhai, Y.; Lin, X.; Weng, Y.; Guo, R.; Zhang, Y.; Feng, J.; et al. Exogenous hydrogen sulfide promotes hepatocellular carcinoma cell growth by activating the STAT3-COX-2 signaling pathway. Oncol. Lett. 2018, 15, 6562–6570. [Google Scholar] [CrossRef]
- Ngowi, E.E.; Afzal, A.; Sarfraz, M.; Khattak, S.; Zaman, S.U.; Khan, N.H.; Li, T.; Jiang, Q.-Y.; Zhang, X.; Duan, S.-F.; et al. Role of hydrogen sulfide donors in cancer development and progression. Int. J. Biol. Sci. 2021, 17, 73–88. [Google Scholar] [CrossRef]
- Lee, Z.W.; Zhou, J.; Chen, C.-S.; Zhao, Y.; Tan, C.-H.; Li, L.; Moore, P.K.; Deng, L.-W. The Slow-Releasing Hydrogen Sulfide Donor, GYY4137, Exhibits Novel Anti-Cancer Effects In Vitro and In Vivo. PLoS ONE 2011, 6, e21077. [Google Scholar] [CrossRef]
- Sakuma, S.; Minamino, S.; Takase, M.; Ishiyama, Y.; Hosokura, H.; Kohda, T.; Ikeda, Y.; Fujimoto, Y. Hydrogen sulfide donor GYY4137 suppresses proliferation of human colorectal cancer Caco-2 cells by inducing both cell cycle arrest and cell death. Heliyon 2019, 5, e02244. [Google Scholar] [CrossRef]
- Oláh, G.; Módis, K.; Törö, G.; Hellmich, M.R.; Szczesny, B.; Szabo, C. Role of endogenous and exogenous nitric oxide, carbon monoxide and hydrogen sulfide in HCT116 colon cancer cell proliferation. Biochem. Pharmacol. 2018, 149, 186–204. [Google Scholar] [CrossRef]
- Wu, D.-D.; Ngowi, E.E.; Zhai, Y.K.; Wang, Y.Z.; Khan, N.H.; Kombo, A.F.; Khattak, S.; Li, T.; Ji, X.-Y. Role of Hydrogen Sulfide in Oral Disease. Oxidative Med. Cell. Longev. 2022, 2022, 1886277. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Bian, H.; Li, X.; Wu, H.; Bi, Q.; Yan, Y.; Wang, Y. Hydrogen sulfide promotes cell proliferation of oral cancer through activation of the COX2/AKT/ERK1/2 axis. Oncol. Rep. 2016, 35, 2825–2832. [Google Scholar] [CrossRef] [PubMed]
- Khattak, S.; Zhang, Q.-Q.; Sarfraz, M.; Muhammad, P.; Ngowi, E.; Khan, N.; Rauf, S.; Wang, Y.-Z.; Qi, H.-W.; Wang, D.; et al. The Role of Hydrogen Sulfide in Respiratory Diseases. Biomolecules 2021, 11, 682. [Google Scholar] [CrossRef]
- Bates, M.N.; Crane, J.; Balmes, J.R.; Garrett, N. Investigation of Hydrogen Sulfide Exposure and Lung Function, Asthma and Chronic Obstructive Pulmonary Disease in a Geothermal Area of New Zealand. PLoS ONE 2015, 10, e0122062. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.-Z.; Ngowi, E.; Wang, D.; Qi, H.-W.; Jing, M.-R.; Zhang, Y.-X.; Cai, C.-B.; He, Q.-L.; Khattak, S.; Khan, N.; et al. The Potential of Hydrogen Sulfide Donors in Treating Cardiovascular Diseases. Int. J. Mol. Sci. 2021, 22, 2194. [Google Scholar] [CrossRef] [PubMed]
- Aghagolzadeh, P.; Radpour, R.; Bachtler, M.; van Goor, H.; Smith, E.R.; Lister, A.; Odermatt, A.; Feelisch, M.; Pasch, A. Hydrogen sulfide attenuates calcification of vascular smooth muscle cells via KEAP1/NRF2/NQO1 activation. Atherosclerosis 2017, 265, 78–86. [Google Scholar] [CrossRef] [PubMed]
- Baskar, R.; Radpour, R.; Bachtler, M.; van Goor, H.; Smith, E.R.; Lister, A.; Odermatt, A.; Feelisch, M.; Pasch, A. Effect of S-diclofenac, a novel hydrogen sulfide releasing derivative inhibit rat vascular smooth muscle cell proliferation. Eur. J. Pharmacol. 2008, 594, 1–8. [Google Scholar] [CrossRef]
- Ngowi, E.E.; Sarfraz, M.; Afzal, A.; Khan, N.H.; Khattak, S.; Zhang, X.; Li, T.; Duan, S.-F.; Ji, X.-Y.; Wu, D.-D. Roles of Hydrogen Sulfide Donors in Common Kidney Diseases. Front. Pharmacol. 2020, 11, 564281. [Google Scholar] [CrossRef]
- Koniukh, S.; Voloshchuk, N.; Melnyk, A.; Domin, I. Hydrogen Sulfide Metabolism and Its Role in Kidney Function in a Rat Model of Chronic Kidney Disease. Health Probl. Civiliz. 2020, 14, 289–297. [Google Scholar] [CrossRef]
- Qian, X.; Li, X.; Ma, F.; Luo, S.; Ge, R.; Zhu, Y. Novel hydrogen sulfide-releasing compound, S-propargyl-cysteine, prevents STZ-induced diabetic nephropathy. Biochem. Biophys. Res. Commun. 2016, 473, 931–938. [Google Scholar] [CrossRef]
- Zhang, L.; Wang, Y.; Li, Y.; Li, L.; Xu, S.; Feng, X.; Liu, S. Hydrogen Sulfide (H2S)-Releasing Compounds: Therapeutic Potential in Cardiovascular Diseases. Front. Pharmacol. 2018, 9, 1066. [Google Scholar] [CrossRef] [PubMed]
- Szabo, C. Gasotransmitters in cancer: From pathophysiology to experimental therapy. Nat. Rev. Drug Discov. 2015, 15, 185–203. [Google Scholar] [CrossRef] [PubMed]
- Hartle, M.D.; Pluth, M.D. A practical guide to working with H2S at the interface of chemistry and biology. Chem. Soc. Rev. 2016, 45, 6108–6117. [Google Scholar] [CrossRef] [PubMed]
- Wang, R. Two’s company, three’s a crowd: Can H2S be the third endogenous gaseous transmitter? FASEB J. 2002, 16, 1792–1798. [Google Scholar] [CrossRef] [PubMed]
- Cai, W.-J.; Wang, M.-J.; Moore, P.K.; Jin, H.-M.; Yao, T.; Zhu, Y.-C. The novel proangiogenic effect of hydrogen sulfide is dependent on Akt phosphorylation. Cardiovasc. Res. 2007, 76, 29–40. [Google Scholar] [CrossRef]
- Katsouda, A.; Bibli, S.-I.; Pyriochou, A.; Szabo, C.; Papapetropoulos, A. Regulation and role of endogenously produced hydrogen sulfide in angiogenesis. Pharmacol. Res. 2016, 113, 175–185. [Google Scholar] [CrossRef]
- Shackelford, R.E.; Mohammad, I.Z.; Meram, A.T.; Kim, D.; Alotaibi, F.; Patel, S.; Ghali, G.E.; Kevil, C.G. Molecular Functions of Hydrogen Sulfide in Cancer. Pathophysiology 2021, 28, 437–456. [Google Scholar] [CrossRef]
- Paul, B.D.; Snyder, S.H.; Kashfi, K. Effects of hydrogen sulfide on mitochondrial function and cellular bioenergetics. Redox Biol. 2021, 38, 101772. [Google Scholar] [CrossRef]
- Wang, M.; Yan, J.; Cao, X.; Hua, P.; Li, Z. Hydrogen sulfide modulates epithelial-mesenchymal transition and angiogenesis in non-small cell lung cancer via HIF-1α activation. Biochem. Pharmacol. 2020, 172, 113775. [Google Scholar] [CrossRef]
- Mao, Z.; Yang, X.; Muzutani, S.; Huang, Y.; Zhang, Z.; Shinmori, H.; Gao, K.; Yao, J. Hydrogen Sulfide Mediates Tumor Cell Resistance to Thioredoxin Inhibitor. Front. Oncol. 2020, 10, 252. [Google Scholar] [CrossRef]
- Shackelford, R.; Ozluk, E.; Islam, M.Z.; Hopper, B.; Meram, A.; Ghali, G.; Kevil, C.G. Hydrogen sulfide and DNA repair. Redox Biol. 2021, 38, 101675. [Google Scholar] [CrossRef] [PubMed]
- Cao, X.; Ding, L.; Xie, Z.-Z.; Yang, Y.; Whiteman, M.; Moore, P.K.; Bian, J.-S. A review of hydrogen sulfide synthesis, metabolism, and measurement: Is modulation of hydrogen sulfide a novel therapeutic for cancer? Antioxid. Redox Signal. 2019, 31, 1–38. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Wang, H.; Teng, T.; Duan, S.; Ji, A.; Li, Y. Hydrogen sulfide and autophagy: A double edged sword. Pharmacol. Res. 2018, 131, 120–127. [Google Scholar] [CrossRef] [PubMed]
- Szabo, C.; Coletta, C.; Chao, C.; Módis, K.; Szczesny, B.; Papapetropoulos, A.; Hellmich, M.R. Tumor-derived hydrogen sulfide, produced by cystathionine-β-synthase, stimulates bioenergetics, cell proliferation, and angiogenesis in colon cancer. Proc. Natl. Acad. Sci. USA 2013, 110, 12474–12479. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Wang, J.; Li, H.; Xue, M.; Ji, A.; Li, Y. Role of Hydrogen Sulfide in Ischemia-Reperfusion Injury. Oxidative Med. Cell. Longev. 2015, 2015, 186908. [Google Scholar] [CrossRef]
- Köhn, C.; Dubrovska, G.; Huang, Y.; Gollasch, M. Hydrogen Sulfide: Potent Regulator of Vascular Tone and Stimulator of Angiogenesis. Int. J. Biomed. Sci. IJBS 2012, 8, 81–86. [Google Scholar]
- Ariyaratnam, P.; Loubani, M.; Morice, A.H. Hydrogen sulphide vasodilates human pulmonary arteries: A possible role in pulmonary hypertension? Microvasc. Res. 2013, 90, 135–137. [Google Scholar] [CrossRef]
- Wallace, J.L. Hydrogen sulfide-releasing anti-inflammatory drugs. Trends Pharmacol. Sci. 2007, 28, 501–505. [Google Scholar] [CrossRef]
- Wallace, J.L.; Vong, L.; McKnight, W.; Dicay, M.; Martin, G.R. Endogenous and exogenous hydrogen sulfide promotes resolution of colitis in rats. Gastroenterology 2009, 137, 569–578.e1. [Google Scholar] [CrossRef]
- Kaium, M.; Liu, Y.; Zhu, Q.; Liu, C.-h.; Duan, J.-L.; Tan, B.K.-H.; Zhu, Y.Z. H2S donor, S-propargyl-cysteine, increases CSE in SGC-7901 and cancer-induced mice: Evidence for a novel anti-cancer effect of endogenous H2S? PLoS ONE 2011, 6, e20525. [Google Scholar]
- Zhao, Y.; Yang, C.; Organ, C.; Li, Z.; Bhushan, S.; Otsuka, H.; Pacheco, A.; Kang, J.; Aguilar, H.C.; Lefer, D.J. Design, synthesis, and cardioprotective effects of N-mercapto-based hydrogen sulfide donors. J. Med. Chem. 2015, 58, 7501–7511. [Google Scholar] [CrossRef] [PubMed]
- Calvert, J.; Coetzee, W.; Lefer, D.J. Novel Insights into Hydrogen Sulfide–Mediated Cytoprotection. Antioxid. Redox Signal. 2010, 12, 1203–1217. [Google Scholar] [CrossRef] [PubMed]
- Faro, M.L.L.; Fox, B.; Whatmore, J.L.; Winyard, P.G.; Whiteman, M. Hydrogen sulfide and nitric oxide interactions in inflammation. Nitric Oxide 2014, 41, 38–47. [Google Scholar] [CrossRef] [PubMed]
- Chiku, T.; Padovani, D.; Zhu, W.; Singh, S.; Vitvitsky, V.; Banerjee, R. H2S Biogenesis by Human Cystathionine γ-Lyase Leads to the Novel Sulfur Metabolites Lanthionine and Homolanthionine and Is Responsive to the Grade of Hyperhomocysteinemia. J. Biol. Chem. 2009, 284, 11601–11612. [Google Scholar] [CrossRef]
- Kabil, O.; Banerjee, R. Redox Biochemistry of Hydrogen Sulfide. J. Biol. Chem. 2010, 285, 21903–21907. [Google Scholar] [CrossRef]
- Bouillaud, F.; Blachier, F. Mitochondria and Sulfide: A Very Old Story of Poisoning, Feeding, and Signaling? Antioxid. Redox Signal. 2011, 15, 379–391. [Google Scholar] [CrossRef]
- Levitt, M.D.; Abdel-Rehim, M.S.; Furne, J. Free and acid-labile hydrogen sulfide concentrations in mouse tissues: Anomalously high free hydrogen sulfide in aortic tissue. Antioxid. Redox Signal. 2011, 15, 373–378. [Google Scholar] [CrossRef]
- Yang, G.; Cao, K.; Wu, L.; Wang, R. Cystathionine γ-lyase overexpression inhibits cell proliferation via a H2-dependent modulation of ERK1/2 phosphorylation and p21Cip/WAK-1. J. Biol. Chem. 2004, 279, 49199–49205. [Google Scholar] [CrossRef]
- Bełtowski, J. Hydrogen sulfide in pharmacology and medicine–An update. Pharmacol. Rep. 2015, 67, 647–658. [Google Scholar] [CrossRef]
- Bankhele, P.; Salvi, A.; Jamil, J.; Njie-Mbye, F.; Ohia, S.; Opere, C.A. Comparative Effects of Hydrogen Sulfide-Releasing Compounds on [3H]D-Aspartate Release from Bovine Isolated Retinae. Neurochem. Res. 2018, 43, 692–701. [Google Scholar] [CrossRef]
- Kondo, K.; Bhushan, S.; King, A.L.; Prabhu, S.D.; Hamid, T.; Koenig, S.; Murohara, T.; Predmore, B.L.; GojonSr, G.; Wang, R.; et al. H2S Protects Against Pressure Overload–Induced Heart Failure via Upregulation of Endothelial Nitric Oxide Synthase. Circulation 2013, 127, 1116–1127. [Google Scholar] [CrossRef] [PubMed]
- Kashfi, K.; Olson, K.R. Biology and therapeutic potential of hydrogen sulfide and hydrogen sulfide-releasing chimeras. Biochem. Pharmacol. 2013, 85, 689–703. [Google Scholar] [CrossRef] [PubMed]
- Salvi, A.; Bankhele, P.; Jamil, J.M.; Kulkarni-Chitnis, M.; Njie-Mbye, Y.F.; Ohia, S.E.; Opere, C.A. Pharmacological Actions of Hydrogen Sulfide Donors on Sympathetic Neurotransmission in the Bovine Anterior Uvea, In Vitro. Neurochem. Res. 2016, 41, 1020–1028. [Google Scholar] [CrossRef] [PubMed]
- Salvi, A.; Bankhele, P.; Jamil, J.; Chitnis, M.K.; Njie-Mbye, Y.F.; Ohia, S.E.; Opere, C.A. Effect of Hydrogen Sulfide Donors on Intraocular Pressure in Rabbits. J. Ocul. Pharmacol. Ther. 2016, 32, 371–375. [Google Scholar] [CrossRef] [PubMed]
- Polhemus, D.J.; Lefer, D.J. Emergence of Hydrogen Sulfide as an Endogenous Gaseous Signaling Molecule in Cardiovascular Disease. Circ. Res. 2014, 114, 730–737. [Google Scholar] [CrossRef] [PubMed]
- Wallace, J.L.; Wang, R. Hydrogen sulfide-based therapeutics: Exploiting a unique but ubiquitous gasotransmitter. Nat. Rev. Drug Discov. 2015, 14, 329–345. [Google Scholar] [CrossRef]
- Szabó, C. Hydrogen sulphide and its therapeutic potential. Nat. Rev. Drug Discov. 2007, 6, 917–935. [Google Scholar] [CrossRef]
- Wang, R. Physiological Implications of Hydrogen Sulfide: A Whiff Exploration That Blossomed. Physiol. Rev. 2012, 92, 791–896. [Google Scholar] [CrossRef]
- Shen, X.; Carlstrom, M.; Borniquel, S.; Jädert, C.; Kevil, C.G.; Lundberg, J.O. Microbial regulation of host hydrogen sulfide bioavailability and metabolism. Free Radic. Biol. Med. 2013, 60, 195–200. [Google Scholar] [CrossRef]
- Shen, X.; Peter, E.A.; Bir, S.; Wang, R.; Kevil, C.G. Analytical measurement of discrete hydrogen sulfide pools in biological specimens. Free Radic. Biol. Med. 2012, 52, 2276–2283. [Google Scholar] [CrossRef]
- Wu, D.-D.; Wang, D.-Y.; Li, H.-M.; Guo, J.-C.; Duan, S.-F.; Ji, X.-Y. Hydrogen Sulfide as a Novel Regulatory Factor in Liver Health and Disease. Oxidative Med. Cell. Longev. 2019, 2019, 3831713. [Google Scholar] [CrossRef] [PubMed]
- Mani, S.; Untereiner, A.; Wu, L.; Wang, R. Hydrogen Sulfide and the Pathogenesis of Atherosclerosis. Antioxid. Redox Signal. 2014, 20, 805–817. [Google Scholar] [CrossRef] [PubMed]
- Kimura, H. Production and Physiological Effects of Hydrogen Sulfide. Antioxid. Redox Signal. 2014, 20, 783–793. [Google Scholar] [CrossRef] [PubMed]
- Donnarumma, E.; Trivedi, R.K.; Lefer, D.J. Protective Actions of H2S in Acute Myocardial Infarction and Heart Failure. Compr. Physiol. 2011, 7, 583–602. [Google Scholar] [CrossRef]
- Searcy, D.G.; Lee, S.H. Sulfur reduction by human erythrocytes. J. Exp. Zool. 1998, 282, 310–322. [Google Scholar] [CrossRef]
- Kolluru, G.K.; Shen, X.; Bir, S.C.; Kevil, C.G. Hydrogen sulfide chemical biology: Pathophysiological roles and detection. Nitric Oxide 2013, 35, 5–20. [Google Scholar] [CrossRef]
- Benavides, G.A.; Squadrito, G.L.; Mills, R.W.; Patel, H.D.; Isbell, T.S.; Patel, R.P.; Darley-Usmar, V.M.; Doeller, J.E.; Kraus, D.W. Hydrogen sulfide mediates the vasoactivity of garlic. Proc. Natl. Acad. Sci. USA 2007, 104, 17977–17982. [Google Scholar] [CrossRef]
- Predmore, B.L.; Lefer, D.J.; Gojon, G. Hydrogen Sulfide in Biochemistry and Medicine. Antioxid. Redox Signal. 2012, 17, 119–140. [Google Scholar] [CrossRef]
- Kimura, H. Hydrogen sulfide: Its production, release and functions. Amino Acids 2011, 41, 113–121. [Google Scholar] [CrossRef]
- Maclean, K.N.; Kraus, E.; Kraus, J.P. The Dominant Role of Sp1 in Regulating the Cystathionine β-Synthase –1a and –1b Promoters Facilitates Potential Tissue-specific Regulation by Kruppel-like Factors. J. Biol. Chem. 2004, 279, 8558–8566. [Google Scholar] [CrossRef]
- Huang, C.W.; Moore, P.K. H2S synthesizing enzymes: Biochemistry and molecular aspects. Chem. Biochem. Pharmacol. Hydrog. Sulfide 2015, 3–25. [Google Scholar]
- Li, L.; Rose, P.; Moore, P.K. Hydrogen sulfide and cell signaling. Annu. Rev. Pharmacol. Toxicol. 2011, 51, 169–187. [Google Scholar] [CrossRef] [PubMed]
- Rose, P.; Moore, P.K.; Zhu, Y.Z. H2S biosynthesis and catabolism: New insights from molecular studies. Cell Mol. Life Sci. 2017, 74, 1391–1412. [Google Scholar] [CrossRef] [PubMed]
- Augsburger, F.; Szabo, C. Potential role of the 3-mercaptopyruvate sulfurtransferase (3-MST)—Hydrogen sulfide (H2S) pathway in cancer cells. Pharmacol. Res. 2020, 154, 104083. [Google Scholar] [CrossRef]
- Nagy, P. Mechanistic Chemical Perspective of Hydrogen Sulfide Signaling. Methods Enzymol. 2015, 554, 3–29. [Google Scholar] [CrossRef]
- Akbari, M.; Sogutdelen, E.; Juriasingani, S.; Sener, A. Hydrogen Sulfide: Emerging Role in Bladder, Kidney, and Prostate Malignancies. Oxidative Med. Cell. Longev. 2019, 2019, 2360945. [Google Scholar] [CrossRef]
- Giuffrè, A.; Tomé, C.S.; Fernandes, D.G.F.H.; Zuhra, K.; Vicente, J.B. Hydrogen Sulfide Metabolism and Signaling in the Tumor Microenvironment. In Advances in Experimental Medicine and Biology; Springer: Berlin/Heidelberg, Germany, 2020; Volume 1219, pp. 335–353. [Google Scholar] [CrossRef]
- Zuhra, K.; Tomé, C.S.; Masi, L.; Giardina, G.; Paulini, G.; Malagrinò, F.; Forte, E.; Vicente, J.B.; Giuffrè, A. N-Acetylcysteine Serves as Substrate of 3-Mercaptopyruvate Sulfurtransferase and Stimulates Sulfide Metabolism in Colon Cancer Cells. Cells 2019, 8, 828. [Google Scholar] [CrossRef]
- Libiad, M.; Vitvitsky, V.; Bostelaar, T.; Bak, D.; Lee, H.-J.; Sakamoto, N.; Fearon, E.; Lyssiotis, C.A.; Weerapana, E.; Banerjee, R. Hydrogen sulfide perturbs mitochondrial bioenergetics and triggers metabolic reprogramming in colon cells. J. Biol. Chem. 2019, 294, 12077–12090. [Google Scholar] [CrossRef]
- Malagrinò, F.; Zuhra, K.; Mascolo, L.; Mastronicola, D.; Vicente, J.B.; Forte, E.; Giuffrè, A. Hydrogen Sulfide Oxidation: Adaptive Changes in Mitochondria of SW480 Colorectal Cancer Cells upon Exposure to Hypoxia. Oxidative Med. Cell. Longev. 2019, 2019, 8102936. [Google Scholar] [CrossRef]
- Augsburger, F.; Randi, E.B.; Jendly, M.; Ascencao, K.; Dilek, N.; Szabo, C. Role of 3-Mercaptopyruvate Sulfurtransferase in the Regulation of Proliferation, Migration, and Bioenergetics in Murine Colon Cancer Cells. Biomolecules 2020, 10, 447. [Google Scholar] [CrossRef]
- Yue, T.; Zuo, S.; Bu, D.; Zhu, J.; Chen, S.; Ma, Y.; Ma, J.; Guo, S.; Wen, L.; Zhang, X.; et al. Aminooxyacetic acid (AOAA) sensitizes colon cancer cells to oxaliplatin via exaggerating apoptosis induced by ROS. J. Cancer 2020, 11, 1828–1838. [Google Scholar] [CrossRef] [PubMed]
- Ye, F.; Li, X.; Sun, K.; Xu, W.; Shi, H.; Bian, J.; Lu, R.; Ye, Y. Inhibition of endogenous hydrogen sulfide biosynthesis enhances the anti-cancer effect of 3,3′-diindolylmethane in human gastric cancer cells. Life Sci. 2020, 261, 118348. [Google Scholar] [CrossRef] [PubMed]
- Karim, Z.; Panagaki, T.; Randi, E.B.; Augsburger, F.; Blondel, M.; Friocourt, G.; Herault, Y.; Szabo, C. Mechanism of cystathionine-β-synthase inhibition by disulfiram: The role of bis (N, N-diethyldithiocarbamate)-copper (II). Biochem. Pharmacol. 2020, 182, 114267. [Google Scholar]
- Ascenção, K.; Dilek, N.; Augsburger, F.; Panagaki, T.; Zuhra, K.; Szabo, C. Pharmacological induction of mesenchymal-epithelial transition via inhibition of H2 biosynthesis and consequent suppression of ACLY activity in colon cancer cells. Pharmacol. Res. 2021, 165, 105393. [Google Scholar] [CrossRef] [PubMed]
- Szabo, C.; Ransy, C.; Módis, K.; Andriamihaja, M.; Murghes, B.; Coletta, C.; Olah, G.; Yanagi, K.; Bouillaud, F. Regulation of mitochondrial bioenergetic function by hydrogen sulfide. Part I. Biochemical and physiological mechanisms. J. Cereb. Blood Flow Metab. 2014, 171, 2099–2122. [Google Scholar] [CrossRef] [PubMed]
- Teng, H.; Wu, B.; Zhao, K.; Yang, G.; Wu, L.; Wang, R. Oxygen-sensitive mitochondrial accumulation of cystathionine β-synthase mediated by Lon protease. Proc. Natl. Acad. Sci. USA 2013, 110, 12679–12684. [Google Scholar] [CrossRef]
- Bhattacharyya, S.; Saha, S.; Giri, K.; Lanza, I.R.; Nair, K.S.; Jennings, N.B.; Rodriguez-Aguayo, C.; Lopez-Berestein, G.; Basal, E.; Weaver, A.L.; et al. Cystathionine Beta-Synthase (CBS) Contributes to Advanced Ovarian Cancer Progression and Drug Resistance. PLoS ONE 2013, 8, e79167. [Google Scholar] [CrossRef]
- Panza, E.; De Cicco, P.; Armogida, C.; Scognamiglio, G.; Gigantino, V.; Botti, G.; Germano, D.; Napolitano, M.; Papapetropoulos, A.; Bucci, M.; et al. Role of the cystathionine γ lyase/hydrogen sulfide pathway in human melanoma progression. Pigment. Cell Melanoma Res. 2015, 28, 61–72. [Google Scholar] [CrossRef]
- Guo, H.; Gai, J.-W.; Wang, Y.; Jin, H.-F.; Du, J.-B.; Jin, J. Characterization of Hydrogen Sulfide and Its Synthases, Cystathionine β-Synthase and Cystathionine γ-Lyase, in Human Prostatic Tissue and Cells. Urology 2012, 79, 483.e1–483.e5. [Google Scholar] [CrossRef]
- De Vos, J.; Thykjær, T.; Tarte, K.; Ensslen, M.; Raynaud, P.; Requirand, G.; Pellet, F.; Pantesco, V.; Rème, T.; Jourdan, M.; et al. Comparison of gene expression profiling between malignant and normal plasma cells with oligonucleotide arrays. Oncogene 2002, 21, 6848–6857. [Google Scholar] [CrossRef]
- Hansel, D.E.; Rahman, A.; Hidalgo, M.; Thuluvath, P.J.; Lillemoe, K.D.; Shulick, R.; Ku, J.-L.; Park, J.-G.; Miyazaki, K.; Ashfaq, R.; et al. Identification of Novel Cellular Targets in Biliary Tract Cancers Using Global Gene Expression Technology. Am. J. Pathol. 2003, 163, 217–229. [Google Scholar] [CrossRef]
- Zhang, W.; Braun, A.; Bauman, Z.; Olteanu, H.; Madzelan, P.; Banerjee, R. Expression Profiling of Homocysteine Junction Enzymes in the NCI60 Panel of Human Cancer Cell Lines. Cancer Res. 2005, 65, 1554–1560. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.; Bi, Q.; Wang, Y. Hydrogen sulfide accelerates cell cycle progression in oral squamous cell carcinoma cell lines. Oral Dis. 2014, 21, 156–162. [Google Scholar] [CrossRef] [PubMed]
- Sen, S.; Kawahara, B.; Gupta, D.; Tsai, R.; Khachatryan, M.; Roy-Chowdhuri, S.; Bose, S.; Yoon, A.; Faull, K.; Farias-Eisner, R.; et al. Role of cystathionine β-synthase in human breast Cancer. Free Radic. Biol. Med. 2015, 86, 228–238. [Google Scholar] [CrossRef]
- Wu, N.; Siow, Y.L.; Karmin, O. Ischemia/Reperfusion Reduces Transcription Factor Sp1-mediated Cystathionine β-Synthase Expression in the Kidney. J. Biol. Chem. 2010, 285, 18225–18233. [Google Scholar] [CrossRef]
- Zhang, J.; Xie, Y.; Xu, Y.; Pan, Y.; Shao, C. Hydrogen sulfide contributes to hypoxia-induced radioresistance on hepatoma cells. J. Radiat. Res. 2011, 52, 622–628. [Google Scholar] [CrossRef]
- Zhao, H.; Li, Q.; Wang, J.; Su, X.; Ng, K.M.; Qiu, T.; Shan, L.; Ling, Y.; Wang, L.; Cai, J.; et al. Frequent Epigenetic Silencing of the Folate-Metabolising Gene Cystathionine-Beta-Synthase in Gastrointestinal Cancer. PLoS ONE 2012, 7, e49683. [Google Scholar] [CrossRef]
- Takano, N.; Sarfraz, Y.; Gilkes, D.M.; Chaturvedi, P.; Xiang, L.; Suematsu, M.; Zagzag, D.; Semenza, G.L. Decreased expression of cystathionine β-synthase promotes glioma tumorigenesis. Mol. Cancer Res. 2014, 12, 1398–1406. [Google Scholar] [CrossRef]
- Hellmich, M.R.; Szabo, C. Hydrogen sulfide and cancer. Chem. Biochem. Pharmacol. Hydrog. Sulfide 2015, 230, 233–241. [Google Scholar]
- Mustafa, A.K.; Gadalla, M.M.; Sen, N.; Kim, S.; Mu, W.; Gazi, S.K.; Barrow, R.K.; Yang, G.; Wang, R.; Snyder, S.H. H2S signals through protein S-sulfhydration. Sci. Signal. 2009, 2, ra72. [Google Scholar] [CrossRef]
- Bohanon, F.J.; Mrazek, A.A.; Porro, L.J.; Spratt, H.; Hellmich, M.R.; Chao, C. Aminooxyacetic Acid in Combination with Oxaliplatin Significantly Decreases Colorectal Liver Metastasis in vivo. J. Am. Coll. Surg. 2014, 219, S132. [Google Scholar] [CrossRef]
- Fan, K.; Li, N.; Qi, J.; Yin, P.; Zhao, C.; Wang, L.; Li, Z.; Zha, X. Wnt/β-catenin signaling induces the transcription of cystathionine-γ-lyase, a stimulator of tumor in colon cancer. Cell. Signal. 2014, 26, 2801–2808. [Google Scholar] [CrossRef] [PubMed]
- Cai, W.; Wang, M.; Ju, L.; Wang, C.; Zhu, Y. Hydrogen sulfide induces human colon cancer cell proliferation: Role of Akt, ERK and p21. Cell Biol. Int. 2010, 34, 565–572. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Ye, S.; Yuan, D.; Zhang, J.; Bai, Y.; Shao, C. Hydrogen sulfide (H2)/cystathionine γ-lyase (CSE) pathway contributes to the proliferation of hepatoma cells. Mutat. Res./Fundam. Mol. Mech. Mutagen. 2014, 763, 10–18. [Google Scholar] [CrossRef] [PubMed]
- Kandil, S.; Brennan, L.; McBean, G.J. Glutathione depletion causes a JNK and p38MAPK-mediated increase in expression of cystathionine-γ-lyase and upregulation of the transsulfuration pathway in C6 glioma cells. Neurochem. Int. 2010, 56, 611–619. [Google Scholar] [CrossRef]
- Pei, Y.; Wu, B.; Cao, Q.; Wu, L.; Yang, G. Hydrogen sulfide mediates the anti-survival effect of sulforaphane on human prostate cancer cells. Toxicol. Appl. Pharmacol. 2011, 257, 420–428. [Google Scholar] [CrossRef]
- Yang, G.; Wu, L.; Jiang, B.; Yang, W.; Qi, J.; Cao, K.; Meng, Q.; Mustafa, A.K.; Mu, W.; Zhang, S.; et al. H2 as a physiologic vasorelaxant: Hypertension in mice with deletion of cystathionine γ-lyase. Science 2008, 322, 587–590. [Google Scholar] [CrossRef]
- Zhao, K.; Ju, Y.; Li, S.; Altaany, Z.; Wang, R.; Yang, G. S-sulfhydration of MEK 1 leads to PARP-1 activation and DNA damage repair. EMBO Rep. 2014, 15, 792–800. [Google Scholar] [CrossRef]
- Pálinkás, Z.; Furtmüller, P.G.; Nagy, A.; Jakopitsch, C.; Pirker, K.F.; Magierowski, M.; Jasnos, K.; Wallace, J.L.; Obinger, C.; Nagy, P. Interactions of hydrogen sulfide with myeloperoxidase. J. Cereb. Blood Flow Metab. 2015, 172, 1516–1532. [Google Scholar] [CrossRef]
- Zhang, L.; Qi, Q.; Yang, J.; Sun, D.; Li, C.; Xue, Y.; Jiang, Q.; Tian, Y.; Xu, C.; Wang, R. An Anticancer Role of Hydrogen Sulfide in Human Gastric Cancer Cells. Oxidative Med. Cell. Longev. 2015, 2015, 1–8. [Google Scholar] [CrossRef]
- Yin, P.; Zhao, C.; Li, Z.; Mei, C.; Yao, W.; Liu, Y.; Li, N.; Qi, J.; Wang, L.; Shi, Y.; et al. Sp1 is involved in regulation of cystathionine γ-lyase gene expression and biological function by PI3K/Akt pathway in human hepatocellular carcinoma cell lines. Cell. Signal. 2012, 24, 1229–1240. [Google Scholar] [CrossRef] [PubMed]
- Gai, J.-W.; Qin, W.; Liu, M.; Wang, H.-F.; Zhang, M.; Li, M.; Zhou, W.-H.; Ma, Q.-T.; Liu, G.-M.; Song, W.-H.; et al. Expression profile of hydrogen sulfide and its synthases correlates with tumor stage and grade in urothelial cell carcinoma of bladder. Urol. Oncol. Semin. Orig. Investig. 2016, 34, 166.e15–166.e20. [Google Scholar] [CrossRef] [PubMed]
- Jurkowska, H.; Placha, W.; Nagahara, N.; Wróbel, M. The expression and activity of cystathionine-γ-lyase and 3-mercaptopyruvate sulfurtransferase in human neoplastic cell lines. Amino Acids 2011, 41, 151–158. [Google Scholar] [CrossRef] [PubMed]
- Wróbel, M.; Bronowicka-Adamska, P.; Bentke, A. Hydrogen sulfide generation from L-cysteine in the human glioblastoma-astrocytoma U-87 MG and neuroblastoma SHSY5Y cell lines. Acta Biochim. Pol. 2017, 64, 171–176. [Google Scholar] [CrossRef]
- Módis, K.; Coletta, C.; Erdélyi, K.; Papapetropoulos, A.; Szabo, C. Intramitochondrial hydrogen sulfide production by 3-mercaptopyruvate sulfurtransferase maintains mitochondrial electron flow and supports cellular bioenergetics. FASEB J. 2013, 27, 601–611. [Google Scholar] [CrossRef]
- Ostrakhovitch, E.A.; Akakura, S.; Sanokawa-Akakura, R.; Goodwin, S.; Tabibzadeh, S. Dedifferentiation of cancer cells following recovery from a potentially lethal damage is mediated by H2–Nampt. Exp. Cell Res. 2015, 330, 135–150. [Google Scholar] [CrossRef]
- Untereiner, A.; Pavlidou, A.; Druzhyna, N.; Papapetropoulos, A.; Hellmich, M.R.; Szabo, C. Drug resistance induces the upregulation of H2-producing enzymes in HCT116 colon cancer cells. Biochem. Pharmacol. 2018, 149, 174–185. [Google Scholar] [CrossRef]
- Breza, J., Jr.; Soltysova, A.; Hudecova, S.; Penesova, A.; Szadvari, I.; Babula, P.; Chovancova, B.; Lencesova, L.; Pos, O.; Ondrias, K.; et al. Endogenous H2S producing enzymes are involved in apoptosis induction in clear cell renal cell carcinoma. BMC Cancer 2018, 18, 591. [Google Scholar] [CrossRef]
- Wahafu, W.; Gai, J.; Song, L.; Ping, H.; Wang, M.; Yang, F.; Niu, Y.; Xing, N. Increased H2 and its synthases in urothelial cell carcinoma of the bladder, and enhanced cisplatin-induced apoptosis following H2 inhibition in EJ cells. Oncol. Lett. 2018, 15, 8484–8490. [Google Scholar] [CrossRef]
- Bai, Y.W.; Ye, M.J.; Yang, D.L.; Yu, M.P.; Zhou, C.F.; Shen, T. Hydrogen sulfide attenuates paraquat—Induced epithelial-mesenchymal transition of human alveolar epithelial cells through regulating transforming growth factor—β1/Smad2/3 signaling pathway. J. Appl. Toxicol. 2019, 39, 432–440. [Google Scholar] [CrossRef]
- Wróbel, M.; Czubak, J.; Bronowicka-Adamska, P.; Jurkowska, H.; Adamek, D.; Papla, B. Is development of high-grade gliomas sulfur-dependent? Molecules 2014, 19, 21350–21362. [Google Scholar] [CrossRef] [PubMed]
- Shiota, M.; Naya, M.; Yamamoto, T.; Hishiki, T.; Tani, T.; Takahashi, H.; Kubo, A.; Koike, D.; Itoh, M.; Ohmura, M.; et al. Gold-nanofève surface-enhanced Raman spectroscopy visualizes hypotaurine as a robust anti-oxidant consumed in cancer survival. Nat. Commun. 2018, 9, 1561. [Google Scholar] [CrossRef] [PubMed]
- Meram, A.T.; Chen, J.; Patel, S.; Kim, D.D.; Shirley, B.; Covello, P.; Coppola, D.; Wei, E.X.; Ghali, G.; Kevil, C.G.; et al. Hydrogen Sulfide Is Increased in Oral Squamous Cell Carcinoma Compared to Adjacent Benign Oral Mucosae. Anticancer Res. 2018, 38, 3843–3852. [Google Scholar] [CrossRef] [PubMed]
- Szczesny, B.; Marcatti, M.; Zatarain, J.R.; Druzhyna, N.; Wiktorowicz, J.E.; Nagy, P.; Hellmich, M.R.; Szabo, C. Inhibition of hydrogen sulfide biosynthesis sensitizes lung adenocarcinoma to chemotherapeutic drugs by inhibiting mitochondrial DNA repair and suppressing cellular bioenergetics. Sci. Rep. 2016, 6, 36125. [Google Scholar] [CrossRef]
- Dongsoo, K.; Chen, J.; Wei, E.; Ansari, J.; Meram, A.; Patel, S.; Ghali, G.; Kevil, C.; Shackelford, R.E. Hydrogen Sulfide and Hydrogen Sulfide-Synthesizing Enzymes Are Altered in a Case of Oral Adenoid Cystic Carcinoma. Case Rep. Oncol. 2018, 11, 585–590. [Google Scholar] [CrossRef]
- Kun, E.; Klausner, C.; Fanshier, D. The rate of cleavage of β-mercaptopyruvate by rapidly dividing cells. Experientia 1960, 16, 55–56. [Google Scholar] [CrossRef]
- Włodek, L.; Wróbel, M.; Czubak, J. Transamination and transsulphuration of l-cysteine in ehrlich ascites tumor cells and mouse liver: The nonenzymatic reaction of l-cysteine with pyruvate. Int. J. Biochem. 1993, 25, 107–112. [Google Scholar] [CrossRef]
- Panagaki, T.; Randi, E.B.; Szabo, C. Role of 3-Mercaptopyruvate Sulfurtransferase in the Regulation of Proliferation and Cellular Bioenergetics in Human Down Syndrome Fibroblasts. Biomolecules 2020, 10, 653. [Google Scholar] [CrossRef]
- Govar, A.A.; Törő, G.; Szaniszlo, P.; Pavlidou, A.; Bibli, S.; Thanki, K.; Resto, V.A.; Chao, C.; Hellmich, M.R.; Szabo, C.; et al. 3-Mercaptopyruvate sulfurtransferase supports endothelial cell angiogenesis and bioenergetics. J. Cereb. Blood Flow Metab. 2020, 177, 866–883. [Google Scholar] [CrossRef]
- Nagahara, N. Multiple role of 3-mercaptopyruvate sulfurtransferase: Antioxidative function, H2 and polysulfide production and possible SOx production. Br. J. Pharmacol. 2018, 175, 577–589. [Google Scholar] [CrossRef]
- Fräsdorf, B.; Radon, C.; Leimkühler, S. Characterization and Interaction Studies of Two Isoforms of the Dual Localized 3-Mercaptopyruvate Sulfurtransferase TUM1 from Humans. J. Biol. Chem. 2014, 289, 34543–34556. [Google Scholar] [CrossRef] [PubMed]
- Goubern, M.; Andriamihaja, M.; Nübel, T.; Blachier, F.; Bouillaud, F. Sulfide, the first inorganic substrate for human cells. FASEB J. 2007, 21, 1699–1706. [Google Scholar] [CrossRef] [PubMed]
- Papapetropoulos, A.; Pyriochou, A.; Altaany, Z.; Yang, G.; Marazioti, A.; Zhou, Z.; Jeschke, M.G.; Branski, L.K.; Herndon, D.N.; Wang, R.; et al. Hydrogen sulfide is an endogenous stimulator of angiogenesis. Proc. Natl. Acad. Sci. USA 2009, 106, 21972–21977. [Google Scholar] [CrossRef] [PubMed]
- Mustafa, A.K.; Sikka, G.; Gazi, S.K.; Steppan, J.; Jung, S.M.; Bhunia, A.K.; Barodka, V.M.; Gazi, F.K.; Barrow, R.K.; Wang, R.; et al. Hydrogen Sulfide as Endothelium-Derived Hyperpolarizing Factor Sulfhydrates Potassium Channels. Circ. Res. 2011, 109, 1259–1268. [Google Scholar] [CrossRef]
- Liu, Y.-H.; Lu, M.; Hu, L.-F.; Wong, P.T.-H.; Webb, G.D.; Bian, J. Hydrogen Sulfide in the Mammalian Cardiovascular System. Antioxid. Redox Signal. 2012, 17, 141–185. [Google Scholar] [CrossRef]
- Wu, D.; Luo, N.; Wang, L.; Zhao, Z.; Bu, H.; Xu, G.; Yan, Y.; Che, X.; Jiao, Z.; Zhao, T.; et al. Hydrogen sulfide ameliorates chronic renal failure in rats by inhibiting apoptosis and inflammation through ROS/MAPK and NF-κB signaling pathways. Sci. Rep. 2017, 7, 455. [Google Scholar] [CrossRef]
- Zhou, H.; Ding, L.; Wu, Z.; Cao, X.; Zhang, Q.; Lin, L.; Bian, J.-S. Hydrogen sulfide reduces RAGE toxicity through inhibition of its dimer formation. Free Radic. Biol. Med. 2017, 104, 262–271. [Google Scholar] [CrossRef]
- Yin, J.; Tu, C.; Zhao, J.; Ou, D.; Chen, G.; Liu, Y.; Xiao, X. Exogenous hydrogen sulfide protects against global cerebral ischemia/reperfusion injury via its anti-oxidative, anti-inflammatory and anti-apoptotic effects in rats. Brain Res. 2013, 1491, 188–196. [Google Scholar] [CrossRef]
- Rose, P.; Moore, P.K.; Ming, S.H.; Nam, O.C.; Armstrong, J.S.; Whiteman, M. Hydrogen sulfide protects colon cancer cells from chemopreventative agent β-phenylethyl isothiocyanate induced apoptosis. World J. Gastroenterol. 2005, 11, 3990–3997. [Google Scholar] [CrossRef]
- Zhen, Y.; Pan, W.; Hu, F.; Wu, H.; Feng, J.; Zhang, Y.; Chen, J. Exogenous hydrogen sulfide exerts proliferation/anti-apoptosis/angiogenesis/migration effects via amplifying the activation of NF-κB pathway in PLC/PRF/5 hepatoma cells. Int. J. Oncol. 2015, 46, 2194–2204. [Google Scholar] [CrossRef]
- Tiong, C.X.; Lu, M.; Bian, J.-S. Protective effect of hydrogen sulphide against 6-OHDA-induced cell injury in SH-SY5Y cells involves PKC/PI3K/Akt pathway. J. Cereb. Blood Flow Metab. 2010, 161, 467–480. [Google Scholar] [CrossRef] [PubMed]
- Sen, N.; Paul, B.D.; Gadalla, M.M.; Mustafa, A.K.; Sen, T.; Xu, R.; Kim, S.; Snyder, S.H. Hydrogen sulfide-linked sulfhydration of NF-κB mediates its antiapoptotic actions. Mol. Cell 2012, 45, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.; Zhao, K.; Ju, Y.; Mani, S.; Cao, Q.; Puukila, S.; Khaper, N.; Wu, L.; Wang, R. Hydrogen Sulfide Protects Against Cellular Senescence via S-Sulfhydration of Keap1 and Activation of Nrf2. Antioxid. Redox Signal. 2013, 18, 1906–1919. [Google Scholar] [CrossRef] [PubMed]
- Francia, M.; Striepen, B. Cell division in apicomplexan parasites. Nat. Rev. Genet. 2014, 12, 125–136. [Google Scholar] [CrossRef]
- Schwartz, G.K.; Shah, M.A. Targeting the Cell Cycle: A New Approach to Cancer Therapy. J. Clin. Oncol. 2005, 23, 9408–9421. [Google Scholar] [CrossRef]
- Malumbres, M.; Barbacid, M. Cell cycle, CDKs and cancer: A changing paradigm. Nat. Cancer 2009, 9, 153–166. [Google Scholar] [CrossRef]
- Bartkova, J.; Hořejší, Z.; Koed, K.; Krämer, A.; Tort, F.; Zieger, K.; Guldberg, P.; Sehested, M.; Nesland, J.M.; Lukas, C.; et al. DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature 2005, 434, 864–870. [Google Scholar] [CrossRef]
- Osaki, M.; Oshimura, M.; Ito, H. PI3K-Akt pathway: Its functions and alterations in human cancer. Apoptosis 2004, 9, 667–676. [Google Scholar] [CrossRef]
- De Cicco, P.; Ercolano, G.; Rubino, V.; Terrazzano, G.; Ruggiero, G.; Cirino, G.; Ianaro, A. Modulation of the functions of myeloid-derived suppressor cells: A new strategy of hydrogen sulfide anti-cancer effects. J. Cereb. Blood Flow Metab. 2020, 177, 884–897. [Google Scholar] [CrossRef]
- Szabo, C. Hydrogen Sulfide, an Endogenous Stimulator of Mitochondrial Function in Cancer Cells. Cells 2021, 10, 220. [Google Scholar] [CrossRef]
- Li, H.; Xu, F.; Gao, G.; Gao, X.; Wu, B.; Zheng, C.; Wang, P.; Li, Z.; Hua, H.; Li, D. Hydrogen sulfide and its donors: Novel antitumor and antimetastatic therapies for triple-negative breast cancer. Redox Biol. 2020, 34, 101564. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Liu, Y.; Deng, Y.; Pan, L.; Fu, H.; Han, X.; Li, Y.; Shi, H.; Wang, T. Therapeutic potential of endogenous hydrogen sulfide inhibition in breast cancer (Review). Oncol. Rep. 2021, 45, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Lechuga, T.J.; Qi, Q.; Magness, R.R.; Chen, D.-B. Ovine uterine artery hydrogen sulfide biosynthesis in vivo: Effects of ovarian cycle and pregnancy. Biol. Reprod. 2019, 100, 1630–1636. [Google Scholar] [CrossRef] [PubMed]
- Panagaki, T.; Lozano-Montes, L.; Janickova, L.; Zuhra, K.; Szabo, M.P.; Majtan, T.; Rainer, G.; Maréchal, D.; Herault, Y.; Szabo, C. Overproduction of hydrogen sulfide, generated by cystathionine β-synthase, disrupts brain wave patterns and contributes to neurobehavioral dysfunction in a rat model of down syndrome. Redox Biol. 2022, 51, 102233. [Google Scholar] [CrossRef] [PubMed]
- Peng, J.; Zhang, D. Potentials of CCL21 and CBS as therapeutic approaches for breast cancer. Eur. Surg. Res. 2022. [Google Scholar] [CrossRef]
- Arif, H.M.; Qian, Z.M.; Wang, R. Signaling Integration of Hydrogen Sulfide and Iron on Cellular Functions. Antioxid. Redox Signal. 2022, 36, 275–293. [Google Scholar] [CrossRef]
- Yao, M.; Lu, Y.; Shi, L.; Huang, Y.; Zhang, Q.; Tan, J.; Hu, P.; Zhang, J.; Luo, G. A ROS-responsive, self-immolative and self-reporting hydrogen sulfide donor with multiple biological activities for the treatment of myocardial infarction. Bioact. Mater. 2022, 9, 168–182. [Google Scholar] [CrossRef]
- Panda, A.K.; Keerthi, M.; Sakthivel, R.; Dhawan, U.; Liu, X.; Chung, R.-J. Biocompatible Electrochemical Sensor Based on Platinum-Nickel Alloy Nanoparticles for In Situ Monitoring of Hydrogen Sulfide in Breast Cancer Cells. Nanomaterials 2022, 12, 258. [Google Scholar] [CrossRef]
- Pozzi, G.; Gobbi, G.; Masselli, E.; Carubbi, C.; Presta, V.; Ambrosini, L.; Vitale, M.; Mirandola, P. Buffering Adaptive Immunity by Hydrogen Sulfide. Cells 2022, 11, 325. [Google Scholar] [CrossRef]
- He, W.; Fu, Y.; Zheng, Y.; Wang, X.; Liu, B.; Zeng, J. Diallyl thiosulfinate enhanced the anti-cancer activity of dexamethasone in the side population cells of multiple myeloma by promoting miR-127-3p and deactivating the PI3K/AKT signaling pathway. BMC Cancer 2021, 21, 125. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, L.; Zhang, X.; Deng, Y.; Pan, L.; Li, H.; Shi, X.; Wang, T. A novel cystathionine γ-lyase inhibitor, I194496, inhibits the growth and metastasis of human TNBC via downregulating multiple signaling pathways. Sci. Rep. 2021, 11, 8963. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Schulze, A.; Harris, A.L. How cancer metabolism is tuned for proliferation and vulnerable to disruption. Nature 2012, 491, 364–373. [Google Scholar] [CrossRef] [PubMed]
- Webb, B.A.; Chimenti, M.; Jacobson, M.P.; Barber, D.L. Dysregulated pH: A perfect storm for cancer progression. Nat. Cancer 2011, 11, 671–677. [Google Scholar] [CrossRef] [PubMed]
- Lee, Z.-W.; Teo, X.-Y.; Tay, E.Y.-W.; Tan, C.-H.; Hagen, T.; Moore, P.K.; Deng, L.-W. Utilizing hydrogen sulfide as a novel anti-cancer agent by targeting cancer glycolysis and pH imbalance. J. Cereb. Blood Flow Metab. 2014, 171, 4322–4336. [Google Scholar] [CrossRef]
- Lee, S.W.; Cheng, Y.; Moore, P.K.; Bian, J.-S. Hydrogen sulphide regulates intracellular pH in vascular smooth muscle cells. Biochem. Biophys. Res. Commun. 2007, 358, 1142–1147. [Google Scholar] [CrossRef]
- Lu, M.; Choo, C.H.; Hu, L.-F.; Tan, B.H.; Hu, G.; Bian, J.-S. Hydrogen sulfide regulates intracellular pH in rat primary cultured glia cells. Neurosci. Res. 2010, 66, 92–98. [Google Scholar] [CrossRef]
- Evan, G.I.; Vousden, K.H. Proliferation, cell cycle and apoptosis in cancer. Nature 2001, 411, 342–348. [Google Scholar] [CrossRef]
- Wu, Y.C.; Wang, X.J.; Yu, L.; Chan, F.K.L.; Cheng, A.; Yu, J.; Sung, J.J.Y.; Wu, W.K.K.; Cho, C.H. Hydrogen Sulfide Lowers Proliferation and Induces Protective Autophagy in Colon Epithelial Cells. PLoS ONE 2012, 7, e37572. [Google Scholar] [CrossRef]
- Lu, S.; Gao, Y.; Huang, X.; Wang, X. GYY4137, a hydrogen sulfide (H2) donor, shows potent anti-hepatocellular carcinoma activity through blocking the STAT3 pathway. Int. J. Oncol. 2014, 44, 1259–1267. [Google Scholar] [CrossRef]
- Bertoli, C.; Skotheim, J.M.; de Bruin, R.A.M. Control of cell cycle transcription during G1 and S phases. Nat. Rev. Mol. Cell Biol. 2013, 14, 518–528. [Google Scholar] [CrossRef] [PubMed]
- Lucarini, E.; Micheli, L.; Trallori, E.; Citi, V.; Martelli, A.; Testai, L.; De Nicola, G.R.; Iori, R.; Calderone, V.; Ghelardini, C.; et al. Effect of glucoraphanin and sulforaphane against chemotherapy-induced neuropathic pain: Kv7 potassium channels modulation by H2 S release in vivo. Phytother. Res. 2018, 32, 2226–2234. [Google Scholar] [CrossRef] [PubMed]
- Hong, M.; Tang, X.; He, K. Effect of hydrogen sulfide on human colon cancer SW480 cell proliferation and migration in vitro. Nan Fang Yi Ke Da Xue Xue Bao = J. South. Med. Univ. 2014, 34, 699–703. [Google Scholar]
- Vandiver, M.S.; Paul, B.D.; Xu, R.; Karuppagounder, S.; Rao, F.; Snowman, A.M.; Ko, H.S.; Lee, Y.I.; Dawson, V.L.; Dawson, T.M.; et al. Sulfhydration mediates neuroprotective actions of parkin. Nat. Commun. 2013, 4, 1626. [Google Scholar] [CrossRef]
- Howard, E.W.; Ling, M.-T.; Chua, C.W.; Cheung, H.W.; Wang, X.-H.; Wong, Y.C. Garlic-Derived S-allylmercaptocysteine Is a Novel In vivo Antimetastatic Agent for Androgen-Independent Prostate Cancer. Clin. Cancer Res. 2007, 13, 1847–1856. [Google Scholar] [CrossRef]
- Nian, H.; Delage, B.; Ho, E.; Dashwood, R.H. Modulation of histone deacetylase activity by dietary isothiocyanates and allyl sulfides: Studies with sulforaphane and garlic organosulfur compounds. Environ. Mol. Mutagen. 2009, 50, 213–221. [Google Scholar] [CrossRef]
- Cotter, T.G. Apoptosis and cancer: The genesis of a research field. Nat. Cancer 2009, 9, 501–507. [Google Scholar] [CrossRef]
- Whiteman, M.; Li, L.; Rose, P.; Tan, C.-H.; Parkinson, D.B.; Moore, P.K. The Effect of Hydrogen Sulfide Donors on Lipopolysaccharide-Induced Formation of Inflammatory Mediators in Macrophages. Antioxid. Redox Signal. 2010, 12, 1147–1154. [Google Scholar] [CrossRef]
- Gong, Q.H.; Wang, Q.; Pan, L.L.; Liu, X.H.; Xin, H.; Zhu, Y.Z. S-propargyl-cysteine, a novel hydrogen sulfide-modulated agent, attenuates lipopolysaccharide-induced spatial learning and memory impairment: Involvement of TNF signaling and NF-κB pathway in rats. Brain Behav. Immun. 2011, 25, 110–119. [Google Scholar] [CrossRef]
- Kashfi, K. Anti-Cancer Activity of New Designer Hydrogen Sulfide-Donating Hybrids. Antioxi. Redox Signal. 2014, 20, 831–846. [Google Scholar] [CrossRef]
- Murata, T.; Sato, T.; Kamoda, T.; Moriyama, H.; Kumazawa, Y.; Hanada, N. Differential susceptibility to hydrogen sulfide-induced apoptosis between PHLDA1-overexpressing oral cancer cell lines and oral keratinocytes: Role of PHLDA1 as an apoptosis suppressor. Exp. Cell Res. 2014, 320, 247–257. [Google Scholar] [CrossRef] [PubMed]
- Manna, P.; Jain, S.K. Hydrogen sulfide and L-cysteine increase phosphatidylinositol 3, 4, 5-trisphosphate (PIP3) and glucose utilization by inhibiting phosphatase and tensin homolog (PTEN) protein and activating phosphoinositide 3-kinase (PI3K)/serine/threonine protein kinase (AKT)/protein kinase Cζ/λ (PKCζ/λ) in 3T3l1 adipocytes. J. Biol. Chem. 2011, 286, 39848–39859. [Google Scholar] [PubMed]
- Xu, Y.; Ma, N.; Wei, P.; Zeng, Z.; Meng, J. Expression of hydrogen sulfide synthases and Hh signaling pathway components correlate with the clinicopathological characteristics of papillary thyroid cancer patients. Int. J. Clin. Exp. Pathol. 2018, 11, 1818–1824. [Google Scholar] [PubMed]
- Wang, S.S.; Chen, Y.H.; Chen, N.; Wang, L.J.; Chen, D.X.; Weng, H.L.; Dooley, S.; Ding, H.G. Hydrogen sulfide promotes autophagy of hepatocellular carcinoma cells through the PI3K/Akt/mTOR signaling pathway. Cell Death Dis. 2017, 8, e2688. [Google Scholar] [CrossRef]
- Kundu, S.; Pushpakumar, S.; Khundmiri, S.J.; Sen, U. Hydrogen sulfide mitigates hyperglycemic remodeling via liver kinase B1-adenosine monophosphate-activated protein kinase signaling. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2014, 1843, 2816–2826. [Google Scholar] [CrossRef]
- Zheng, D.; Chen, Z.; Chen, J.; Zhuang, X.; Feng, J.; Li, J. Exogenous hydrogen sulfide exerts proliferation, anti-apoptosis, migration effects and accelerates cell cycle progression in multiple myeloma cells via activating the Akt pathway. Oncol. Rep. 2016, 36, 1909–1916. [Google Scholar] [CrossRef]
- Honda, K.; Hishiki, T.; Yamamoto, S.; Yamamoto, T.; Miura, N.; Kubo, A.; Itoh, M.; Chen, W.-Y.; Takano, M.; Yoshikawa, T.; et al. On-tissue polysulfide visualization by surface-enhanced Raman spectroscopy benefits patients with ovarian cancer to predict post-operative chemosensitivity. Redox Biol. 2021, 41, 101926. [Google Scholar] [CrossRef]
- Kumar Chakraborty, P.; Murphy, B.; Banerjee Mustafi, S.; Dey, A.; Xiong, X.; Rao, G.; Naz, S.; Zhang, M.; Yang, D.; Dhanasekaran, D.N.; et al. Cystathionine β-synthase regulates mitochondrial morphogenesis in ovarian cancer. FASEB J. 2018, 32, 4145–4157. [Google Scholar] [CrossRef]
- Greiner, R.; Pálinkás, Z.; Bäsell, K.; Becher, D.; Antelmann, H.; Nagy, P.; Dick, T.P. Polysulfides link H2 to protein thiol oxidation. Antioxid. Redox Signal. 2013, 19, 1749–1765. [Google Scholar] [CrossRef]
- Mitchell, S.; Vargas, J.; Hoffmann, A. Signaling via the NFκB system. Wiley Interdiscip. Rev. Syst. Biol. Med. 2016, 8, 227–241. [Google Scholar] [CrossRef]
- Wang, Y.; Huang, J.; Chen, W.; Wang, R.; Kao, M.; Pan, Y.; Chan, S.-H.; Tsai, K.-W.; Kung, H.; Lin, K.; et al. Dysregulation of cystathionine γ-lyase promotes prostate cancer progression and metastasis. EMBO Rep. 2019, 20, e45986. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, L.A.; Kaltschmidt, C. NF-kB: A crucial transcription factor for glial and neuronal cell function. Trends Neurosci. 1997, 20, 252–258. [Google Scholar] [CrossRef]
- Calabrese, E.J.; Dhawan, G.; Kapoor, R.; Iavicoli, I.; Calabrese, V. HORMESIS: A Fundamental Concept with Widespread Biological and Biomedical Applications. Gerontology 2015, 62, 530–535. [Google Scholar] [CrossRef] [PubMed]
- Calabrese, V.; Giordano, J.; Signorile, A.; Ontario, M.L.; Castorina, S.; De Pasquale, C.; Eckert, G.; Calabrese, E.J. Major pathogenic mechanisms in vascular dementia: Roles of cellular stress response and hormesis in neuroprotection. J. Neurosci. Res. 2016, 94, 1588–1603. [Google Scholar] [CrossRef]
- Baskar, R.; Bian, J. Hydrogen sulfide gas has cell growth regulatory role. Eur. J. Pharmacol. 2011, 656, 5–9. [Google Scholar] [CrossRef]
- Chen, J.I.; Shen, X.; Pardue, S.; Meram, A.T.; Rajendran, S.; Ghali, G.E.; Kevil, C.G.; Shackelford, R.E. The Ataxia telangiectasia-mutated and Rad3-related protein kinase regulates cellular hydrogen sulfide concentrations. DNA Repair 2019, 73, 55–63. [Google Scholar] [CrossRef]
- Parikh, R.; Appleman, L.; Bauman, J.E.; Sankunny, M.; Lewis, D.W.; Vlad, A.; Gollin, S.M. Upregulation of the ATR-CHEK1 pathway in oral squamous cell carcinomas. Genes Chromosom. Cancer 2014, 53, 25–37. [Google Scholar] [CrossRef]
- Li, C.-C.; Yang, J.-C.; Lu, M.-C.; Lee, C.-L.; Peng, C.-Y.; Hsu, W.-Y.; Dai, Y.-H.; Chang, F.-R.; Zhang, D.-Y.; Wu, W.-J.; et al. ATR-Chk1 signaling inhibition as a therapeutic strategy to enhance cisplatin chemosensitivity in urothelial bladder cancer. Oncotarget 2015, 7, 1947–1959. [Google Scholar] [CrossRef]
- Abdel-Fatah, T.M.; Middleton, F.K.; Arora, A.; Agarwal, D.; Chen, T.; Moseley, P.M.; Perry, C.; Doherty, R.; Chan, S.; Green, A.R.; et al. Untangling the ATR-CHEK1 network for prognostication, prediction and therapeutic target validation in breast cancer. Mol. Oncol. 2015, 9, 569–585. [Google Scholar] [CrossRef]
- Feng, W.; Dean, D.C.; Hornicek, F.J.; Wang, J.; Jia, Y.; Duan, Z.; Shi, H. ATR and p-ATR are emerging prognostic biomarkers and DNA damage response targets in ovarian cancer. Ther. Adv. Med. Oncol. 2020, 12. [Google Scholar] [CrossRef]
- Sundar, R.; Brown, J.; Russo, A.I.; Yap, T.A. Targeting ATR in cancer medicine. Curr. Probl. Cancer 2017, 41, 302–315. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Zhang, J.; Xi, Z.; Li, L.-Y.; Gu, X.; Zhang, Q.-Z.; Yi, L. A new H2S-specific near-infrared fluorescence-enhanced probe that can visualize the H2S level in colorectal cancer cells in mice. Chem. Sci. 2017, 8, 2776–2781. [Google Scholar] [CrossRef] [PubMed]
- Dilek, N.; Papapetropoulos, A.; Toliver-Kinsky, T.; Szabo, C. Hydrogen sulfide: An endogenous regulator of the immune system. Pharmacol. Res. 2020, 161, 105119. [Google Scholar] [CrossRef]
- Kim, M.; Yun, G.; Kim, S. Metabolic Regulation of Ferroptosis in Cancer. Biology 2021, 10, 83. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Huang, Y. Ferroptosis: An iron-dependent cell death form linking metabolism, diseases, immune cell and targeted therapy. Clin. Transl. Oncol. 2021, 24, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Aguirre, J.; Castillo, E.; Kimmel, D. Biologic and pathologic aspects of osteocytes in the setting of medication-related osteonecrosis of the jaw (MRONJ). Bone 2021, 153, 116168. [Google Scholar] [CrossRef] [PubMed]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An Iron-Dependent Form of Nonapoptotic Cell Death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef]
- Kinowaki, Y.; Taguchi, T.; Onishi, I.; Kirimura, S.; Kitagawa, M.; Yamamoto, K. Overview of Ferroptosis and Synthetic Lethality Strategies. Int. J. Mol. Sci. 2021, 22, 9271. [Google Scholar] [CrossRef]
- Mazhar, M.; Din, A.U.; Ali, H.; Yang, G.; Ren, W.; Wang, L.; Fan, X.; Yang, S. Implication of ferroptosis in aging. Cell Death Discov. 2021, 7, 149. [Google Scholar] [CrossRef]
- Li, J.; Cao, F.; Yin, H.L.; Huang, Z.J.; Lin, Z.T.; Mao, N.; Sun, B.; Wang, G. Ferroptosis: Past, present and future. Cell Death Dis. 2020, 11, 88. [Google Scholar] [CrossRef]
- Lu, R.; Jiang, Y.; Lai, X.; Liu, S.; Sun, L.; Zhou, Z.-W. A Shortage of FTH Induces ROS and Sensitizes RAS-Proficient Neuroblastoma N2A Cells to Ferroptosis. Int. J. Mol. Sci. 2021, 22, 8898. [Google Scholar] [CrossRef] [PubMed]
- Yao, F.; Cui, X.; Zhang, Y.; Bei, Z.; Wang, H.; Zhao, D.; Wang, H.; Yang, Y. Iron regulatory protein 1 promotes ferroptosis by sustaining cellular iron homeostasis in melanoma. Oncol. Lett. 2021, 22, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Kimura, H. Hydrogen sulfide (H2) and polysulfide (H2n) signaling: The first 25 years. Biomolecules 2021, 11, 896. [Google Scholar] [CrossRef] [PubMed]
- Tabassum, R.; Jeong, N.Y. Potential for therapeutic use of hydrogen sulfide in oxidative stress-induced neurodegenerative diseases. Int. J. Med. Sci. 2019, 16, 1386–1396. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Bu, D.; Zhu, J.; Yue, T.; Guo, S.; Wang, X.; Pan, Y.; Liu, Y.; Wang, P. Endogenous hydrogen sulfide regulates xCT stability through persulfidation of OTUB1 at cysteine 91 in colon cancer cells. Neoplasia 2021, 23, 461–472. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wang, S.; Xin, Y.; Zhang, J.; Wang, S.; Yang, Z.; Liu, C. Hydrogen sulfide alleviates the anxiety-like and depressive-like behaviors of type 1 diabetic mice via inhibiting inflammation and ferroptosis. Life Sci. 2021, 278, 119551. [Google Scholar] [CrossRef]
- Rodrigues, C.; Percival, S.S. Immunomodulatory Effects of Glutathione, Garlic Derivatives, and Hydrogen Sulfide. Nutrients 2019, 11, 295. [Google Scholar] [CrossRef]
- Kimura, Y.; Goto, Y.-I.; Kimura, H. Hydrogen Sulfide Increases Glutathione Production and Suppresses Oxidative Stress in Mitochondria. Antioxid. Redox Signal. 2010, 12, 1–13. [Google Scholar] [CrossRef]
- Parsanathan, R.; Jain, S.K. Hydrogen sulfide increases glutathione biosynthesis, and glucose uptake and utilisation in C2C12 mouse myotubes. Free. Radic. Res. 2018, 52, 288–303. [Google Scholar] [CrossRef]
- Jin, R.; Yang, R.; Cui, C.; Zhang, H.; Cai, J.; Geng, B.; Chen, Z. Ferroptosis due to Cystathionine γ Lyase/Hydrogen Sulfide Downregulation Under High Hydrostatic Pressure Exacerbates VSMC Dysfunction. Front. Cell Dev. Biol. 2022, 10, 79. [Google Scholar] [CrossRef]
- Wang, Y.; Yu, R.; Wu, L.; Yang, G. Hydrogen sulfide guards myoblasts from ferroptosis by inhibiting ALOX12 acetylation. Cell. Signal. 2021, 78, 109870. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Li, M.; Li, L.; Ma, J.; Yao, C.; Yao, S. Hydrogen sulfide attenuates ferroptosis and stimulates autophagy by blocking mTOR signaling in sepsis-induced acute lung injury. Mol. Immunol. 2022, 141, 318–327. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Cai, H.; Hu, Y.; Liu, F.; Huang, S.; Zhou, Y.; Yu, J.; Xu, J.; Wu, F. A pharmacological probe identifies cystathionine β-synthase as a new negative regulator for ferroptosis. Cell Death Dis. 2018, 9, 1005. [Google Scholar] [CrossRef] [PubMed]
- Pan, X.; Qi, Y.; Du, Z.; He, J.; Yao, S.; Lu, W.; Ding, K.; Zhou, M. Zinc oxide nanosphere for hydrogen sulfide scavenging and ferroptosis of colorectal cancer. J. Nanobiotechnol. 2021, 19, 392. [Google Scholar] [CrossRef]
- Chai, M.; Li, X.; Zhang, Y.; Tang, Y.; Shu, P.; Lin, J.; Shi, K.; Wang, L.; Huang, X. A Nomogram Integrating Ferroptosis- and Immune-Related Biomarkers for Prediction of Overall Survival in Lung Adenocarcinoma. Front. Genet. 2021, 12, 706814. [Google Scholar] [CrossRef]
- Liu, T.; Yang, Q.; Zheng, H.; Jia, H.; He, Y.; Zhang, X.; Zheng, J.; Xi, Y.; Zhang, H.; Sun, R.; et al. Multifaceted roles of a bioengineered nanoreactor in repressing radiation-induced lung injury. Biomaterials 2021, 277, 121103. [Google Scholar] [CrossRef]
- Tsubura, A.; Lai, Y.C.; Kuwata, M.; Uehara, N.; Yoshizawa, K. Anticancer effects of garlic and garlic-derived compounds for breast cancer control. Anti-Cancer Agents Med. Chem. (Former. Curr. Med. Chem. Anti-Cancer Agents) 2011, 11, 249–253. [Google Scholar] [CrossRef]
- Calabrese, V.; Cornelius, C.; Dinkova-Kostova, A.; Calabrese, E.J.; Mattson, M.P. Cellular Stress Responses, The Hormesis Paradigm, and Vitagenes: Novel Targets for Therapeutic Intervention in Neurodegenerative Disorders. Antioxid. Redox Signal. 2010, 13, 1763–1811. [Google Scholar] [CrossRef]
- Mukherjee, S.; Lekli, I.; Ray, D.; Gangopadhyay, H.; Raychaudhuri, U.; Das, D.K. Comparison of the protective effects of steamed and cooked broccolis on ischaemia–reperfusion-induced cardiac injury. Br. J. Nutr. 2010, 103, 815–823. [Google Scholar] [CrossRef]
- Zhao, Y.; Wang, H.; Xian, M. Cysteine-activated hydrogen sulfide (H2) donors. J. Am. Chem. Soc. 2011, 133, 15–17. [Google Scholar] [CrossRef]
- Wang, Q.; Wang, X.-L.; Liu, H.-R.; Rose, P.; Zhu, Y.-Z. Protective Effects of Cysteine Analogues on Acute Myocardial Ischemia: Novel Modulators of Endogenous H2 Production. Antioxid. Redox Signal. 2010, 12, 1155–1165. [Google Scholar] [CrossRef] [PubMed]
- Switzer, C.H.; Cheng, R.Y.S.; Ridnour, L.A.; Murray, M.C.; Tazzari, V.; Sparatore, A.; Del Soldato, P.; Hines, H.B.; Glynn, S.A.; Ambs, S.; et al. Dithiolethiones inhibit NF-κB activity via covalent modification in human estrogen receptor–negative breast cancer. Cancer Res. 2012, 72, 2394–2404. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Moore, P.K. Could hydrogen sulfide be the next blockbuster treatment for inflammatory disease? Expert Rev. Clin. Pharmacol. 2013, 6, 593–595. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Li, L.; Salto-Tellez, M.; Tan, C.H.; Whiteman, M.; Moore, P.K. GYY4137, a novel hydrogen sulfide-releasing molecule, protects against endotoxic shock in the rat. Free Radic. Biol. Med. 2009, 47, 103–113. [Google Scholar] [CrossRef]
- Li, L.; Whiteman, M.; Guan, Y.Y.; Neo, K.L.; Cheng, Y.; Lee, S.W.; Zhao, Y.; Baskar, R.; Tan, C.H.; Moore, P.K. Characterization of a novel, water-soluble hydrogen sulfide-releasing molecule (GYY4137). Circulation 2008, 117, 2351–2360. [Google Scholar]
- Busquet, M.; Calsamiglia, S.; Ferret, A.; Carro, M.D.; Kamel, C. Effect of Garlic Oil and Four of its Compounds on Rumen Microbial Fermentation. J. Dairy Sci. 2005, 88, 4393–4404. [Google Scholar] [CrossRef]
- Lai, K.C.; Hsu, S.C.; Kuo, C.L.; Yang, J.S.; Ma, C.Y.; Lu, H.F.; Tang, N.Y.; Hsia, T.C.; Ho, H.C.; Chung, J.G. Diallyl sulfide, diallyl disulfide, and diallyl trisulfide inhibit migration and invasion in human colon cancer colo 205 cells through the inhibition of matrix metalloproteinase-2, -7, and -9 expressions. Environ. Toxicol. 2013, 28, 479–488. [Google Scholar] [CrossRef]
- Hasegawa, U.; van der Vlies, A.J. Design and Synthesis of Polymeric Hydrogen Sulfide Donors. Bioconjug. Chem. 2014, 25, 1290–1300. [Google Scholar] [CrossRef]
- Wallace, J.L.; Caliendo, G.; Santagada, V.; Cirino, G. Markedly reduced toxicity of a hydrogen sulphide-releasing derivative of naproxen (ATB-346). J. Cereb. Blood Flow Metab. 2010, 159, 1236–1246. [Google Scholar] [CrossRef]
- Cenac, N.; Castro, M.; Desormeaux, C.; Colin, P.; Sie, M.; Ranger, M.; Vergnolle, N. A novel orally administered trimebutine compound (GIC-1001) is anti-nociceptive and features peripheral opioid agonistic activity and Hydrogen Sulphide-releasing capacity in mice. Eur. J. Pain 2016, 20, 723–730. [Google Scholar] [CrossRef]
- Nagpure, B.; Bian, J.-S. Brain, learning, and memory: Role of H 2 S in neurodegenerative diseases. Chem. Biochem. Pharmacol. Hydrog. Sulfide 2015, 230, 193–215. [Google Scholar]
- Levinn, C.M.; Cerda, M.M.; Pluth, M.D. Activatable Small-Molecule Hydrogen Sulfide Donors. Antioxid. Redox Signal. 2020, 32, 96–109. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.; Tazzari, V.; Giustarini, D.; Rossi, R.; Sparatore, A.; Del Soldato, P.; McGeer, E.; McGeer, P.L. Effects of Hydrogen Sulfide-releasing l-DOPA Derivatives on Glial Activation: Potential for treating Parkinson disease. J. Biol. Chem. 2010, 285, 17318–17328. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.; Sparatore, A.; Del Soldato, P.; Mcgeer, E.; McGeer, P.L. Hydrogen sulfide-releasing NSAIDs attenuate neuroinflammation induced by microglial and astrocytic activation. Glia 2010, 58, 103–113. [Google Scholar] [CrossRef] [PubMed]
- Guo, W.; Cheng, Z.-Y.; Zhu, Y.-Z. Hydrogen sulfide and translational medicine. Acta Pharmacol. Sin. 2013, 34, 1284–1291. [Google Scholar] [CrossRef]
- Yang, N.; Liu, Y.; Li, T.; Tuo, Q. Role of Hydrogen Sulfide in Chronic Diseases. DNA Cell Biol. 2020, 39, 187–196. [Google Scholar] [CrossRef]
- Wu, D.; Si, W.; Wang, M.; Lv, S.; Ji, A.; Li, Y. Hydrogen sulfide in cancer: Friend or foe? Nitric Oxide 2015, 50, 38–45. [Google Scholar] [CrossRef]
- Reis, A.K.C.A.; Stern, A.; Monteiro, H.P. S-nitrosothiols and H2 donors: Potential chemo-therapeutic agents in cancer. Redox Biol. 2019, 27, 101190. [Google Scholar] [CrossRef]
- Wu, D.; Li, M.; Tian, W.; Wang, S.; Cui, L.; Li, H.; Wang, H.; Ji, A.; Li, Y. Hydrogen sulfide acts as a double-edged sword in human hepatocellular carcinoma cells through EGFR/ERK/MMP-2 and PTEN/AKT signaling pathways. Sci. Rep. 2017, 7, 5134. [Google Scholar] [CrossRef]
- Citi, V.; Piragine, E.; Pagnotta, E.; Ugolini, L.; Di Cesare Mannelli, L.; Testai, L.; Ghelardini, C.; Lazzeri, L.; Calderone, V.; Martelli, A. Anticancer properties of erucin, an H2-releasing isothiocyanate, on human pancreatic adenocarcinoma cells (AsPC-1). Phytother. Res. 2019, 33, 845–855. [Google Scholar] [CrossRef]
- Lam, T.K.; Gallicchio, L.; Lindsley, K.; Shiels, M.; Hammond, E.; Tao, X.; Chen, L.; Robinson, K.A.; Caulfield, L.E.; Herman, J.G.; et al. Cruciferous Vegetable Consumption and Lung Cancer Risk: A Systematic Review. Cancer Epidemiol. Biomark. Prev. 2009, 18, 184–195. [Google Scholar] [CrossRef] [PubMed]
- Wolf, M.A.; Claudio, P.P. Benzyl Isothiocyanate Inhibits HNSCC Cell Migration and Invasion, and Sensitizes HNSCC Cells to Cisplatin. Nutr. Cancer 2013, 66, 285–294. [Google Scholar] [CrossRef] [PubMed]
- De Gianni, E.; Fimognari, C. Anticancer Mechanism of Sulfur-Containing Compounds. In The Enzymes; Elsevier: Amsterdam, The Netherlands, 2015; pp. 167–192. [Google Scholar]
- Tsai, S.C.; Huang, W.W.; Huang, W.C.; Lu, C.C.; Chiang, J.H.; Peng, S.F.; Chung, J.G.; Lin, Y.H.; Hsu, Y.M.; Amagaya, S.; et al. ERK-modulated intrinsic signaling and G2/M phase arrest contribute to the induction of apoptotic death by allyl isothiocyanate in MDA-MB-468 human breast adenocarcinoma cells. Int. J. Oncol. 2012, 41, 2065–2072. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.L.; Huang, A.C.; Yang, J.S.; Liao, C.L.; Lu, H.F.; Chou, S.T.; Ma, C.Y.; Hsia, T.C.; Ko, Y.C.; Chung, J.G. Benzyl isothiocyanate (BITC) and phenethyl isothiocyanate (PEITC)-mediated generation of reactive oxygen species causes cell cycle arrest and induces apoptosis via activation of caspase-3, mitochondria dysfunction and nitric oxide (NO) in human osteogenic sarcoma U-2 OS cells. J. Orthop. Res. 2011, 29, 1199–1209. [Google Scholar]
- Xiao, D.; Vogel, V.; Singh, S.V. Benzyl isothiocyanate–induced apoptosis in human breast cancer cells is initiated by reactive oxygen species and regulated by Bax and Bak. Mol. Cancer Ther. 2006, 5, 2931–2945. [Google Scholar] [CrossRef]
- Chen, P.Y.; Lin, K.C.; Lin, J.P.; Tang, N.Y.; Yang, J.S.; Lu, K.W.; Chung, J.G. Phenethyl Isothiocyanate (PEITC) inhibits the growth of human oral squamous carcinoma HSC-3 cells through G0/G1 phase arrest and mitochondria-mediated apoptotic cell death. Evid. Based Complement. Altern. Med. 2012, 2012, 718320. [Google Scholar]
- Tripathi, K.; Hussein, U.K.; Anupalli, R.; Barnett, R.; Bachaboina, L.; Scalici, J.; Rocconi, R.P.; Owen, L.B.; Piazza, G.; Palle, K. Allyl isothiocyanate induces replication-associated DNA damage response in NSCLC cells and sensitizes to ionizing radiation. Oncotarget 2015, 6, 5237–5252. [Google Scholar] [CrossRef]
- Hwang, E.-S.; Lee, H.J. Effects of phenylethyl isothiocyanate and its metabolite on cell-cycle arrest and apoptosis in LNCaP human prostate cancer cells. Int. J. Food Sci. Nutr. 2010, 61, 324–336. [Google Scholar] [CrossRef]
- Pappa, G.; Lichtenberg, M.; Iori, R.; Barillari, J.; Bartsch, H.; Gerhäuser, C. Comparison of growth inhibition profiles and mechanisms of apoptosis induction in human colon cancer cell lines by isothiocyanates and indoles from Brassicaceae. Mutat. Res. Mol. Mech. Mutagen. 2006, 599, 76–87. [Google Scholar] [CrossRef]
- Ma, X.; Fang, Y.; Beklemisheva, A.; Dai, W.; Feng, J.; Ahmed, T.; Liu, D.; Chiao, J.W. Phenylhexyl isothiocyanate inhibits histone deacetylases and remodels chromatins to induce growth arrest in human leukemia cells. Int. J. Oncol. 2006, 28, 1287–1293. [Google Scholar] [CrossRef][Green Version]
- Rajendran, P.; Delage, B.; Dashwood, W.M.; Yu, T.W.; Wuth, B.; Williams, D.E.; Ho, E.; Dashwood, R.H. Histone deacetylase turnover and recovery in sulforaphane-treated colon cancer cells: Competing actions of 14-3-3 and Pin1 in HDAC3/SMRT corepressor complex dissociation/reassembly. Mol. Cancer 2011, 10, 68. [Google Scholar] [CrossRef] [PubMed]
- Myzak, M.C.; Karplus, P.A.; Chung, F.L.; Dashwood, R.H. A novel mechanism of chemoprotection by sulforaphane: Inhibition of histone deacetylase. Cancer Res. 2004, 64, 5767–5774. [Google Scholar] [CrossRef] [PubMed]
- Myzak, M.C.; Hardin, K.; Wang, R.; Dashwood, R.H.; Ho, E. Sulforaphane inhibits histone deacetylase activity in BPH-1, LnCaP and PC-3 prostate epithelial cells. Carcinogenesis 2006, 27, 811–819. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.G.; Beklemisheva, A.; Liu, X.M.; Ferrari, A.C.; Feng, J.; Chiao, J.W. Dual action on promoter demethylation and chromatin by an isothiocyanate restored GSTP1 silenced in prostate cancer. Mol. Carcinog. 2007, 46, 24–31. [Google Scholar] [CrossRef]
- Xu, C.; Shen, G.; Chen, C.; Gelinas, C.; Kong, A.N.T. Suppression of NF-κB and NF-κB-regulated gene expression by sulforaphane and PEITC through IκBα, IKK pathway in human prostate cancer PC-3 cells. Oncogene 2005, 24, 4486–4495. [Google Scholar] [CrossRef]
- Boreddy, S.R.; Sahu, R.P.; Srivastava, S.K. Benzyl Isothiocyanate Suppresses Pancreatic Tumor Angiogenesis and Invasion by Inhibiting HIF-α/VEGF/Rho-GTPases: Pivotal Role of STAT-3. PLoS ONE 2011, 6, e25799. [Google Scholar] [CrossRef]
- Lawson, A.P.; Long, M.; Coffey, R.T.; Qian, Y.; Weerapana, E.; El Oualid, F.; Hedstrom, L. Naturally Occurring Isothiocyanates Exert Anticancer Effects by Inhibiting Deubiquitinating Enzymes. Cancer Res. 2015, 75, 5130–5142. [Google Scholar] [CrossRef]
- Mi, L.; Xiao, Z.; Hood, B.L.; Dakshanamurthy, S.; Wang, X.; Govind, S.; Conrads, T.P.; Veenstra, T.D.; Chung, F.-L. Covalent binding to tubulin by isothiocyanates: A mechanism of cell growth arrest and apoptosis. J. Biol. Chem. 2008, 283, 22136–22146. [Google Scholar] [CrossRef]
- Smith, T.K.; Lund, E.K.; Parker, M.L.; Clarke, R.G.; Johnson, I.T. Allyl-isothiocyanate causes mitotic block, loss of cell adhesion and disrupted cytoskeletal structure in HT29 cells. Carcinogenesis 2004, 25, 1409–1415. [Google Scholar] [CrossRef]
- Jackson, S.J.T.; Singletary, K.W. Sulforaphane Inhibits Human MCF-7 Mammary Cancer Cell Mitotic Progression and Tubulin Polymerization. J. Nutr. 2004, 134, 2229–2236. [Google Scholar] [CrossRef]
- Bryant, C.S.; Kumar, S.; Chamala, S.; Shah, J.; Pal, J.; Haider, M.; Seward, S.; Qazi, A.M.; Morris, R.; Semaan, A.; et al. Sulforaphane induces cell cycle arrest by protecting RB-E2F-1 complex in epithelial ovarian cancer cells. Mol. Cancer 2010, 9, 47. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.-S.; Cho, H.-J.; Jeong, Y.-J.; Shin, J.-M.; Park, K.-K.; Park, Y.-Y.; Bae, Y.-S.; Chung, I.-K.; Kim, M.; Kim, C.-H.; et al. Isothiocyanates inhibit the invasion and migration of C6 glioma cells by blocking FAK/JNK-mediated MMP-9 expression. Oncol. Rep. 2015, 34, 2901–2908. [Google Scholar] [CrossRef] [PubMed]
- Tang, L.; Zhang, Y. Dietary Isothiocyanates Inhibit the Growth of Human Bladder Carcinoma Cells. J. Nutr. 2004, 134, 2004–2010. [Google Scholar] [CrossRef] [PubMed]
- Pledgie-Tracy, A.; Sobolewski, M.D.; Davidson, N.E. Sulforaphane induces cell type–specific apoptosis in human breast cancer cell lines. Mol. Cancer Ther. 2007, 6, 1013–1021. [Google Scholar] [CrossRef]
- Xiao, D.; Powolny, A.A.; Singh, S.V. Benzyl Isothiocyanate Targets Mitochondrial Respiratory Chain to Trigger Reactive Oxygen Species-dependent Apoptosis in Human Breast Cancer Cells. J. Biol. Chem. 2008, 283, 30151–30163. [Google Scholar] [CrossRef]
- Lu, Q.; Lin, X.; Feng, J.; Zhao, X.; Gallagher, R.; Lee, M.Y.; Chiao, J.W.; Liu, D. Phenylhexyl isothiocyanate has dual function as histone deacetylase inhibitor and hypomethylating agent and can inhibit myeloma cell growth by targeting critical pathways. J. Hematol. Oncol. 2008, 1, 1–10. [Google Scholar] [CrossRef]
- Chen, H.-E.; Lin, J.-F.; Lin, Y.-C.; Tsai, T.-F.; Chou, K.-Y.; Hwang, T. 44 Allyl isothiocyanate induces reactive oxygen species-mediated autophagy through beclin-1 in human prostate cancer cells. Eur. Urol. Suppl. 2016, 3, e44. [Google Scholar] [CrossRef]
- Kim, J.H.; Xu, C.; Keum, Y.-S.; Reddy, B.; Conney, A.; Kong, A.-N.T. Inhibition of EGFR signaling in human prostate cancer PC-3 cells by combination treatment with β-phenylethyl isothiocyanate and curcumin. Carcinogenesis 2006, 27, 475–482. [Google Scholar] [CrossRef]
- Gamet-Payrastre, L.; Li, P.; Lumeau, S.; Cassar, G.; Dupont, M.A.; Chevolleau, S.; Gasc, N.; Tulliez, J.; Tercé, F. Sulforaphane, a naturally occurring isothiocyanate, induces cell cycle arrest and apoptosis in HT29 human colon cancer cells. Cancer Res. 2000, 60, 1426–1433. [Google Scholar]
- Huang, S.-H.; Wu, L.-W.; Huang, A.-C.; Yu, C.-C.; Lien, J.-C.; Huang, Y.-P.; Yang, J.-S.; Yang, J.-H.; Hsiao, Y.-P.; Wood, W.G.; et al. Benzyl Isothiocyanate (BITC) Induces G2/M Phase Arrest and Apoptosis in Human Melanoma A375.S2 Cells through Reactive Oxygen Species (ROS) and both Mitochondria-Dependent and Death Receptor-Mediated Multiple Signaling Pathways. J. Agric. Food Chem. 2012, 60, 665–675. [Google Scholar] [CrossRef]
- Lai, K.-C.; Hsiao, Y.-T.; Yang, J.-L.; Ma, Y.-S.; Huang, Y.-P.; Chiang, T.-A.; Chung, J.-G. Benzyl isothiocyanate and phenethyl isothiocyanate inhibit murine melanoma B16F10 cell migration and invasion in vitro. Int. J. Oncol. 2017, 51, 832–840. [Google Scholar] [CrossRef] [PubMed]
- Abe, N.; Hou, D.X.; Munemasa, S.; Murata, Y.; Nakamura, Y. Nuclear factor-kappaB sensitizes to benzyl isothiocyanate-induced antiproliferation in p53-deficient colorectal cancer cells. Cell Death Dis. 2014, 5, e1534. [Google Scholar] [CrossRef] [PubMed]
- Kasiappan, R.; Jutooru, I.; Karki, K.; Hedrick, E.; Safe, S.H. Benzyl Isothiocyanate (BITC) Induces Reactive Oxygen Species-dependent Repression of STAT3 Protein by Down-regulation of Specificity Proteins in Pancreatic Cancer. J. Biol. Chem. 2016, 291, 27122–27133. [Google Scholar] [CrossRef] [PubMed]
- Tang, N.-Y.; Chueh, F.-S.; Yu, C.-C.; Liao, C.-L.; Lin, J.-J.; Hsia, T.-C.; Wu, K.-C.; Liu, H.-C.; Lu, K.-W.; Chung, J.-G. Benzyl isothiocyanate alters the gene expression with cell cycle regulation and cell death in human brain glioblastoma GBM 8401 cells. Oncol. Rep. 2016, 35, 2089–2096. [Google Scholar] [CrossRef]
- Kim, S.-H.; Singh, S.V. p53-Independent Apoptosis by Benzyl Isothiocyanate in Human Breast Cancer Cells Is Mediated by Suppression of XIAP Expression. Cancer Prev. Res. 2010, 3, 718–726. [Google Scholar] [CrossRef]
- Zhou, T.; Li, G.; Cao, B.; Liu, L.; Cheng, Q.; Kong, H.; Shan, C.; Huang, X.; Chen, J.; Gao, N. Downregulation of Mcl-1 through inhibition of translation contributes to benzyl isothiocyanate-induced cell cycle arrest and apoptosis in human leukemia cells. Cell Death Dis. 2013, 4, e515. [Google Scholar] [CrossRef]
- Zhu, M.; Li, W.; Guo, J.; Lu, Y.; Dong, X.; Lin, B.; Chen, Y.; Zhang, X.; Li, M. Alpha fetoprotein antagonises benzyl isothiocyanate inhibition of the malignant behaviors of hepatocellular carcinoma cells. Oncotarget 2016, 7, 75749–75762. [Google Scholar] [CrossRef]
- Tsou, M.-F.; Peng, C.-T.; Shih, M.-C.; Yang, J.-S.; Lu, C.-C.; Chiang, J.-H.; Wu, C.-L.; Lin, J.-P.; Lo, C.; Fan, M.-J.; et al. Benzyl isothiocyanate inhibits murine WEHI-3 leukemia cells in vitro and promotes phagocytosis in BALB/c mice in vivo. Leuk. Res. 2009, 33, 1505–1511. [Google Scholar] [CrossRef]
- Han, K.W.W.; Po, W.W.; Sohn, U.D.; Kim, H.-J. Benzyl Isothiocyanate Induces Apoptosis via Reactive Oxygen Species-Initiated Mitochondrial Dysfunction and DR4 and DR5 Death Receptor Activation in Gastric Adenocarcinoma Cells. Biomolecules 2019, 9, 839. [Google Scholar] [CrossRef]
- Sehrawat, A.; Croix, C.S.; Baty, C.J.; Watkins, S.; Tailor, D.; Singh, R.P.; Singh, S.V. Inhibition of mitochondrial fusion is an early and critical event in breast cancer cell apoptosis by dietary chemopreventative benzyl isothiocyanate. Mitochondrion 2016, 30, 67–77. [Google Scholar] [CrossRef]
- Ma, Y.-S.; Lin, J.-J.; Lin, C.-C.; Lien, J.-C.; Peng, S.-F.; Fan, M.-J.; Hsu, F.-T.; Chung, J.-G. Benzyl isothiocyanate inhibits human brain glioblastoma multiforme GBM 8401 cell xenograft tumor in nude mice in vivo. Environ. Toxicol. 2018, 33, 1097–1104. [Google Scholar] [CrossRef] [PubMed]
- Xie, B.; Nagalingam, A.; Kuppusamy, P.; Muniraj, N.; Langford, P.; Győrffy, B.; Saxena, N.K.; Sharma, D. Benzyl Isothiocyanate potentiates p53 signaling and antitumor effects against breast cancer through activation of p53-LKB1 and p73-LKB1 axes. Sci. Rep. 2017, 7, 40070. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Zhuang, J.-X.; Wang, Q.; Zhang, H.-Y.; Yang, P. Inhibitory effect of benzyl isothiocyanate on proliferation in vitro of human glioma cells. Asian Pac. J. Cancer Prev. 2013, 14, 2607–2610. [Google Scholar] [CrossRef] [PubMed]
- Tsai, T.F.; Chen, P.C.; Lin, Y.C.; Chou, K.Y.; Chen, H.E.; Ho, C.Y.; Lin, J.F.; Hwang, T.I.S. Benzyl isothiocyanate promotes miR-99a expression through ERK/AP-1-dependent pathway in bladder cancer cells. Environ. Toxicol. 2020, 35, 47–54. [Google Scholar] [CrossRef]
- Rajan, T.S.; De Nicola, G.R.; Iori, R.; Rollin, P.; Bramanti, P.; Mazzon, E. Anticancer activity of glucomoringin isothiocyanate in human malignant astrocytoma cells. Fitoterapia 2016, 110, 1–7. [Google Scholar] [CrossRef]
- Yeh, Y.-T.; Hsu, Y.-N.; Huang, S.-Y.; Lin, J.-S.; Chen, Z.-F.; Chow, N.-H.; Su, S.-H.; Shyu, H.-W.; Lin, C.-C.; Huang, W.-T.; et al. Benzyl isothiocyanate promotes apoptosis of oral cancer cells via an acute redox stress-mediated DNA damage response. Food Chem. Toxicol. 2016, 97, 336–345. [Google Scholar] [CrossRef]
- Lee, Z.-W.; Deng, L.-W. Role of H2S donors in cancer biology. Chem. Biochem. Pharmacol. Hydrog. Sulfide 2015, 230, 243–265. [Google Scholar]
- Lee, Z.-W.; Teo, X.-Y.; Song, Z.J.; Nin, D.S.; Novera, W.; Choo, B.A.; Dymock, B.W.; Moore, P.K.; Huang, R.Y.-J.; Deng, L.-W. Intracellular Hyper-Acidification Potentiated by Hydrogen Sulfide Mediates Invasive and Therapy Resistant Cancer Cell Death. Front. Pharmacol. 2017, 8, 763. [Google Scholar] [CrossRef]
- Kalkunte, S.; Swamy, N.; Dizon, D.S.; Brard, L. Benzyl isothiocyanate (BITC) induces apoptosis in ovarian cancer cells in vitro. J. Exp. Ther. Oncol. 2006, 5, 287–300. [Google Scholar]
- Lv, M.; Li, Y.; Ji, M.-H.; Zhuang, M.; Tang, J.-H. Inhibition of invasion and epithelial-mesenchymal transition of human breast cancer cells by hydrogen sulfide through decreased phospho-p38 expression. Mol. Med. Rep. 2014, 10, 341–346. [Google Scholar] [CrossRef]
- Kim, E.J.; Hong, J.E.; Eom, S.J.; Lee, J.-Y.; Park, J.H.Y. Oral administration of benzyl-isothiocyanate inhibits solid tumor growth and lung metastasis of 4T1 murine mammary carcinoma cells in BALB/c mice. Breast Cancer Res. Treat. 2010, 130, 61–71. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.-H.; Nagalingam, A.; Saxena, N.K.; Singh, S.V.; Sharma, D. Benzyl isothiocyanate inhibits oncogenic actions of leptin in human breast cancer cells by suppressing activation of signal transducer and activator of transcription 3. Carcinogenesis 2010, 32, 359–367. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Yan, Z.; Deng, X.; Guo, J.; Hu, J.; Yu, Y.; Jiao, F. Anticancer effect of exogenous hydrogen sulfide in cisplatin-resistant A549/DDP cells. Oncol. Rep. 2018, 39, 2969–2977. [Google Scholar] [CrossRef] [PubMed]
- Ye, M.; Yu, M.; Yang, D.; Li, J.; Wang, H.; Chen, F.; Yu, H.; Shen, T.; Zhu, Q.; Zhou, C. Exogenous hydrogen sulfide donor NaHS alleviates nickel-induced epithelial-mesenchymal transition and the migration of A549 cells by regulating TGF-β1/Smad2/Smad3 signaling. Ecotoxicol. Environ. Saf. 2020, 195, 110464. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.-C.; Pan, Z.; Liu, B.-N.; Meng, Z.-W.; Wu, X.; Zhou, Q.-H.; Xu, K. Benzyl isothiocyanate induces protective autophagy in human lung cancer cells through an endoplasmic reticulum stress-mediated mechanism. Acta Pharmacol. Sin. 2017, 38, 539–550. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.-P.; Jiang, Y.-W.; Chen, H.-Y.; Hsiao, Y.-T.; Peng, S.-F.; Chou, Y.-C.; Yang, J.-L.; Hsia, T.-C.; Chung, J.-G. Benzyl Isothiocyanate Induces Apoptotic Cell Death through Mitochondria-dependent Pathway in Gefitinib-resistant NCI-H460 Human Lung Cancer Cells In Vitro. Anticancer Res. 2018, 38, 5165–5176. [Google Scholar] [CrossRef]
- Wu, D.; Li, J.; Zhang, Q.; Tian, W.; Zhong, P.; Liu, Z.; Wang, H.; Wang, H.; Ji, A.; Li, Y. Exogenous Hydrogen Sulfide Regulates the Growth of Human Thyroid Carcinoma Cells. Oxidative Med. Cell. Longev. 2019, 2019, 6927298. [Google Scholar] [CrossRef]
- Xu, S.; Pan, J.; Cheng, X.; Zheng, J.; Wang, X.; Guan, H.; Yu, H.; Bao, J.; Zhang, L. Diallyl trisulfide, a H2 donor, inhibits cell growth of human papillary thyroid carcinoma KTC-1 cells through a positive feedback loop between H2 and cystathionine-gamma-lyase. Phytother. Res. 2020, 34, 1154–1165. [Google Scholar] [CrossRef]
- Zhao, L.; Wang, Y.; Yan, Q.; Lv, W.; Zhang, Y.; He, S. Exogenous hydrogen sulfide exhibits anti-cancer effects though p38 MAPK signaling pathway in C6 glioma cells. Biol. Chem. 2015, 396, 1247–1253. [Google Scholar] [CrossRef]
- Zhen, Y.; Zhang, W.; Liu, C.; He, J.; Lu, Y.; Guo, R.; Feng, J.; Zhang, Y.; Chen, J. Exogenous hydrogen sulfide promotes C6 glioma cell growth through activation of the p38 MAPK/ERK1/2-COX-2 pathways. Oncol. Rep. 2015, 34, 2413–2422. [Google Scholar] [CrossRef]
- Zhu, Y.; Zhang, L.; Zhang, G.-D.; Wang, H.-O.; Liu, M.-Y.; Jiang, Y.; Qi, L.-S.; Ling, Z.; Yang, P. Potential Mechanisms of Benzyl Isothiocyanate Suppression of Invasion and Angiogenesis by the U87MG Human Glioma Cell Line. Asian Pac. J. Cancer Prev. 2014, 15, 8225–8228. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Liu, H.; Chang, J.; Zhao, Z.; Hou, J. Effects of exogenous hydrogen sulfide on the proliferation and invasion of human Bladder cancer cells. J. Cancer Res. Ther. 2017, 13, 829–832. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, V.S.; Xin, W.; Petkov, G.V. Novel mechanism of hydrogen sulfide-induced guinea pig urinary bladder smooth muscle contraction: Role of BK channels and cholinergic neurotransmission. Am. J. Physiol. Physiol. 2015, 309, C107–C116. [Google Scholar] [CrossRef] [PubMed]
- Lazarević, M.; Mazzon, E.; Momčilović, M.; Basile, M.S.; Colletti, G.; Petralia, M.C.; Bramanti, P.; Nicoletti, F.; Miljković, D. The H2S Donor GYY4137 Stimulates Reactive Oxygen Species Generation in BV2 Cells While Suppressing the Secretion of TNF and Nitric Oxide. Molecules 2018, 23, 2966. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Chen, Y.; Han, R.; Wang, S. Benzyl isothiocyanate inhibits invasion and induces apoptosis via reducing S100A4 expression and increases PUMA expression in oral squamous cell carcinoma cells. Braz. J. Med. Biol. Res. 2019, 52, e8409. [Google Scholar] [CrossRef] [PubMed]
- Lei, Y.Y.; Feng, Y.F.; Zeng, B.; Zhang, W.; Xu, Q.; Cheng, F.; Lan, J.; Luo, H.H.; Zou, J.Y.; Chen, Z.G.; et al. Exogenous H2S promotes cancer progression by activating JAK2/STAT3 signaling pathway in esophageal EC109 cells. Int. J. Clin. Exp. Pathol. 2018, 11, 3247. [Google Scholar]
- Boyd, M.R.; Paull, K.D. Some practical considerations and applications of the national cancer institute in vitro anticancer drug discovery screen. Drug Dev. Res. 1995, 34, 91–109. [Google Scholar] [CrossRef]
- Cornell, S. Differentiating among incretin therapies: A multiple-target approach to type 2 diabetes. J. Clin. Pharm. Ther. 2012, 37, 510–524. [Google Scholar] [CrossRef]
- Vanneman, M.; Dranoff, G. Combining immunotherapy and targeted therapies in cancer treatment. Nat. Cancer 2012, 12, 237–251. [Google Scholar] [CrossRef]
- Grothey, A.; Van Cutsem, E.; Sobrero, A.; Siena, S.; Falcone, A.; Ychou, M.; Humblet, Y.; Bouché, O.; Mineur, L.; Barone, C.; et al. Regorafenib monotherapy for previously treated metastatic colorectal cancer (CORRECT): An international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet 2013, 381, 303–312. [Google Scholar] [CrossRef]
- Bekaii-Saab, T.; Kim, R.; Kim, T.W.; O’Connor, J.M.; Strickler, J.H.; Malka, D.; Sartore-Bianchi, A.; Bi, F.; Yamaguchi, K.; Yoshino, T.; et al. Third- or Later-line Therapy for Metastatic Colorectal Cancer: Reviewing Best Practice. Clin. Color. Cancer 2019, 18, e117–e129. [Google Scholar] [CrossRef] [PubMed]
- Shiri, P.; Ramezanpour, S.; Amani, A.M.; Dehaen, W. A patent review on efficient strategies for the total synthesis of pazopanib, regorafenib and lenvatinib as novel anti-angiogenesis receptor tyrosine kinase inhibitors for cancer therapy. Mol. Divers. 2022, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Bekaii-Saab, T.S.; Ou, F.-S.; Ahn, D.H.; Boland, P.M.; Ciombor, K.K.; Heying, E.N.; Dockter, T.J.; Jacobs, N.L.; Pasche, B.C.; Cleary, J.M.; et al. Regorafenib dose-optimisation in patients with refractory metastatic colorectal cancer (ReDOS): A randomised, multicentre, open-label, phase 2 study. Lancet Oncol. 2019, 20, 1070–1082. [Google Scholar] [CrossRef]
- Rosenbaum, S.E.; Wu, S.; Newman, M.A.; West, D.; Kuzel, T.; Lacouture, M.E. Dermatological reactions to the multitargeted tyrosine kinase inhibitor sunitinib. Support. Care Cancer 2008, 16, 557–566. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Gu, J. Hand-foot skin reaction with vascular endothelial growth factor receptor tyrosine kinase inhibitors in cancer patients: A systematic review and meta-analysis. Crit. Rev. Oncol. 2017, 119, 50–58. [Google Scholar] [CrossRef] [PubMed]
- Díaz-González, A.; Sanduzzi-Zamparelli, M.; Sapena, V.; Torres, F.; Llarch, N.; Iserte, G.; Forner, A.; Da Fonseca, L.; Ríos, J.; Bruix, J.; et al. Systematic review with meta-analysis: The critical role of dermatological events in patients with hepatocellular carcinoma treated with sorafenib. Aliment. Pharmacol. Ther. 2019, 49, 482–491. [Google Scholar] [CrossRef]
- McLellan, B.; Ciardiello, F.; Lacouture, M.E.; Segaert, S.; Van Cutsem, E. Regorafenib-associated hand–foot skin reaction: Practical advice on diagnosis, prevention, and management. Ann. Oncol. 2015, 26, 2017–2026. [Google Scholar] [CrossRef]
- Grothey, A.; George, S.; van Cutsem, E.; Blay, J.-Y.; Sobrero, A.; Demetri, G.D. Optimizing Treatment Outcomes with Regorafenib: Personalized Dosing and Other Strategies to Support Patient Care. Oncologist 2014, 19, 669–680. [Google Scholar] [CrossRef][Green Version]
- Bruix, J.; Qin, S.; Merle, P.; Granito, A.; Huang, Y.-H.; Bodoky, G.; Pracht, M.; Yokosuka, O.; Rosmorduc, O.; Breder, V.; et al. Regorafenib for patients with hepatocellular carcinoma who progressed on sorafenib treatment (RESORCE): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2017, 389, 56–66. [Google Scholar] [CrossRef]
- Li, J.; Qin, S.; Xu, R.; Yau, T.C.C.; Ma, B.; Pan, H.; Xu, J.; Bai, Y.; Chi, Y.; Wang, L.; et al. Regorafenib plus best supportive care versus placebo plus best supportive care in Asian patients with previously treated metastatic colorectal cancer (CONCUR): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2015, 16, 619–629. [Google Scholar] [CrossRef]
- Grothey, A.; Huang, L.; Wagner, A.; Van Cutsem, E. Hand-foot skin reaction (HFSR) and outcomes in the phase 3 CORRECT trial of regorafenib for metastatic colorectal cancer (mCRC). J. Clin. Oncol. 2017, 35, 3551. [Google Scholar] [CrossRef]
- Bruix, J.; Merle, P.; Granito, A.; Huang, Y.H.; Bodoky, G.; Yokosuka, O.; Rosmorduc, O.; Breder, V.V.; Gerolami, R.; Masi, G.; et al. Hand-foot skin reaction (HFSR) and overall survival (OS) in the phase 3 RESORCE trial of regorafenib for treatment of hepatocellular carcinoma (HCC) progressing on sorafenib. Am. Soc. Clin. Oncol. 2018. [Google Scholar] [CrossRef]
- Adenis, A.; de la Fouchardiere, C.; Paule, B.; Burtin, P.; Tougeron, D.; Wallet, J.; Dourthe, L.M.; Etienne, P.L.; Mineur, L.; Clisant, S.; et al. Survival, safety, and prognostic factors for outcome with Regorafenib in patients with metastatic colorectal cancer refractory to standard therapies: Results from a multicenter study (REBECCA) nested within a compassionate use program. BMC Cancer 2016, 16, 1–8. [Google Scholar]
- Yamaguchi, K.; Komatsu, Y.; Satoh, T.; Uetake, H.; Yoshino, T.; Nishida, T.; Yamazaki, N.; Takikawa, H.; Morimoto, T.; Chosa, M.; et al. Large-Scale, Prospective Observational Study of Regorafenib in Japanese Patients with Metastatic Colorectal Cancer in a Real-World Clinical Setting. Oncologist 2019, 24, e450–e457. [Google Scholar] [CrossRef] [PubMed]
- Reig, M.; Torres, F.; Rodriguez-Lope, C.; Forner, A.; Llarch, N.; Rimola, J.; Darnell, A.; Ríos, J.; Ayuso, C.; Bruix, J. Early dermatologic adverse events predict better outcome in HCC patients treated with sorafenib. J. Hepatol. 2014, 61, 318–324. [Google Scholar] [CrossRef]
- Rimola, J.; Díaz-González, Á.; Darnell, A.; Varela, M.; Pons, F.; Hernandez-Guerra, M.; Delgado, M.; Castroagudin, J.; Matilla, A.; Sangro, B.; et al. Complete response under sorafenib in patients with hepatocellular carcinoma: Relationship with dermatologic adverse events. Hepatology 2018, 67, 612–622. [Google Scholar] [CrossRef]
- Faruque, L.I.; Lin, M.; Battistella, M.; Wiebe, N.; Reiman, T.; Hemmelgarn, B.; Thomas, C.; Tonelli, M. Systematic Review of the Risk of Adverse Outcomes Associated with Vascular Endothelial Growth Factor Inhibitors for the Treatment of Cancer. PLoS ONE 2014, 9, e101145. [Google Scholar] [CrossRef]
- Mousa, S.A.; Mousa, S.S. Current status of vascular endothelial growth factor inhibition in age-related macular degeneration. BioDrugs 2010, 24, 183–194. [Google Scholar] [CrossRef]
- Schmidinger, M. Understanding and managing toxicities of vascular endothelial growth factor (VEGF) inhibitors. Eur. J. Cancer Suppl. 2013, 11, 172–191. [Google Scholar] [CrossRef]
- Vrdoljak, E.; Ciuleanu, T.E.; Kharkevich, G.; Mardiak, J.; Mego, M.; Padrik, P.; Petruzelka, L.; Purkalne, G.; Shparyk, Y.; Škrbinc, B.; et al. Optimizing treatment for patients with metastatic renal cell carcinoma in the central and Eastern European region. Expert Opin. Pharmacother. 2012, 13, 159–174. [Google Scholar] [CrossRef]
- Schutz, F.A.; Je, Y.; Richards, C.J.; Choueiri, T.K. Meta-Analysis of Randomized Controlled Trials for the Incidence and Risk of Treatment-Related Mortality in Patients with Cancer Treated with Vascular Endothelial Growth Factor Tyrosine Kinase Inhibitors. J. Clin. Oncol. 2012, 30, 871–877. [Google Scholar] [CrossRef] [PubMed]
- Crawford, E.D.; Schally, A.V.; Pinthus, J.H.; Block, N.L.; Rick, F.G.; Garnick, M.B.; Eckel, R.H.; Keane, T.E.; Shore, N.D.; Dahdal, D.N.; et al. The potential role of follicle-stimulating hormone in the cardiovascular, metabolic, skeletal, and cognitive effects associated with androgen deprivation therapy. Urol. Oncol. Semin. Orig. Investig. 2017, 35, 183–191. [Google Scholar] [CrossRef] [PubMed]
- Patel, P.; Srinivas, S. Toxicities of targeted agents in advanced renal cell carcinoma. Curr. Clin. Pharmacol. 2011, 6, 181–188. [Google Scholar] [CrossRef]
- Svoboda, M.; Poprach, A.; Dobes, S.; Kiss, I.; Vyzula, R. Cardiac Toxicity of Targeted Therapies Used in the Treatment for Solid Tumours: A Review. Cardiovasc. Toxicol. 2012, 12, 191–207. [Google Scholar] [CrossRef] [PubMed]
- Dickler, M.N.; Rugo, H.S.; Eberle, C.A.; Brogi, E.; Caravelli, J.F.; Panageas, K.S.; Boyd, J.; Yeh, B.; Lake, D.E.; Dang, C.T.; et al. A Phase II Trial of Erlotinib in Combination with Bevacizumab in Patients with Metastatic Breast Cancer. Clin. Cancer Res. 2008, 14, 7878–7883. [Google Scholar] [CrossRef]
- Rees, M.L.; Khakoo, A.Y. Molecular Mechanisms of Hypertension and Heart Failure Due to Antiangiogenic Cancer Therapies. Hear. Fail. Clin. 2011, 7, 299–311. [Google Scholar] [CrossRef] [PubMed]
- Qi, W.-X.; Lin, F.; Sun, Y.-J.; Tang, L.-N.; He, A.-N.; Yao, Y.; Shen, Z. Incidence and risk of hypertension with pazopanib in patients with cancer: A meta-analysis. Cancer Chemother. Pharmacol. 2012, 71, 431–439. [Google Scholar] [CrossRef]
- Ghatalia, P.; Je, Y.; Kaymakcalan, M.; Sonpavde, G.; Choueiri, T.K. QTc interval prolongation with vascular endothelial growth factor receptor tyrosine kinase inhibitors. Br. J. Cancer 2015, 112, 296–305. [Google Scholar] [CrossRef]
- De Marinis, F.; Bria, E.; Baas, P.; Tiseo, M.; Camerini, A.; Favaretto, A.G.; Gridelli, C. Treatment of Unfit Patients with Advanced Non–Small-Cell Lung Cancer: Definition Criteria According an Expert Panel. Clin. Lung Cancer 2015, 16, 399–405. [Google Scholar] [CrossRef]
- Teo, Y.L.; Ho, H.K.; Chan, A. Risk of tyrosine kinase inhibitors-induced hepatotoxicity in cancer patients: A meta-analysis. Cancer Treat. Rev. 2013, 39, 199–206. [Google Scholar] [CrossRef]
- Xu, C.-F.; Xue, Z.; Bing, N.; King, K.S.; McCann, L.A.; de Souza, P.L.; Goodman, V.L.; Spraggs, C.F.; Mooser, V.E.; Pandite, L.N. Concomitant use of pazopanib and simvastatin increases the risk of transaminase elevations in patients with cancer. Ann. Oncol. 2012, 23, 2470–2471. [Google Scholar] [CrossRef] [PubMed]
- Abramson, R.G.; Abramson, V.G.; Chan, E.; Horn, L.; Keedy, V.L.; Pao, W.; Sosman, J.A. Complications of Targeted Drug Therapies for Solid Malignancies: Manifestations and Mechanisms. Am. J. Roentgenol. 2013, 200, 475–483. [Google Scholar] [CrossRef] [PubMed]
- Kandula, P.; Agarwal, R. Proteinuria and hypertension with tyrosine kinase inhibitors. Kidney Int. 2011, 80, 1271–1277. [Google Scholar] [CrossRef] [PubMed]
- Escudier, B.; Szczylik, C.; Porta, C.; Gore, M. Treatment selection in metastatic renal cell carcinoma: Expert consensus. Nat. Rev. Clin. Oncol. 2012, 9, 327–337. [Google Scholar] [CrossRef]
- Vogelzang, N.J.; Hackshaw, M.D.; Hutson, T.E.; Bhowmik, D.; Yap, M.; Rembert, D.; Jonasch, E. First-Line and Sequential Use of Pazopanib Followed by Mammalian Target of Rapamycin Inhibitor Therapy Among Patients with Advanced Renal Cell Carcinoma in a US Community Oncology Setting. Clin. Genitourin. Cancer 2015, 13, 210–217. [Google Scholar] [CrossRef]
- Ward, J.E.; Karrison, T.; Chatta, G.; Hussain, M.; Shevrin, D.; Szmulewitz, R.Z.; O’Donnell, P.H.; Stadler, W.M.; Posadas, E.M. A randomized, phase II study of pazopanib in castrate-sensitive prostate cancer: A University of Chicago Phase II Consortium/Department of Defense Prostate Cancer Clinical Trials Consortium study. Prostate Cancer Prostatic Dis. 2012, 15, 87–92. [Google Scholar] [CrossRef]
- Cella, D.; Jensen, S.E.; Hahn, E.A.; Beaumont, J.; Korytowsky, B.; Bhattacharyya, H.; Motzer, R. Fatigue in patients with advanced renal cell carcinoma receiving sunitinib on an intermittent versus continuous dosing schedule in a randomized phase II trial. Cancer Med. 2014, 3, 1353–1358. [Google Scholar] [CrossRef]
- Santoni, M.; Conti, A.; Massari, F.; Arnaldi, G.; Iacovelli, R.; Rizzo, M.; De Giorgi, U.; Trementino, L.; Procopio, G.; Tortora, G.; et al. Treatment-related fatigue with sorafenib, sunitinib and pazopanib in patients with advanced solid tumors: An up-to-date review and meta-analysis of clinical trials. Int. J. Cancer 2015, 136, 1–10. [Google Scholar] [CrossRef]
- Diéras, V.; Bachelot, T.; Campone, M.; Isambert, N.; Joly, F.; Le Tourneau, C.; Cassier, P.; Bompas, E.; Fumoleau, P.; Noal, S.; et al. A Phase I, Dose-Escalation Trial of Pazopanib in Combination with Cisplatin in Patients with Advanced Solid Tumors: A UNICANCER Study. Oncol. Ther. 2016, 4, 211–223. [Google Scholar] [CrossRef]
- Hamberg, P.; Mathijssen, R.H.J.; De Bruijn, P.; Leonowens, C.; Van Der Biessen, D.; Eskens, F.A.L.M.; Sleijfer, S.; Verweij, J.; De Jonge, M.J.A. Impact of pazopanib on docetaxel exposure: Results of a phase I combination study with two different docetaxel schedules. Cancer Chemother. Pharmacol. 2015, 75, 365–371. [Google Scholar] [CrossRef]
- Matsui, J.; Yamamoto, Y.; Funahashi, Y.; Tsuruoka, A.; Watanabe, T.; Wakabayashi, T.; Uenaka, T.; Asada, M. E7080, a novel inhibitor that targets multiple kinases, has potent antitumor activities against stem cell factor producing human small cell lung cancer H146, based on angiogenesis inhibition. Int. J. Cancer 2008, 122, 664–671. [Google Scholar] [CrossRef] [PubMed]
- Matsui, J.; Funahashi, Y.; Uenaka, T.; Watanabe, T.; Tsuruoka, A.; Asada, M. Multi-Kinase Inhibitor E7080 Suppresses Lymph Node and Lung Metastases of Human Mammary Breast Tumor MDA-MB-231 via Inhibition of Vascular Endothelial Growth Factor-Receptor (VEGF-R) 2 and VEGF-R3 Kinase. Clin. Cancer Res. 2008, 14, 5459–5465. [Google Scholar] [CrossRef] [PubMed]
- Ikuta, K.; Yano, S.; Trung, V.T.; Hanibuchi, M.; Goto, H.; Li, Q.; Wang, W.; Yamada, T.; Ogino, H.; Kakiuchi, S.; et al. E7080, a Multi-Tyrosine Kinase Inhibitor, Suppresses the Progression of Malignant Pleural Mesothelioma with Different Proangiogenic Cytokine Production Profiles. Clin. Cancer Res. 2009, 15, 7229–7237. [Google Scholar] [CrossRef] [PubMed]
- Bruheim, S.; Kristian, A.; Uenaka, T.; Suo, Z.; Tsuruoka, A.; Nesland, J.M.; Fodstad, Ø. Antitumour activity of oral E7080, a novel inhibitor of multiple tyrosine kinases, in human sarcoma xenografts. Int. J. Cancer 2011, 129, 742–750. [Google Scholar] [CrossRef] [PubMed]
- Shumaker, R.; Aluri, J.; Fan, J.; Martinez, G.; Ren, M.; Chen, K. Evaluation of the effects of formulation and food on the pharmacokinetics of lenvatinib (E7080) in healthy volunteers. Int. J. Clin. Pharmacol. Ther. 2014, 52, 284–291. [Google Scholar] [CrossRef]
- Gupta, A.; Jarząb, B.; Capdevila, J.; Shumaker, R.; Hussein, Z. Population pharmacokinetic analysis of lenvatinib in healthy subjects and patients with cancer. Br. J. Clin. Pharmacol. 2016, 81, 1124–1133. [Google Scholar] [CrossRef]
- Nakamichi, S.; Nokihara, H.; Yamamoto, N.; Yamada, Y.; Honda, K.; Tamura, Y.; Wakui, H.; Sasaki, T.; Yusa, W.; Fujino, K.; et al. A phase 1 study of lenvatinib, multiple receptor tyrosine kinase inhibitor, in Japanese patients with advanced solid tumors. Cancer Chemother. Pharmacol. 2015, 76, 1153–1161. [Google Scholar] [CrossRef]
- Boss, D.S.; Glen, H.; Beijnen, J.H.; Keesen, M.; Morrison, R.; Tait, B.; Copalu, W.; Mazur, A.; Wanders, J.; O’Brien, J.P.; et al. A phase I study of E7080, a multitargeted tyrosine kinase inhibitor, in patients with advanced solid tumours. Br. J. Cancer 2012, 106, 1598–1604. [Google Scholar] [CrossRef]
- Molina, A.M.; Hutson, T.E.; Larkin, J.; Gold, A.M.; Wood, K.; Carter, D.; Motzer, R.; Michaelson, M.D. A phase 1b clinical trial of the multi-targeted tyrosine kinase inhibitor lenvatinib (E7080) in combination with everolimus for treatment of metastatic renal cell carcinoma (RCC). Cancer Chemother. Pharmacol. 2014, 73, 181–189. [Google Scholar] [CrossRef]
- Jasim, S.; Iniguez-Ariza, N.M.; Hilger, C.R.; Chintakuntlawar, A.V.; Ryder, M.M.; Morris, J.C.; Bible, K.C. Optimizing Lenvatinib Therapy in Patients with Metastatic Radioactive Iodine-resistant Differentiated Thyroid Cancers. Endocr. Pract. 2017, 23, 1254–1261. [Google Scholar] [CrossRef]
- Tahara, M.; Brose, M.S.; Wirth, L.J.; Suzuki, T.; Miyagishi, H.; Fujino, K.; Dutcus, C.E.; Gianoukakis, A. Impact of dose interruption on the efficacy of lenvatinib in a phase 3 study in patients with radioiodine-refractory differentiated thyroid cancer. Eur. J. Cancer 2019, 106, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, H.; Iwasaki, H.; Takasaki, H.; Suganuma, N.; Sakai, R.; Masudo, K.; Nakayama, H.; Rino, Y.; Masuda, M. Efficacy and tolerability of initial low-dose lenvatinib to treat differentiated thyroid cancer. Medicine 2019, 98, e14774. [Google Scholar] [CrossRef] [PubMed]
- Arai, N.; Sasaki, H.; Tamura, R.; Ohara, K.; Yoshida, K. Unusual Magnetic Resonance Imaging Findings of a Glioblastoma Arising During Treatment with Lenvatinib for Thyroid Cancer. World Neurosurg. 2017, 107, 1047.e9–1047.e15. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Yamada, T.; Arai, S.; Fukuda, K.; Taniguchi, H.; Tanimoto, A.; Nishiyama, A.; Takeuchi, S.; Yamashita, K.; Ohtsubo, K.; et al. Distribution and Activity of Lenvatinib in Brain Tumor Models of Human Anaplastic Thyroid Cancer Cells in Severe Combined Immune Deficient Mice. Mol. Cancer Ther. 2019, 18, 947–956. [Google Scholar] [CrossRef] [PubMed]
- Meng, G.; Wang, J.; Xiao, Y.; Bai, W.; Xie, L.; Shan, L.; Moore, P.K.; Ji, Y. GYY4137 protects against myocardial ischemia and reperfusion injury by attenuating oxidative stress and apoptosis in rats. J. Biomed. Res. 2015, 29, 203–213. [Google Scholar] [CrossRef]
- Meng, G.; Zhu, J.; Xiao, Y.; Huang, Z.; Zhang, Y.; Tang, X.; Xie, L.; Chen, Y.; Shao, Y.; Ferro, A.; et al. Hydrogen Sulfide Donor GYY4137 Protects against Myocardial Fibrosis. Oxidative Med. Cell. Longev. 2015, 2015, 1–14. [Google Scholar] [CrossRef]
- Xie, Z.-Z.; Liu, Y.; Bian, J.-S. Hydrogen Sulfide and Cellular Redox Homeostasis. Oxidative Med. Cell. Longev. 2016, 2016, 6043038. [Google Scholar] [CrossRef]
- Giustarini, D.; Del Soldato, P.; Sparatore, A.; Rossi, R. Modulation of thiol homeostasis induced by H2-releasing aspirin. Free Radic. Biol. Med. 2010, 48, 1263–1272. [Google Scholar] [CrossRef]
- Cao, X.; Bian, J.-S. The Role of Hydrogen Sulfide in Renal System. Front. Pharmacol. 2016, 7, 385. [Google Scholar] [CrossRef]
- Trédan, O.; Galmarini, C.M.; Patel, K.; Tannock, I.F. Drug Resistance and the Solid Tumor Microenvironment. J. Natl. Cancer Inst. 2007, 99, 1441–1454. [Google Scholar] [CrossRef]
- Kumar, S.; Huang, J.; Cushnir, J.R.; Španěl, P.; Smith, D.; Hanna, G.B. Selected Ion Flow Tube-MS Analysis of Headspace Vapor from Gastric Content for the Diagnosis of Gastro-Esophageal Cancer. Anal. Chem. 2012, 84, 9550–9557. [Google Scholar] [CrossRef] [PubMed]
- Bhatt, A.; Parsi, M.A.; Stevens, T.; Gabbard, S.; Kumaravel, A.; Jang, S.; Grove, D.; Lopez, R.; Murthy, S.; Vargo, J.J.; et al. Volatile organic compounds in plasma for the diagnosis of esophageal adenocarcinoma: A pilot study. Gastrointest. Endosc. 2015, 84, 597–603. [Google Scholar] [CrossRef] [PubMed]
- Yamagishi, K.; Onuma, K.; Chiba, Y.; Yagi, S.; Aoki, S.; Sato, T.; Sugawara, Y.; Hosoya, N.; Saeki, Y.; Takahashi, M.; et al. Generation of gaseous sulfur-containing compounds in tumour tissue and suppression of gas diffusion as an antitumour treatment. Gut 2012, 61, 554–561. [Google Scholar] [CrossRef] [PubMed]
- Chwatko, G.; Forma, E.; Wilkosz, J.; Głowacki, R.; Jóźwiak, P.; Różański, W.; Bryś, M.; Krześlak, A. Thiosulfate in urine as a facilitator in the diagnosis of prostate cancer for patients with prostate-specific antigen less or equal 10 ng/mL. Clin. Chem. Lab. Med. (CCLM) 2013, 51, 1825–1831. [Google Scholar] [CrossRef] [PubMed]
- Stabler, S.; Koyama, T.; Zhao, Z.; Martinez-Ferrer, M.; Allen, R.H.; Luka, Z.; Loukachevitch, L.V.; Clark, P.E.; Wagner, C.; Bhowmick, N.A. Serum Methionine Metabolites Are Risk Factors for Metastatic Prostate Cancer Progression. PLoS ONE 2011, 6, e22486. [Google Scholar] [CrossRef]
- Stephan, C.; Jung, K.; Miller, K.; Ralla, B. New biomarkers in serum and urine for detection of prostate cancer. Aktuelle Urol. 2015, 46, 129–143. [Google Scholar]
- Hasan, T.; Arora, R.; Bansal, A.; Bhattacharya, R.; Sharma, G.S.; Singh, L.R. Disturbed homocysteine metabolism is associated with cancer. Exp. Mol. Med. 2019, 51, 1–13. [Google Scholar] [CrossRef]
- Zhang, C.; Zhang, Q.-Z.; Zhang, K.; Li, L.-Y.; Pluth, M.D.; Yi, L.; Xi, Z. Dual-biomarker-triggered fluorescence probes for differentiating cancer cells and revealing synergistic antioxidant effects under oxidative stress. Chem. Sci. 2019, 10, 1945–1952. [Google Scholar] [CrossRef]






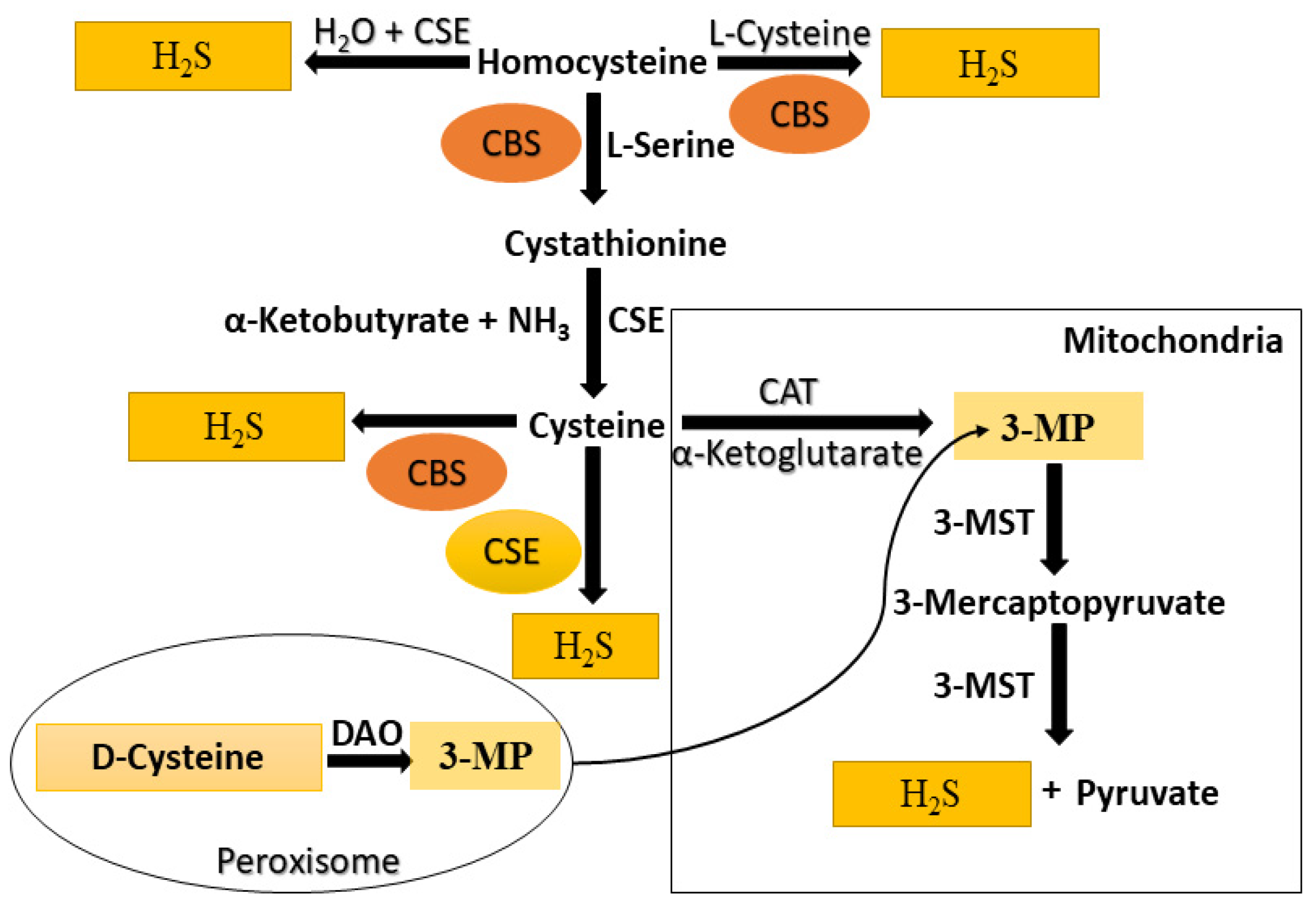{kind=link}
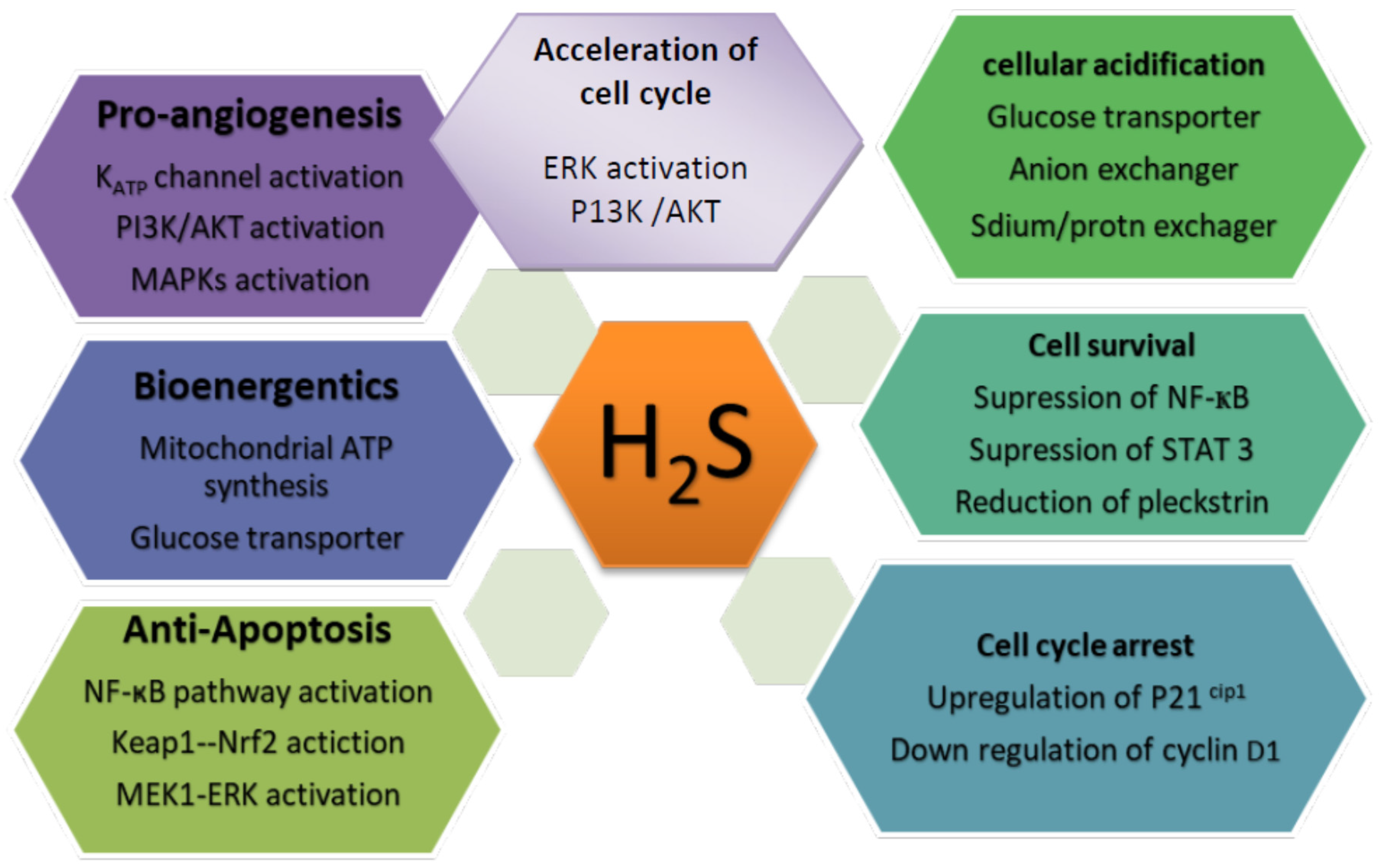{kind=link}
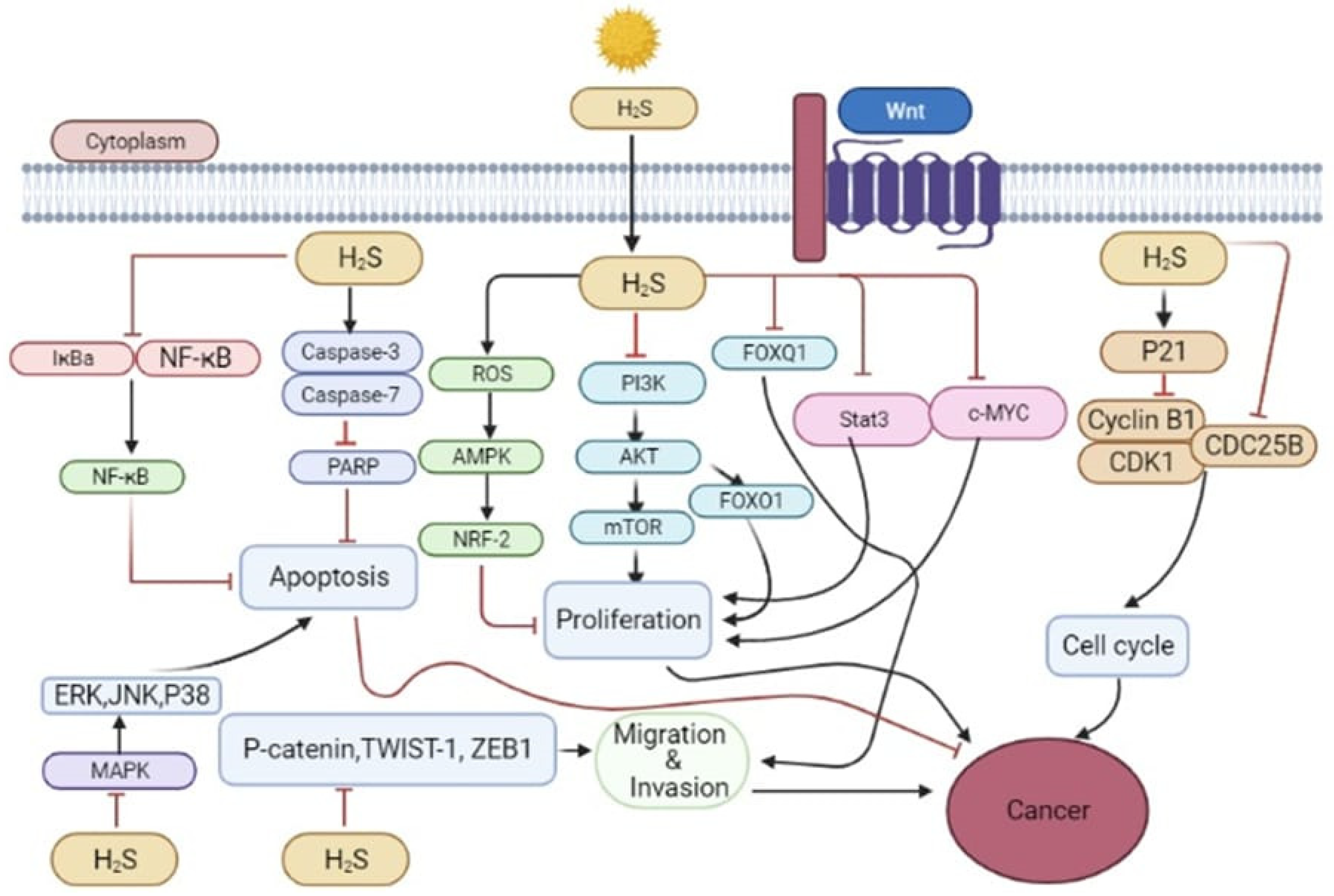{kind=link}
{kind=link}
{kind=link}
| S/No. | Cancer Types | Cell Lines | H2S Producing Enzyme | ||
|---|---|---|---|---|---|
| CSE | CBS | 3-MST | |||
| 1 | Melanoma | A375, WM35, SK-Mel-5, Sk-Mel-28, PES 43 | ↑ | NT | ↑ |
| 2 | Colon cancer | HCT116, HT29 | ↑ | ↑ | ↑ |
| 3 | Prostate cancer | LNCaP, PC3, LNCaP-B | ↑ | ↑ | NT |
| 4 | Gastric cancer | SGC-7901 | ↑ | ↑ | NT |
| 5 | Ovarian | OV202, SKOV3, A2780, OVCAR3, OVCAR4, OVCAR5 | NC | ↑ | NT |
| 6 | Breast | Hs578T, MCF7, MDA-MB-428 | ↑ | ↑ | NT |
| 7 | Renal | RCC4 | ↑ | ↑ | ↑ |
| 8 | Thyroid | TPC1, TT, ARO | ↑ | ↑ | NC |
| 9 | Gliomas | C6, U87MG | NT | NT | ↑ |
| 10 | Hepatocellular Carcinoma | HepG2, PLC/PRF/5 | ↑ | ↑ | NT |
| 11 | Urothelial carcinoma | 5637, EJ, UM-UC-3 | ↑ | ↑ | ↑ |
| 12 | Astrocytoma | U373 | NT | NT | ↑ |
| 13 | Neuroblastoma | SH-SY5Y | NT | NT | ↑ |
| 14 | Oral squamous cell carcinoma | OSCC | ↑ | ↑ | ↑ |
| 15 | Leukaemia | HL-60, MV4-11 | NT | ↑ | NT |
| 16 | Biliary tract carcinoma | EDI-1, TFK-1, HUCCT-1, SNU308, GB-D1, GB-H3 | NT | ↑ | NT |
| S/No | Cancer Types | Cell Lines | H2S Donors | Effects on Cancer | References |
|---|---|---|---|---|---|
| 1 | Melanoma | NCI-H929 | NaHS | Promotion | [187] |
| SKMel 5, SKMel 28 B16F10 A375 S2 | GYY4137 BITC | Promotion Inhibition | [89] [282,283] | ||
| 2 | Colon cancer | HCT 116 SW480, | NaHS | Promotion Inhibition | [104] [174] |
| HCT 116 | GYY4137 | Inhibition | [8] | ||
| BITC | Inhibition | [284] | |||
| 3 | Prostate cancer | PC-3 | NaHS | Inhibition | [107,109] |
| PC-3, Rv1, DU145 | BITC | Inhibition | [285] | ||
| LnCaP, DU145 | GYY4137 | Inhibition | [300] | ||
| 4 | Gastric cancer | SGC 7901 | NaHS | Inhibition | [111] |
| AGS | Inhibition | [291] | |||
| 5 | Ovarian | A2780, HeyA8, PEA1, PEA2 | GYY4137 | Inhibition | [300] |
| OC | BITC | Inhibition | [301] | ||
| 6 | Breast | MCF-7 | NaHS | Inhibition | [8,302] |
| MCF-7, MDA-MB-231 | GYY4137 | Inhibition | [8,300] | ||
| MCF-7, MDA-MB-231 | BITC | Inhibition | [303,304] | ||
| 7 | Lung | A549 | NaHS | Inhibition | [305,306] |
| IMR90, WI-38, A549, H1299 | GYY4137 | Inhibition | [8,300] | ||
| A549, H661, NCI-H460/G | BITC | Inhibition | [307,308] | ||
| 8 | Thyroid | TPC-1 ARO KTC-1 | NaHS NaHS | Promotion Inhibition Inhibition | [309] [310] |
| KTC-1 | GYY4137 | Inhibition | [310] | ||
| 9 | Gliomas | C6 | NaHS | Promotion/Inhibition | [311,312] |
| U87MG | BITC | Inhibition | [313] | ||
| 10 | Hepatocellular Carcinoma | HepG2, HLE PLC/PRF/5, SMMC-7721 | NaHS | Inhibition Promotion | [8,185] [6] |
| HepG2 | GYY4137 | Inhibition | [8] | ||
| Bel 7402,HLE | BITC | Inhibition | [289] | ||
| 11 | Urothelial carcinoma | EJ | NaHS | Promotion | [314] |
| DSM cell | GYY4137 | Promotion | [315] | ||
| 5637,T24 | BITC | Promotion | [296] | ||
| 12 | Astrocytoma | U373 | NaHS | Inhibition | [244] |
| BV2 Cell | GYY4137 | Inhibition | [316] | ||
| CCF-STTG1 | BITC | Inhibition | [297] | ||
| 13 | Neuroblastoma | SH-SY5Y | NaHS | Inhibition | [244] |
| 14 | Oral squamous cell carcinoma | Cal-27, WSU-HN6 | NaHS | Promotion | [12,94] |
| OG2 | BITC | Inhibition | [298] | ||
| SCC9 | BITC | Inhibition | [317] | ||
| 15 | Leukemia | MV4-11 | NaHS | Inhibition | [244] |
| HL-60, MV4-11 | GYY4137 | Inhibition | [8] | ||
| WEHI-3 | BITC | Inhibition | [290] | ||
| 16 | Esophageal carcinoma | EC-109 | NaHS | Promotion | [318] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khattak, S.; Rauf, M.A.; Khan, N.H.; Zhang, Q.-Q.; Chen, H.-J.; Muhammad, P.; Ansari, M.A.; Alomary, M.N.; Jahangir, M.; Zhang, C.-Y.; et al. Hydrogen Sulfide Biology and Its Role in Cancer. Molecules 2022, 27, 3389. https://doi.org/10.3390/molecules27113389
Khattak S, Rauf MA, Khan NH, Zhang Q-Q, Chen H-J, Muhammad P, Ansari MA, Alomary MN, Jahangir M, Zhang C-Y, et al. Hydrogen Sulfide Biology and Its Role in Cancer. Molecules. 2022; 27(11):3389. https://doi.org/10.3390/molecules27113389
Chicago/Turabian StyleKhattak, Saadullah, Mohd Ahmar Rauf, Nazeer Hussain Khan, Qian-Qian Zhang, Hao-Jie Chen, Pir Muhammad, Mohammad Azam Ansari, Mohammad N. Alomary, Muhammad Jahangir, Chun-Yang Zhang, and et al. 2022. "Hydrogen Sulfide Biology and Its Role in Cancer" Molecules 27, no. 11: 3389. https://doi.org/10.3390/molecules27113389
APA StyleKhattak, S., Rauf, M. A., Khan, N. H., Zhang, Q.-Q., Chen, H.-J., Muhammad, P., Ansari, M. A., Alomary, M. N., Jahangir, M., Zhang, C.-Y., Ji, X.-Y., & Wu, D.-D. (2022). Hydrogen Sulfide Biology and Its Role in Cancer. Molecules, 27(11), 3389. https://doi.org/10.3390/molecules27113389