
Structural Investigation of the Interaction Mechanism between Chlorogenic Acid and AMPA Receptor via In Silico Approaches

Abstract
:1. Introduction
2. Materials and Methods
2.1. Reagents and Cell
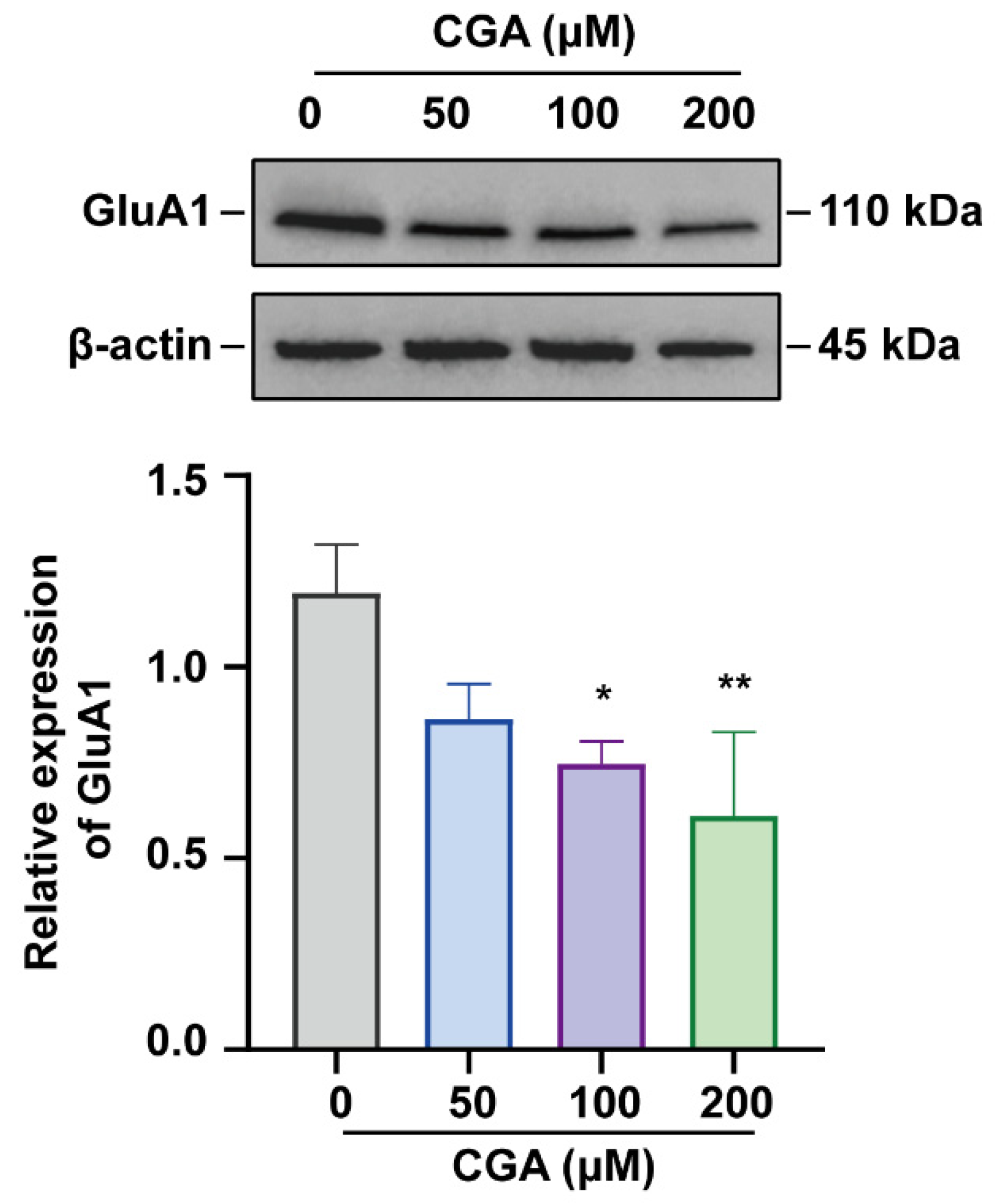
2.2. Western Blot Analysis
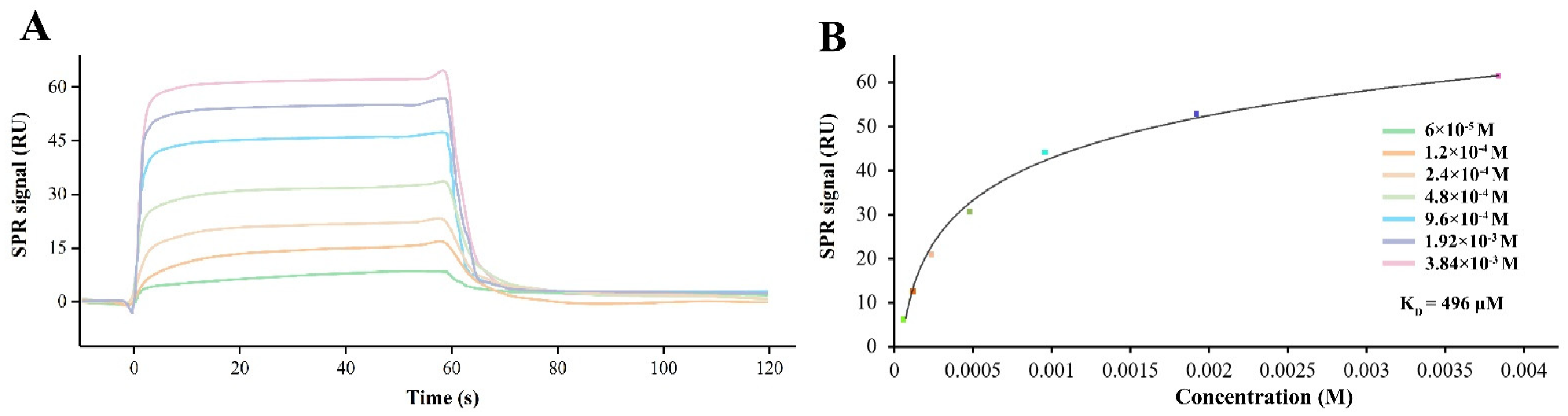
2.3. SPR Assay
2.4. Molecular Docking
2.4.1. Structure Preparation
2.4.2. Molecular Docking Simulation of GluA1 and CGA
2.5. MD Simulation
2.6. MMPBSA Binding Free Energy Calculation and Decomposition
2.7. Statistical Analysis
3. Results
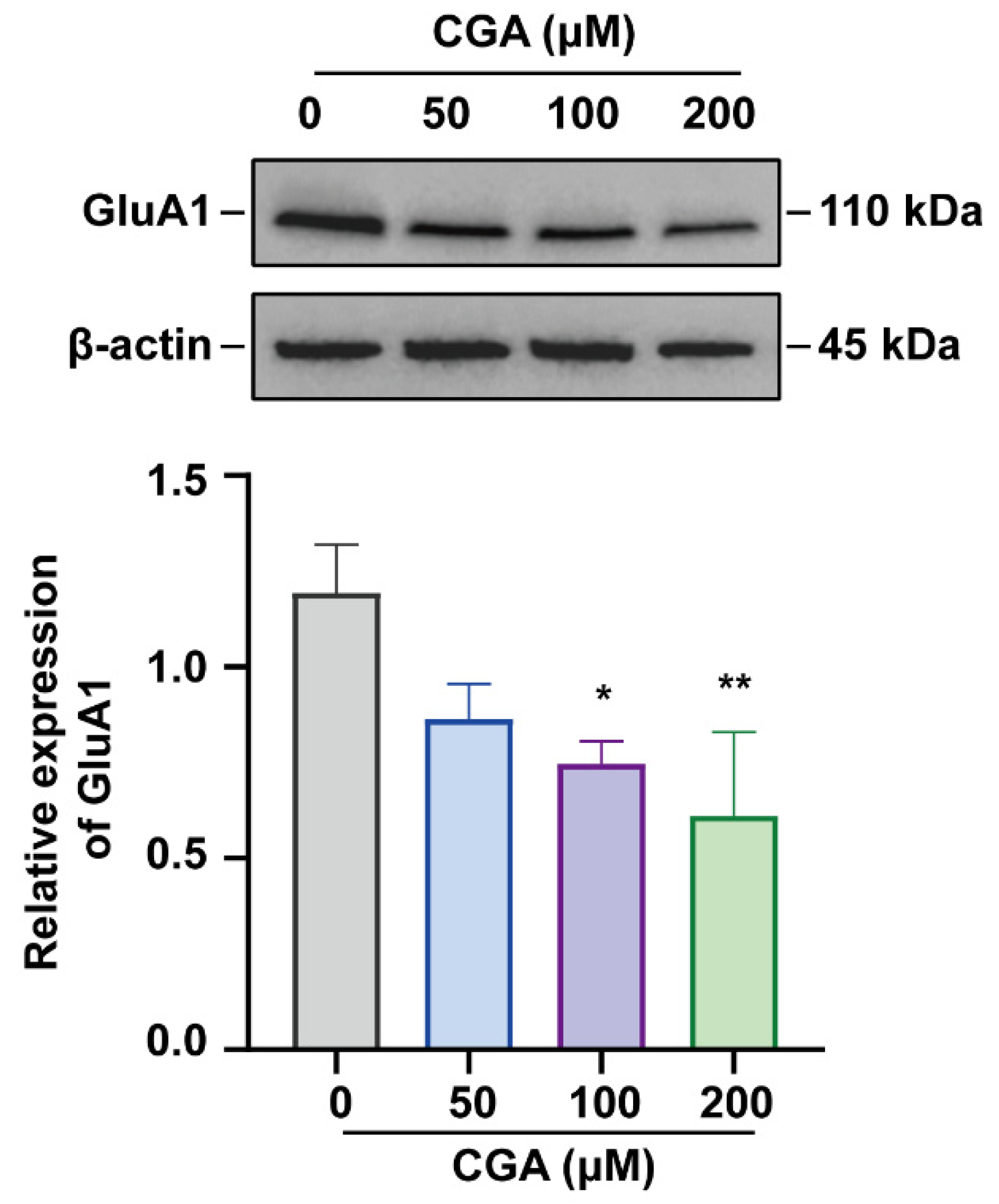
3.1. Effect of CGA on GluA1 in DRG Cells
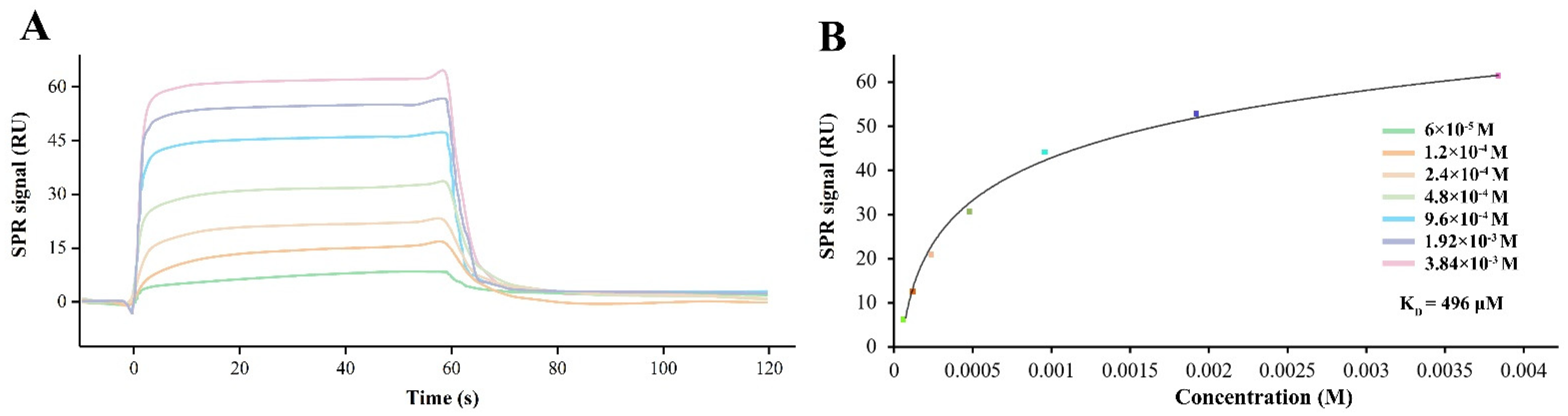
3.2. CGA Directly Binds to Human GluA1
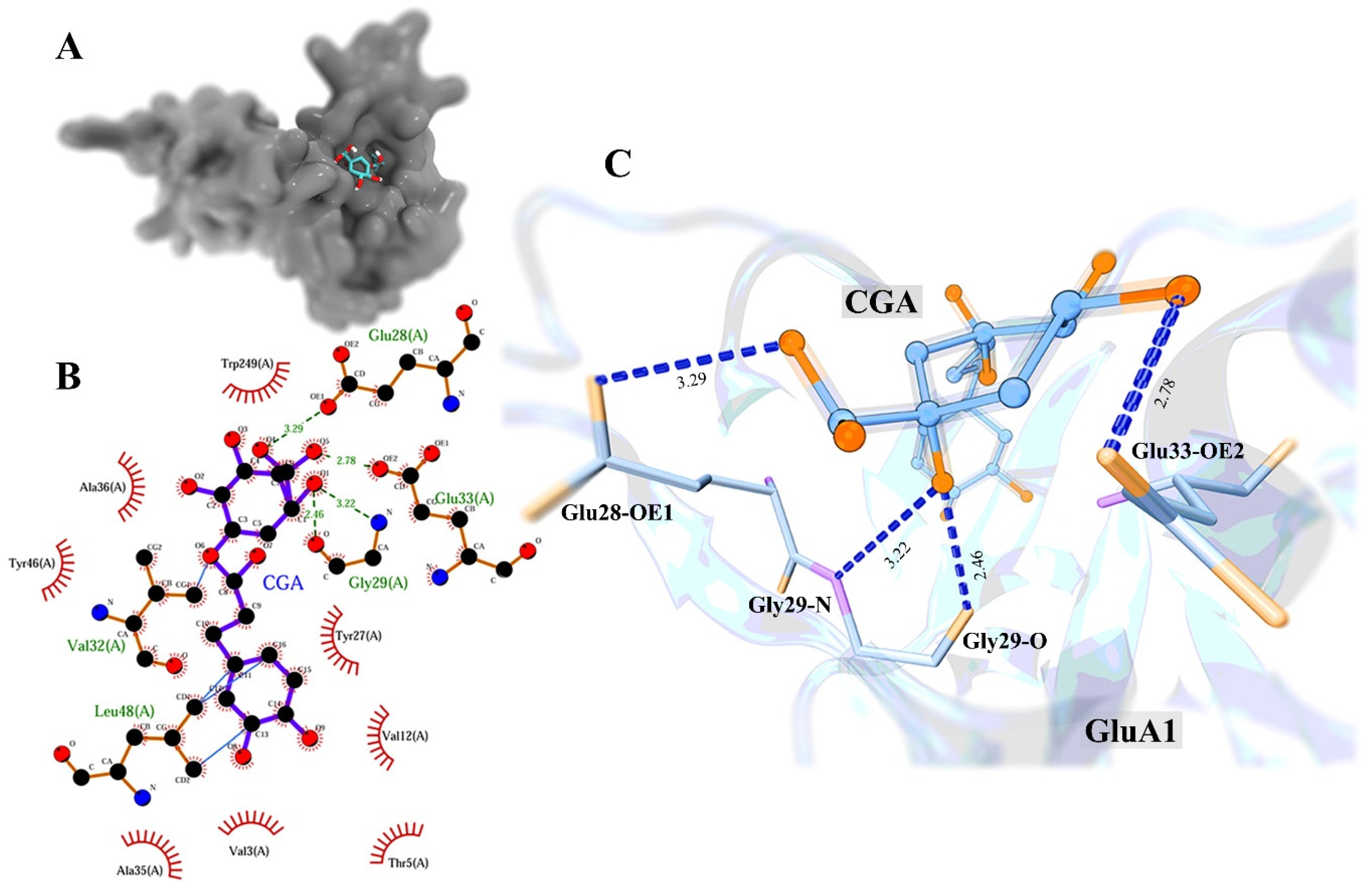
3.3. Molecular Docking Study
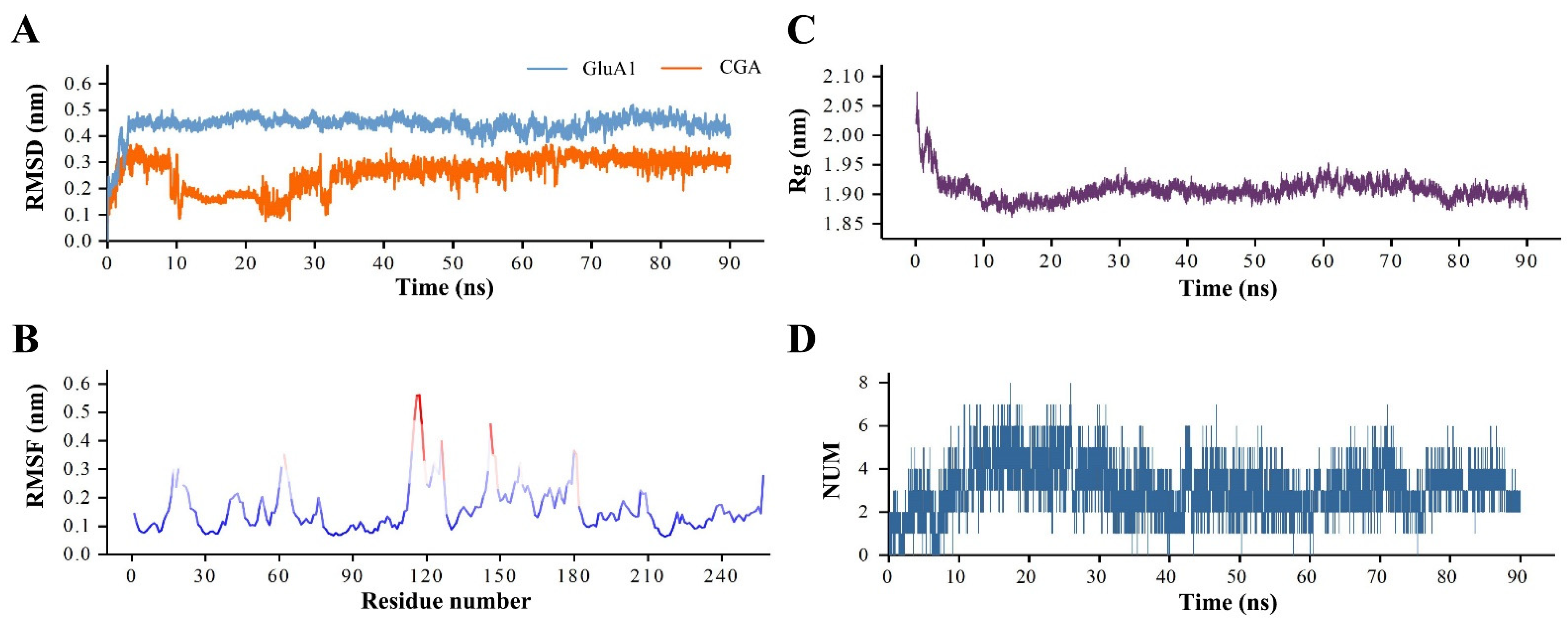
3.4. The Analysis of GluA1–CGA Complex MD Simulation
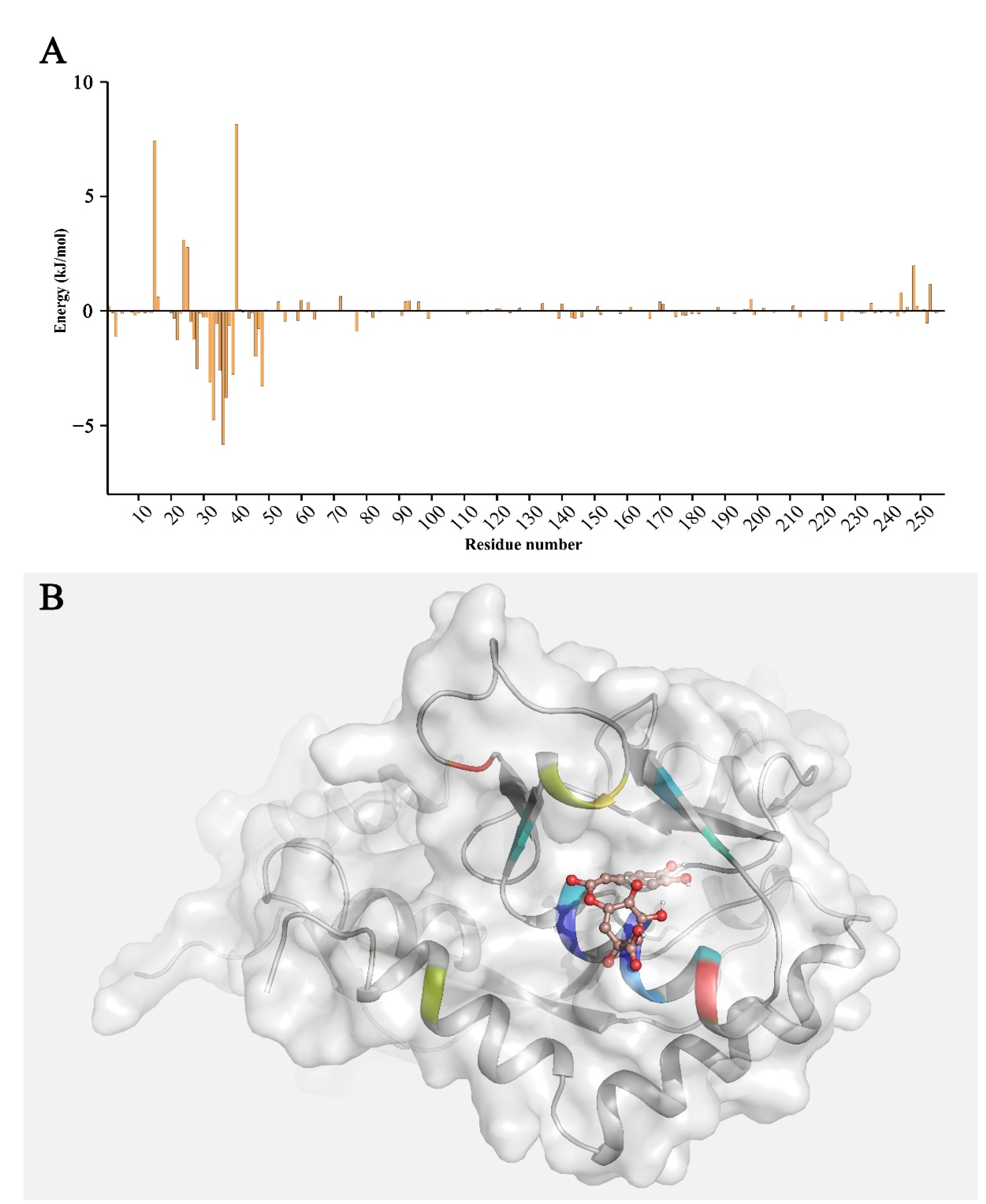
3.5. Binding Free Energy Calculation and Decomposition
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Raja, S.N.; Carr, D.B.; Cohen, M.; Finnerup, N.B.; Flor, H.; Gibson, S.; Keefe, F.; Mogil, J.S.; Ringkamp, M.; Sluka, K.A.; et al. The revised International Association for the Study of Pain definition of pain: Concepts, challenges, and compromises. Pain 2020, 161, 1976–1982. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.-J.; Zhang, T.-J.; Yang, X.-Q.; Feng, Z.-Y.; Qiu, F.; Xin, G.-K.; Liu, J.-F.; Nie, F.-C.; Jin, X.-H.; Liu, Y.-Q. A survey of chronic pain in China. Libyan J. Med. 2020, 15, 1730550. [Google Scholar]
- Uta, D.; Kato, G.; Doi, A.; Andoh, T.; Kume, T.; Yoshimura, M.; Koga, K. Animal models of chronic pain increase spontaneous glutamatergic transmission in adult rat spinal dorsal horn in vitro and in vivo. Biochem. Biophys. Res. Commun. 2019, 512, 352–359. [Google Scholar] [CrossRef] [PubMed]
- James, M.H.; Quinn, R.K.; Ong, L.K.; Levi, E.M.; Smith, D.W.; Dickson, P.W.; Dayas, C.V. Rapamycin reduces motivated responding for cocaine and alters GluA1 expression in the ventral but not dorsal striatum. Eur. J. Pharmacol. 2016, 784, 147–154. [Google Scholar] [CrossRef]
- Jiang, W.-N.; Jing, X.; Li, M.; Deng, H.; Jiang, T.; Xiong, K.-Z.; Chen, Y.; Wang, X.-F.; Wang, Q.-J. Corydaline and l-tetrahydropalmatine attenuate morphine-induced conditioned place preference and the changes in dopamine D2 and GluA1 AMPA receptor expression in rats. Eur. J. Pharmacol. 2020, 884, 173397. [Google Scholar] [CrossRef]
- Zhang, K.; Yamaki, V.N.; Wei, Z.; Zheng, Y.; Cai, X. Differential regulation of GluA1 expression by ketamine and memantine. Behav. Brain Res. 2017, 316, 152–159. [Google Scholar] [CrossRef]
- Wigerblad, G.; Huie, J.R.; Yin, H.Z.; Leinders, M.; Pritchard, R.A.; Koehrn, F.J.; Xiao, W.-H.; Bennett, G.J.; Huganir, R.L.; Ferguson, A.R. Inflammation-induced GluA1 trafficking and membrane insertion of Ca2+ permeable AMPA receptors in dorsal horn neurons is dependent on spinal tumor necrosis factor, PI3 kinase and protein kinase A. Exp. Neurol. 2017, 293, 144–158. [Google Scholar] [CrossRef]
- Hou, Y.-Y.; Cai, Y.-Q.; Pan, Z.Z. GluA1 in Central Amygdala Promotes Opioid Use and Reverses Inhibitory Effect of Pain. Neuroscience 2020, 426, 141–153. [Google Scholar] [CrossRef]
- Ballantyne, J.C. Opioids for the Treatment of Chronic Pain: Mistakes Made, Lessons Learned, and Future Directions. Anesth. Analg. 2017, 125, 1769–1778. [Google Scholar] [CrossRef]
- Li, Y.-X.; Gao, M.; Bai, X.; Liu, Y.-N.; Zhang, Y.-H.; Suo, Z.-W. Chlorogenic acid alleviates chronic inflammatory pain via inhibiting synaptic expression of AMPA receptor. Chin. Pharmacol. Bull. 2021, 37, 1251–1256. [Google Scholar]
- Naveed, M.; Hejazi, V.; Abbas, M.; Kamboh, A.A.; Khan, G.J.; Shumzaid, M.; Ahmad, F.; Babazadeh, D.; FangFang, X.; Modarresi-Ghazani, F. Chlorogenic acid (CGA): A pharmacological review and call for further research. Biomed. Pharm. 2018, 97, 67–74. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharyya, S.; Majhi, S.; Saha, B.P.; Mukherjee, P.K. Chlorogenic acid-phospholipid complex improve protection against UVA induced oxidative stress. J. Photochem. Photobiol. B Biol. 2014, 130, 293–298. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.; Chang, C.; Zhang, L.; Liu, Y.; Huang, X.; Chen, Z. Chlorogenic acid improves late diabetes through adiponectin receptor signaling pathways in db/db mice. PLoS ONE 2015, 10, e0120842. [Google Scholar] [CrossRef] [Green Version]
- Valduga, A.T.; Gonçalves, I.L.; Magri, E.; Finzer, J.R.D. Chemistry, pharmacology and new trends in traditional functional and medicinal beverages. Food Res. Int. 2019, 120, 478–503. [Google Scholar] [CrossRef] [PubMed]
- Yonathan, M.; Asres, K.; Assefa, A.; Bucar, F. In vivo anti-inflammatory and anti-nociceptive activities of Cheilanthes farinosa. J. Ethnopharmacol. 2006, 108, 462–470. [Google Scholar] [CrossRef] [PubMed]
- Hara, K.; Haranishi, Y.; Kataoka, K.; Takahashi, Y.; Terada, T.; Nakamura, M.; Sata, T. Chlorogenic acid administered intrathecally alleviates mechanical and cold hyperalgesia in a rat neuropathic pain model. Eur. J. Pharmacol. 2014, 723, 459–464. [Google Scholar] [CrossRef] [PubMed]
- Hung, Y.C.; Kuo, Y.H.; Hsieh, P.W.; Hsieh, T.Y.; Kuo, J.R.; Wang, S.J. Chlorogenic Acid Decreases Glutamate Release from Rat Cortical Nerve Terminals by P/Q-Type Ca2+ Channel Suppression: A Possible Neuroprotective Mechanism. Int. J. Mol. Sci. 2021, 22, 11447. [Google Scholar] [CrossRef]
- Rich, M.T.; Huang, Y.H.; Torregrossa, M.M. Calcineurin Promotes Neuroplastic Changes in the Amygdala Associated with Weakened Cocaine-Cue Memories. J. Neurosci. 2020, 40, 1344–1354. [Google Scholar] [CrossRef]
- Kamalova, A.; Nakagawa, T. AMPA receptor structure and auxiliary subunits. J. Physiol. 2021, 599, 453–469. [Google Scholar] [CrossRef]
- Zhang, D.; Watson, J.F.; Matthews, P.M.; Cais, O.; Greger, I.H. Gating and modulation of a hetero-octameric AMPA glutamate receptor. Nature 2021, 594, 454–458. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pronk, S.; Páll, S.; Schulz, R.; Larsson, P.; Bjelkmar, P.; Apostolov, R.; Shirts, M.R.; Smith, J.C.; Kasson, P.M.; Van Der Spoel, D. GROMACS 4.5: A high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics 2013, 29, 845–854. [Google Scholar] [CrossRef] [PubMed]
- Stroet, M.; Caron, B.; Visscher, K.M.; Geerke, D.P.; Malde, A.K.; Mark, A.E. Automated Topology Builder Version 3.0: Prediction of Solvation Free Enthalpies in Water and Hexane. J. Chem. Theory. Comput. 2018, 14, 5834–5845. [Google Scholar] [CrossRef] [PubMed]
- Kumari, R.; Kumar, R. Open Source Drug Discovery Consortium, Lynn, A. g_mmpbsa--a GROMACS tool for high-throughput MM-PBSA calculations. J. Chem. Inf. Model. 2014, 54, 1951–1962. [Google Scholar] [CrossRef]
- Sun, H.; Duan, L.; Chen, F.; Liu, H.; Wang, Z.; Pan, P.; Zhu, F.; Zhang, J.Z.H.; Hou, T. Assessing the performance of MM/PBSA and MM/GBSA methods. 7. Entropy effects on the performance of end-point binding free energy calculation approaches. Phys. Chem. Chem. Phys. 2018, 20, 14450–14460. [Google Scholar] [CrossRef]
- Sciú, M.L.; Sebastián-Pérez, V.; Martinez-Gonzalez, L.; Benitez, R.; Perez, D.I.; Pérez, C.; Campillo, N.E.; Martinez, A.; Moyano, E.L. Computer-aided molecular design of pyrazolotriazines targeting glycogen synthase kinase 3. J. Enzym. Inhib. Med. Chem. 2019, 34, 87–96. [Google Scholar] [CrossRef] [Green Version]
- Guo, N.; Liu, Z.; Yan, Z.; Liu, Z.; Hao, K.; Liu, C.; Wang, J. Subinhibitory concentrations of Honokiol reduce α-Hemolysin (Hla) secretion by Staphylococcus aureus and the Hla-induced inflammatory response by inactivating the NLRP3 inflammasome. Emerg. Microbes Infect. 2019, 8, 707–716. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.; Wang, J.; Li, Y.; Wang, Z.; Chu, M.; Wang, Y. Tuftsin: A Natural Molecule Against SARS-CoV-2 Infection. Front. Mol. Biosci. 2022, 9, 859162. [Google Scholar] [CrossRef]
- Ge, Y.; Wang, Y.T. GluA1-homomeric AMPA receptor in synaptic plasticity and neurological diseases. Neuropharmacology 2021, 197, 108708. [Google Scholar] [CrossRef]
- Qu, W.; Yuan, B.; Liu, J.; Liu, Q.; Zhang, X.; Cui, R.; Yang, W.; Li, B. Emerging role of AMPA receptor subunit GluA1 in synaptic plasticity: Implications for Alzheimer’s disease. Cell Prolif. 2021, 54, e12959. [Google Scholar] [CrossRef]
- Sanderson, D.J.; Good, M.A.; Skelton, K.; Sprengel, R.; Seeburg, P.H.; Rawlins, J.N.P.; Bannerman, D.M. Enhanced long-term and impaired short-term spatial memory in GluA1 AMPA receptor subunit knockout mice: Evidence for a dual-process memory model. Learn. Mem. 2009, 16, 379–386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, Y.Q.; Hou, Y.Y.; Pan, Z.Z. GluA1 in central amygdala increases pain but inhibits opioid withdrawal-induced aversion. Mol. Pain 2020, 16, 1744806920911543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uritu, C.M.; Mihai, C.T.; Stanciu, G.-D.; Dodi, G.; Alexa-Stratulat, T.; Luca, A.; Leon-Constantin, M.-M.; Stefanescu, R.; Bild, V.; Melnic, S.; et al. Medicinal Plants of the Family Lamiaceae in Pain Therapy: A Review. Pain Res. Manag. 2018, 2018, 7801543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lazarou, R.; Heinrich, M. Herbal medicine: Who cares? The changing views on medicinal plants and their roles in British lifestyle. Phytother. Res. 2019, 33, 2409–2420. [Google Scholar] [CrossRef]
- Riondato, I.; Donno, D.; Roman, A.; Razafintsalama, V.E.; Petit, T.; Mellano, M.G.; Torti, V.; De Biaggi, M.; Rakotoniaina, E.N.; Giacoma, C.; et al. First ethnobotanical inventory and phytochemical analysis of plant species used by indigenous people living in the Maromizaha forest, Madagascar. J. Ethnopharmacol. 2019, 232, 73–89. [Google Scholar] [CrossRef]
- Castellanos, J.P.; Woolley, C.; Bruno, K.A.; Zeidan, F.; Halberstadt, A.; Furnish, T. Chronic pain and psychedelics: A review and proposed mechanism of action. Reg. Anesth. Pain Med. 2020, 45, 486–494. [Google Scholar] [CrossRef]
- Khumalo, G.P.; Vacn Wyk, B.E.; Feng, Y.; Cock, I.E. A review of the traditional use of southern African medicinal plants for the treatment of inflammation and inflammatory pain. J. Ethnopharmacol. 2022, 283, 114436. [Google Scholar] [CrossRef]
- Liu, D.; Wang, H.; Zhang, Y.; Zhang, Z. Protective Effects of Chlorogenic Acid on Cerebral Ischemia/Reperfusion Injury Rats by Regulating Oxidative Stress-Related Nrf2 Pathway. Drug. Des. Devel. Ther. 2020, 14, 51–60. [Google Scholar] [CrossRef] [Green Version]
- Ochiai, R.; Saitou, K.; Suzukamo, C.; Osaki, N.; Asada, T. Effect of Chlorogenic Acids on Cognitive Function in Mild Cognitive Impairment: A Randomized Controlled Crossover Trial. J. Alzheimers Dis. 2019, 72, 1209–1216. [Google Scholar] [CrossRef] [Green Version]
- Fernandes, M.Y.D.; Dobrachinski, F.; Silva, H.B.; Lopes, J.P.; Gonçalves, F.Q.; Soares, F.A.; Porciúncula, L.O.; Andrade, G.M.; Cunha, R.A.; Tomé, A.R. Neuromodulation and neuroprotective effects of chlorogenic acids in excitatory synapses of mouse hippocampal slices. Sci. Rep. 2021, 11, 10488. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Energy Item | Energy (kJ/mol) |
|---|---|
| −95.763 ± 13.707 | |
| −63.083 ± 9.975 | |
| 132.238 ± 16.630 | |
| −13.194 ± 1.277 | |
| −39.803 ± 14.772 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhu, W.; Wu, F.; Hu, J.; Wang, W.; Zhang, J.; Guo, G. Structural Investigation of the Interaction Mechanism between Chlorogenic Acid and AMPA Receptor via In Silico Approaches. Molecules 2022, 27, 3394. https://doi.org/10.3390/molecules27113394
Zhu W, Wu F, Hu J, Wang W, Zhang J, Guo G. Structural Investigation of the Interaction Mechanism between Chlorogenic Acid and AMPA Receptor via In Silico Approaches. Molecules. 2022; 27(11):3394. https://doi.org/10.3390/molecules27113394
Chicago/Turabian StyleZhu, Wei, Fengming Wu, Jindie Hu, Wenjing Wang, Jifeng Zhang, and Guoqing Guo. 2022. "Structural Investigation of the Interaction Mechanism between Chlorogenic Acid and AMPA Receptor via In Silico Approaches" Molecules 27, no. 11: 3394. https://doi.org/10.3390/molecules27113394
APA StyleZhu, W., Wu, F., Hu, J., Wang, W., Zhang, J., & Guo, G. (2022). Structural Investigation of the Interaction Mechanism between Chlorogenic Acid and AMPA Receptor via In Silico Approaches. Molecules, 27(11), 3394. https://doi.org/10.3390/molecules27113394