
An Investigation towards Coupling Molecular Dynamics with Computational Fluid Dynamics for Modelling Polymer Pyrolysis

 ,
, 
 ,
,  ,
, 
 ,
,  and
and 
Abstract
:1. Introduction
2. Results and Discussion
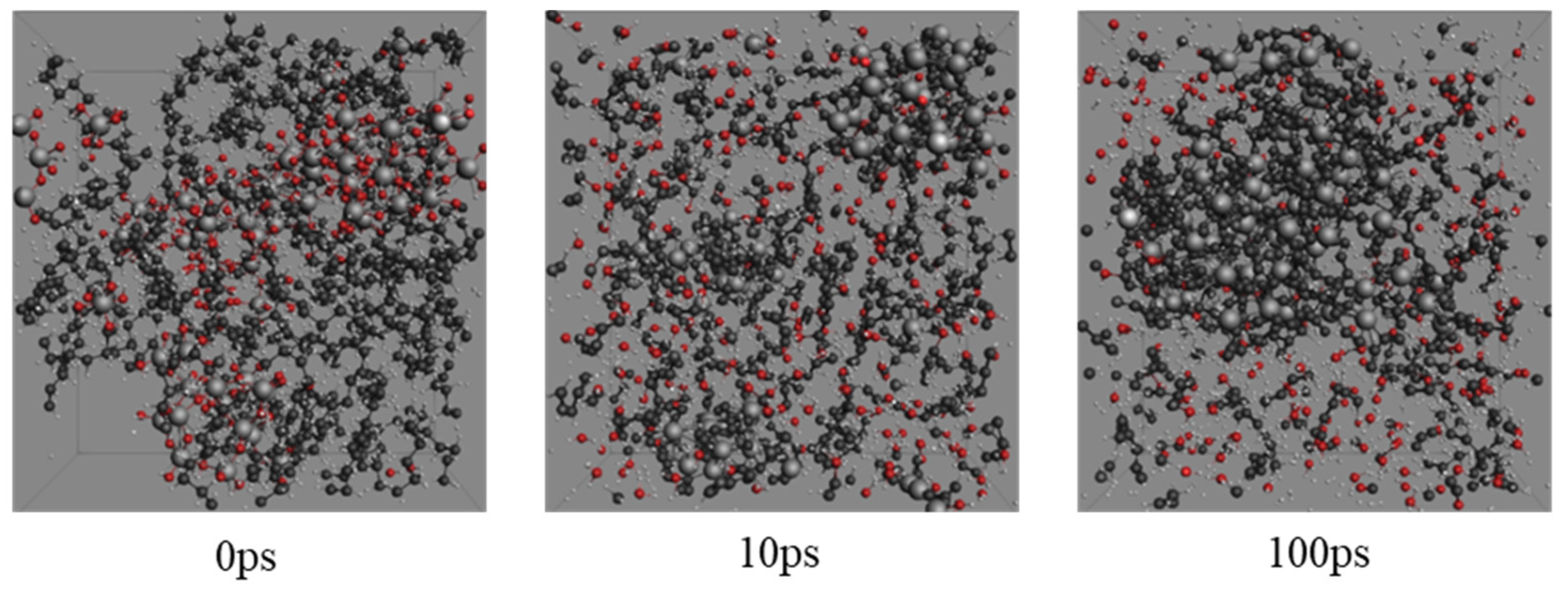
2.1. Molecular Systems
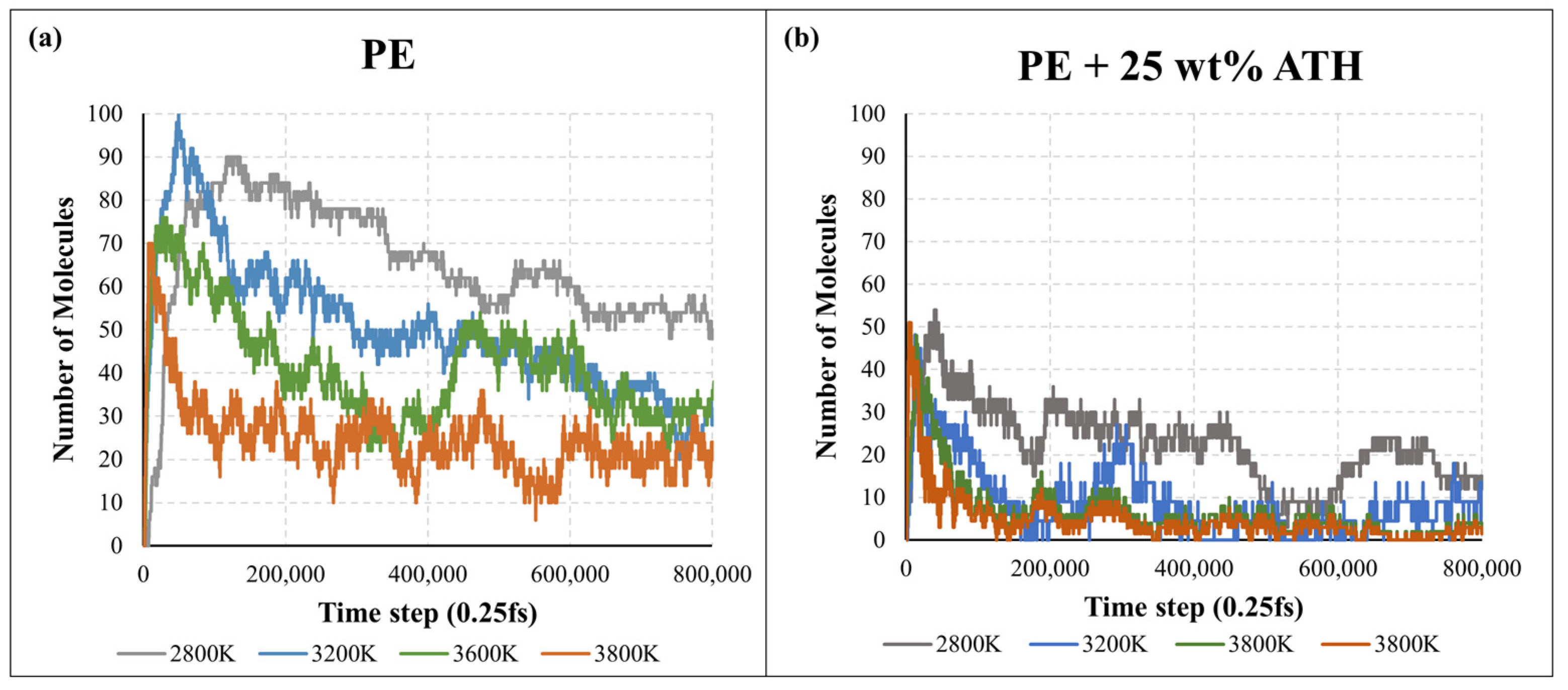
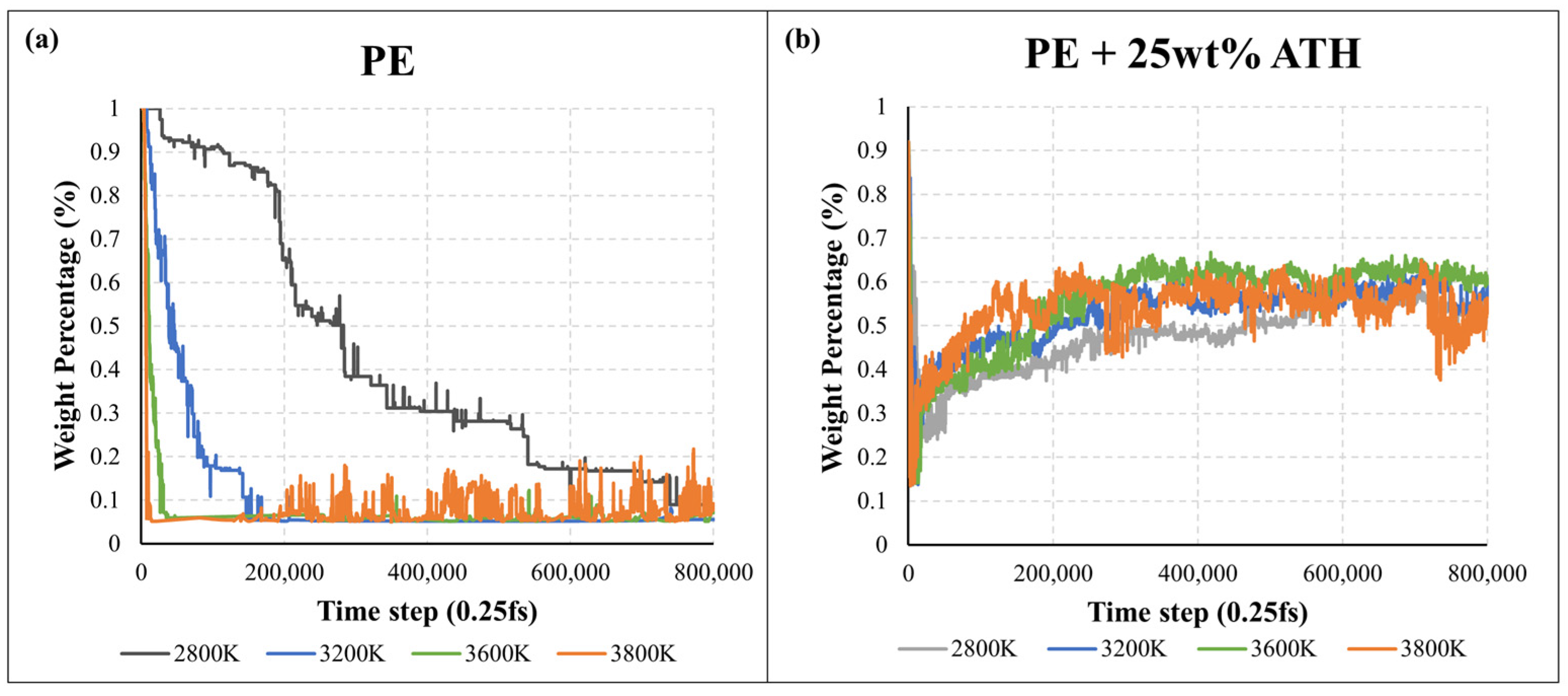
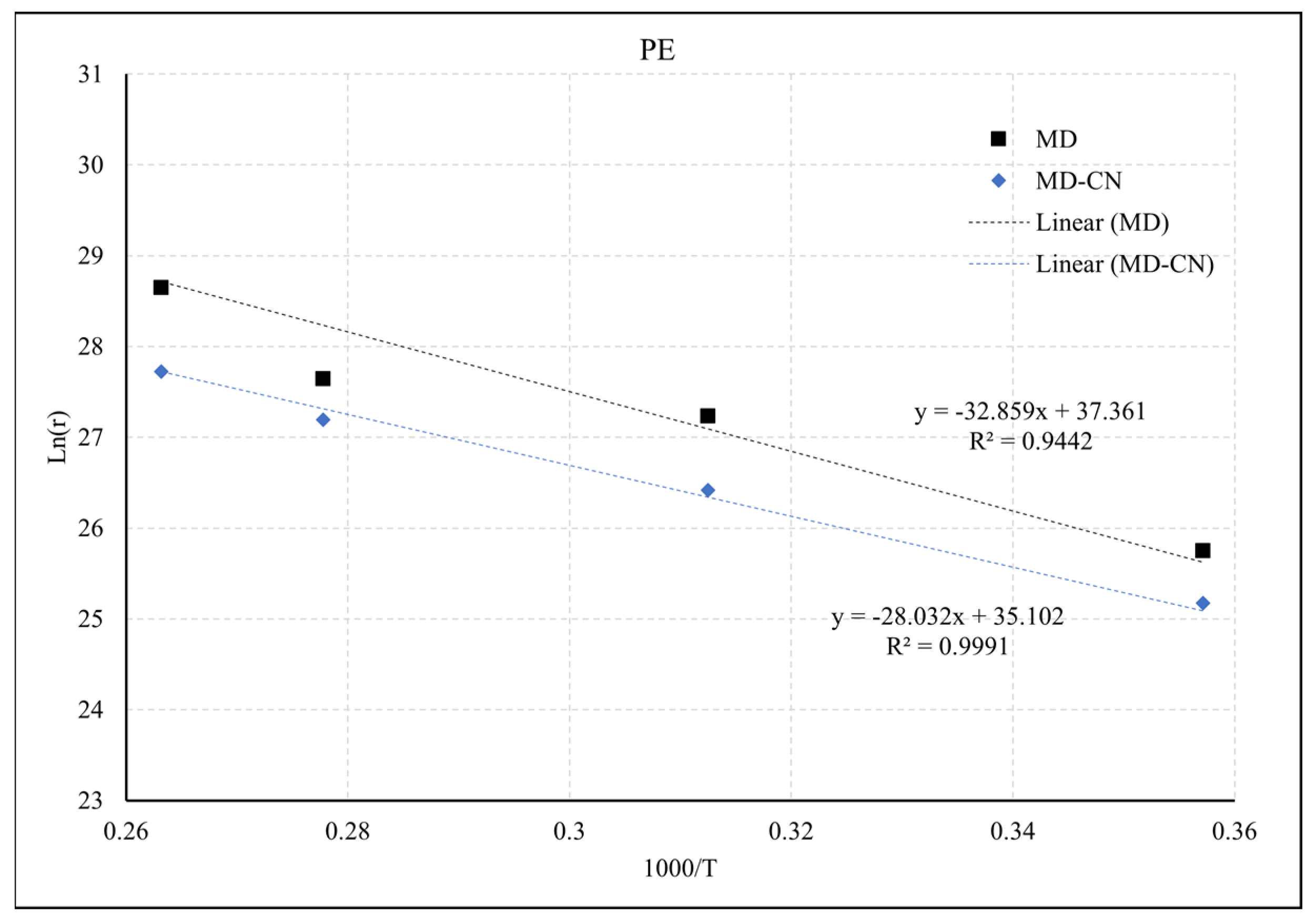
2.2. Pyrolysis Kinetic Analysis
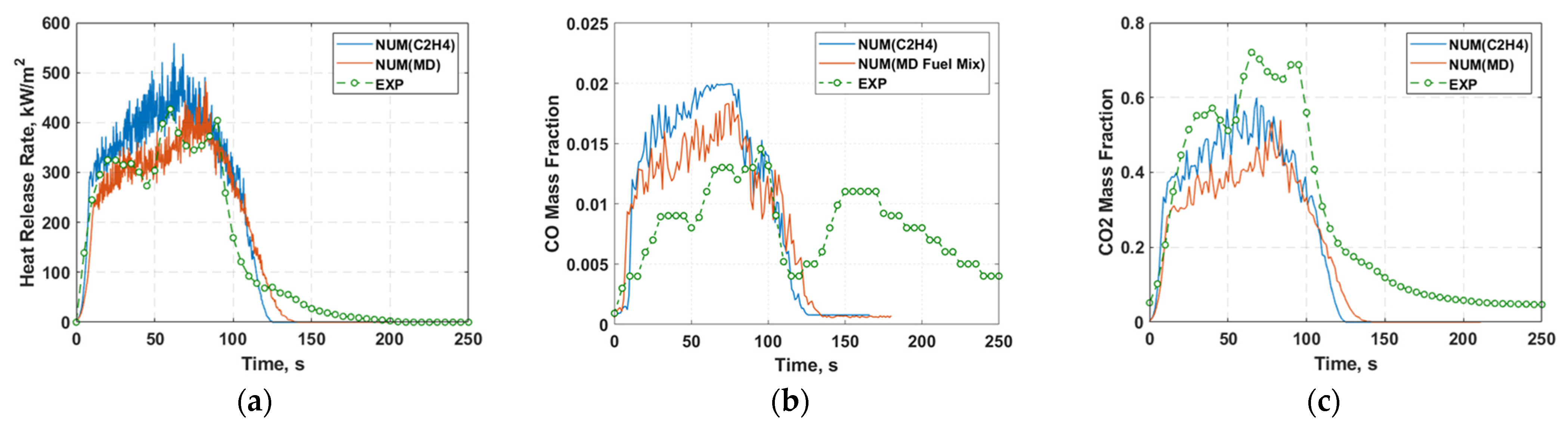
2.3. Incorporating MD into Detailed Chemistry Kinetics
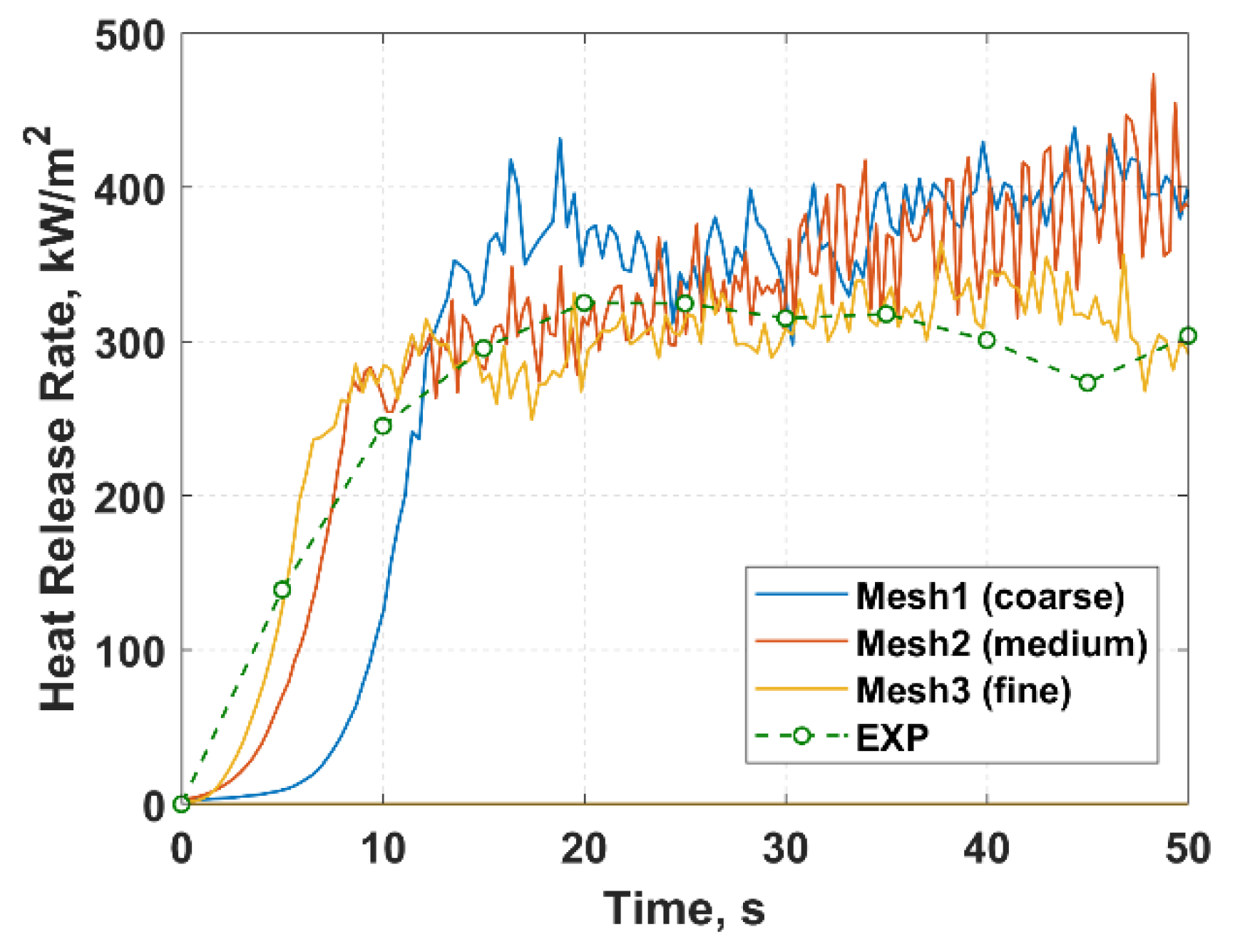
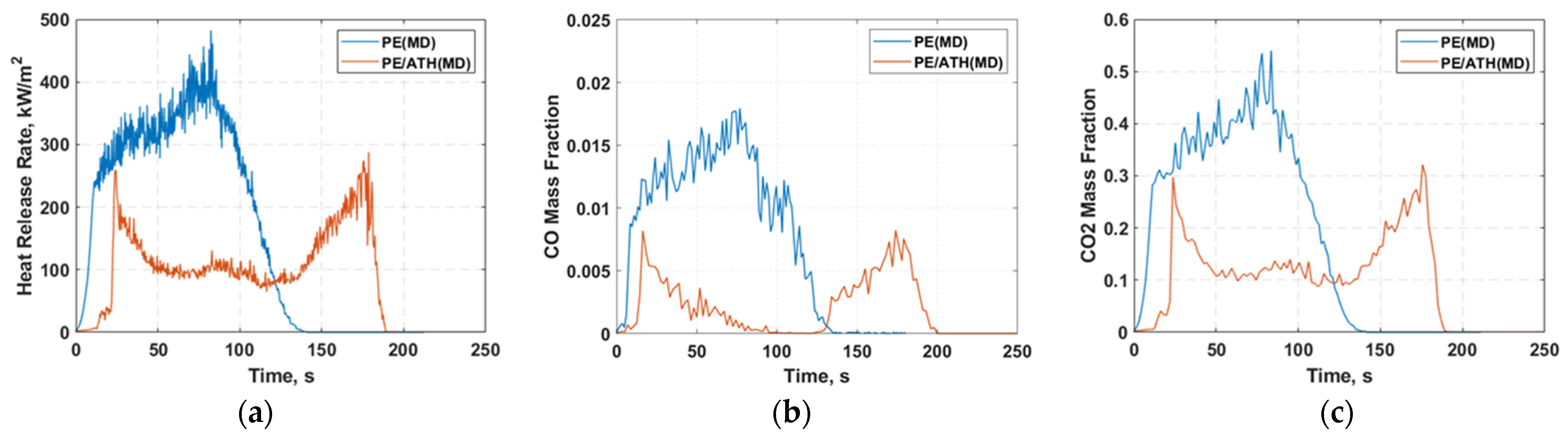
2.4. Cone Calorimeter Simulation
3. Materials and Methods
3.1. Molecular Dynamics
Molecular System Configuration and Simulation Details
3.2. Computational Fluid Dynamics
3.2.1. Pyrolysis Model
3.2.2. Turbulence
3.2.3. Detailed Chemistry Combustion
3.2.4. Soot Formation
3.2.5. Radiation
3.3. Experiment
3.3.1. Thermogravimetry
3.3.2. Cone Calorimetry
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Nomenclature
| A | Exponential factor |
| a | Acceleration |
| Normalised radical nuclei concentration | |
| ci | Mass fraction |
| WALE model constant | |
| D | Diffusivity |
| Activation energy | |
| Esystem | System energy |
| Ebond | Bond energy |
| Eover | Over-coordinated atom energy |
| Eunder | Under-coordinated atom energy |
| Eval | Valence angle term |
| Epen | Penalty energy |
| Etors | Torsion energy |
| Econj | Conjugation effects to molecular energy |
| EvdWaals | Nonbonded van der Waals interaction |
| ECoulomb | Coulomb interaction |
| F | Instantaneous force |
| Mixture fraction variance | |
| g | Gravity |
| H | Enthalpy |
| Heat of reaction | |
| Heat of formation | |
| Radiation intensities | |
| K | Thermal conductivity |
| Summation of the gas absorption coefficient | |
| Mixing length for sub-grid scales | |
| Soot mass concentration | |
| m | Mass |
| Fuel released at material surface | |
| Number density of soot particles | |
| ni | Reaction order of the reaction |
| P | Pressure |
| Q | Heat released by the fluid combustion |
| Net heat gain due to chemical reactions during pyrolysis | |
| Net heat gain due to chemical reactions via radiation absorption | |
| Flux equation of thermal radiation | |
| R | Gas constant |
| Pyrolysis reaction rates | |
| Rate-of-strain tensor | |
| T | Absolute temperature |
| Ts | Solid fuel temperature |
| t | time |
| u | velocity |
| Scalar dissipation | |
| x | Instantaneous particle position |
| Ysoot | Mass fraction of soot |
| Z | Elemental mass fraction |
| Density | |
| Subgrid-scale stresses | |
| Turbulent viscosity | |
| Von Kármán constant |
Abbreviations
| ATH | Aluminium (tri)hydroxide |
| C | Carbon |
| CFD | Computational Fluid Dynamics |
| CO | Carbon Monoxide |
| CO2 | Carbon Dioxide |
| C2H4 | Ethylene |
| FDS | Fire Dynamic Simulator |
| HRR | Heat Release Rate |
| H2O | Water |
| LES | Large Eddy Simulation |
| MD | Molecular Dynamics |
| MD-CN | Moleculary Dynamics Carbon Number Approach |
| PE | Polyethylene |
| ReaxFF | Reactive Forcefield |
| TGA | Thermogravimetric Analysis |
| WALE | Wall Adapting Local Eddy-viscosity |
| wt% | weight percentage |
References
- Evegren, F.; Hertzberg, T. Fire safety regulations and performance of fibre-reinforced polymer composite ship structures. Proc. Inst. Mech. Eng. Part M J. Eng. Marit. Environ. 2017, 231, 46–56. [Google Scholar] [CrossRef]
- Morgan, A.B.; Gilman, J.W. An overview of flame retardancy of polymeric materials: Application, technology, and future directions. Fire Mater. 2013, 37, 259–279. [Google Scholar] [CrossRef] [Green Version]
- Yuen, A.C.Y.; Chen, T.B.Y.; Yeoh, G.H.; Yang, W.; Cheung, S.C.-P.; Cook, M.; Yu, B.; Chan, Q.N.; Yip, H.L. Establishing pyrolysis kinetics for the modelling of the flammability and burning characteristics of solid combustible materials. J. Fire Sci. 2018, 36, 494–517. [Google Scholar] [CrossRef] [Green Version]
- Yuen, A.C.Y.; Chen, T.B.Y.; Wang, C.; Wei, W.; Kabir, I.; Vargas, J.B.; Chan, Q.N.; Kook, S.; Yeoh, G.H. Utilising genetic algorithm to optimise pyrolysis kinetics for fire modelling and characterisation of chitosan/graphene oxide polyurethane composites. Compos. Part B Eng. 2020, 182, 107619. [Google Scholar] [CrossRef]
- Mallet, V.; Keyes, D.E.; Fendell, F.E. Modeling wildland fire propagation with level set methods. Comput. Math. Appl. 2009, 57, 1089–1101. [Google Scholar] [CrossRef] [Green Version]
- Rochoux, M.C.; Delmotte, B.; Cuenot, B.; Ricci, S.; Trouvé, A. Regional-scale simulations of wildland fire spread informed by real-time flame front observations. Proc. Combust. Inst. 2013, 34, 2641–2647. [Google Scholar] [CrossRef]
- Chen, T.B.Y.; Yuen, A.C.Y.; Yeoh, G.H.; Timchenko, V.; Cheung, S.C.P.; Chan, Q.N.; Yang, W.; Lu, H. Numerical study of fire spread using the level-set method with large eddy simulation incorporating detailed chemical kinetics gas-phase combustion model. J. Comput. Sci. 2018, 24, 8–23. [Google Scholar] [CrossRef]
- McGrattan, K.B.; McDermott, R.J.; Weinschenk, C.G.; Forney, G.P. Fire Dynamics Simulator Technical Reference Guide; National Institute of Standards and Technology: Gaithersburg, MD, USA, 2017.
- Ding, Y.; Wang, C.; Lu, S. Modeling the pyrolysis of wet wood using FireFOAM. Energy Convers. Manag. 2015, 98, 500–506. [Google Scholar] [CrossRef]
- Yuen, R.K.K.; Yeoh, G.H.; de Vahl Davis, G.; Leonardi, E. Modelling the pyrolysis of wet wood—I. Three-dimensional formulation and analysis. Int. J. Heat Mass Transf. 2007, 50, 4371–4386. [Google Scholar] [CrossRef]
- Chen, T.B.Y.; Yuen, A.C.Y.; Wang, C.; Yeoh, G.H.; Timchenko, V.; Cheung, S.C.P.; Chan, Q.N.; Yang, W. Predicting the fire spread rate of a sloped pine needle board utilizing pyrolysis modelling with detailed gas-phase combustion. Int. J. Heat Mass Transf. 2018, 125, 310–322. [Google Scholar] [CrossRef]
- Rein, G.; Lautenberger, C.; Fernandez-Pello, A.C.; Torero, J.L.; Urban, D.L. Application of genetic algorithms and thermogravimetry to determine the kinetics of polyurethane foam in smoldering combustion. Combust. Flame 2006, 146, 95–108. [Google Scholar] [CrossRef] [Green Version]
- Mishra, G.; Kumar, J.; Bhaskar, T. Kinetic studies on the pyrolysis of pinewood. Bioresour. Technol. 2015, 182, 282–288. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.; Chen, D.; Gu, J.; Bao, B.; Zhang, Q. Determination of pyrolysis characteristics and kinetics of palm kernel shell using TGA–FTIR and model-free integral methods. Energy Convers. Manag. 2015, 89, 251–259. [Google Scholar] [CrossRef]
- Yang, H.; Wang, X.; Yuan, H.; Song, L.; Hu, Y.; Yuen, R.K.K. Fire performance and mechanical properties of phenolic foams modified by phosphorus-containing polyethers. J. Polym. Res. 2012, 19, 9831. [Google Scholar] [CrossRef]
- Zhao, Q.; Zhang, B.; Quan, H.; Yam, R.C.M.; Yuen, R.K.K.; Li, R.K.Y. Flame retardancy of rice husk-filled high-density polyethylene ecocomposites. Compos. Sci. Technol. 2009, 69, 2675–2681. [Google Scholar] [CrossRef]
- Yuen, A.C.Y.; Chen, T.B.Y.; Lin, B.; Yang, W.; Kabir, I.I.; De Cachinho Cordeiro, I.M.; Whitten, A.E.; Mata, J.; Yu, B.; Lu, H.-D.; et al. Study of structure morphology and layer thickness of Ti3C2 MXene with Small-Angle Neutron Scattering (SANS). Compos. Part C Open Access 2021, 5, 100155. [Google Scholar] [CrossRef]
- Nguyen, Q.T.; Tran, P.; Ngo, T.D.; Tran, P.A.; Mendis, P. Experimental and computational investigations on fire resistance of GFRP composite for building façade. Compos. Part B Eng. 2014, 62, 218–229. [Google Scholar] [CrossRef]
- Dutta, S.; Kim, N.K.; Das, R.; Bhattacharyya, D. Effects of sample orientation on the fire reaction properties of natural fibre composites. Compos. Part B Eng. 2019, 157, 195–206. [Google Scholar] [CrossRef]
- Lin, B.; Yuen, A.C.Y.; Chen, T.B.Y.; Yu, B.; Yang, W.; Zhang, J.; Yao, Y.; Wu, S.; Wang, C.H.; Yeoh, G.H. Experimental and numerical perspective on the fire performance of MXene/Chitosan/Phytic acid coated flexible polyurethane foam. Sci. Rep. 2021, 11, 4684. [Google Scholar] [CrossRef]
- Di Martino, G.D.; Carmicino, C.; Mungiguerra, S.; Savino, R. The Application of Computational Thermo-Fluid-Dynamics to the Simulation of Hybrid Rocket Internal Ballistics with Classical or Liquefying Fuels: A Review. Aerospace 2019, 6, 56. [Google Scholar] [CrossRef] [Green Version]
- Tyurenkova, V.; Smirnova, M.J. Material combustion in oxidant flows: Self-similar solutions. Acta Astronaut. 2016, 120, 129–137. [Google Scholar] [CrossRef]
- Betelin, V.; Kushnirenko, A.; Smirnov, N.; Nikitin, V.; Tyurenkova, V.; Stamov, L. Numerical investigations of hybrid rocket engines. Acta Astronaut. 2018, 144, 363–370. [Google Scholar] [CrossRef]
- Tyurenkova, V.V.; Stamov, L.I. Flame propagation in weightlessness above the burning surface of material. Acta Astronaut. 2019, 159, 342–348. [Google Scholar] [CrossRef]
- Kushnirenko, A.; Stamov, L.; Tyurenkova, V.; Smirnova, M.; Mikhalchenko, E. Three-dimensional numerical modeling of a rocket engine with solid fuel. Acta Astronaut. 2021, 181, 544–551. [Google Scholar] [CrossRef]
- Yuen, A.C.Y.; Chen, T.B.Y.; Li, A.; De Cachinho Cordeiro, I.M.; Liu, L.; Liu, H.; Lo, A.L.P.; Chan, Q.N.; Yeoh, G.H. Evaluating the fire risk associated with cladding panels: An overview of fire incidents, policies, and future perspective in fire standards. Fire Mater. 2021, 45, 663–689. [Google Scholar] [CrossRef]
- Ciottoli, P.P.; Malpica Galassi, R.; Lapenna, P.E.; Leccese, G.; Bianchi, D.; Nasuti, F.; Creta, F.; Valorani, M. CSP-based chemical kinetics mechanisms simplification strategy for non-premixed combustion: An application to hybrid rocket propulsion. Combust. Flame 2017, 186, 83–93. [Google Scholar] [CrossRef]
- Van Duin, A.C.; Dasgupta, S.; Lorant, F.; Goddard, W.A. ReaxFF: A reactive force field for hydrocarbons. J. Phys. Chem. A 2001, 105, 9396–9409. [Google Scholar] [CrossRef] [Green Version]
- Chen, T.B.Y.; Yuen, A.C.Y.; Lin, B.; Liu, L.; Lo, A.; Chan, Q.N.; Zhang, J.; Cheung, S.C.P.; Yeoh, G.H. Characterisation of pyrolysis kinetics and detailed gas species formations of engineering polymers via reactive molecular dynamics (ReaxFF). J. Anal. Appl. Pyrolysis 2020, 153, 104931. [Google Scholar] [CrossRef]
- Vaari, J.; Paajanen, A. Evaluation of the reactive molecular dynamics method for Research on flame retardants: ATH-filled polyethylene. Comput. Mater. Sci. 2018, 153, 103–112. [Google Scholar] [CrossRef]
- Rahmani, F.; Mahdavi, M.; Nouranian, S.; Al-Ostaz, A. Confinement effects on the thermal stability of poly (ethylene oxide)/graphene nanocomposites: A reactive molecular dynamics simulation study. J. Polym. Sci. Part B Polym. Phys. 2017, 55, 1026–1035. [Google Scholar] [CrossRef]
- Lan, Y.; Li, D.; Yang, R.; Liang, W.; Zhou, L.; Chen, Z. Computer simulation study on the compatibility of cyclotriphosphazene containing aminopropylsilicone functional group in flame retarded polypropylene/ammonium polyphosphate composites. Compos. Sci. Technol. 2013, 88, 9–15. [Google Scholar] [CrossRef]
- Stoliarov, S.I.; Westmoreland, P.R.; Nyden, M.R.; Forney, G.P. A reactive molecular dynamics model of thermal decomposition in polymers: I. Poly(methyl methacrylate). Polymer 2003, 44, 883–894. [Google Scholar] [CrossRef]
- Stoliarov, S.I.; Lyon, R.E.; Nyden, M.R. A reactive molecular dynamics model of thermal decomposition in polymers. II. Polyisobutylene. Polymer 2004, 45, 8613–8621. [Google Scholar] [CrossRef]
- Liu, X.; Li, X.; Liu, J.; Wang, Z.; Kong, B.; Gong, X.; Yang, X.; Lin, W.; Guo, L. Study of high density polyethylene (HDPE) pyrolysis with reactive molecular dynamics. Polym. Degrad. Stab. 2014, 104, 62–70. [Google Scholar] [CrossRef]
- Wan, Y.; Yu, S.; Jiang, S.; Pei, Q.; Xu, S.; Cao, W.; Liu, X.; Lan, Y. Microscopic pyrolysis mechanism on the octyphenylsiloxane flame retarded polycarbonate by reactive molecular dynamics. J. Anal. Appl. Pyrolysis 2021, 158, 105274. [Google Scholar] [CrossRef]
- Li, G.-Y.; Li, A.-Q.; Zhang, H.; Wang, J.-P.; Chen, S.-Y.; Liang, Y.-H. Theoretical study of the CO formation mechanism in the CO2 gasification of lignite. Fuel 2018, 211, 353–362. [Google Scholar] [CrossRef]
- Xu, F.; Liu, H.; Wang, Q.; Pan, S.; Zhao, D.; Liu, Q.; Liu, Y. ReaxFF-based molecular dynamics simulation of the initial pyrolysis mechanism of lignite. Fuel Process. Technol. 2019, 195, 106147. [Google Scholar] [CrossRef]
- Hong, D.; Li, P.; Si, T.; Guo, X.J.E. ReaxFF simulations of the synergistic effect mechanisms during co-pyrolysis of coal and polyethylene/polystyrene. Energy 2021, 218, 119553. [Google Scholar] [CrossRef]
- Fletcher, T.H.; Kerstein, A.R.; Pugmire, R.J.; Solum, M.S.; Grant, D.M. Chemical percolation model for devolatilization. 3. Direct use of carbon-13 NMR data to predict effects of coal type. Energy Fuels 1992, 6, 414–431. [Google Scholar] [CrossRef]
- Sinfrônio, F.S.M.; Santos, J.C.O.; Pereira, L.G.; Souza, A.G.; Conceiçăo, M.; Fernandes, V.J., Jr.; Fonseca, V.M. calorimetry, Kinetic of thermal degradation of low-density and high-density polyethylene by non-isothermal thermogravimetry. J. Therm. Anal. Calorim. 2005, 79, 393–399. [Google Scholar] [CrossRef]
- DiNenno, P.J.; Drysdale, D.; Beyler, C.L.; Walton, D.W. SFPE Handbook of Fire Protection Engineering, 3rd ed.; National Fire Protection Association: Quincy, MA, USA, 2002. [Google Scholar]
- Plimpton, S. Fast Parallel Algorithms for Short-Range Molecular Dynamics. J. Comput. Phys. 1995, 117, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Aktulga, H.M.; Fogarty, J.C.; Pandit, S.A.; Grama, A.Y. Parallel reactive molecular dynamics: Numerical methods and algorithmic techniques. Parallel Comput. 2012, 38, 245–259. [Google Scholar] [CrossRef] [Green Version]
- Hong, S.; Van Duin, A.C. Atomistic-scale analysis of carbon coating and its effect on the oxidation of aluminum nanoparticles by ReaxFF-molecular dynamics simulations. J. Phys. Chem. C 2016, 120, 9464–9474. [Google Scholar] [CrossRef]
- Martínez, L.; Andrade, R.; Birgin, E.G.; Martínez, J.M. PACKMOL: A package for building initial configurations for molecular dynamics simulations. J. Comput. Chem. 2009, 30, 2157–2164. [Google Scholar] [CrossRef]
- Hoover, W.G. Canonical dynamics: Equilibrium phase-space distributions. Phys. Rev. A 1985, 31, 1695–1697. [Google Scholar] [CrossRef] [Green Version]
- Salmon, E.; van Duin, A.C.; Lorant, F.; Marquaire, P.-M.; Goddard, W.A., III. Early maturation processes in coal. Part 2: Reactive dynamics simulations using the ReaxFF reactive force field on Morwell Brown coal structures. Org. Geochem. 2009, 40, 1195–1209. [Google Scholar] [CrossRef]
- Salmon, E.; van Duin, A.C.; Lorant, F.; Marquaire, P.-M.; Goddard, W.A., III. Thermal decomposition process in algaenan of Botryococcus braunii race L. Part 2: Molecular dynamics simulations using the ReaxFF reactive force field. Org. Geochem. 2009, 40, 416–427. [Google Scholar] [CrossRef]
- Chenoweth, K.; Cheung, S.; Van Duin, A.C.; Goddard, W.A.; Kober, E.M. Simulations on the thermal decomposition of a poly (dimethylsiloxane) polymer using the ReaxFF reactive force field. J. Am. Chem. Soc. 2005, 127, 7192–7202. [Google Scholar] [CrossRef] [Green Version]
- Diao, Z.; Zhao, Y.; Chen, B.; Duan, C.; Song, S. ReaxFF reactive force field for molecular dynamics simulations of epoxy resin thermal decomposition with model compound. J. Anal. Appl. Pyrolysis 2013, 104, 618–624. [Google Scholar] [CrossRef]
- Risberg, D.; Risberg, M.; Westerlund, L. CFD modelling of radiators in buildings with user-defined wall functions. Appl. Therm. Eng. 2016, 94, 266–273. [Google Scholar] [CrossRef]
- De Cachinho Cordeiro, I.M.; Liu, H.; Yuen, A.C.Y.; Chen, T.B.Y.; Li, A.; Cao, R.F.; Yeoh, G.H. Numerical investigation of expandable graphite suppression on metal-based fire. Heat Mass Transf. 2021. [Google Scholar] [CrossRef]
- De Cachinho Cordeiro, I.M.; Liu, H.; Yuen, A.C.Y.; Chen, T.B.Y.; Li, A.; Yeoh, G.H. Numerical assessment of LES subgrid-scale turbulence models for expandable particles in fire suppression. Exp. Comput. Multiph. Flow 2021. [Google Scholar] [CrossRef]
- e Silva, V.B.R.; Cardoso, J. Computational Fluid Dynamics Applied to Waste-to-Energy Processes: A Hands-on Approach; Butterworth-Heinemann: Cambridge, MA, USA, 2020. [Google Scholar]
- Mochizuki, H. Verification of neutronics and thermal-hydraulics coupling method for FLUENT code using the MSRE pump startup, trip data. Nucl. Eng. Des. 2021, 378, 111191. [Google Scholar] [CrossRef]
- Brookes, S.J.; Moss, J.B. Predictions of soot and thermal radiation properties in confined turbulent jet diffusion flames. Combust. Flame 1999, 116, 486–503. [Google Scholar] [CrossRef]
- Yuen, A.C.Y.; Yeoh, G.H. Numerical simulation of an enclosure fire in a large test hall. Comput. Therm. Sci. 2013, 5, 459–471. [Google Scholar] [CrossRef]
- Yuen, A.C.Y.; Yeoh, G.H.; Yuen, R.K.K.; Chen, T. Numerical Simulation of a Ceiling Jet Fire in a Large Compartment. Procedia Eng. 2013, 52, 3–12. [Google Scholar]
- Nicoud, F.; Ducros, F. Subgrid-Scale Stress Modelling Based on the Square of the Velocity Gradient Tensor. Flow Turbul. Combust. 1999, 62, 183–200. [Google Scholar] [CrossRef]
- Smagorinsky, J. General circulation experiments with the primitive equations, i. the basic experiment. Mon. Weather Rev. 1963, 91, 99–164. [Google Scholar] [CrossRef]
- Yuen, A.C.Y.; Yeoh, G.H.; Timchenko, V.; Cheung, S.C.P.; Chen, T. Study of three LES subgrid-scale turbulence models for predictions of heat and mass transfer in large-scale compartment fires. Numer. Heat Transf. Part A 2016, 69, 1223–1241. [Google Scholar] [CrossRef]
- Chen, Q.; Chen, T.B.Y.; Yuen, A.C.Y.; Wang, C.; Chan, Q.N.; Yeoh, G.H. Investigation of door width towards flame tilting behaviours and combustion species in compartment fire scenarios using large eddy simulation. Int. J. Heat Mass Transf. 2020, 150, 119373. [Google Scholar] [CrossRef]
- Smith, G.P.; Golden, D.M.; Frenklach, M.; Moriarty, N.W.; Eiteneer, B.; Goldenberg, M.; Bowman, C.T.; Hanson, R.K.; Song, S.; Gardiner, W.C., Jr.; et al. GRI-Mech, Version 3.0. 2000. Available online: http://www.me.berkeley.edu/gri_mech/ (accessed on 11 June 2021).
- Kee, R.J.; Rupley, F.M.; Miller, J.A.; Coltrin, M.E.; Grcar, J.F.; Meeks, E. CHEMKIN Collection, Release 3.6; Reaction Design, Inc.: San Diego, CA, USA, 2000. [Google Scholar]
- Yuen, A.C.Y.; Yeoh, G.H.; Timchenko, V.; Chen, T.B.Y.; Chan, Q.N.; Wang, C.; Li, D.D. Comparison of detailed soot formation models for sooty and non-sooty flames in an under-ventilated ISO room. Int. J. Heat Mass Transf. 2017, 115, 717–729. [Google Scholar] [CrossRef]
- Yuen, A.C.Y.; Yeoh, G.H.; Timchenko, V.; Cheung, S.C.P.; Barber, T.J. Importance of detailed chemical kinetics on combustion and soot modelling of ventilated and under-ventilated fires in compartment. Int. J. Heat Mass Transf. 2016, 96, 171–188. [Google Scholar] [CrossRef]
- Jamaluddin, A.S.; Smith, P.J. Predicting Radiative Transfer in Rectangular Enclosures Using the Discrete Ordinates Method. Combust. Sci. Technol. 1988, 59, 321–340. [Google Scholar] [CrossRef]
- Beer, J.M.; Foster, P.J.; Siddall, R.G. Calculation Methods of Radiation Heat Transfer; HFTS Design Report No. 22; AEA Technology: Burlington, MA, USA, 1971. [Google Scholar]
- Kent, J.; Honnery, D. A soot formation rate map for a laminar ethylene diffusion flame. Combust. Flame 1990, 79, 287–298. [Google Scholar] [CrossRef]


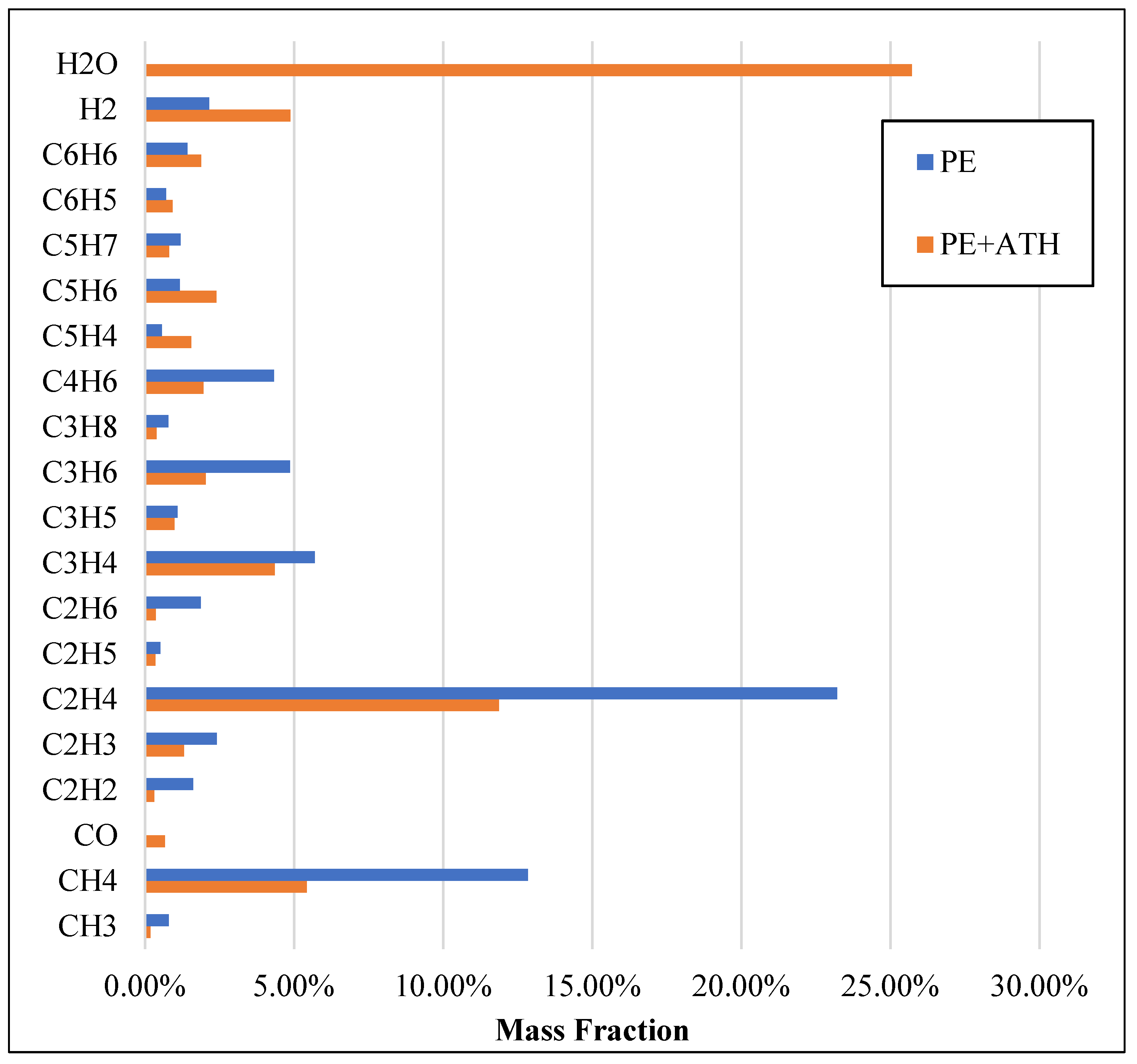




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Polymer Type | Ea (kJ/mol) | A (1/s) | |
|---|---|---|---|
| PE | MD | 273.25 | 1.798 × 1016 |
| MD-CN | 233.06 | 1.76 × 1013 | |
| TGA Experiment | 266.74 | 1.52 × 1016 | |
| PE + ATH | MD | 325.43 | 5.0291 × 1011 |
| MD-CN | 285.41 | 5.03 × 1013 |
| Mesh | Mesh Size (mm) | Total Number of Cells |
|---|---|---|
| Mesh 1 (coarse) | 5 | 676,875 |
| Mesh 2 (medium) | 2 | 1,353,750 |
| Mesh 3 (fine) | 1.5 | 3,210,000 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, T.B.Y.; De Cachinho Cordeiro, I.M.; Yuen, A.C.Y.; Yang, W.; Chan, Q.N.; Zhang, J.; Cheung, S.C.P.; Yeoh, G.H. An Investigation towards Coupling Molecular Dynamics with Computational Fluid Dynamics for Modelling Polymer Pyrolysis. Molecules 2022, 27, 292. https://doi.org/10.3390/molecules27010292
Chen TBY, De Cachinho Cordeiro IM, Yuen ACY, Yang W, Chan QN, Zhang J, Cheung SCP, Yeoh GH. An Investigation towards Coupling Molecular Dynamics with Computational Fluid Dynamics for Modelling Polymer Pyrolysis. Molecules. 2022; 27(1):292. https://doi.org/10.3390/molecules27010292
Chicago/Turabian StyleChen, Timothy Bo Yuan, Ivan Miguel De Cachinho Cordeiro, Anthony Chun Yin Yuen, Wei Yang, Qing Nian Chan, Jin Zhang, Sherman C. P. Cheung, and Guan Heng Yeoh. 2022. "An Investigation towards Coupling Molecular Dynamics with Computational Fluid Dynamics for Modelling Polymer Pyrolysis" Molecules 27, no. 1: 292. https://doi.org/10.3390/molecules27010292
APA StyleChen, T. B. Y., De Cachinho Cordeiro, I. M., Yuen, A. C. Y., Yang, W., Chan, Q. N., Zhang, J., Cheung, S. C. P., & Yeoh, G. H. (2022). An Investigation towards Coupling Molecular Dynamics with Computational Fluid Dynamics for Modelling Polymer Pyrolysis. Molecules, 27(1), 292. https://doi.org/10.3390/molecules27010292