
The Kynurenine Pathway and Kynurenine 3-Monooxygenase Inhibitors

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
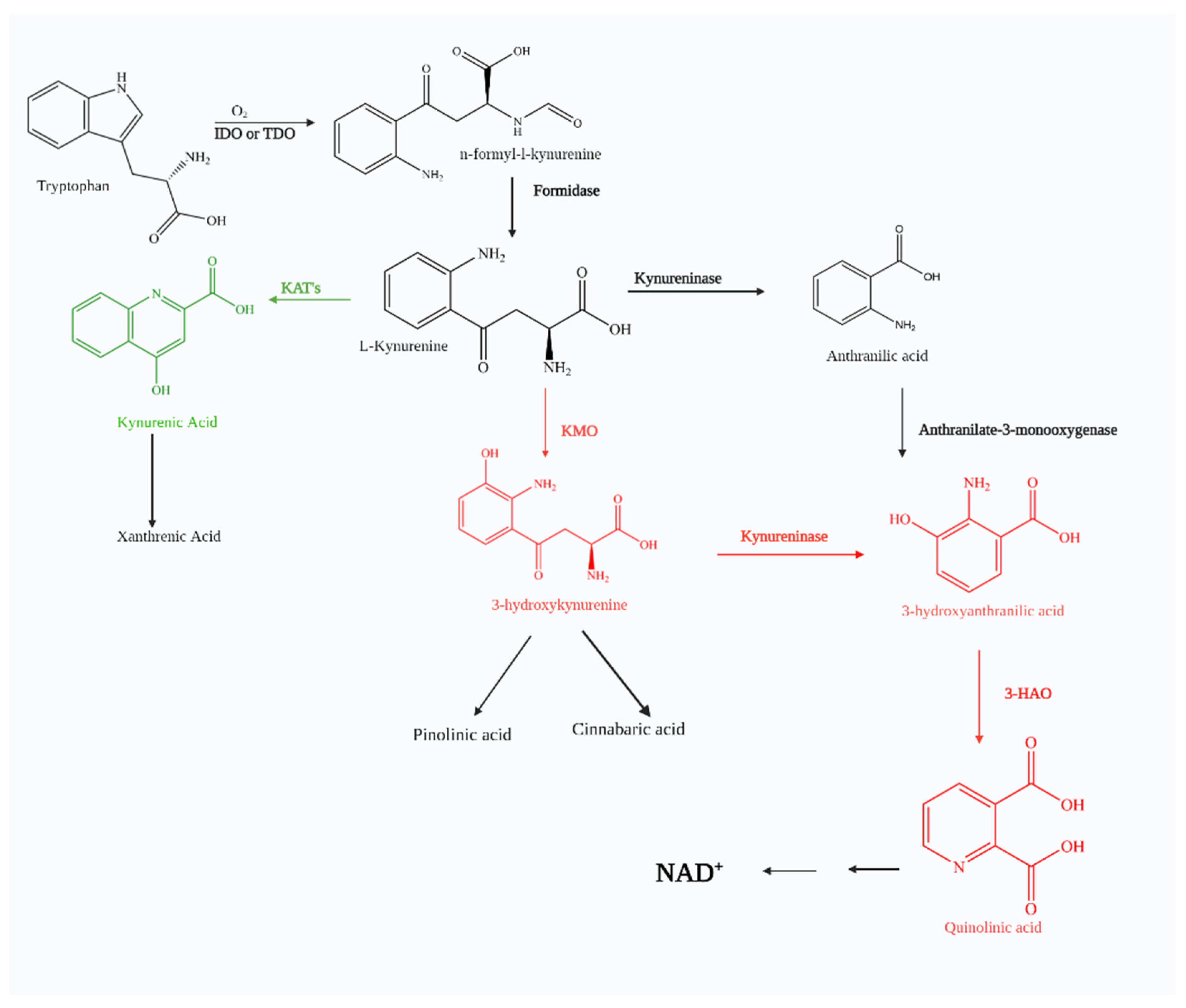
1. The Kynurenine Pathway
1.1. Kynurenic Acid
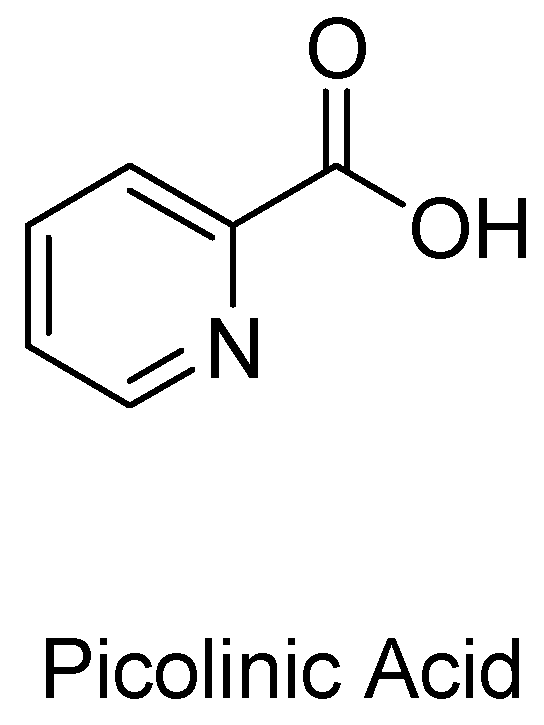
1.2. Picolinic Acid and Other Possible Neuroprotective Metabolites
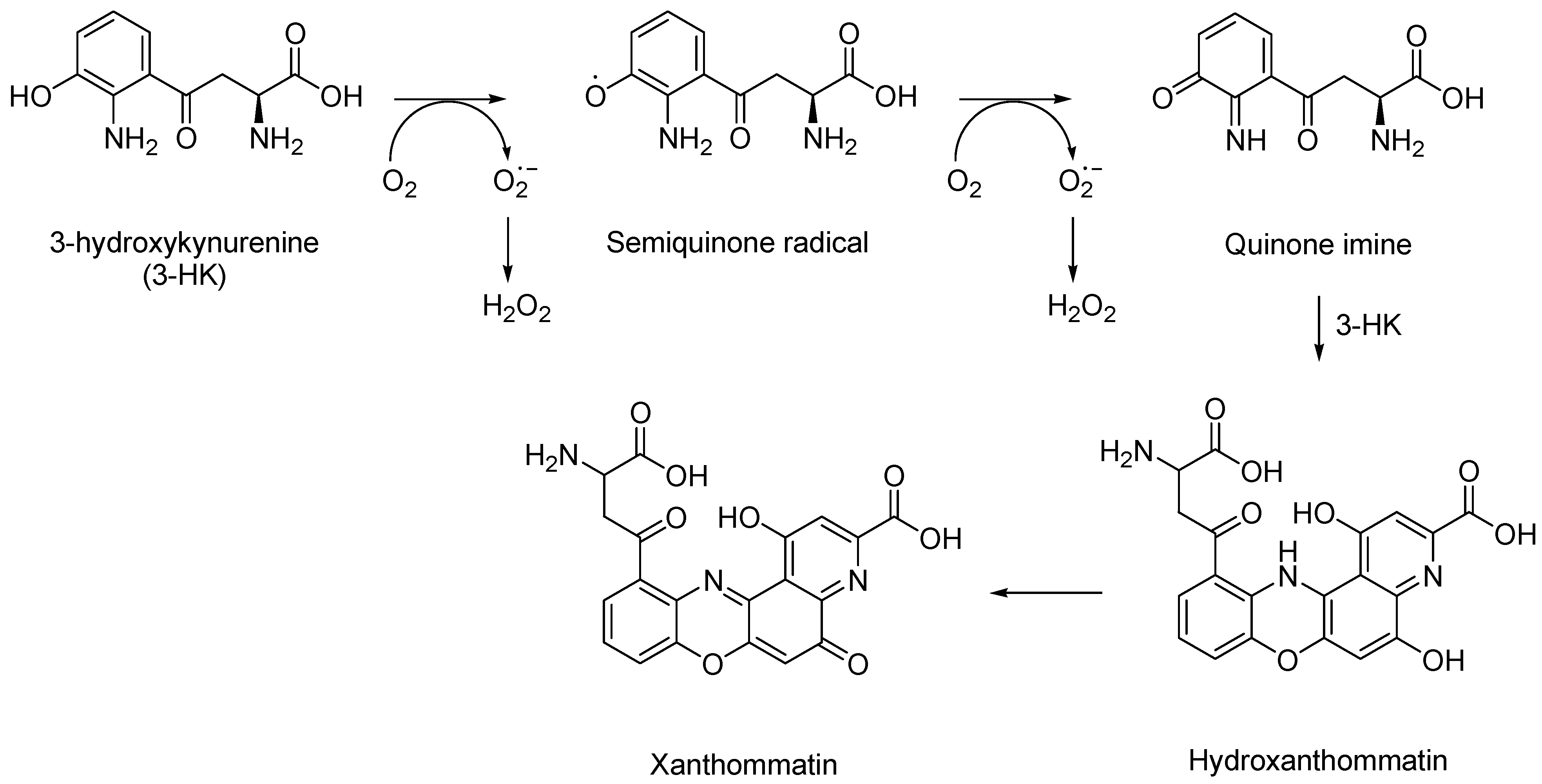
1.3. 3-Hydroxykynurenine
1.4. 3-Hydroxyanthranilic Acid
1.5. Quinolinic Acid
1.6. Kynurenine 3-Monooxygenase (KMO)
2. KMO in Disease
2.1. Huntington’s Disease
2.2. Alzheimer’s Disease
2.3. Parkinson’s Disease
2.4. Epilepsy
2.5. Neuropsychiatric Disorders
2.6. Stroke
2.7. Other Immune-Related Illnesses
3. KMO Mechanisms and Properties
3.1. Efforts to Isolate and Characterize KMO
3.2. Structures
3.3. Pseudomonas Fluorescens
3.4. Human KMO
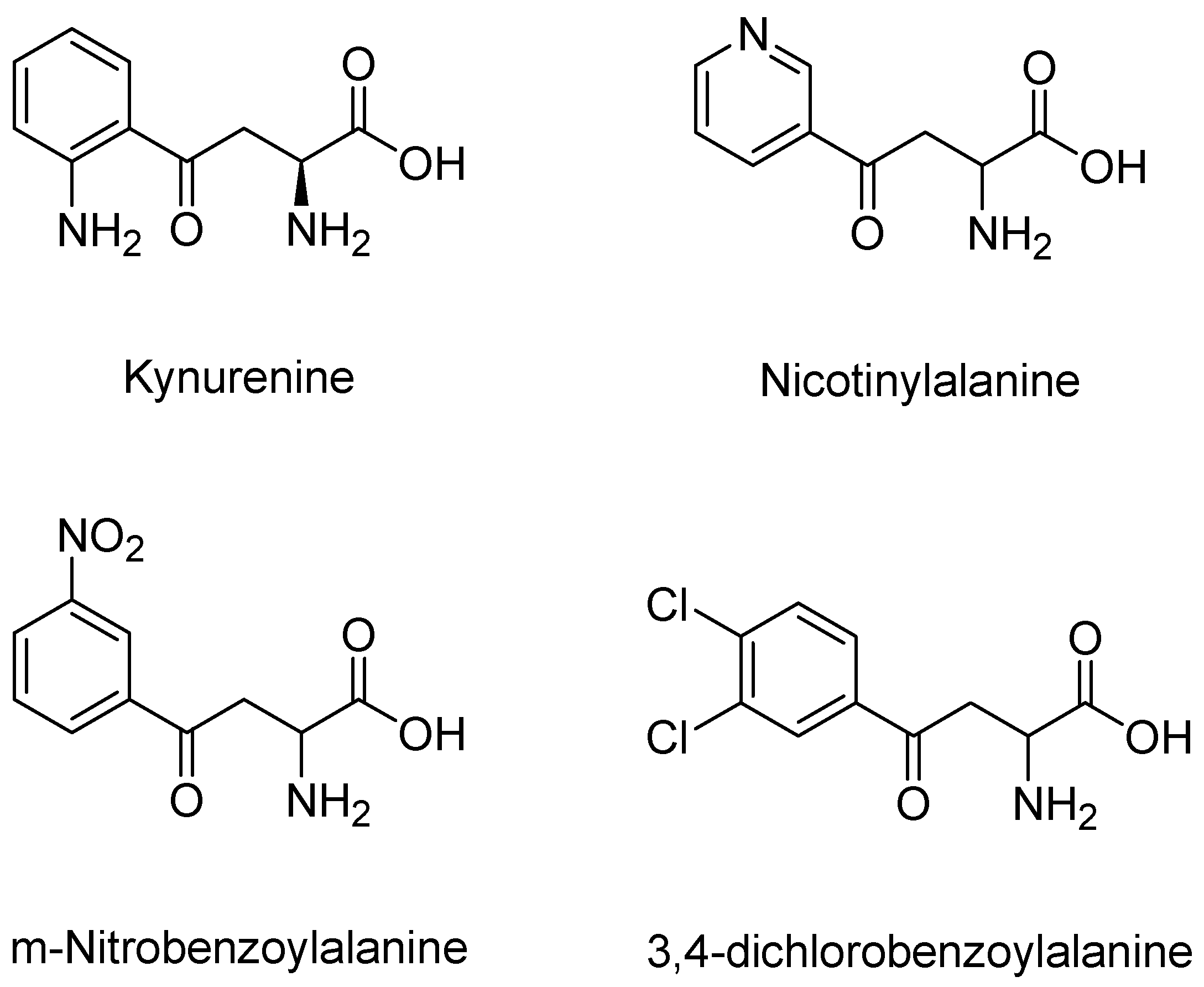
3.5. KMO Inhibitors
4. Pharmacophore Development
4.1. KMO Inhibitor Pharmacophore Development
4.2. Inhibition Studies
5. Discussion and Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Fila, M.; Chojnacki, J.; Pawlowska, E.; Szczepanska, J.; Chojnacki, C.; Blasiak, J. Kynurenine Pathway of Tryptophan Metabolism in Migraine and Functional Gastrointestinal Disorders. Int. J. Mol. Sci. 2021, 22, 10134. [Google Scholar] [CrossRef]
- Kaur, H.; Bose, C.; Mande, S.S. Tryptophan Metabolism by Gut Microbiome and Gut-Brain-Axis: An in silico Analysis. Front. Neurosci. 2019, 13, 1365. [Google Scholar] [CrossRef]
- Moroni, F.; Russi, P.; Lombardi, G.; Beni, M.; Carlà, V. Presence of Kynurenic Acid in the Mammalian Brain. J. Neurochem. 1988, 51, 177–180. [Google Scholar] [CrossRef] [PubMed]
- Bertolino, M.; Vicini, S.; Costa, E. Kynurenic acid inhibits the activation of kainic and N-methyl-D-aspartic acid-sensitive ionotropic receptors by a different mechanism. Neuropharmacology 1989, 28, 453–457. [Google Scholar] [CrossRef]
- Hilmas, C.; Pereira, E.F.R.; Alkondon, M.; Rassoulpour, A.; Schwarcz, R.; Albuquerque, E.X. The Brain Metabolite Kynurenic Acid Inhibits α7 Nicotinic Receptor Activity and Increases Non-α7 Nicotinic Receptor Expression: Physiopathological Implications. J. Neurosci. 2001, 21, 7463. [Google Scholar] [CrossRef] [PubMed]
- Cozzi, A.; Carpenedo, R.; Moroni, F. Kynurenine Hydroxylase Inhibitors Reduce Ischemic Brain Damage: Studies with (m-Nitrobenzoyl)-Alanine (mNBA) and 3,4-Dimethoxy-[-N-4-(Nitrophenyl)Thiazol-2YL]-Benzenesulfonamide (Ro 61-8048) in Models of Focal or Global Brain Ischemia. J. Cereb. Blood Flow Metab. 1999, 19, 771–777. [Google Scholar] [CrossRef] [PubMed]
- Myint, A.M.; Kim, Y.K.; Verkerk, R.; Scharpé, S.; Steinbusch, H.; Leonard, B. Kynurenine pathway in major depression: Evidence of impaired neuroprotection. J. Affect. Disord. 2007, 98, 143–151. [Google Scholar] [CrossRef]
- Hartai, Z.; Juhász, A.; Rimanóczy, A.; Janáky, T.; Donkó, T.; Dux, L.; Penke, B.; Tóth, G.K.; Janka, Z.; Kálmán, J. Decreased serum and red blood cell kynurenic acid levels in Alzheimer’s disease. Neurochem. Int. 2007, 50, 308–313. [Google Scholar] [CrossRef]
- Beal, M.F.; Matson, W.R.; Storey, E.; Milbury, P.; Ryan, E.A.; Ogawa, T.; Bird, E.D. Kynurenic acid concentrations are reduced in Huntington’s disease cerebral cortex. J. Neurol. Sci. 1992, 108, 80–87. [Google Scholar] [CrossRef]
- Ogawa, T.; Matson, W.R.; Beal, M.F.; Myers, R.H.; Bird, E.D.; Milbury, P.; Saso, S. Kynurenine pathway abnormalities in Parkinson’s disease. Neurology 1992, 42, 1702–1706. [Google Scholar] [CrossRef]
- Kwok, J.B.J.; Kapoor, R.; Gotoda, T.; Iwamoto, Y.; Iizuka, Y.; Yamada, N.; Isaacs, K.E.; Kushwaha, V.V.; Church, W.B.; Schofield, P.R.; et al. A Missense Mutation in Kynurenine Aminotransferase-1 in Spontaneously Hypertensive Rats. J. Biol. Chem. 2002, 277, 35779–35782. [Google Scholar] [CrossRef] [PubMed]
- Walczak, K.; Żurawska, M.; Kiś, J.; Starownik, R.; Zgrajka, W.; Bar, K.; Turski, W.A.; Rzeski, W. Kynurenic acid in human renal cell carcinoma: Its antiproliferative and antimigrative action on Caki-2 cells. Amino Acids 2012, 43, 1663–1670. [Google Scholar] [CrossRef] [PubMed]
- Walczak, K.; Turski, W.A.; Rajtar, G. Kynurenic acid inhibits colon cancer proliferation in vitro: Effects on signaling pathways. Amino Acids 2014, 46, 2393–2401. [Google Scholar] [CrossRef] [PubMed]
- Lugo-Huitrón, R.; Blanco-Ayala, T.; Ugalde-Muñiz, P.; Carrillo-Mora, P.; Pedraza-Chaverrí, J.; Silva-Adaya, D.; Maldonado, P.D.; Torres, I.; Pinzón, E.; Ortiz-Islas, E.; et al. On the antioxidant properties of kynurenic acid: Free radical scavenging activity and inhibition of oxidative stress. Neurotoxicol. Teratol. 2011, 33, 538–547. [Google Scholar] [CrossRef]
- Grace, A.A. Phasic versus tonic dopamine release and the modulation of dopamine system responsivity: A hypothesis for the etiology of schizophrenia. Neuroscience 1991, 41, 1–24. [Google Scholar] [CrossRef]
- Erhardt, S.; Schwieler, L.; Nilsson, L.; Linderholm, K.; Engberg, G. The kynurenic acid hypothesis of schizophrenia. Physiol. Behav. 2007, 92, 203–209. [Google Scholar] [CrossRef]
- Sagan, D.; Kocki, T.; Kocki, J.; Szumilo, J. Serum kynurenic acid: Possible association with invasiveness of non-small cell lung cancer. Asian Pac. J. Cancer Prev. 2012, 13, 4241–4244. [Google Scholar] [CrossRef]
- Baran, H.; Hainfellner, J.A.; Kepplinger, B.; Mazal, P.R.; Schmid, H.; Budka, H. Kynurenic acid metabolism in the brain of HIV-1 infected patients. J. Neural. Transm. 2000, 107, 1127–1138. [Google Scholar] [CrossRef]
- Zarnowski, T.; Rejdak, R.; Zielinska-Rzecka, E.; Zrenner, E.; Grieb, P.; Zagórski, Z.; Junemann, A.; Turski, W.A. Elevated concentrations of kynurenic acid, a tryptophan derivative, in dense nuclear cataracts. Curr. Eye Res. 2007, 32, 27–32. [Google Scholar] [CrossRef]
- Holtze, M.; Mickiené, A.; Atlas, A.; Lindquist, L.; Schwieler, L. Elevated cerebrospinal fluid kynurenic acid levels in patients with tick-borne encephalitis. J. Intern. Med. 2012, 272, 394–401. [Google Scholar] [CrossRef]
- Tanaka, M.; Bohár, Z.; Martos, D.; Telegdy, G.; Vécsei, L. Antidepressant-like effects of kynurenic acid in a modified forced swim test. Pharmacol. Rep. 2020, 72, 449–455. [Google Scholar] [CrossRef] [PubMed]
- Spekker, E.; Laborc, K.F.; Bohár, Z.; Nagy-Grócz, G.; Fejes-Szabó, A.; Szűcs, M.; Vécsei, L.; Párdutz, Á. Effect of dural inflammatory soup application on activation and sensitization markers in the caudal trigeminal nucleus of the rat and the modulatory effects of sumatriptan and kynurenic acid. J. Headache Pain 2021, 22, 17. [Google Scholar] [CrossRef]
- Suzuki, K.; Yasuda, M.; Yamasaki, K. Stability Constants of Picolinic and Quinaldic Acid Chelates of Bivalent Metals. J. Phys. Chem. 1957, 61, 229–231. [Google Scholar] [CrossRef]
- Broadhurst, C.L.; Domenico, P. Clinical studies on chromium picolinate supplementation in diabetes mellitus—A review. Diabetes Technol. 2006, 8, 677–687. [Google Scholar] [CrossRef]
- Ruffmann, R.; Schlick, R.; Chirigos, M.A.; Budzynsky, W.; Varesio, L. Antiproliferative activity of picolinic acid due to macrophage activation. Drugs Exp. Clin. Res. 1987, 13, 607–614. [Google Scholar]
- Abe, S.; Hu, W.; Ishibashi, H.; Hasumi, K.; Yamaguchi, H. Augmented inhibition of Candida albicans growth by murine neutrophils in the presence of a tryptophan metabolite, picolinic acid. J. Infect. Chemother. 2004, 10, 181–184. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Pol, J.A.; Klos, D.J.; Hamilton, P.D. Cytotoxic activity of fusaric acid on human adenocarcinoma cells in tissue culture. Anticancer Res. 1993, 13, 57–64. [Google Scholar]
- Beninger, R.J.; Colton, A.M.; Ingles, J.L.; Jhamandas, K.; Boegman, R.J. Picolinic acid blocks the neurotoxic but not the neuroexcitant properties of quinolinic acid in the rat brain: Evidence from turning behaviour and tyrosine hydroxylase immunohistochemistry. Neuroscience 1994, 61, 603–612. [Google Scholar] [CrossRef]
- Jhamandas, K.H.; Boegman, R.J.; Beninger, R.J.; Flesher, S. Role of zinc in blockade of excitotoxic action of quinolinic acid by picolinic acid. Amino Acids 1998, 14, 257–261. [Google Scholar] [CrossRef]
- Vrooman, L.; Jhamandas, K.; Boegman, R.J.; Beninger, R.J. Picolinic acid modulates kainic acid-evoked glutamate release from the striatum in vitro. Brain Res. 1993, 627, 193–198. [Google Scholar] [CrossRef]
- Fazio, F.; Lionetto, L.; Molinaro, G.; Bertrand, H.O.; Acher, F.; Ngomba, R.T.; Notartomaso, S.; Curini, M.; Rosati, O.; Scarselli, P.; et al. Cinnabarinic acid, an endogenous metabolite of the kynurenine pathway, activates type 4 metabotropic glutamate receptors. Mol. Pharm. 2012, 81, 643–656. [Google Scholar] [CrossRef] [PubMed]
- Fazio, F.; Lionetto, L.; Curto, M.; Iacovelli, L.; Copeland, C.S.; Neale, S.A.; Bruno, V.; Battaglia, G.; Salt, T.E.; Nicoletti, F. Cinnabarinic acid and xanthurenic acid: Two kynurenine metabolites that interact with metabotropic glutamate receptors. Neuropharmacology 2017, 112, 365–372. [Google Scholar] [CrossRef]
- Malina, H.Z.; Martin, X.D. Deamination of 3-hydroxykynurenine in bovine lenses: A possible mechanism of cataract formation in general. Graefe’s Arch. Clin. Exp. Ophthalmol. 1995, 233, 38–44. [Google Scholar] [CrossRef] [PubMed]
- Colín-González, A.L.; Maldonado, P.D.; Santamaría, A. 3-Hydroxykynurenine: An intriguing molecule exerting dual actions in the central nervous system. Neurotoxicology 2013, 34, 189–204. [Google Scholar] [CrossRef]
- Vazquez, S.; Garner, B.; Sheil, M.M.; Truscott, R.J. Characterisation of the major autoxidation products of 3-hydroxykynurenine under physiological conditions. Free Radic Res. 2000, 32, 11–23. [Google Scholar] [CrossRef]
- Eastman, C.L.; Guilarte, T.R. The role of hydrogen peroxide in the in vitro cytotoxicity of 3-hydroxykynurenine. Neurochem. Res. 1990, 15, 1101–1107. [Google Scholar] [CrossRef] [PubMed]
- Okuda, S.; Nishiyama, N.; Saito, H.; Katsuki, H. Hydrogen peroxide-mediated neuronal cell death induced by an endogenous neurotoxin, 3-hydroxykynurenine. Proc. Natl. Acad. Sci. USA 1996, 93, 12553. [Google Scholar] [CrossRef]
- Eastman, C.L.; Guilarte, T.R. Cytotoxicity of 3-hydroxykynurenine in a neuronal hybrid cell line. Brain Res. 1989, 495, 225–231. [Google Scholar] [CrossRef]
- Pawlak, K.; Domaniewski, T.; Mysliwiec, M.; Pawlak, D. The kynurenines are associated with oxidative stress, inflammation and the prevalence of cardiovascular disease in patients with end-stage renal disease. Atherosclerosis 2009, 204, 309–314. [Google Scholar] [CrossRef]
- Topczewska-Bruns, J.; Pawlak, D.; Chabielska, E.; Tankiewicz, A.; Buczko, W. Increased levels of 3-hydroxykynurenine in different brain regions of rats with chronic renal insufficiency. Brain Res. Bull. 2002, 58, 423–428. [Google Scholar] [CrossRef]
- Schwarz, M.J.; Guillemin, G.J.; Teipel, S.J.; Buerger, K.; Hampel, H. Increased 3-hydroxykynurenine serum concentrations differentiate Alzheimer’s disease patients from controls. Eur. Arch. Psychiatry Clin. Neurosci. 2013, 263, 345–352. [Google Scholar] [CrossRef] [PubMed]
- Guidetti, P.; Bates, G.P.; Graham, R.K.; Hayden, M.R.; Leavitt, B.R.; MacDonald, M.E.; Slow, E.J.; Wheeler, V.C.; Woodman, B.; Schwarcz, R. Elevated brain 3-hydroxykynurenine and quinolinate levels in Huntington disease mice. Neurobiol. Dis. 2006, 23, 190–197. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, F.; Tsubouchi, R.; Izuta, S.; Shibata, Y. Kynurenine metabolism and xanthurenic acid formation in vitamin B6-deficient rat after tryptophan injection. J. Nutr. Sci. Vitam. 1989, 35, 111–122. [Google Scholar] [CrossRef] [PubMed]
- Dietrich-Muszalska, A.; Chauhan, V.; Grignon, S. Studies on Psychiatric Disorders; Dietrich-Muszalska, A., Chauhan, V., Grignon, S., Eds.; Humana Press: Totowa, NJ, USA, 2015. [Google Scholar] [CrossRef]
- Manthey, M.K.; Pyne, S.G.; Truscott, R.J.W. Structural elucidation and independent synthesis of the radical-radical coupling products of 3-hydroxyanthranilic acid with tyrosine and phenols. J. Org. Chem. 1990, 55, 4581–4585. [Google Scholar] [CrossRef]
- Boyland, E.; Wallace, D.M.; Williams, D.C. The activity of the enzymes sulphatase and beta-glucuronidase in the urine, serum and bladder tissue. Br. J. Cancer 1955, 9, 62–79. [Google Scholar] [CrossRef] [PubMed]
- Stone, T.W. Kynurenines in the CNS: From endogenous obscurity to therapeutic importance. Prog. Neurobiol. 2001, 64, 185–218. [Google Scholar] [CrossRef]
- Carpenedo, R.; Chiarugi, A.; Russi, P.; Lombardi, G.; Carlà, V.; Pellicciari, R.; Mattoli, L.; Moroni, F. Inhibitors of kynurenine hydroxylase and kynureninase increase cerebral formation of kynurenate and have sedative and anticonvulsant activities. Neuroscience 1994, 61, 237–244. [Google Scholar] [CrossRef]
- Heyes, M.P.; Saito, K.; Crowley, J.S.; Davis, L.E.; Demitrack, M.A.; Der, M.; Dilling, L.A.; Elia, J.; Kruesi, M.J.; Lackner, A.; et al. Quinolinic acid and kynurenine pathway metabolism in inflammatory and non-inflammatory neurological disease. Brain 1992, 115 Pt 5, 1249–1273. [Google Scholar] [CrossRef]
- Chen, Y.; Guillemin, G.J. Kynurenine pathway metabolites in humans: Disease and healthy States. Int. J. Tryptophan Res. 2009, 2, 1–19. [Google Scholar] [CrossRef]
- Guillemin, G.J. Quinolinic acid, the inescapable neurotoxin. FEBS J. 2012, 279, 1356–1365. [Google Scholar] [CrossRef]
- Aguilera, P.; Chánez-Cárdenas, M.E.; Floriano-Sánchez, E.; Barrera, D.; Santamaría, A.; Sánchez-González, D.J.; Pérez-Severiano, F.; Pedraza-Chaverrí, J.; Jiménez, P.D.M. Time-related changes in constitutive and inducible nitric oxide synthases in the rat striatum in a model of Huntington’s disease. Neurotoxicology 2007, 28, 1200–1207. [Google Scholar] [CrossRef]
- Lee, M.-C.; Ting, K.K.; Adams, S.; Brew, B.J.; Chung, R.; Guillemin, G.J. Characterisation of the Expression of NMDA Receptors in Human Astrocytes. PLoS ONE 2010, 5, e14123. [Google Scholar] [CrossRef] [PubMed]
- Pierozan, P.; Zamoner, A.; Soska, A.K.; Silvestrin, R.B.; Loureiro, S.O.; Heimfarth, L.; Mello e Souza, T.; Wajner, M.; Pessoa-Pureur, R. Acute intrastriatal administration of quinolinic acid provokes hyperphosphorylation of cytoskeletal intermediate filament proteins in astrocytes and neurons of rats. Exp. Neurol. 2010, 224, 188–196. [Google Scholar] [CrossRef]
- Rahman, A.; Ting, K.; Cullen, K.M.; Braidy, N.; Brew, B.J.; Guillemin, G.J. The Excitotoxin Quinolinic Acid Induces Tau Phosphorylation in Human Neurons. PLoS ONE 2009, 4, e6344. [Google Scholar] [CrossRef]
- Allegri, G.; Costa, C.V.L.; Bertazzo, A. Recent Advances in Tryptophan Research: Tryptophan and Serotonin Pathways; Springer Science & Business Media: Berlin/Heidelberg, Germany, 2012. [Google Scholar]
- Aeinehband, S.; Brenner, P.; Ståhl, S.; Bhat, M.; Fidock, M.D.; Khademi, M.; Olsson, T.; Engberg, G.; Jokinen, J.; Erhardt, S.; et al. Cerebrospinal fluid kynurenines in multiple sclerosis; relation to disease course and neurocognitive symptoms. Brain Behav. Immun. 2016, 51, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Saito, K.; Nowak, T.S., Jr.; Markey, S.P.; Heyes, M.P. Mechanism of delayed increases in kynurenine pathway metabolism in damaged brain regions following transient cerebral ischemia. J. Neurochem. 1993, 60, 180–192. [Google Scholar] [CrossRef]
- Dobbie, M.; Crawley, J.; Waruiru, C.; Marsh, K.; Surtees, R. Cerebrospinal fluid studies in children with cerebral malaria: An excitotoxic mechanism? Am. J. Trop. Med. Hyg. 2000, 62, 284–290. [Google Scholar] [CrossRef][Green Version]
- Heyes, M.P.; Saito, K.; Devinsky, O.; Nadi, N.S. Kynurenine pathway metabolites in cerebrospinal fluid and serum in complex partial seizures. Epilepsia 1994, 35, 251–257. [Google Scholar] [CrossRef]
- Okamoto, H.; Yamamoto, S.; Nozaki, M.; Hayaishi, O. On the submitochondrial localization of l-kynurenine-3-hydroxylase. Biochem. Biophys. Res. Commun. 1967, 26, 309–314. [Google Scholar] [CrossRef]
- Crozier, K.R.; Moran, G.R. Heterologous expression and purification of kynurenine-3-monooxygenase from Pseudomonas fluorescens strain 17400. Protein Expr. Purif. 2007, 51, 324–333. [Google Scholar] [CrossRef] [PubMed]
- Van Berkel, W.J.; Kamerbeek, N.M.; Fraaije, M.W. Flavoprotein monooxygenases, a diverse class of oxidative biocatalysts. J. Biotechnol. 2006, 124, 670–689. [Google Scholar] [CrossRef]
- Alberati-Giani, D.; Cesura, A.M.; Broger, C.; Warren, W.D.; Röver, S.; Malherbe, P. Cloning and functional expression of human kynurenine 3-monooxygenase. FEBS Lett. 1997, 410, 407–412. [Google Scholar] [CrossRef]
- Breton, J.; Avanzi, N.; Magagnin, S.; Covini, N.; Magistrelli, G.; Cozzi, L.; Isacchi, A. Functional characterization and mechanism of action of recombinant human kynurenine 3-hydroxylase. Eur. J. Biochem. 2000, 267, 1092–1099. [Google Scholar] [CrossRef] [PubMed]
- Schwarcz, R.; Köhler, C. Differential vulnerability of central neurons of the rat to quinolinic acid. Neurosci. Lett. 1983, 38, 85–90. [Google Scholar] [CrossRef]
- Goodman, L.S.; Gilman, A.; Hardman, J.G.; Gilman, A.G.; Limbird, L.E. Goodman & Gilman’s the Pharmacological Basis of Therapeutics, 9th ed.; Hardman, J.G., Limbird, L.E., Molinoff, P.B., Ruddon, R.W., Gilman, A.G., Kunkel, E., Eds.; McGraw-Hill, Health Professions Division: New York, NY, USA, 1996. [Google Scholar]
- Sari, Y. Huntington’s Disease: From Mutant Huntingtin Protein to Neurotrophic Factor Therapy. Int. J. Biomed. Sci. 2011, 7, 89–100. [Google Scholar]
- Carlock, L.; Walker, P.D.; Shan, Y.; Gutridge, K. Transcription of the Huntington disease gene during the quinolinic acid excitotoxic cascade. Neuroreport 1995, 6, 1121–1124. [Google Scholar] [CrossRef]
- Ramaswamy, S.; McBride, J.L.; Kordower, J.H. Animal models of Huntington’s disease. Ilar J. 2007, 48, 356–373. [Google Scholar] [CrossRef] [PubMed]
- Zeron, M.M.; Hansson, O.; Chen, N.; Wellington, C.L.; Leavitt, B.R.; Brundin, P.; Hayden, M.R.; Raymond, L.A. Increased sensitivity to N-methyl-D-aspartate receptor-mediated excitotoxicity in a mouse model of Huntington’s disease. Neuron 2002, 33, 849–860. [Google Scholar] [CrossRef]
- Sapko, M.T.; Guidetti, P.; Yu, P.; Tagle, D.A.; Pellicciari, R.; Schwarcz, R. Endogenous kynurenate controls the vulnerability of striatal neurons to quinolinate: Implications for Huntington’s disease. Exp. Neurol. 2006, 197, 31–40. [Google Scholar] [CrossRef]
- Reynolds, G.P.; Pearson, S.J.; Halket, J.; Sandier, M. Brain Quinolinic Acid in Huntington’s Disease. J. Neurochem. 1988, 50, 1959–1968. [Google Scholar] [CrossRef]
- Guidetti, P.; Luthi-Carter, R.E.; Augood, S.J.; Schwarcz, R. Neostriatal and cortical quinolinate levels are increased in early grade Huntington’s disease. Neurobiol. Dis. 2004, 17, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Pearson, S.J.; Reynolds, G.P. Increased brain concentrations of a neurotoxin, 3-hydroxykynurenine, in Huntington’s disease. Neurosci. Lett. 1992, 144, 199–201. [Google Scholar] [CrossRef]
- Perez-De La Cruz, V.; Santamaria, A. Integrative hypothesis for Huntington’s disease: A brief review of experimental evidence. Physiol. Res. 2007, 56, 513–526. [Google Scholar] [CrossRef]
- Yero, T.; Rey, J.A. Tetrabenazine (Xenazine), An FDA-Approved Treatment Option For Huntington’s Disease-Related Chorea. Pharm. Ther. 2008, 33, 690–694. [Google Scholar]
- Caron, N.S.; Wright, G.E.B.; Hayden, M.R. Huntington Disease. In GeneReviews(®); Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Mirzaa, G., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Maccioni, R.B.; Farías, G.; Morales, I.; Navarrete, L. The revitalized tau hypothesis on Alzheimer’s disease. Arch. Med. Res. 2010, 41, 226–231. [Google Scholar] [CrossRef] [PubMed]
- Cummings, J.L.; Morstorf, T.; Zhong, K. Alzheimer’s disease drug-development pipeline: Few candidates, frequent failures. Alzheimers Res. 2014, 6, 37. [Google Scholar] [CrossRef]
- Bautista-Aguilera, O.M.; Esteban, G.; Bolea, I.; Nikolic, K.; Agbaba, D.; Moraleda, I.; Iriepa, I.; Samadi, A.; Soriano, E.; Unzeta, M.; et al. Design, synthesis, pharmacological evaluation, QSAR analysis, molecular modeling and ADMET of novel donepezil-indolyl hybrids as multipotent cholinesterase/monoamine oxidase inhibitors for the potential treatment of Alzheimer’s disease. Eur. J. Med. Chem. 2014, 75, 82–95. [Google Scholar] [CrossRef] [PubMed]
- Yamada, A.; Akimoto, H.; Kagawa, S.; Guillemin, G.J.; Takikawa, O. Proinflammatory cytokine interferon-gamma increases induction of indoleamine 2,3-dioxygenase in monocytic cells primed with amyloid beta peptide 1-42: Implications for the pathogenesis of Alzheimer’s disease. J. Neurochem. 2009, 110, 791–800. [Google Scholar] [CrossRef]
- Toledo-Sherman, L.M.; Prime, M.E.; Mrzljak, L.; Beconi, M.G.; Beresford, A.; Brookfield, F.A.; Brown, C.J.; Cardaun, I.; Courtney, S.M.; Dijkman, U.; et al. Development of a Series of Aryl Pyrimidine Kynurenine Monooxygenase Inhibitors as Potential Therapeutic Agents for the Treatment of Huntington’s Disease. J. Med. Chem. 2015, 58, 1159–1183. [Google Scholar] [CrossRef]
- Chen, J.J.; Swope, D.M. Parkinson’s Disease and Its Management. In Pharmacotherapy: A Pathophysiologic Approach, 9e (Vols. 1–Book, Section); McGraw-Hill Education: New York, NY, USA, 2014; Volume 1. [Google Scholar]
- Sulzer, D. Multiple hit hypotheses for dopamine neuron loss in Parkinson’s disease. Trends Neurosci. 2007, 30, 244–250. [Google Scholar] [CrossRef]
- Tan, L.; Yu, J.-T.; Tan, L. The kynurenine pathway in neurodegenerative diseases: Mechanistic and therapeutic considerations. J. Neurol. Sci. 2012, 323, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Zinger, A.; Barcia, C.; Herrero, M.T.; Guillemin, G.J. The involvement of neuroinflammation and kynurenine pathway in Parkinson’s disease. Parkinsons Dis. 2011, 2011, 716859. [Google Scholar] [CrossRef]
- Talbert, R.L.; Yee, G.C.; Matzke, G.R.; Wells, B.G.; Posey, L.M. 2014. Available online: http://vlib.kmu.ac.ir/kmu/handle/kmu/88825 (accessed on 20 December 2021).
- Krauss, G. Epilepsy is Not Resolved: Epilepsy Is Not Resolved. Epilepsy Curr. 2014, 14, 339–340. [Google Scholar] [CrossRef]
- DiPiro, J.T.; Talbert, R.L.; Yee, G.C.; Matzke, G.R.; Wells, B.G.; Posey, L. Chapter 56: Epilepsy. In Pharmacotherapy: A Pathophysiologic Approach, 10e; McGraw-Hill Education: New York, NY, USA, 2017. [Google Scholar]
- Myint, A.M. Kynurenines: From the perspective of major psychiatric disorders. FEBS J. 2012, 279, 1375–1385. [Google Scholar] [CrossRef]
- Slominski, A.; Semak, I.; Pisarchik, A.; Sweatman, T.; Szczesniewski, A.; Wortsman, J. Conversion of L-tryptophan to serotonin and melatonin in human melanoma cells. FEBS Lett. 2002, 511, 102–106. [Google Scholar] [CrossRef]
- Hunt, C.; Macedo, E.C.T.; Suchting, R.; de Dios, C.; Cuellar Leal, V.A.; Soares, J.C.; Dantzer, R.; Teixeira, A.L.; Selvaraj, S. Effect of immune activation on the kynurenine pathway and depression symptoms—A systematic review and meta-analysis. Neurosci. Biobehav. Rev. 2020, 118, 514–523. [Google Scholar] [CrossRef] [PubMed]
- Erabi, H.; Okada, G.; Shibasaki, C.; Setoyama, D.; Kang, D.; Takamura, M.; Yoshino, A.; Fuchikami, M.; Kurata, A.; Kato, T.A.; et al. Kynurenic acid is a potential overlapped biomarker between diagnosis and treatment response for depression from metabolome analysis. Sci. Rep. 2020, 10, 16822. [Google Scholar] [CrossRef]
- Uher, R.; Payne, J.L.; Pavlova, B.; Perlis, R.H. Major depressive disorder in DSM-5: Implications for clinical practice and research of changes from DSM-IV. Depress. Anxiety 2014, 31, 459–471. [Google Scholar] [CrossRef]
- Myint, A.M.; Kim, Y.K. Cytokine-serotonin interaction through IDO: A neurodegeneration hypothesis of depression. Med. Hypotheses 2003, 61, 519–525. [Google Scholar] [CrossRef]
- Laumet, G.; Zhou, W.; Dantzer, R.; Edralin, J.D.; Huo, X.; Budac, D.P.; O’Connor, J.C.; Lee, A.W.; Heijnen, C.J.; Kavelaars, A. Upregulation of neuronal kynurenine 3-monooxygenase mediates depression-like behavior in a mouse model of neuropathic pain. Brain Behav. Immun. 2017, 66, 94–102. [Google Scholar] [CrossRef] [PubMed]
- Gabbay, V.; Klein, R.G.; Katz, Y.; Mendoza, S.; Guttman, L.E.; Alonso, C.M.; Babb, J.S.; Hirsch, G.S.; Liebes, L. The possible role of the kynurenine pathway in adolescent depression with melancholic features. J. Child. Psychol. Psychiatry 2010, 51, 935–943. [Google Scholar] [CrossRef] [PubMed]
- Tandon, R. Schizophrenia and Other Psychotic Disorders in Diagnostic and Statistical Manual of Mental Disorders (DSM)-5: Clinical Implications of Revisions from DSM-IV. Indian J. Psychol. Med. 2014, 36, 223–225. [Google Scholar] [CrossRef]
- Dipiro, J.T.; Talbert, R.L.; Yee, G.C.; Matzke, G.R.; Wells, B.G.; Posey, L.M. Pharmacotherapy: A Pathophysiologic Approach. In Pharmacotherapy A Pathophysiologic Approach 9/E, 9th ed.; McGraw-Hill Education: New York, NY, USA, 2014; Chapter 50. [Google Scholar]
- Schwarcz, R.; Rassoulpour, A.; Wu, H.Q.; Medoff, D.; Tamminga, C.A.; Roberts, R.C. Increased cortical kynurenate content in schizophrenia. Biol. Psychiatry 2001, 50, 521–530. [Google Scholar] [CrossRef]
- Lloyd-Jones, D.; Adams, R.; Carnethon, M.; De Simone, G.; Ferguson, T.B.; Flegal, K.; Ford, E.; Furie, K.; Go, A.; Greenlund, K.; et al. Heart disease and stroke statistics—2009 update: A report from the American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Circulation 2009, 119, e21–e181. [Google Scholar] [CrossRef] [PubMed]
- Ormstad, H.; Verkerk, R. Role of the Kynurenine Pathway in Stroke. In Targeting the Broadly Pathogenic Kynurenine Pathway; Springer: Cham, Switzerland, 2016; pp. 215–232. [Google Scholar] [CrossRef]
- Csányi, G.; Miller, F.J., Jr. Oxidative stress in cardiovascular disease. Int. J. Mol. Sci. 2014, 15, 6002–6008. [Google Scholar] [CrossRef]
- Schnabel, R.; Blankenberg, S. Oxidative Stress in Cardiovascular Disease. Circulation 2007, 116, 1338–1340. [Google Scholar] [CrossRef]
- Barone, F.C.; Feuerstein, G.Z. Inflammatory mediators and stroke: New opportunities for novel therapeutics. J. Cereb. Blood Flow Metab. 1999, 19, 819–834. [Google Scholar] [CrossRef] [PubMed]
- Brouns, R.; Verkerk, R.; Aerts, T.; De Surgeloose, D.; Wauters, A.; Scharpé, S.; De Deyn, P.P. The role of tryptophan catabolism along the kynurenine pathway in acute ischemic stroke. Neurochem. Res. 2010, 35, 1315–1322. [Google Scholar] [CrossRef]
- Darlington, L.G.; Mackay, G.M.; Forrest, C.M.; Stoy, N.; George, C.; Stone, T.W. Altered kynurenine metabolism correlates with infarct volume in stroke. Eur. J. Neurosci. 2007, 26, 2211–2221. [Google Scholar] [CrossRef]
- Jin, H.; Zhang, Y.; You, H.; Tao, X.; Wang, C.; Jin, G.; Wang, N.; Ruan, H.; Gu, D.; Huo, X.; et al. Prognostic significance of kynurenine 3-monooxygenase and effects on proliferation, migration and invasion of human hepatocellular carcinoma. Sci. Rep. 2015, 5, 10466. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.Y.; Huang, T.T.; Lee, C.H.; Wang, W.L.; Lee, H.C.; Tseng, L.M. 9P—Kynurenine-3-monooxygenase (KMO) protein promotes triple negative breast cancer progression. Ann. Oncol. 2017, 28, 3. [Google Scholar] [CrossRef]
- Mole, D.J.; Webster, S.P.; Uings, I.; Zheng, X.; Binnie, M.; Wilson, K.; Hutchinson, J.P.; Mirguet, O.; Walker, A.; Beaufils, B.; et al. Kynurenine-3-monooxygenase inhibition prevents multiple organ failure in rodent models of acute pancreatitis. Nat. Med. 2016, 22, 202–209. [Google Scholar] [CrossRef]
- Crozier-Reabe, K.R.; Phillips, R.S.; Moran, G.R. Kynurenine 3-Monooxygenase from Pseudomonas fluorescens: Substrate-like Inhibitors both Stimulate Flavin Reduction and Stabilize the Flavin−Peroxo Intermediate yet Result in the Production of Hydrogen Peroxide. Biochemistry 2008, 47, 12420–12433. [Google Scholar] [CrossRef]
- Saito, Y.; Hayaishi, O.; Rothberg, S. Studies on oxygenases; enzymatic formation of 3-hydroxy-L-kynurenine from L-kynurenine. J. Biol. Chem. 1957, 229, 921–934. [Google Scholar] [CrossRef]
- Courtney, S.; Scheel, A. Modulation of the Kynurenine Pathway for the Potential Treatment of Neurodegenerative Diseases. Neurodegener. Dis. 2010, 6, 149–176. [Google Scholar]
- Amaral, M.; Levy, C.; Heyes, D.J.; Lafite, P.; Outeiro, T.F.; Giorgini, F.; Leys, D.; Scrutton, N.S. Structural basis of kynurenine 3-monooxygenase inhibition. Nature 2013, 496, 382–385. [Google Scholar] [CrossRef] [PubMed]
- Huijbers, M.M.E.; Montersino, S.; Westphal, A.H.; Tischler, D.; van Berkel, W.J.H. Flavin dependent monooxygenases. Arch. Biochem. Biophys. 2014, 544, 2–17. [Google Scholar] [CrossRef] [PubMed]
- Kurnasov, O.; Goral, V.; Colabroy, K.; Gerdes, S.; Anantha, S.; Osterman, A.; Begley, T.P. NAD Biosynthesis: Identification of the Tryptophan to Quinolinate Pathway in Bacteria. Chem. Biol. 2003, 10, 1195–1204. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, M. Structural Dynamics and Ligand Binding in Kynurenine-3- Monooxygenase; ERA: Edinburgh, UK, 2013. [Google Scholar]
- Hirai, K.; Kuroyanagi, H.; Tatebayashi, Y.; Hayashi, Y.; Hirabayashi-Takahashi, K.; Saito, K.; Haga, S.; Uemura, T.; Izumi, S. Dual role of the carboxyl-terminal region of pig liver L-kynurenine 3-monooxygenase: Mitochondrial-targeting signal and enzymatic activity. J. Biochem. 2010, 148, 639–650. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.T.; Na, B.K.; Chung, J.; Kim, S.; Kwon, S.K.; Cha, H.; Son, J.; Cho, J.M.; Hwang, K.Y. Structural Basis for Inhibitor-Induced Hydrogen Peroxide Production by Kynurenine 3-Monooxygenase. Cell Chem. Biol. 2018, 25, 426–438. [Google Scholar] [CrossRef]
- Moroni, F.; Russi, P.; Gallo-Mezo, M.A.; Moneti, G.; Pellicciari, R. Modulation of Quinolinic and Kynurenic Acid Content in the Rat Brain: Effects of Endotoxins and Nicotinylalanine. J. Neurochem. 1991, 57, 1630–1635. [Google Scholar] [CrossRef] [PubMed]
- Pellicciari, R.; Natalini, B.; Costantino, G.; Mahmoud, M.R.; Mattoli, L.; Sadeghpour, B.M.; Moroni, F.; Chiarugi, A.; Carpenedo, R. Modulation of the kynurenine pathway in search for new neuroprotective agents. Synthesis and preliminary evaluation of (m-nitrobenzoyl)alanine, a potent inhibitor of kynurenine-3-hydroxylase. J. Med. Chem. 1994, 37, 647–655. [Google Scholar] [CrossRef] [PubMed]
- Chiarugi, A.; Carpenedo, R.; Moroni, F. Kynurenine Disposition in Blood and Brain of Mice: Effects of Selective Inhibitors of Kynurenine Hydroxylase and of Kynureninase. J. Neurochem. 1996, 67, 692–698. [Google Scholar] [CrossRef] [PubMed]
- Giordani, A.; Pevarello, P.; Cini, M.; Bormetti, R.; Greco, F.; Toma, S.; Speciale, C.; Varasi, M. 4-Phenyl-4-oxo-butanoic acid derivatives inhibitors of kynurenine 3-hydroxylase. Bioorg. Med. Chem. Lett. 1998, 8, 2907–2912. [Google Scholar] [CrossRef]
- Drysdale, M.J.; Hind, S.L.; Jansen, M.; Reinhard, J.F., Jr. Synthesis and SAR of 4-aryl-2-hydroxy-4-oxobut-2-enoic acids and esters and 2-amino-4-aryl-4-oxobut-2-enoic acids and esters: Potent inhibitors of kynurenine-3-hydroxylase as potential neuroprotective agents. J. Med. Chem. 2000, 43, 123–127. [Google Scholar] [CrossRef] [PubMed]
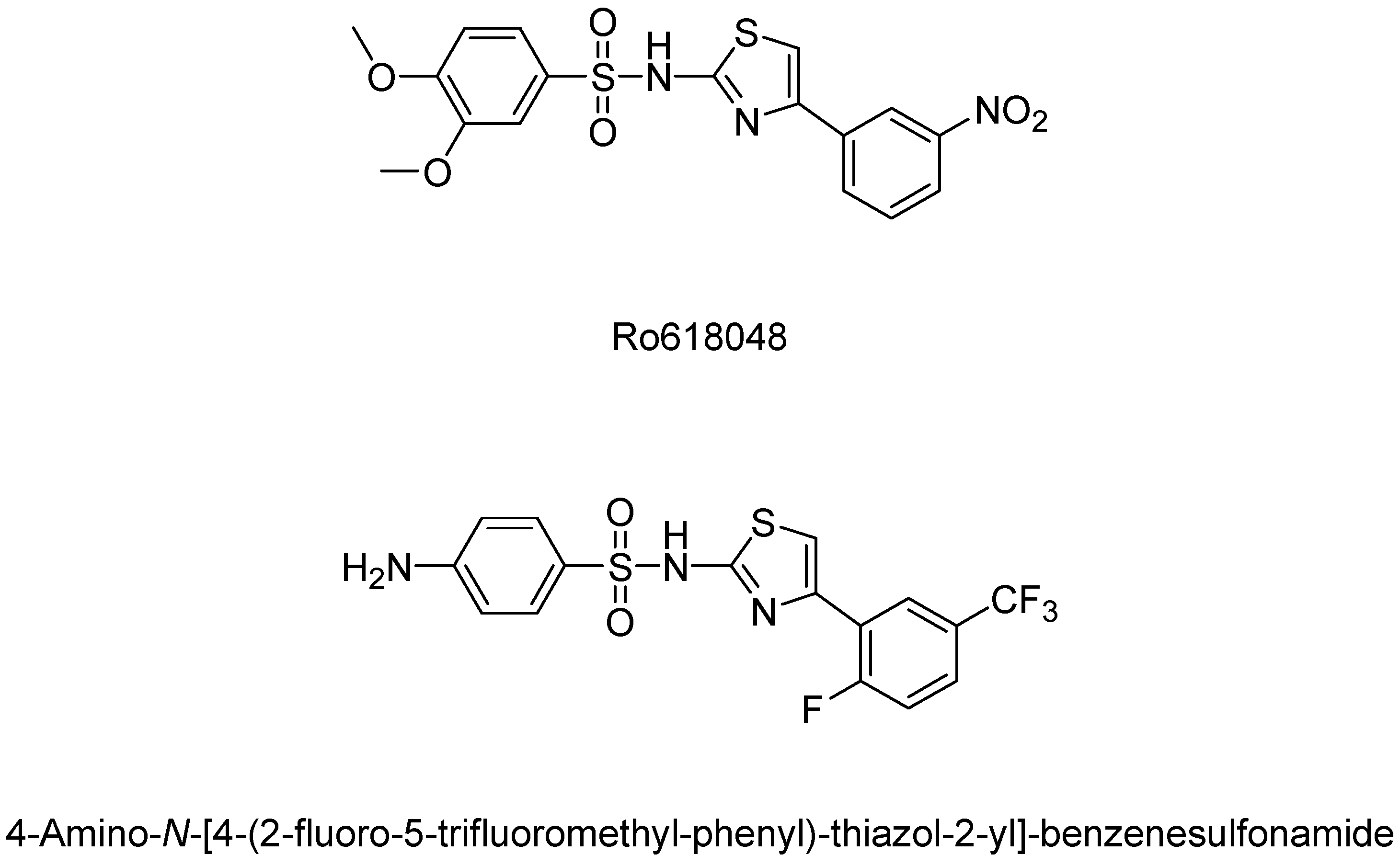
- Röver, S.; Cesura, A.M.; Huguenin, P.; Kettler, R.; Szente, A. Synthesis and biochemical evaluation of N-(4-phenylthiazol-2-yl)benzenesulfonamides as high-affinity inhibitors of kynurenine 3-hydroxylase. J. Med. Chem. 1997, 40, 4378–4385. [Google Scholar] [CrossRef] [PubMed]
- Ceresoli-Borroni, G.; Guidetti, P.; Amori, L.; Pellicciari, R.; Schwarcz, R. Perinatal kynurenine 3-hydroxylase inhibition in rodents: Pathophysiological implications. J. Neurosci. Res. 2007, 85, 845–854. [Google Scholar] [CrossRef]
- Feng, Y.; Bowden, B.F.; Kapoor, V. Ianthellamide A, a selective kynurenine-3-hydroxylase inhibitor from the Australian marine sponge Ianthella quadrangulata. Bioorg. Med. Chem. Lett. 2012, 22, 3398–3401. [Google Scholar] [CrossRef]
- Güner, O.F.; Henry, D.R. Metrics for Analyzing Hit Lists and Pharmacophores. In Pharmacophore Perception, Development, and Use for Drug Design; International University Line: San Diego, CA, USA, 2000. [Google Scholar]
- Güner, O.F.; Henry, D.R. Strategies in Database Mining and Pharmacophore Development; International University Line: San Diego, CA, USA, 2000; pp. 213–232. [Google Scholar]
- Sprague, P.W. Automated chemical hypothesis generation and database searching with Catalyst. Perspect. Drug Discov. Des. 1995, 3, 1–20. [Google Scholar] [CrossRef]
- Dixon, S.L.; Smondyrev, A.M.; Rao, S.N. PHASE: A novel approach to pharmacophore modeling and 3D database searching. Chem. Biol. Drug. Des. 2006, 67, 370–372. [Google Scholar] [CrossRef]
- Guner, O.F.; Henry, D.R.; Pearlman, R.S. Use of flexible queries for searching conformationally flexible molecules in databases of three-dimensional structures. J. Chem. Inf. Comput. Sci. 1992, 32, 101–109. [Google Scholar] [CrossRef]
- Güner, O.F.; Henry, D.R.; Moock, T.E.; Pearlman, R.S. Flexible queries in 3D searching. 2. Techniques in 3D query formulation. Tetrahedron Comput. Methodol. 1990, 3, 557–563. [Google Scholar] [CrossRef]
- Guner, O.F. The impact of pharmacophore modeling in drug design. IDrugs 2005, 8, 567–572. [Google Scholar] [PubMed]
- Phillips, R.S.; Anderson, A.D.; Gentry, H.G.; Güner, O.F.; Bowen, J.P. Substrate and inhibitor specificity of kynurenine monooxygenase from Cytophaga hutchinsonii. Bioorg. Med. Chem. Lett. 2017, 27, 1705–1708. [Google Scholar] [CrossRef]
- Gao, J.; Yao, L.; Xia, T.; Liao, X.; Zhu, D.; Xiang, Y. Biochemistry and structural studies of kynurenine 3-monooxygenase reveal allosteric inhibition by Ro 61-8048. FASEB J. 2018, 32, 2036–2045. [Google Scholar] [CrossRef] [PubMed]
- Garavito, R.M.; Picot, D.; Loll, P.J. Strategies for crystallizing membrane proteins. J. Bioenerg. Biomembr. 1996, 28, 13–27. [Google Scholar] [CrossRef]













Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hughes, T.D.; Güner, O.F.; Iradukunda, E.C.; Phillips, R.S.; Bowen, J.P. The Kynurenine Pathway and Kynurenine 3-Monooxygenase Inhibitors. Molecules 2022, 27, 273. https://doi.org/10.3390/molecules27010273
Hughes TD, Güner OF, Iradukunda EC, Phillips RS, Bowen JP. The Kynurenine Pathway and Kynurenine 3-Monooxygenase Inhibitors. Molecules. 2022; 27(1):273. https://doi.org/10.3390/molecules27010273
Chicago/Turabian StyleHughes, Tamera D., Osman F. Güner, Emma Carine Iradukunda, Robert S. Phillips, and J. Phillip Bowen. 2022. "The Kynurenine Pathway and Kynurenine 3-Monooxygenase Inhibitors" Molecules 27, no. 1: 273. https://doi.org/10.3390/molecules27010273
APA StyleHughes, T. D., Güner, O. F., Iradukunda, E. C., Phillips, R. S., & Bowen, J. P. (2022). The Kynurenine Pathway and Kynurenine 3-Monooxygenase Inhibitors. Molecules, 27(1), 273. https://doi.org/10.3390/molecules27010273