
Enzyme Models—From Catalysis to Prodrugs



Abstract
1. Introduction
2. Computational Methods
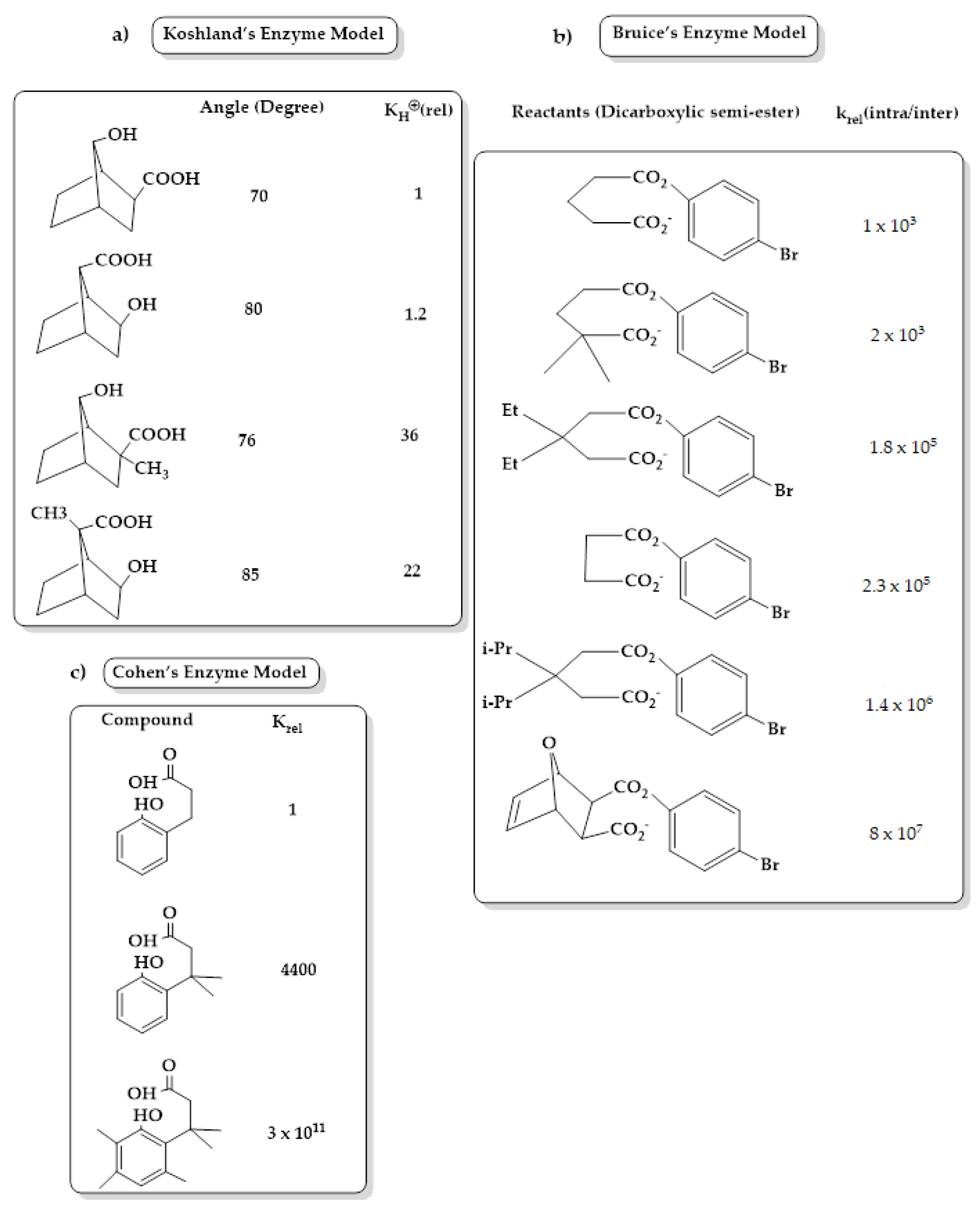
3. Enzyme Catalytic Models
4. The Effective Molarity (EM)
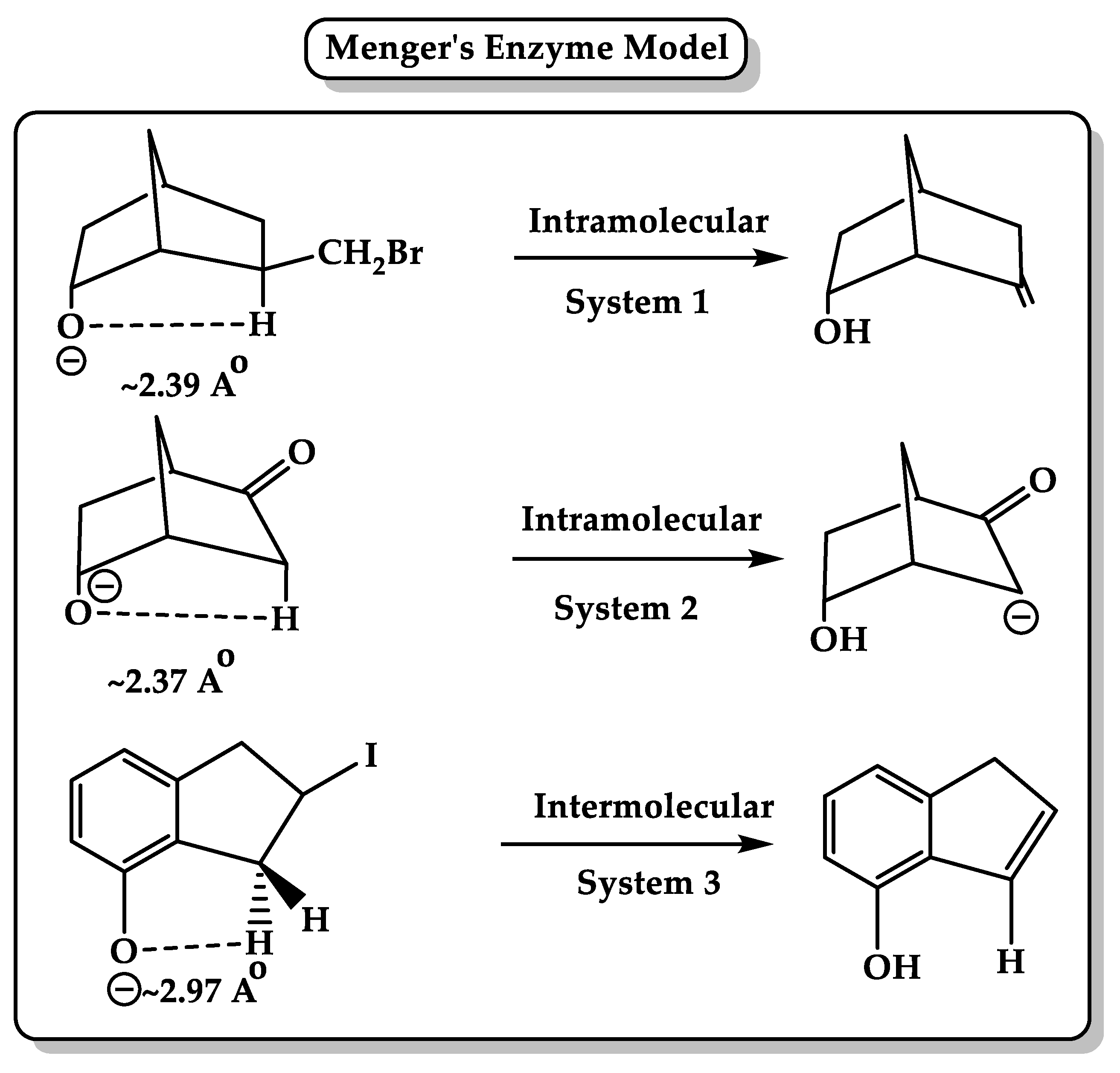
5. Driving Forces for Rate Accelerations in Some Intramolecular Processes
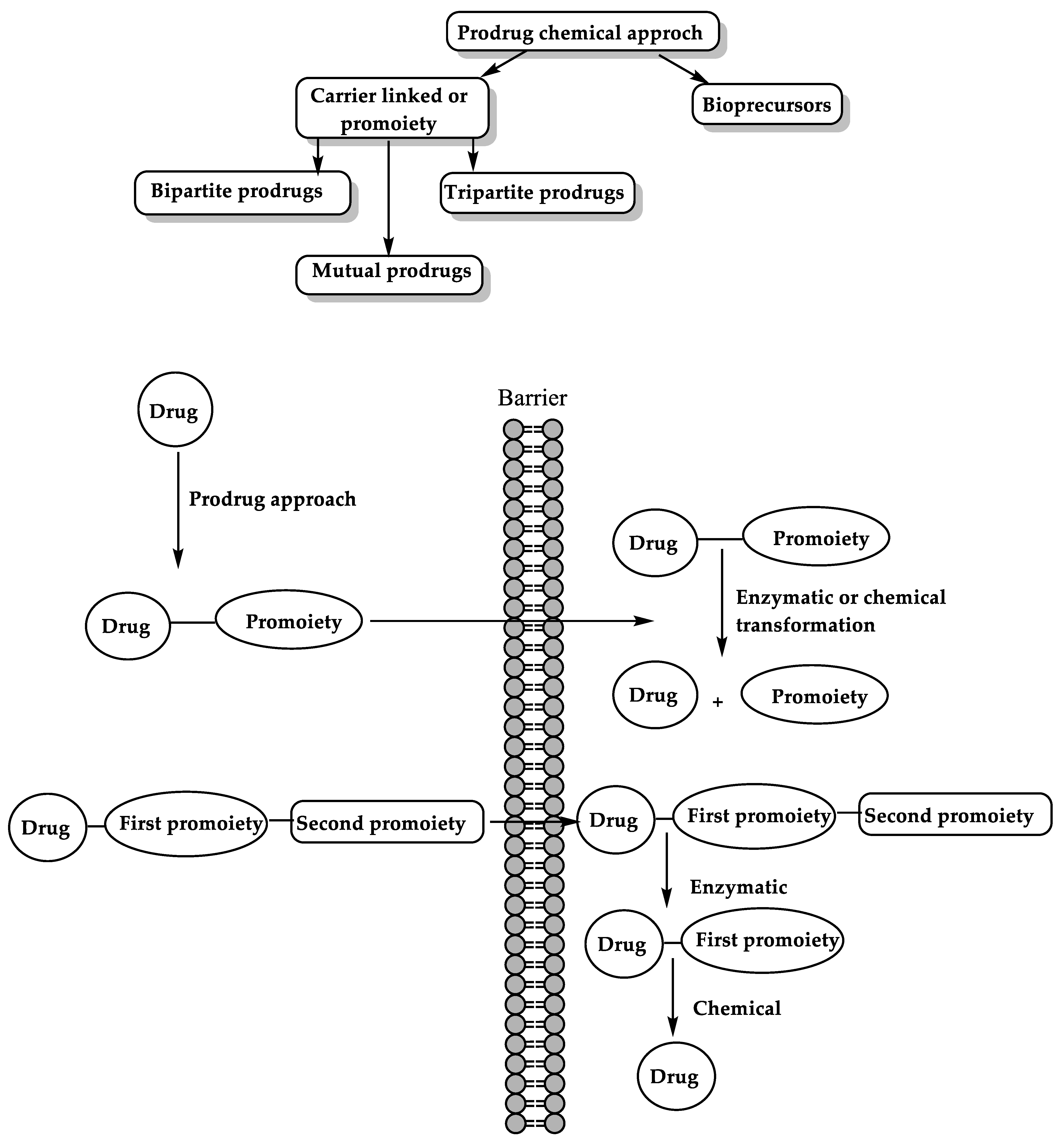
6. The Prodrug Approach
6.1. Enzyme-Mediated Prodrug Activation
6.2. Chemical Approach for Prodrug Activation
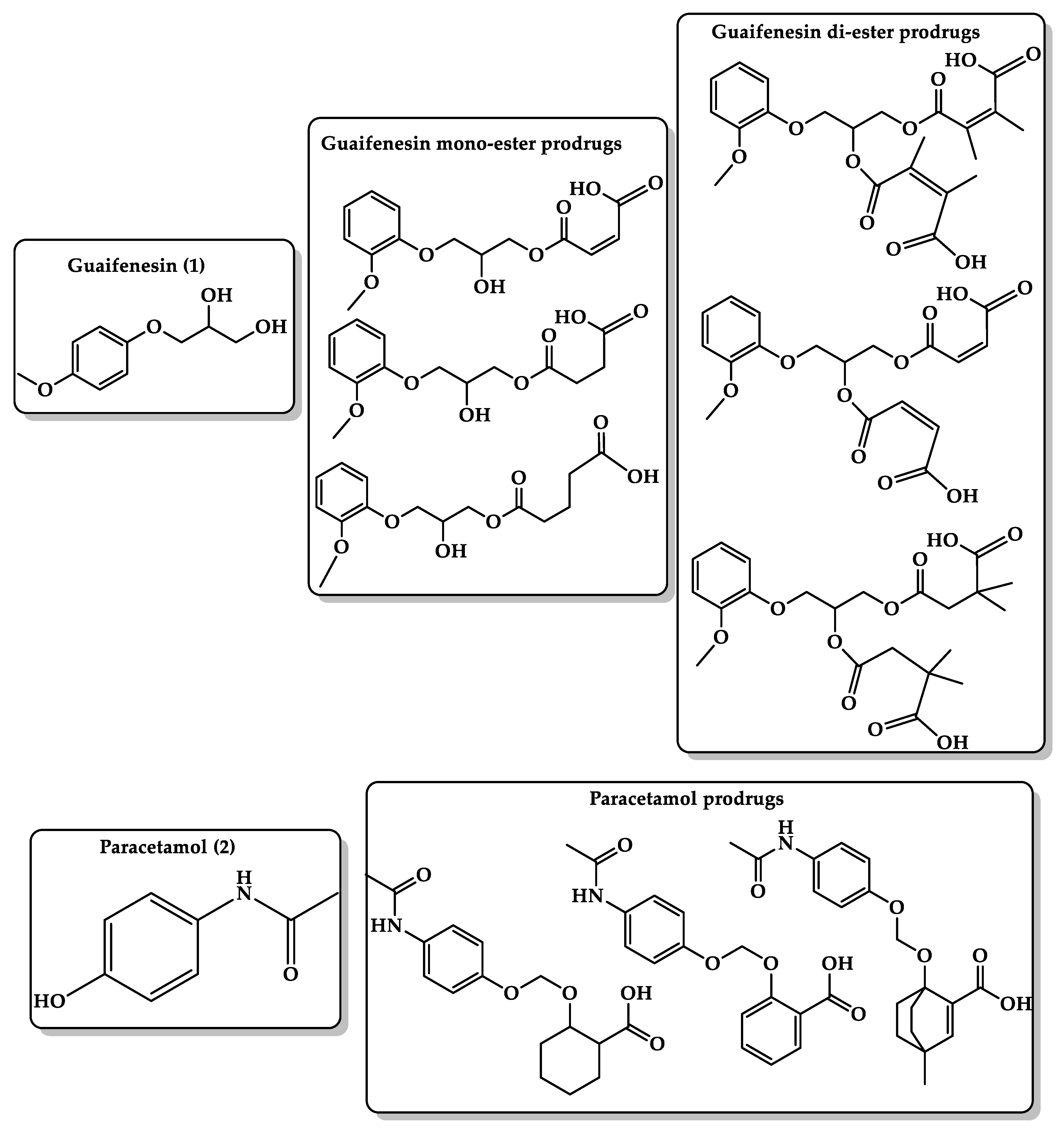
6.3. Masking Bitterness of Drugs
6.4. Bioavailability Enhancement
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Concu, R.; Cordeiro, M. Alignment-Free Method to Predict Enzyme Classes and Subclasses. Int. J. Mol. Sci. 2019, 20, 5389. [Google Scholar] [CrossRef] [PubMed]
- Schomburg, I.; Chang, A.; Schomburg, D. Standardization in enzymology—Data integration in the world’s enzyme information system BRENDA. Perspect. Sci. 2014, 1, 15–23. [Google Scholar] [CrossRef][Green Version]
- Boeckx, J.; Hertog, M.; Geeraerd, A.; Nicolai, B. Kinetic modelling: An integrated approach to analyze enzyme activity assays. Plant Methods 2017, 13, 69. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, A.; Bankaitis, V.A. Molecular Docking: From Lock and Key to Combination Lock. J. Mol. Med. Clin. Appl. 2017, 2. [Google Scholar] [CrossRef]
- Lewis, T.; Stone, W.L. Biochemistry, Proteins Enzymes. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2020. [Google Scholar]
- Menard, R.; Storer, A.C. Oxyanion hole interactions in serine and cysteine proteases. Biol. Chem. Hoppe-Seyler 1992, 373, 393–400. [Google Scholar] [CrossRef] [PubMed]
- Buller, A.R.; Townsend, C.A. Intrinsic evolutionary constraints on protease structure, enzyme acylation, and the identity of the catalytic triad. Proc. Natl. Acad. Sci. USA 2013, 110, E653–E661. [Google Scholar] [CrossRef]
- Berg, J.M.; Tymoczko, J.L.; Stryer, L.; Stryer, L. Biochemistry, 5th ed.; W.H. Freeman: New York, NY, USA, 2002. [Google Scholar]
- Agarwal, P.K. A Biophysical Perspective on Enzyme Catalysis. Biochemistry 2019, 58, 438–449. [Google Scholar] [CrossRef]
- Cooper, G. The Central Role of Enzymes as Biological Catalysts. In The Cell: A Molecular Approach, 2nd ed.; Sinauer Associates: Sunderland, MA, USA, 2000. [Google Scholar]
- Lonsdale, R.; Harvey, J.N.; Mulholland, A.J. A practical guide to modelling enzyme-catalysed reactions. Chem. Soc. Rev. 2012, 41, 3025–3038. [Google Scholar] [CrossRef]
- Aminpour, M.; Montemagno, C.; Tuszynski, J.A. An Overview of Molecular Modeling for Drug Discovery with Specific Illustrative Examples of Applications. Molecules 2019, 24, 1693. [Google Scholar] [CrossRef]
- Frushicheva, M.P.; Mills, M.J.; Schopf, P.; Singh, M.K.; Prasad, R.B.; Warshel, A. Computer aided enzyme design and catalytic concepts. Curr. Opin. Chem. Biol. 2014, 21, 56–62. [Google Scholar] [CrossRef]
- Warshel, A.; Sharma, P.K.; Kato, M.; Xiang, Y.; Liu, H.; Olsson, M.H. Electrostatic basis for enzyme catalysis. Chem. Rev. 2006, 106, 3210–3235. [Google Scholar] [CrossRef] [PubMed]
- Warshel, A. Energetics of enzyme catalysis. Proc. Natl. Acad. Sci. USA 1978, 75, 5250–5254. [Google Scholar] [CrossRef]
- Jindal, G.; Warshel, A. Misunderstanding the preorganization concept can lead to confusions about the origin of enzyme catalysis. Proteins 2017, 85, 2157–2161. [Google Scholar] [CrossRef] [PubMed]
- Menger, F.M.; Nome, F. Interaction vs Preorganization in Enzyme Catalysis. A Dispute That Calls for Resolution. ACS Chem. Biol. 2019, 14, 1386–1392. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.; Yang, W. Development and application of ab initio QM/MM methods for mechanistic simulation of reactions in solution and in enzymes. Theochem 2009, 898, 17–30. [Google Scholar] [CrossRef]
- Kraut, D.A.; Carroll, K.S.; Herschlag, D. Challenges in enzyme mechanism and energetics. Annu. Rev. Biochem. 2003, 72, 517–571. [Google Scholar] [CrossRef] [PubMed]
- Rami Reddy, M.; Erion, M.D. Free Energy Calculations in Rational Drug Design; Kluwer Academic/Plenum Publishers: New York, NY, USA, 2001; p. 22. [Google Scholar]
- Warshel, A.; Levitt, M. Theoretical studies of enzymic reactions: Dielectric, electrostatic and steric stabilization of the carbonium ion in the reaction of lysozyme. J. Mol. Biol. 1976, 103, 227–249. [Google Scholar] [CrossRef]
- Hu, H.; Yang, W. Free energies of chemical reactions in solution and in enzymes with ab initio quantum mechanics/molecular mechanics methods. Annu. Rev. Phys. Chem. 2008, 59, 573–601. [Google Scholar] [CrossRef]
- Markovic, M.; Ben-Shabat, S.; Dahan, A. Computational Simulations to Guide Enzyme-Mediated Prodrug Activation. Int. J. Mol. Sci. 2020, 21, 3621. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Ko, H.Y.; Remsing, R.C.; Calegari Andrade, M.F.; Santra, B.; Sun, Z.; Selloni, A.; Car, R.; Klein, M.L.; Perdew, J.P.; et al. Ab initio theory and modeling of water. Proc. Natl. Acad. Sci. USA 2017, 114, 10846–10851. [Google Scholar] [CrossRef]
- Schrodinger, E. Quantisierung als eigenwertproblem. Ann. Phys. 1926, 384, 361–376. [Google Scholar] [CrossRef]
- Christensen, A.S.; Kubar, T.; Cui, Q.; Elstner, M. Semiempirical Quantum Mechanical Methods for Noncovalent Interactions for Chemical and Biochemical Applications. Chem. Rev. 2016, 116, 5301–5337. [Google Scholar] [CrossRef] [PubMed]
- Dewar, M.J.S.; Thiel, W. Ground states of molecules. 38. The MNDO method. Approximations and parameters J. Am. Chem. Soc. 1977, 99, 4899–4907. [Google Scholar] [CrossRef]
- Roos, G.; Geerlings, P.; Messens, J. Enzymatic catalysis: The emerging role of conceptual density functional theory. J. Phys. Chem. B 2009, 113, 13465–13475. [Google Scholar] [CrossRef] [PubMed]
- Vanommeslaeghe, K.; Guvench, O.; MacKerell, A.D., Jr. Molecular mechanics. Curr. Pharm. Des. 2014, 20, 3281–3292. [Google Scholar] [CrossRef]
- Burkert, U.; Allinger, N.L. Molecular Mechanics; American Chemical Society: Washington, DC, USA, 1982; p. 21. 339p. [Google Scholar]
- van der Kamp, M.W.; Mulholland, A.J. Combined quantum mechanics/molecular mechanics (QM/MM) methods in computational enzymology. Biochemistry 2013, 52, 2708–2728. [Google Scholar] [CrossRef]
- Kulik, H.J. Large-scale QM/MM free energy simulations of enzyme catalysis reveal the influence of charge transfer. Phys. Chem. Chem. Phys. PCCP 2018, 20, 20650–20660. [Google Scholar] [CrossRef]
- Field, M.J.; Bash, P.A.; Karplus, M. A combined quantum mechanical and molecular mechanical potential for molecular dynamics simulations. J. Comput. Chem. 1990, 11, 700–733. [Google Scholar] [CrossRef]
- Arodola, O.A.; Soliman, M.E. Quantum mechanics implementation in drug-design workflows: Does it really help? Drug Des. Devel. Ther. 2017, 11, 2551–2564. [Google Scholar] [CrossRef]
- Hofer, T.S.; de Visser, S.P. Editorial: Quantum Mechanical/Molecular Mechanical Approaches for the Investigation of Chemical Systems—Recent Developments and Advanced Applications. Front. Chem. 2018, 6, 357. [Google Scholar] [CrossRef]
- Lin, X.; Li, X.; Lin, X. A Review on Applications of Computational Methods in Drug Screening and Design. Molecules 2020, 25, 1375. [Google Scholar] [CrossRef] [PubMed]
- Latour, R.A. Molecular simulation of protein-surface interactions: Benefits, problems, solutions, and future directions. Biointerphases 2008, 3, FC2–FC12. [Google Scholar] [CrossRef] [PubMed]
- Rathore, R.S.; Sumakanth, M.; Reddy, M.S.; Reddanna, P.; Rao, A.A.; Erion, M.D.; Reddy, M.R. Advances in binding free energies calculations: QM/MM-based free energy perturbation method for drug design. Curr. Pharm. Des. 2013, 19, 4674–4686. [Google Scholar] [CrossRef] [PubMed]
- Asadi, P.; Khodarahmi, G.; Farrokhpour, H.; Hassanzadeh, F.; Saghaei, L. Quantum mechanical/molecular mechanical and docking study of the novel analogues based on hybridization of common pharmacophores as potential anti-breast cancer agents. Res. Pharm. Sci. 2017, 12, 233–240. [Google Scholar] [CrossRef]
- Dafforn, A.; Koshland, D.E., Jr. Theoretical aspects of orbital steering. Proc. Natl. Acad. Sci. USA 1971, 68, 2463–2467. [Google Scholar] [CrossRef] [PubMed]
- BRUICE, T.C.; LIGHTSTONE, F.C. Ground State and Transition State Contributions to the Rates of Intramolecular and Enzymatic Reactions. Acc. Chem. Res. 1999, 32, 127–136. [Google Scholar] [CrossRef]
- BRUICE, T.C.; BENKOVIC, S.J. The Compensation in AH’ and AS* Accompanying the Conversion of Lower Order Nucleophilic Displacement Reactions to Higher Order Catalytic Processes. The Temperature Dependence of the Hydrazinolysis and Imidazole-Catalyzed Hydrolysis of Substituted Phenyl Acetates. J. Am. Chem. Soc. 1964, 86, 418–426. [Google Scholar] [CrossRef]
- Hillery, P.S.; Cohen, L.A. Stereopopulation Control. 9. Rate and Equilibrium Enhancement in the Lactonization of (0 -Hydroxyphenyl)acetic Acids. J. Org. Chem. 1983, 48, 3465–3471. [Google Scholar] [CrossRef]
- Milstien, S.; Cohen, L.A. Concurrent General-Acid and General-Base Catalysis of Esterification’. J. Am. Chem. Soc. 1970, 92, 4377–4382. [Google Scholar] [CrossRef]
- Milstien, S.; Cohen, L.A. Rate Acceleration by Stereopopulation Control: Models for Enzyme Action. Proc. Natl. Acad. Sci. USA 1970, 67, 1143–1147. [Google Scholar] [CrossRef]
- Milstien, S.; Cohen, L.A. Stereopopulation control. I. Rate enhancement in the lactonizations of o-hydroxyhydrocinnamic acids. J. Am. Chem. Soc. 1972, 94, 9158–9165. [Google Scholar] [CrossRef] [PubMed]
- Karaman, R. A new mathematical equation relating activation energy to bond angle and distance: A key for understanding the role of acceleration in lactonization of the trimethyl lock system. Bioorg. Chem. 2009, 37, 11–25. [Google Scholar] [CrossRef] [PubMed]
- Karaman, R. Reevaluation of Bruice’s proximity orientation. Tetrahedron Lett. 2009, 50, 452–456. [Google Scholar] [CrossRef]
- Kirby, A.J. Effective Molarities for Intramolecular Reactions. Adv. Phys. Org. Chem. 1980, 17, 183–278. [Google Scholar] [CrossRef]
- KIRBY, A.J. Efficiency of Proton Transfer Catalysis in Models and Enzymes. Acc. Chem. Res. 1997, 30, 290–296. [Google Scholar] [CrossRef]
- Kirby, A.J.; Lancaster, P.W. Structure and efficiency in intramolecular and enzymic catalysis. Catalysis of amide hydrolysis by the carboxy-group of substituted maleamic acids. J. Chem. Soc. Perkin Trans. I 1972, 2, 1206–1214. [Google Scholar] [CrossRef]
- Brown, C.J.; Kirby*, A.J. Efficiency of proton transfer catalysis. Intramolecular general acid catalysis of the hydrolysis of dialkyl acetals of benzaldehyde. J. Chem. Soc. Perkin Trans. I 1997, 2, 1081–1094. [Google Scholar] [CrossRef]
- Barber, S.E.; Dean, K.E.S.; Kirby, A.J. A mechanism for efficient proton-transfer catalysis. Intramolecular general acid catalysis of the hydrolysis of 1-arylethyl ethers of salicylic acid. Can. J. Chem. 1999, 77, 792–801. [Google Scholar] [CrossRef]
- Kirby, A.J.; Williams, N.H. Efficient intramolecular general acid catalysis of enol ether hydrolysis. Hydrogen-bonding stabilisation of the transition state for proton transfer to carbon. J. Chem. Soc. Perkin Trans. I 1994, 2, 643–648. [Google Scholar] [CrossRef]
- Kirby, A.J.; Williams, N.H. Efficient intramolecular general acid catalysis of vinyl ether hydrolysis by the neighbouring carboxylic acid group. J. Chem. Soc. Chem. Comm. 1991, 1643–1644. [Google Scholar] [CrossRef]
- Kirby, A.J.; Lima, M.F.; Silva, D.d.; Roussev, C.D.; Nome, F. Efficient Intramolecular General Acid Catalysis of Nucleophilic Attack on a Phosphodiester. J. Am. Chem. Soc. 2006, 128, 16944–16952. [Google Scholar] [CrossRef]
- Karaman, R. The efficiency of proton transfer in Kirby’s enzyme model, a computational approach. Tetrahedron Lett. 2010, 51, 2130–2135. [Google Scholar] [CrossRef]
- Menger, F.M. On the Source of Intramolecular and Enzymatic Reactivity. Acc. Chem. Res. 1985, 18, 128–134. [Google Scholar] [CrossRef]
- Menger, F.M. Nucleophilicity and Distance. Adv. Chem. 1987, 215, 209–218. [Google Scholar] [CrossRef]
- Menger, F.M.; Galloway, A.L.; Musaev, D.G. Relationship between rate and distance. Chem. Comm. 2003, 2370–2371. [Google Scholar] [CrossRef] [PubMed]
- Menger, F.M. Analysis of ground-state and transition-state effects in enzyme catalysis. Biochemistry 1992, 31, 5368–5373. [Google Scholar] [CrossRef]
- Menger, F.M. Enzyme reactivity from an organic perspective. Acc. Chem. Res. 1993, 26, 206–212. [Google Scholar] [CrossRef]
- Menger, F.M.; Ladika, M. Fast hydrolysis of an aliphatic amide at neutral pH and ambient temperature. A peptidase model. J. Am. Chem. Soc. 1988, 110, 6794–6796. [Google Scholar] [CrossRef]
- Souza, B.S.; Mora, J.R.; Wanderlind, E.H.; Clementin, R.M.; Gesser, J.C.; Fiedler, H.D.; Nome, F.; Menger, F.M. Transforming a Stable Amide into a Highly Reactive One: Capturing the Essence of Enzymatic Catalysis. Angew. Chem. Int. Ed. 2017, 56, 5345–5348. [Google Scholar] [CrossRef]
- Menger, F.M.; Karaman, R. A singularity model for chemical reactivity. Chemistry 2010, 16, 1420–1427. [Google Scholar] [CrossRef]
- Karaman, R. Analysis of Menger’s ‘spatiotemporal hypothesis’. Tetrahedron Lett. 2008, 49, 5998–6002. [Google Scholar] [CrossRef]
- Karaman, R. The effective molarity (EM)--a computational approach. Bioorg. Chem. 2010, 38, 165–172. [Google Scholar] [CrossRef] [PubMed]
- Karaman, R. The effective molarity (EM) puzzle in intramolecular ring-closing reactions. J. Mol. Struct. THEOCHEM 2009, 940, 70–75. [Google Scholar] [CrossRef]
- Karaman, R. Effects of substitution on the effective molarity (EM) for five membered ring-closure reactions—A computational approach. J. Mol. Struct. THEOCHEM 2010, 939, 69–74. [Google Scholar] [CrossRef]
- Karaman, R. The gem-disubstituent effect—a computational study that exposes the relevance of existing theoretical models. Tetrahedron Lett. 2009, 50, 6083–6087. [Google Scholar] [CrossRef]
- Karaman, R. The effective molarity (EM) puzzle in proton transfer reactions. Bioorg. Chem. 2009, 37, 106–110. [Google Scholar] [CrossRef] [PubMed]
- Karaman, R. Analyzing the efficiency of proton transfer to carbon in Kirby’s enzyme model—A computational approach. Tetrahedron Lett. 2011, 52, 699–704. [Google Scholar] [CrossRef]
- Karaman, R. Analyzing Kirby’s amine olefin—A model for amino acid ammonia lyases. Tetrahedron Lett. 2009, 50, 7304–7309. [Google Scholar] [CrossRef]
- Karaman, R. Analyzing the efficiency in intramolecular amide hydrolysis of Kirby’s N-alkylmaleamic acids—A computational approach. Comput. Theor. Chem. 2011, 974, 133–142. [Google Scholar] [CrossRef]
- Karaman, R.; Pascal, R. A computational analysis of intramolecularity in proton transfer reactions. Org. Biomol. Chem. 2010, 8, 5174–5178. [Google Scholar] [CrossRef]
- Karaman, R. The role of proximity orientation in intramolecular proton transfer reactions. Comput. Theor. Chem. 2011, 966, 311–321. [Google Scholar] [CrossRef]
- Scorsin, L.; Affeldt, R.F.; Oliveira, B.S.; Silveira, E.V.; Ferraz, M.S.; de Souza, F.P.S.; Caramori, G.F.; Menger, F.M.; Souza, B.S.; Nome, F. Coordination among Bond Formation/Cleavage in a Bifunctional-Catalyzed Fast Amide Hydrolysis: Evidence for an Optimized Intramolecular N-Protonation Event. J. Org. Chem. 2020, 85, 4663–4671. [Google Scholar] [CrossRef]
- Karaman, R. A general equation correlating intramolecular rates with ‘attack’ parameters: Distance and angle. Tetrahedron Lett. 2010, 51, 5185–5190. [Google Scholar] [CrossRef]
- Karaman, R. Accelerations in the Lactonization of Trimethyl Lock Systems Are due to Proximity Orientation and not to Strain Effects. Org. Chem. Int. 2009, 2009, 5. [Google Scholar] [CrossRef][Green Version]
- Karaman, R. Analyzing Kemp’s amide cleavage: A model for amidase enzymes. Comput. Theor. Chem. 2010, 963, 427–434. [Google Scholar] [CrossRef]
- Karaman, R. Cleavage of Menger’s aliphatic amide: A model for peptidase enzyme solely explained by proximity orientation in intramolecular proton transfer. J. Mol. Struct. THEOCHEM 2009, 910, 27–33. [Google Scholar] [CrossRef]
- Stella, V.J. Prodrugs: Challenges and Rewards; Springer; AAPS Press: New York, NY, USA, 2007. [Google Scholar]
- Stella, V.J.; Charman, W.N.; Naringrekar, V.H. Prodrugs. Do they have advantages in clinical practice? Drugs 1985, 29, 455–473. [Google Scholar] [CrossRef]
- Huttunen, K.M.; Raunio, H.; Rautio, J. Prodrugs--from serendipity to rational design. Pharmacol. Rev. 2011, 63, 750–771. [Google Scholar] [CrossRef] [PubMed]
- Stella, V.J.; Nti-Addae, K.W. Prodrug strategies to overcome poor water solubility. Adv. Drug Deliv. Rev. 2007, 59, 677–694. [Google Scholar] [CrossRef]
- Testa, B.; Kramer, S.D. The biochemistry of drug metabolism--an introduction: Part 2. Redox reactions and their enzymes. Chem. Biodivers. 2007, 4, 257–405. [Google Scholar] [CrossRef]
- Surya, Y.A.; Rosenfeld, J.M.; Hillcoat, B.L. Cross-linking of DNA in L1210 cells and nuclei treated with cyclophosphamide and phosphoramide mustard. Cancer Treat. Rep. 1978, 62, 23–29. [Google Scholar]
- Bunting, K.D.; Townsend, A.J. Protection by transfected rat or human class 3 aldehyde dehydrogenase against the cytotoxic effects of oxazaphosphorine alkylating agents in hamster V79 cell lines. Demonstration of aldophosphamide metabolism by the human cytosolic class 3 isozyme. J. Biol. Chem. 1996, 271, 11891–11896. [Google Scholar] [CrossRef]
- Nguyen, T.A.; Tychopoulos, M.; Bichat, F.; Zimmermann, C.; Flinois, J.P.; Diry, M.; Ahlberg, E.; Delaforge, M.; Corcos, L.; Beaune, P.; et al. Improvement of cyclophosphamide activation by CYP2B6 mutants: From in silico to ex vivo. Mol. Pharmacol. 2008, 73, 1122–1133. [Google Scholar] [CrossRef]
- Fleming, R.A. An overview of cyclophosphamide and ifosfamide pharmacology. Pharmacotherapy 1997, 17, 146S–154S. [Google Scholar]
- Sladek, N.E. Metabolism of oxazaphosphorines. Pharmacol. Ther. 1988, 37, 301–355. [Google Scholar] [CrossRef]
- Roy, P.; Yu, L.J.; Crespi, C.L.; Waxman, D.J. Development of a substrate-activity based approach to identify the major human liver P-450 catalysts of cyclophosphamide and ifosfamide activation based on cDNA-expressed activities and liver microsomal P-450 profiles. Drug Metab. Dispos. 1999, 27, 655–666. [Google Scholar]
- Ortiz de Montellano, P.R. Cytochrome P450-activated prodrugs. Future Med. Chem. 2013, 5, 213–228. [Google Scholar] [CrossRef]
- Yin, T.; Maekawa, K.; Kamide, K.; Saito, Y.; Hanada, H.; Miyashita, K.; Kokubo, Y.; Akaiwa, Y.; Otsubo, R.; Nagatsuka, K.; et al. Genetic variations of CYP2C9 in 724 Japanese individuals and their impact on the antihypertensive effects of losartan. Hypertens. Res. 2008, 31, 1549–1557. [Google Scholar] [CrossRef] [PubMed]
- Dodani, S.C.; Kiss, G.; Cahn, J.K.; Su, Y.; Pande, V.S.; Arnold, F.H. Discovery of a regioselectivity switch in nitrating P450s guided by molecular dynamics simulations and Markov models. Nat. Chem. 2016, 8, 419–425. [Google Scholar] [CrossRef] [PubMed]
- Louet, M.; Labbe, C.M.; Fagnen, C.; Aono, C.M.; Homem-de-Mello, P.; Villoutreix, B.O.; Miteva, M.A. Insights into molecular mechanisms of drug metabolism dysfunction of human CYP2C9*30. PLoS ONE 2018, 13, e0197249. [Google Scholar] [CrossRef]
- Dahan, A.; Markovic, M.; Keinan, S.; Kurnikov, I.; Aponick, A.; Zimmermann, E.M.; Ben-Shabat, S. Computational modeling and in-vitro/in-silico correlation of phospholipid-based prodrugs for targeted drug delivery in inflammatory bowel disease. J. Comput. Aided Mol. Des. 2017, 31, 1021–1028. [Google Scholar] [CrossRef]
- Markovic, M.; Ben-Shabat, S.; Keinan, S.; Aponick, A.; Zimmermann, E.M.; Dahan, A. Molecular Modeling-Guided Design of Phospholipid-Based Prodrugs. Int. J. Mol. Sci. 2019, 20, 2210. [Google Scholar] [CrossRef]
- Vyas, B.; Choudhary, S.; Singh, P.K.; Singh, A.; Singh, M.; Verma, H.; Singh, H.; Bahadur, R.; Singh, B.; Silakari, O. Molecular dynamics/quantum mechanics guided designing of natural products based prodrugs of Epalrestat. J. Mol. Struct. 2018, 1171, 556–563. [Google Scholar] [CrossRef]
- Fukui, K.; Yonezawa, T.; Shingu, H. A Molecular Orbital Theory of Reactivity in Aromatic Hydrocarbons. J. Chem. Phys. 1952, 20, 722–725. [Google Scholar] [CrossRef]
- Karaman, R. Prodrugs Design Based on Interand Intramolecular Processes. In Prodrugs Design—A New Era; Nova Science Publishers Inc.: New York, NY, USA, 2014. [Google Scholar] [CrossRef]
- Zhang, J.; Kale, V.; Chen, M. Gene-directed enzyme prodrug therapy. AAPS J. 2015, 17, 102–110. [Google Scholar] [CrossRef] [PubMed]
- Mishra, A.P.; Chandra, S.; Tiwari, R.; Srivastava, A.; Tiwari, G. Therapeutic Potential of Prodrugs Towards Targeted Drug Delivery. Open J. Med. Chem. 2018, 12, 111–123. [Google Scholar] [CrossRef]
- Dahan, A.; Khamis, M.; Agbaria, R.; Karaman, R. Targeted prodrugs in oral drug delivery: The modern molecular biopharmaceutical approach. Expert Opin. Drug Deliv. 2012, 9, 1001–1013. [Google Scholar] [CrossRef]
- Jornada, D.H.; dos Santos Fernandes, G.F.; Chiba, D.E.; de Melo, T.R.; dos Santos, J.L.; Chung, M.C. The Prodrug Approach: A Successful Tool for Improving Drug Solubility. Molecules 2015, 21, 42. [Google Scholar] [CrossRef]
- Rautio, J.; Kumpulainen, H.; Heimbach, T.; Oliyai, R.; Oh, D.; Jarvinen, T.; Savolainen, J. Prodrugs: Design and clinical applications. Nat. Rev. Drug Discov. 2008, 7, 255–270. [Google Scholar] [CrossRef]
- Abu-Jaish, A.; Jumaa, S.; Karaman, R. Prodrugs Overview. In Prodrugs Design—A New Era; Nova Science Publishers Inc.: New York, NY, USA, 2014; pp. 77–102. [Google Scholar]
- Testa, B. Prodrugs: Bridging pharmacodynamic/pharmacokinetic gaps. Curr. Opin. Chem. Biol. 2009, 13, 338–344. [Google Scholar] [CrossRef] [PubMed]
- Cioli, V.; Putzolu, S.; Rossi, V.; Corradino, C. A toxicological and pharmacological study of ibuprofen guaiacol ester (AF 2259) in the rat. Toxicol. Appl. Pharmacol. 1980, 54, 332–339. [Google Scholar] [CrossRef]
- Shah, K.; Gupta, J.K.; Chauhan, N.S.; Upmanyu, N.; Shrivastava, S.K.; Mishra, P. Prodrugs of NSAIDs: A Review. Open J. Med. Chem. 2017, 11, 146–195. [Google Scholar] [CrossRef] [PubMed]
- Lesniewska, M.A.; Ostrowski, T.; Zeidler, J.; Muszalska, I. Ester groups as carriers of antivirally active tricyclic analogue of acyclovir in prodrugs designing: Synthesis, lipophilicity--comparative statistical study of the chromatographic and theoretical methods, validation of the HPLC method. Comb. Chem. High Throughput Screen. 2014, 17, 639–650. [Google Scholar] [CrossRef]
- Hughes, P.M.; Mitra, A.K. Effect of acylation on the ocular disposition of acyclovir. II: Corneal permeability and anti-HSV 1 activity of 2’-esters in rabbit epithelial keratitis. J. Ocul. Pharmacol. 1993, 9, 299–309. [Google Scholar] [CrossRef]
- 113. Bhosle, D.; Bharambe, S.; Gairola, N.; Dhaneshwar, S.S. Mutual prodrug concept: Fundamentals and applications. Indian J. Pharm. Sci. 2006, 68, 286–294. [Google Scholar] [CrossRef]
- Forist, A.A.; DeHaan, R.M.; Metzler, C.M. Clindamycin bioavailability from clindamycin-2-palmitate and clindamycin-2-hexadecylcarbonate in man. J. Pharmacokinet. Biopharm. 1973, 1, 89–98. [Google Scholar] [CrossRef]
- Simplicio, A.L.; Clancy, J.M.; Gilmer, J.F. Prodrugs for amines. Molecules 2008, 13, 519–547. [Google Scholar] [CrossRef]
- Kearney, A.S. Prodrugs and targeted drug delivery. Adv. Drug Deliv. Rev. 1996, 19, 225–239. [Google Scholar] [CrossRef]
- Alexander, J.; Cargill, R.; Michelson, S.R.; Schwam, H. (Acyloxy)alkyl carbamates as novel bioreversible prodrugs for amines: Increased permeation through biological membranes. J. Med. Chem. 1988, 31, 318–322. [Google Scholar] [CrossRef] [PubMed]
- de Groot, F.M.; van Berkom, L.W.; Scheeren, H.W. Synthesis and biological evaluation of 2’-carbamate-linked and 2’-carbonate-linked prodrugs of paclitaxel: Selective activation by the tumor-associated protease plasmin. J. Med. Chem. 2000, 43, 3093–3102. [Google Scholar] [CrossRef] [PubMed]
- Venhuis, B.J.; Dijkstra, D.; Wustrow, D.; Meltzer, L.T.; Wise, L.D.; Johnson, S.J.; Wikstrom, H.V. Orally active oxime derivatives of the dopaminergic prodrug 6-(N,N-di-n-propylamino)-3,4,5,6,7,8-hexahydro-2H-naphthalen-1-one. Synthesis and pharmacological activity. J. Med. Chem. 2003, 46, 4136–4140. [Google Scholar] [CrossRef] [PubMed]
- Kumpulainen, H.; Mahonen, N.; Laitinen, M.L.; Jaurakkajarvi, M.; Raunio, H.; Juvonen, R.O.; Vepsalainen, J.; Jarvinen, T.; Rautio, J. Evaluation of hydroxyimine as cytochrome P450-selective prodrug structure. J. Med. Chem. 2006, 49, 1207–1211. [Google Scholar] [CrossRef]
- Frey, B.M.; Seeberger, M.; Frey, F.J. Pharmacokinetics of 3 prednisolone prodrugs. Evidence of therapeutic inequivalence in renal transplant patients with rejection. Transplantation 1985, 39, 270–274. [Google Scholar] [CrossRef] [PubMed]
- Brouwers, J.; Tack, J.; Augustijns, P. In vitro behavior of a phosphate ester prodrug of amprenavir in human intestinal fluids and in the Caco-2 system: Illustration of intraluminal supersaturation. Int. J. Pharm. 2007, 336, 302–309. [Google Scholar] [CrossRef]
- Wire, M.B.; Shelton, M.J.; Studenberg, S. Fosamprenavir: Clinical pharmacokinetics and drug interactions of the amprenavir prodrug. Clin. Pharmacokinet. 2006, 45, 137–168. [Google Scholar] [CrossRef]
- Fawley, J.; Gourlay, D.M. Intestinal alkaline phosphatase: A summary of its role in clinical disease. J. Surg. Res. 2016, 202, 225–234. [Google Scholar] [CrossRef] [PubMed]
- Jain, A.; Gupta, Y.; Jain, S.K. Azo chemistry and its potential for colonic delivery. Crit. Rev. Ther. Drug Carrier Syst. 2006, 23, 349–400. [Google Scholar] [CrossRef]
- Roldo, M.; Barbu, E.; Brown, J.F.; Laight, D.W.; Smart, J.D.; Tsibouklis, J. Azo compounds in colon-specific drug delivery. Expert Opin. Drug Deliv. 2007, 4, 547–560. [Google Scholar] [CrossRef]
- Banerjee, S.S.; Aher, N.; Patil, R.; Khandare, J. Poly(ethylene glycol)-Prodrug Conjugates: Concept, Design, and Applications. J. Drug Deliv. 2012, 2012, 103973. [Google Scholar] [CrossRef]
- Rodrigues, P.C.; Beyer, U.; Schumacher, P.; Roth, T.; Fiebig, H.H.; Unger, C.; Messori, L.; Orioli, P.; Paper, D.H.; Mulhaupt, R.; et al. Acid-sensitive polyethylene glycol conjugates of doxorubicin: Preparation, in vitro efficacy and intracellular distribution. Bioorg. Med. Chem. 1999, 7, 2517–2524. [Google Scholar] [CrossRef]
- Karaman, R. Computationally Designed Enzyme Models to Replace Natural Enzymes In Prodrug Approaches. Drug Des. 2012, 2. [Google Scholar] [CrossRef]
- Karaman, R. Computationally Designed Prodrugs Based on Enzyme Models. Aperito J. Drug Des. Pharmacol. 2015, 2, 1–6. [Google Scholar] [CrossRef]
- Karaman, R. From Conventional Prodrugs to Prodrugs Designed by Molecular Orbital Methods. In Frontiers in Computational Chemistry, 1st ed.; Ul-Haq, Z., Madura, J.D., Eds.; Bentham Science Publishers: Sharjah, United Arab Emirates, 2015; Volume 2, Chapter 5; pp. 187–249. [Google Scholar] [CrossRef]
- Karaman, R. Prodrug Design by Computation Methods: A New Era. Drug Des. 2013, 2, 1–5. [Google Scholar] [CrossRef]
- Karaman, R.; Jumaa, S.; Awwadallah, H.; Salah, S.; Khawaja, Y.; Karaman, D. Intramolecular Processes and Their Applications in Prodrugs Approaches- Experimental and Computational Studies. Curr. Org. Chem. 2015, 19. [Google Scholar] [CrossRef]
- Karaman, R. The Future of Prodrugs Designed by Computational Chemistry. Drug Des. 2012, 1. [Google Scholar] [CrossRef]
- Karaman, R.; Fattash, B.; Qtait, A. The future of prodrugs—Design by quantum mechanics methods. Expert Opin. Drug Deliv. 2013, 10, 713–729. [Google Scholar] [CrossRef]
- Karaman, R. Design of prodrugs to replace commonly used drugs having bitter sensation. World J. Pharm. Res. 2015, 4, 49–58. [Google Scholar] [CrossRef]
- Karaman, R. Proximity vs. strain in intramolecular ring-closing reactions. Mol. Phys. 2010, 108, 1723–1730. [Google Scholar] [CrossRef]
- Gravina, S.A.; Yep, G.L.; Khan, M. Human biology of taste. Ann. Saudi Med. 2013, 33, 217–222. [Google Scholar] [CrossRef]
- Karaman, R. A Solution to Aversive Tasting Drugs for Pediatric and Geriatric Patients. Drug Des. 2013, 2, 1–4. [Google Scholar] [CrossRef]
- Coupland, J.N.; Hayes, J.E. Physical approaches to masking bitter taste: Lessons from food and pharmaceuticals. Pharm. Res. 2014, 31, 2921–2939. [Google Scholar] [CrossRef]
- Higgins, M.J.; Hayes, J.E. Regional Variation of Bitter Taste and Aftertaste in Humans. Chem. Senses 2019, 44, 721–732. [Google Scholar] [CrossRef] [PubMed]
- Schiffman, S.S. Influence of medications on taste and smell. World J. Otorhinolaryngol. Head Neck Surg. 2018, 4, 84–91. [Google Scholar] [CrossRef] [PubMed]
- Mennella, J.A.; Bobowski, N.K. The sweetness and bitterness of childhood: Insights from basic research on taste preferences. Physiol. Behav. 2015, 152, 502–507. [Google Scholar] [CrossRef] [PubMed]
- Mennella, J.A.; Beauchamp, G.K. Optimizing oral medications for children. Clin. Ther. 2008, 30, 2120–2132. [Google Scholar] [CrossRef] [PubMed]
- Mennella, J.A.; Spector, A.C.; Reed, D.R.; Coldwell, S.E. The bad taste of medicines: Overview of basic research on bitter taste. Clin. Ther. 2013, 35, 1225–1246. [Google Scholar] [CrossRef]
- Sohi, H.; Sultana, Y.; Khar, R.K. Taste masking technologies in oral pharmaceuticals: Recent developments and approaches. Drug Devel. Ind. Pharm. 2004, 30, 429–448. [Google Scholar] [CrossRef]
- Gowthamarajan, K.; Kulkarni, G.T.; Kumar, M.N. Pop the pills without bitterness. Resonance 2004, 9, 25–32. [Google Scholar] [CrossRef]
- Karaman, R. Prodrugs for Masking the Bitter Taste of Drugs. In Application of Nanotechnology in Drug Delivery; IntechOpen: London, UK, 2014. [Google Scholar] [CrossRef]
- Karaman, R. Computationally Designed Prodrugs for Masking the Bitter Taste of Drugs. Drug Des. 2012, 02. [Google Scholar] [CrossRef]
- Albrecht, H.H.; Dicpinigaitis, P.V.; Guenin, E.P. Role of guaifenesin in the management of chronic bronchitis and upper respiratory tract infections. Multidiscip. Respir. Med. 2017, 12, 31. [Google Scholar] [CrossRef]
- Thawabteh, A.; Lelario, F.; Scrano, L.; Bufo, S.A.; Nowak, S.; Behrens, M.; Di Pizio, A.; Niv, M.Y.; Karaman, R. Bitterless guaifenesin prodrugs-design, synthesis, characterization, in vitro kinetics, and bitterness studies. Chem. Biol. Drug Des. 2019, 93, 262–271. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, H.; Onishi, H.; Takahashi, Y.; Iwata, M.; Machida, Y. Development of oral acetaminophen chewable tablets with inhibited bitter taste. Int. J. Pharm. 2003, 251, 123–132. [Google Scholar] [CrossRef]
- Almurisi, S.H.; Doolaanea, A.A.; Akkawi, M.E.; Chatterjee, B.; Ahmed Saeed Aljapairai, K.; Islam Sarker, M.Z. Formulation development of paracetamol instant jelly for pediatric use. Drug Devel. Ind. Pharm. 2020, 46, 1373–1383. [Google Scholar] [CrossRef]
- Hoang Thi, T.H.; Lemdani, M.; Flament, M.P. Optimizing the taste-masked formulation of acetaminophen using sodium caseinate and lecithin by experimental design. Int. J. Pharm. 2013, 453, 408–415. [Google Scholar] [CrossRef] [PubMed]
- Hejaz, H.; Karaman, R.; Khamis, M. Computer-assisted design for paracetamol masking bitter taste prodrugs. J. Mol. Model. 2012, 18, 103–114. [Google Scholar] [CrossRef]
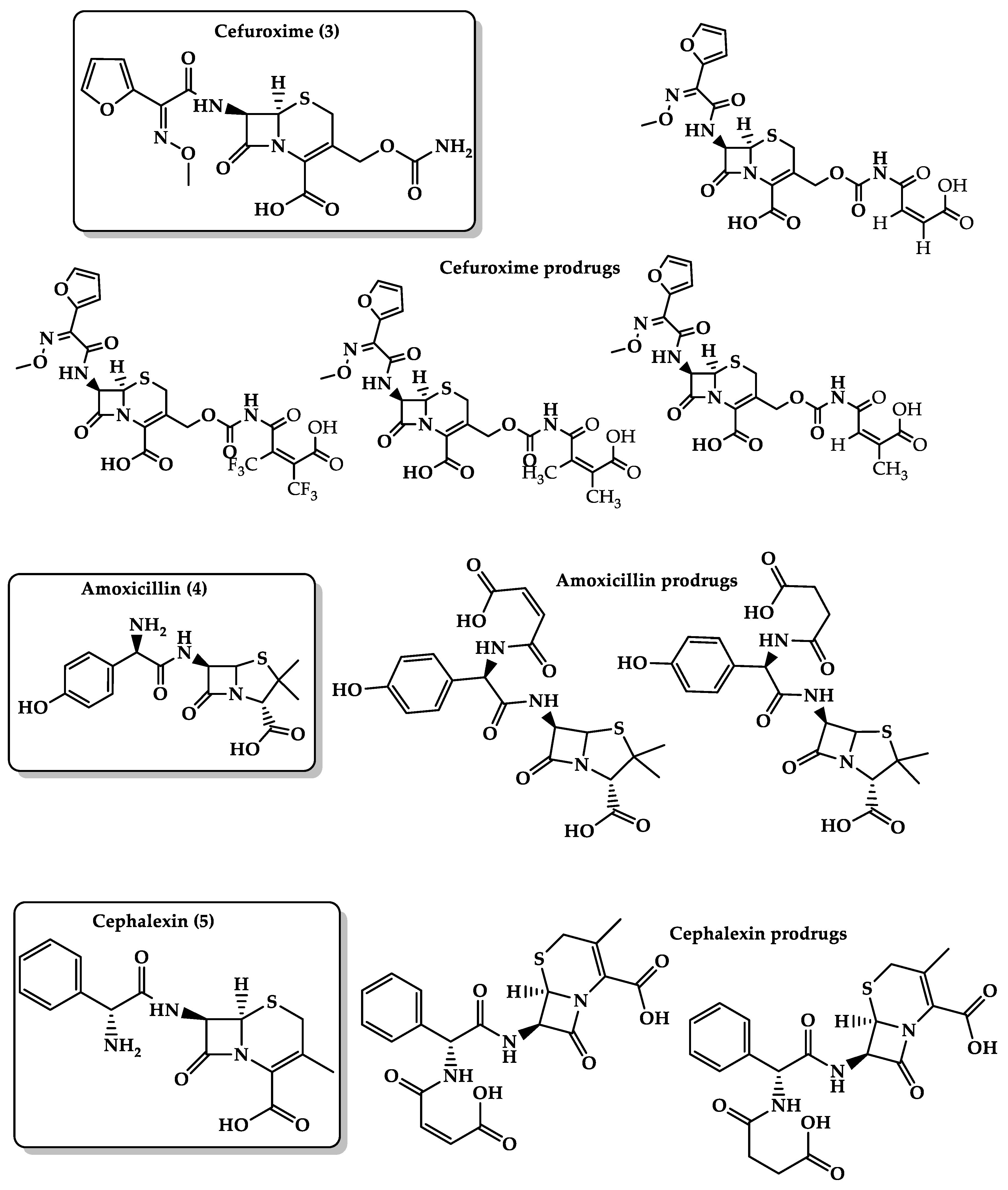
- Karaman, R. Prodrugs for masking bitter taste of antibacterial drugs—A computational approach. J. Mol. Model. 2013, 19, 2399–2412. [Google Scholar] [CrossRef]
- Karaman, R.; Dokmak, G.; Hamarsheh, O.; Karaman, D. Design, synthesis, characterization and in vitro kinetic study of NOVEL antibacterials prodrugs. World J. Pharm. Res. 2015, 4, 2817–2845. [Google Scholar] [CrossRef]
- Karaman, R.; Al-Kurd, S.; Yaghmour, R.; Amro, A.; Karaman, D. ANTIBACTERIAL ACTIVITY OF NOVEL PRODRUGS OF AMOXICILLIN AND CEPHALEXIN. World J. Pharm. Res. 2015, 4, 334–360. [Google Scholar] [CrossRef]
- Foppa, T.; Murakami, F.S.; Silva, M.A. Development, validation and stability study of pediatric atenolol syrup. Pharmazie 2007, 62, 519–521. [Google Scholar] [PubMed]
- Rehman, B.; Sanchez, D.P.; Shah, S. Atenolol. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2020. [Google Scholar]
- Karaman, R.; Dajani, K.; Hallak, H. Computer-assisted design for atenolol prodrugs for the use in aqueous formulations. J. Mol. Model. 2011, 18, 1523–1540. [Google Scholar] [CrossRef]
- Mishra, A.; Singh, S.; Shukla, S. Physiological and Functional Basis of Dopamine Receptors and Their Role in Neurogenesis: Possible Implication for Parkinson’s disease. J. Exp.Neurosci. 2018, 12. [Google Scholar] [CrossRef]
- Haddad, F.; Sawalha, M.; Khawaja, Y.; Najjar, A.; Karaman, R. Dopamine and Levodopa Prodrugs for the Treatment of Parkinson’s Disease. Molecules 2017, 23, 40. [Google Scholar] [CrossRef]
- Karaman, R. Computational-aided design for dopamine prodrugs based on novel chemical approach. Chem. Biol. Drug Des. 2011, 78, 853–863. [Google Scholar] [CrossRef] [PubMed]
- Sizar, O.; Khare, S.; Jamil, R.T.; Talati, R. Statin Medications. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2020. [Google Scholar]
- Stancu, C.; Sima, A. Statins: Mechanism of action and effects. J. Cell. Mol. Med. 2001, 5, 378–387. [Google Scholar] [CrossRef] [PubMed]
- Karaman, R.; Amly, W.; Scrano, L.; Mecca, G.; Bufo, S.A. Computationally designed prodrugs of statins based on Kirby’s enzyme model. J. Mol. Model. 2013, 19, 3969–3982. [Google Scholar] [CrossRef]
- Hengstmann, J.H.; Goronzy, J. Pharmacokinetics of 3H-phenylephrine in man. Eur. J. Clin. Pharmacol. 1982, 21, 335–341. [Google Scholar] [CrossRef]
- Empey, D.W.; Medder, K.T. Nasal decongestants. Drugs 1981, 21, 438–443. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.A.; Hricik, J.G. The pharmacology of alpha-adrenergic decongestants. Pharmacotherapy 1993, 13, 110S–115S;discussion 143S–146S. [Google Scholar] [PubMed]
- Gelotte, C.K.; Zimmerman, B.A. Pharmacokinetics, safety, and cardiovascular tolerability of phenylephrine HCl 10, 20, and 30 mg after a single oral administration in healthy volunteers. Clin. Drug Investig. 2015, 35, 547–558. [Google Scholar] [CrossRef] [PubMed]
- Richards, E.; Lopez, M.J.; Maani, C.V. Phenylephrine. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2020. [Google Scholar]
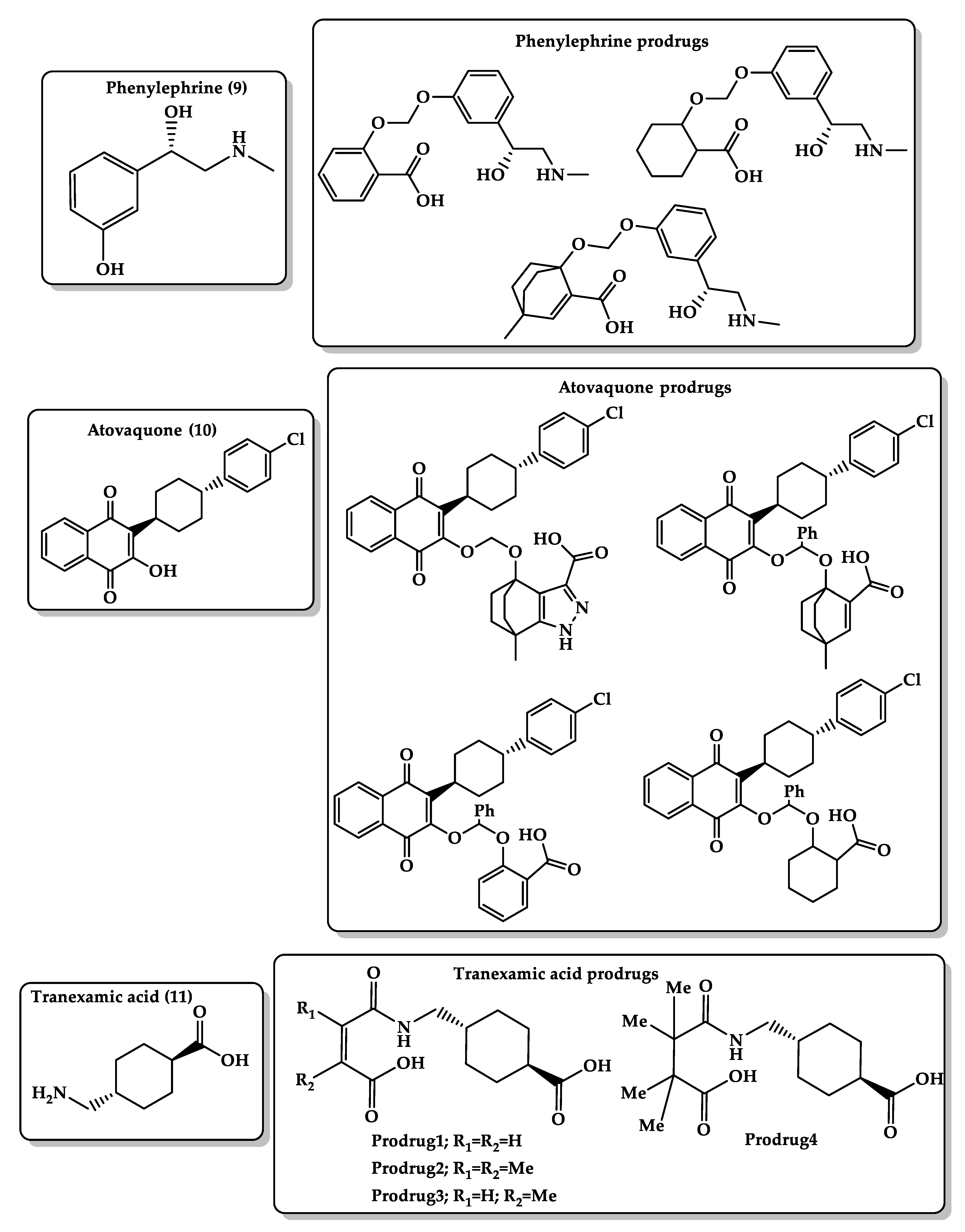
- Karaman, R.; Karaman, D.; Zeiadeh, I. Computationally-designed phenylephrine prodrugs—a model for enhancing bioavailability. Mol. Phys. 2013, 111. [Google Scholar] [CrossRef]
- Karaman, R. Antimalarial Atovaquone Prodrugs Based on Enzyme Models—Molecular Orbital Calculations Approach; Nova Science Publishers Inc.: New York, NY, USA, 2013. [Google Scholar] [CrossRef]
- Karaman, R.; Fattash, B.; Karaman, D. Design, synthesis and in-vitro kinetic study of atovaquone prodrug for the treatment of malaria. World J. Pharm. Res. 2015, 4, 361–390. [Google Scholar] [CrossRef]
- Karaman, R.; Hallak, H. Computer-assisted design of pro-drugs for antimalarial atovaquone. Chem. Biol. Drug Des. 2010, 76, 350–360. [Google Scholar] [CrossRef] [PubMed]
- Karaman, R.; Fattash, B.; Mecca, G.; Bader, M. Computationally designed atovaquone prodrugs based on Bruice’s enzyme model. Curr. Comput. Aided Drug Des. 2014, 10, 15–27. [Google Scholar] [CrossRef] [PubMed]
- Cai, J.; Ribkoff, J.; Olson, S.; Raghunathan, V.; Al-Samkari, H.; DeLoughery, T.G.; Shatzel, J.J. The many roles of tranexamic acid: An overview of the clinical indications for TXA in medical and surgical patients. Eur. J. Haematol. 2020, 104, 79–87. [Google Scholar] [CrossRef] [PubMed]
- Karaman, R.; Ghareeb, H.; Dajani, K.K.; Scrano, L.; Hallak, H.; Abu-Lafi, S.; Mecca, G.; Bufo, S.A. Design, synthesis and in vitro kinetic study of tranexamic acid prodrugs for the treatment of bleeding conditions. J. Comput. Aided Mol. Des. 2013, 27, 615–635. [Google Scholar] [CrossRef]
- Diesch, J.; Zwick, A.; Garz, A.K.; Palau, A.; Buschbeck, M.; Gotze, K.S. A clinical-molecular update on azanucleoside-based therapy for the treatment of hematologic cancers. Clin. Epigenetics 2016, 8, 71. [Google Scholar] [CrossRef] [PubMed]
- Momparler, R.L. Optimization of cytarabine (ARA-C) therapy for acute myeloid leukemia. Exp. Hematol. Oncol. 2013, 2, 20. [Google Scholar] [CrossRef]
- Karahoca, M.; Momparler, R.L. Pharmacokinetic and pharmacodynamic analysis of 5-aza-2’-deoxycytidine (decitabine) in the design of its dose-schedule for cancer therapy. Clin. Epigenetics 2013, 5, 3. [Google Scholar] [CrossRef]
- Hamada, A.; Kawaguchi, T.; Nakano, M. Clinical pharmacokinetics of cytarabine formulations. Clin. Pharmacokinet. 2002, 41, 705–718. [Google Scholar] [CrossRef]
- Garcia-Manero, G.; Stoltz, M.L.; Ward, M.R.; Kantarjian, H.; Sharma, S. A pilot pharmacokinetic study of oral azacitidine. Leukemia 2008, 22, 1680–1684. [Google Scholar] [CrossRef]
- Karaman, R. Prodrugs of aza nucleosides based on proton transfer reaction. J. Comput. Aided Mol. Des. 2010, 24, 961–970. [Google Scholar] [CrossRef] [PubMed]
- Taylor, M.; Gerriets, V. Acyclovir. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2020. [Google Scholar]
- Steingrimsdottir, H.; Gruber, A.; Palm, C.; Grimfors, G.; Kalin, M.; Eksborg, S. Bioavailability of aciclovir after oral administration of aciclovir and its prodrug valaciclovir to patients with leukopenia after chemotherapy. Antimicrob. Agents Chemother. 2000, 44, 207–209. [Google Scholar] [CrossRef] [PubMed]
- Ormrod, D.; Goa, K. Valaciclovir: A review of its use in the management of herpes zoster. Drugs 2000, 59, 1317–1340. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, J.J.; Campoli-Richards, D.M. Acyclovir. An updated review of its antiviral activity, pharmacokinetic properties and therapeutic efficacy. Drugs 1989, 37, 233–309. [Google Scholar] [CrossRef] [PubMed]
- Karaman, R.; Dajani, K.K.; Qtait, A.; Khamis, M. Prodrugs of acyclovir—A computational approach. Chem. Biol. Drug Des. 2012, 79, 819–834. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Quantum Mechanics | Molecular Mechanics | Molecular Docking |
|---|---|---|
| Used to calculate electronic behavior of atoms and molecules such as electron density [34]. | Used for studying physical properties such as structure, energy, and dipole moment by using molecular force fields (FFs) to provide an efficient description of chemical system but cannot calculate electronic behavior [35]. | Used to predict interaction between proteins/proteins or proteins/small molecules to evaluate the binding between them. It is used widely in the field of drug screening and design [36]. |
| Used for small molecule, around hundreds of atoms [18]. | Used for large molecule, more than ten thousands of atoms [37]. | Allows virtual screening of thousands of small molecules [38]. |
| Time and money consuming and requires high computational effort [35]. | Time and money consuming with low computational effort [35]. | Fast and inexpensive method [39]. |
| FF approaches cannot describe the formation and cleavage of covalent chemical bonds, while reactive force fields can make such processes accessible [35]. | Uses force-field-based fixed dielectric charges for both protein and ligand atoms, which gives false-positive or false-negative protein–ligand binding energy [39]. Ignores many inherent factors underlying ligand–receptor interactions such as solvation [38]. Unfitting target binding site and provides only an approximate assessment of binding affinities [38]. Low accuracy, 65%–75% [38,39] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Breijyeh, Z.; Karaman, R. Enzyme Models—From Catalysis to Prodrugs. Molecules 2021, 26, 3248. https://doi.org/10.3390/molecules26113248
Breijyeh Z, Karaman R. Enzyme Models—From Catalysis to Prodrugs. Molecules. 2021; 26(11):3248. https://doi.org/10.3390/molecules26113248
Chicago/Turabian StyleBreijyeh, Zeinab, and Rafik Karaman. 2021. "Enzyme Models—From Catalysis to Prodrugs" Molecules 26, no. 11: 3248. https://doi.org/10.3390/molecules26113248
APA StyleBreijyeh, Z., & Karaman, R. (2021). Enzyme Models—From Catalysis to Prodrugs. Molecules, 26(11), 3248. https://doi.org/10.3390/molecules26113248