
Human Serum Albumin Labelling with a New BODIPY Dye Having a Large Stokes Shift

 , and
, and
Abstract
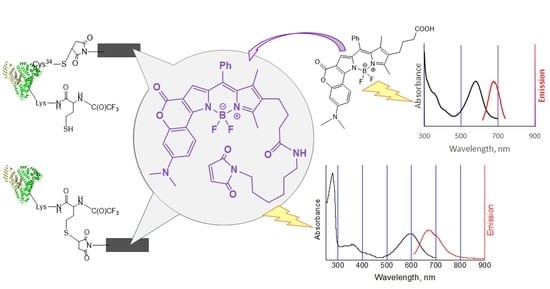
1. Introduction
2. Results and Discussion
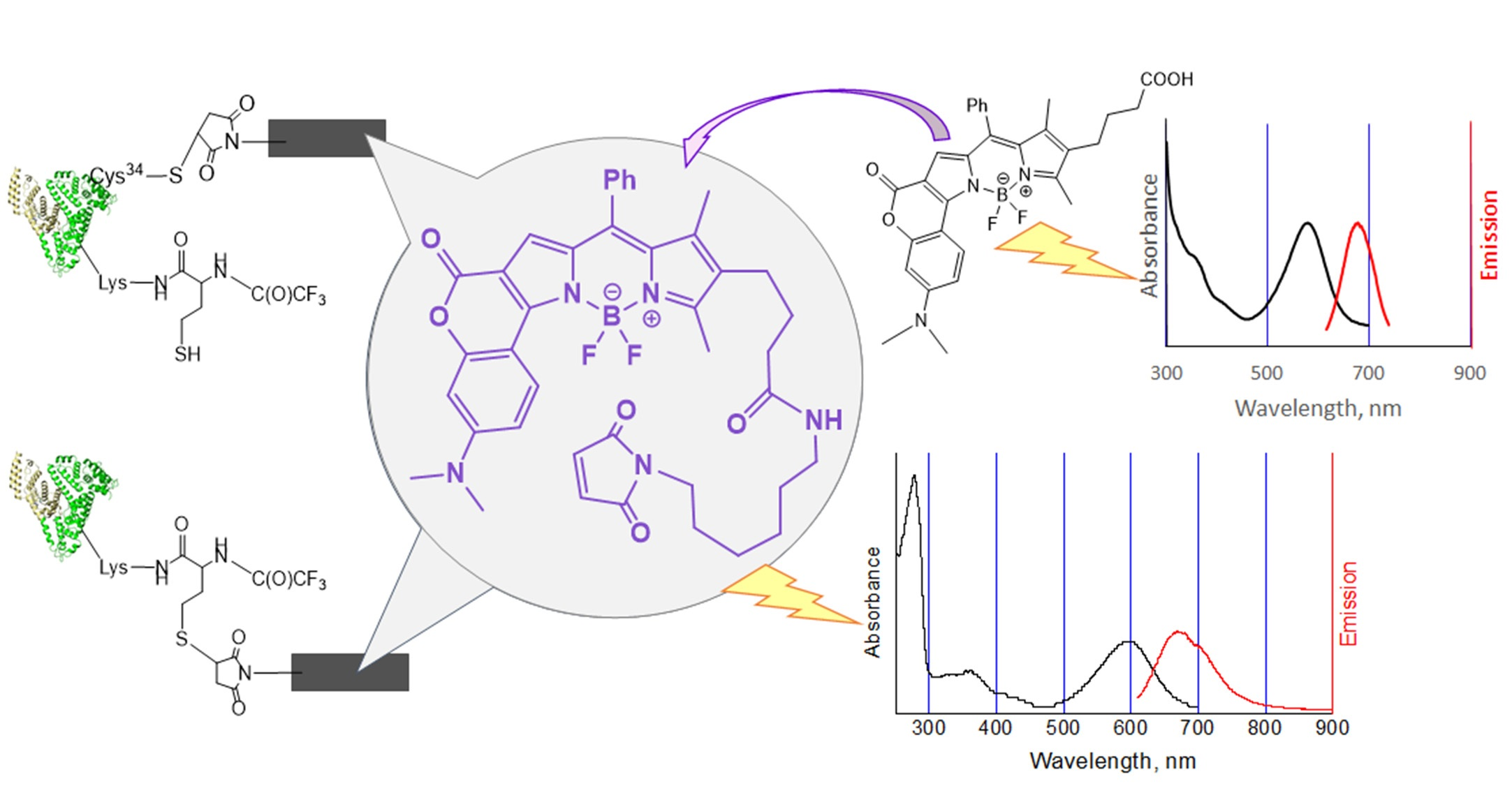
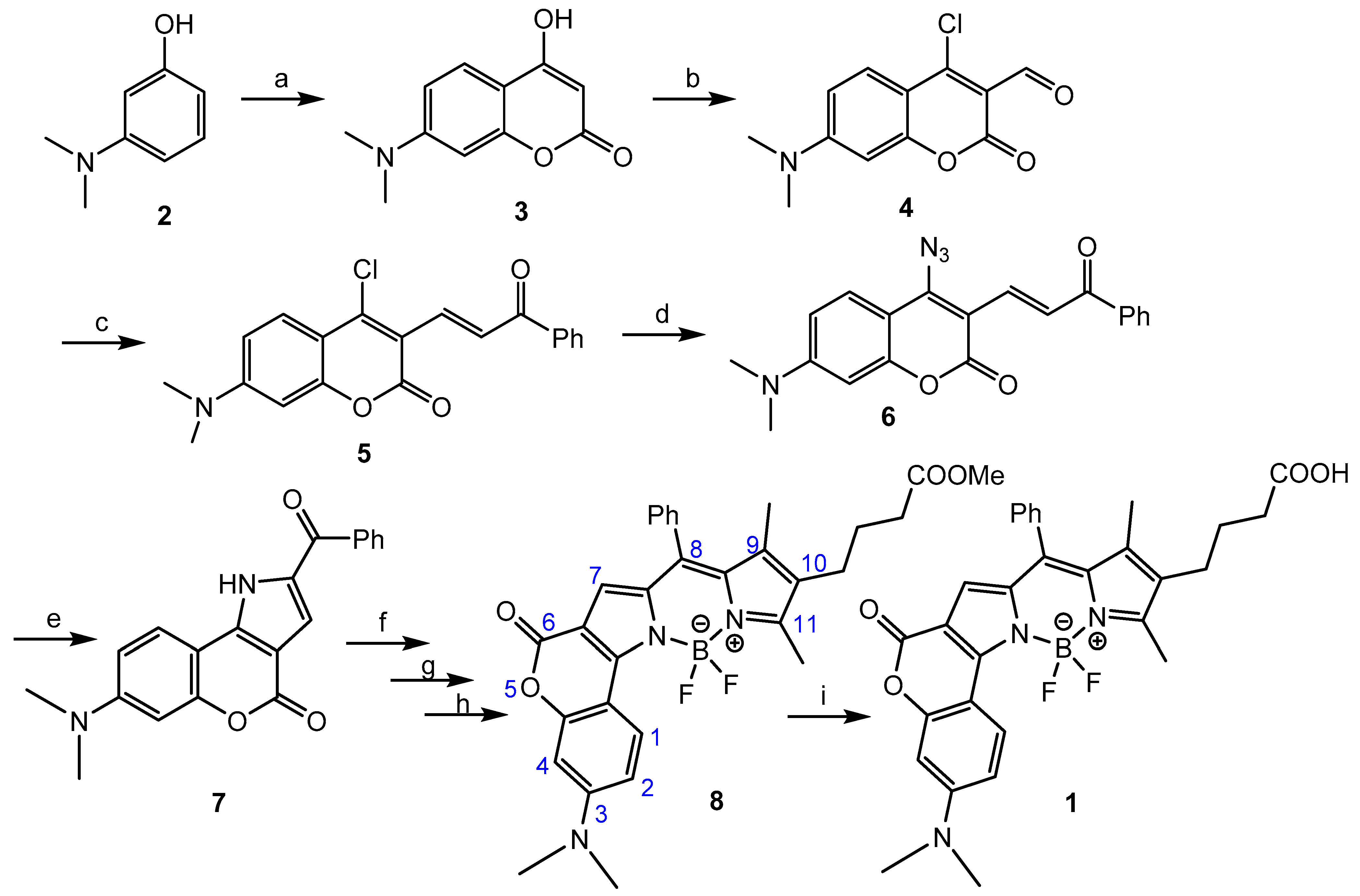
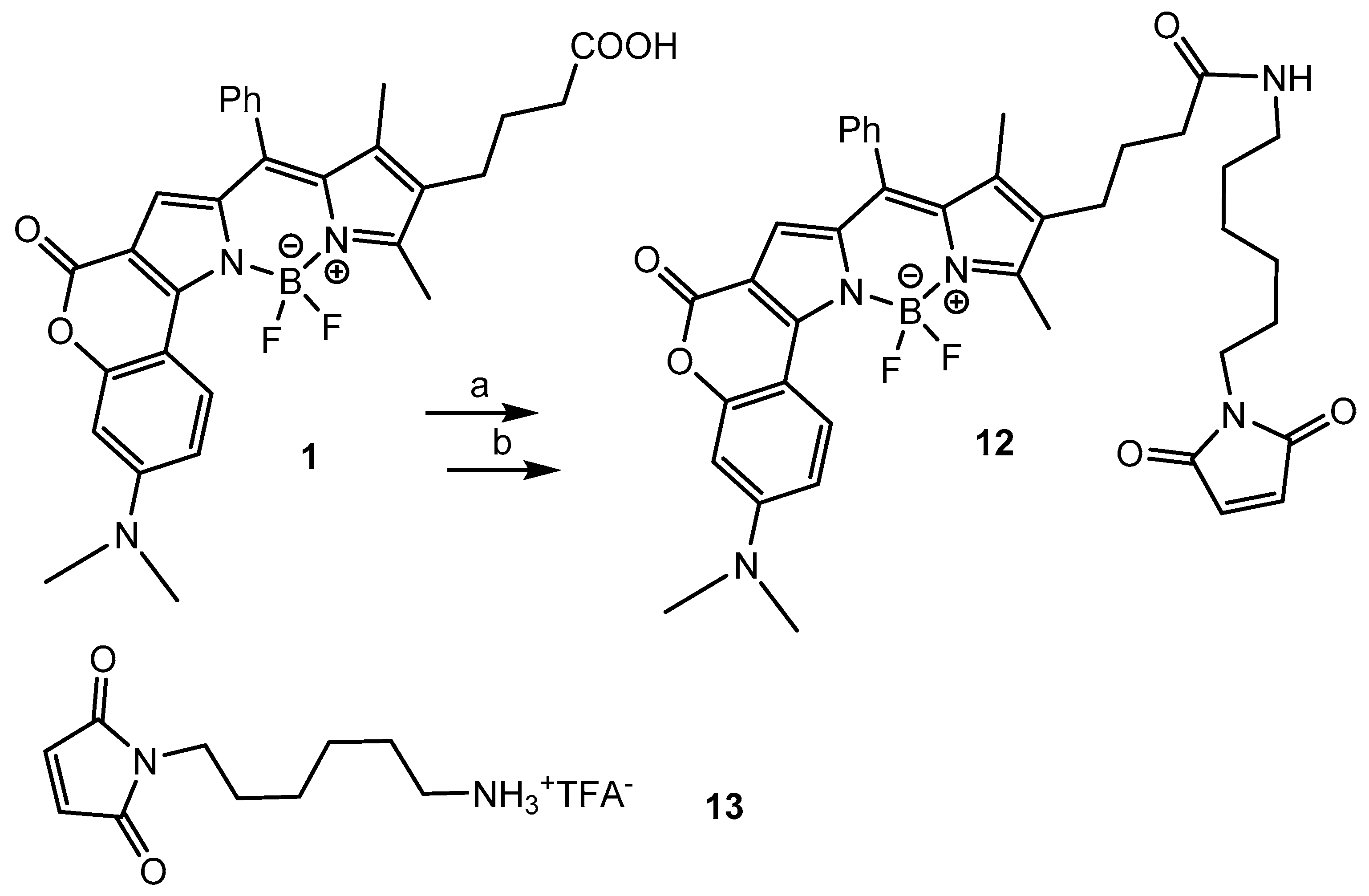
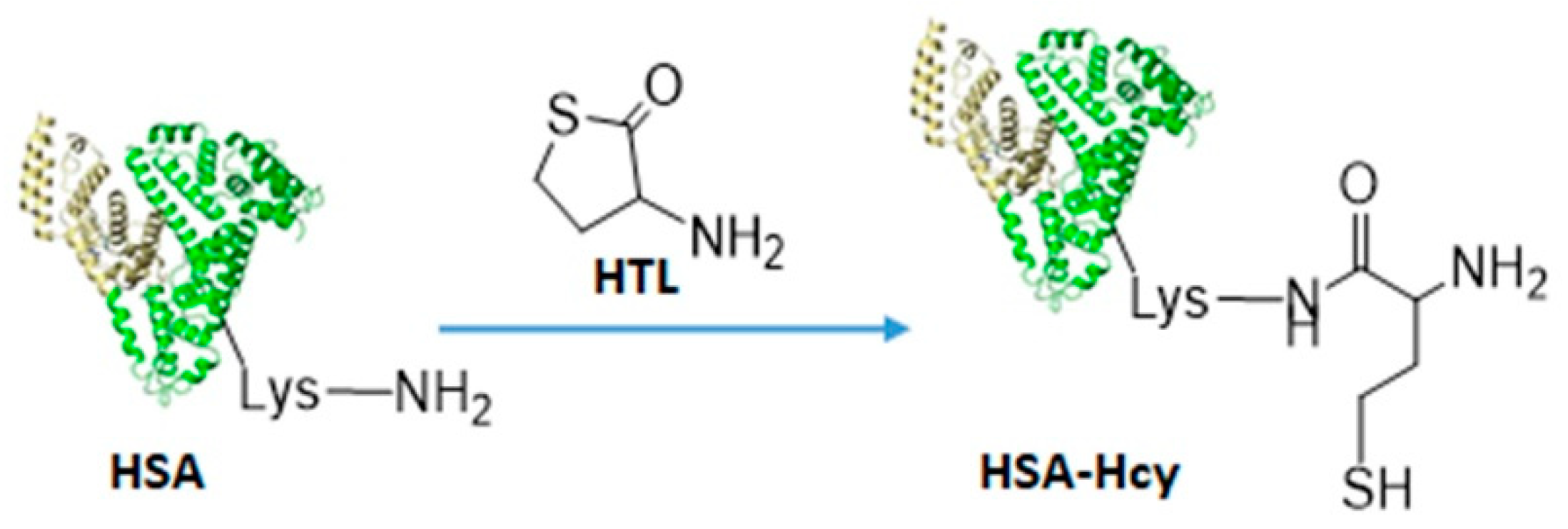
2.1. Chemistry
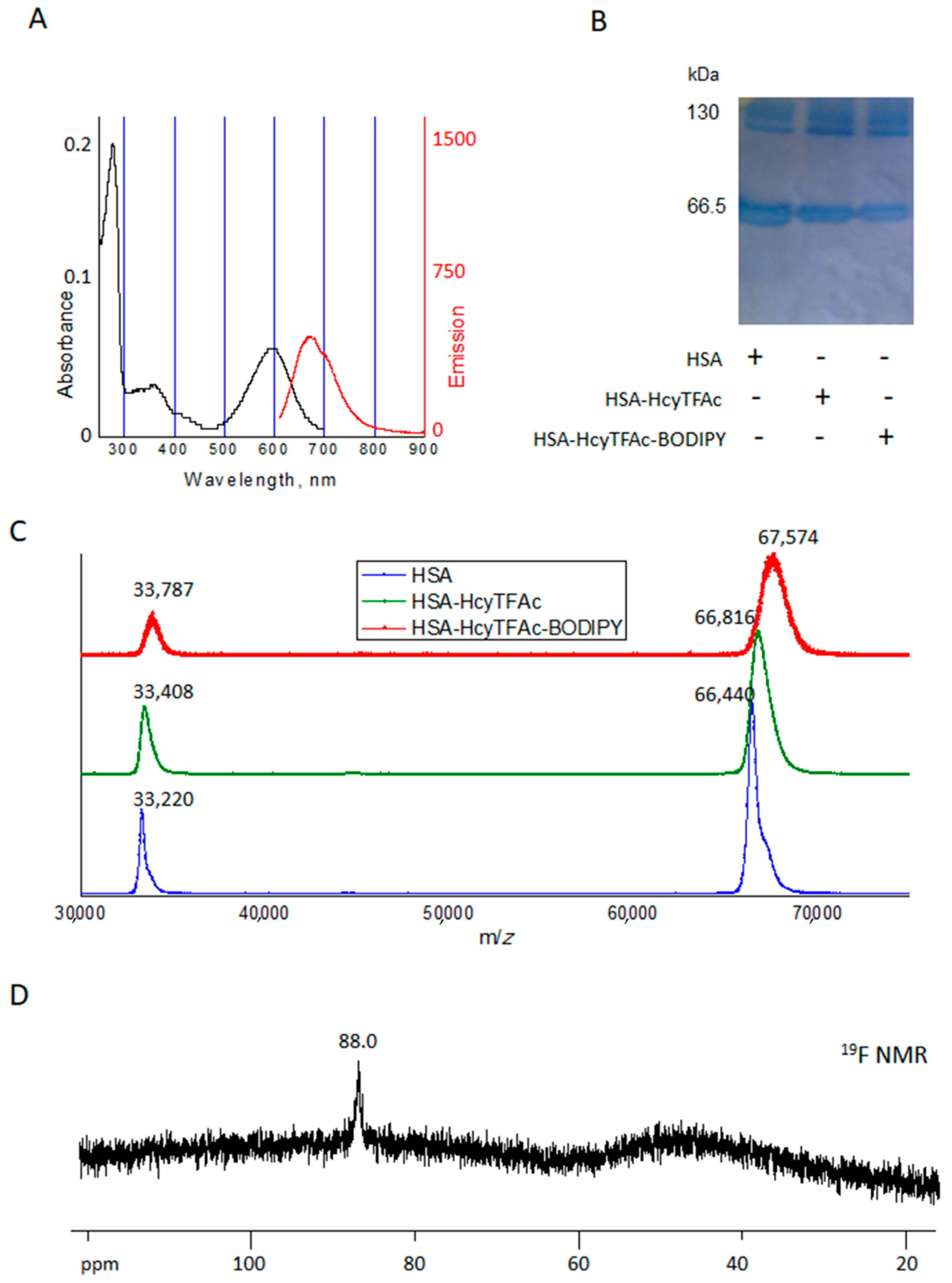
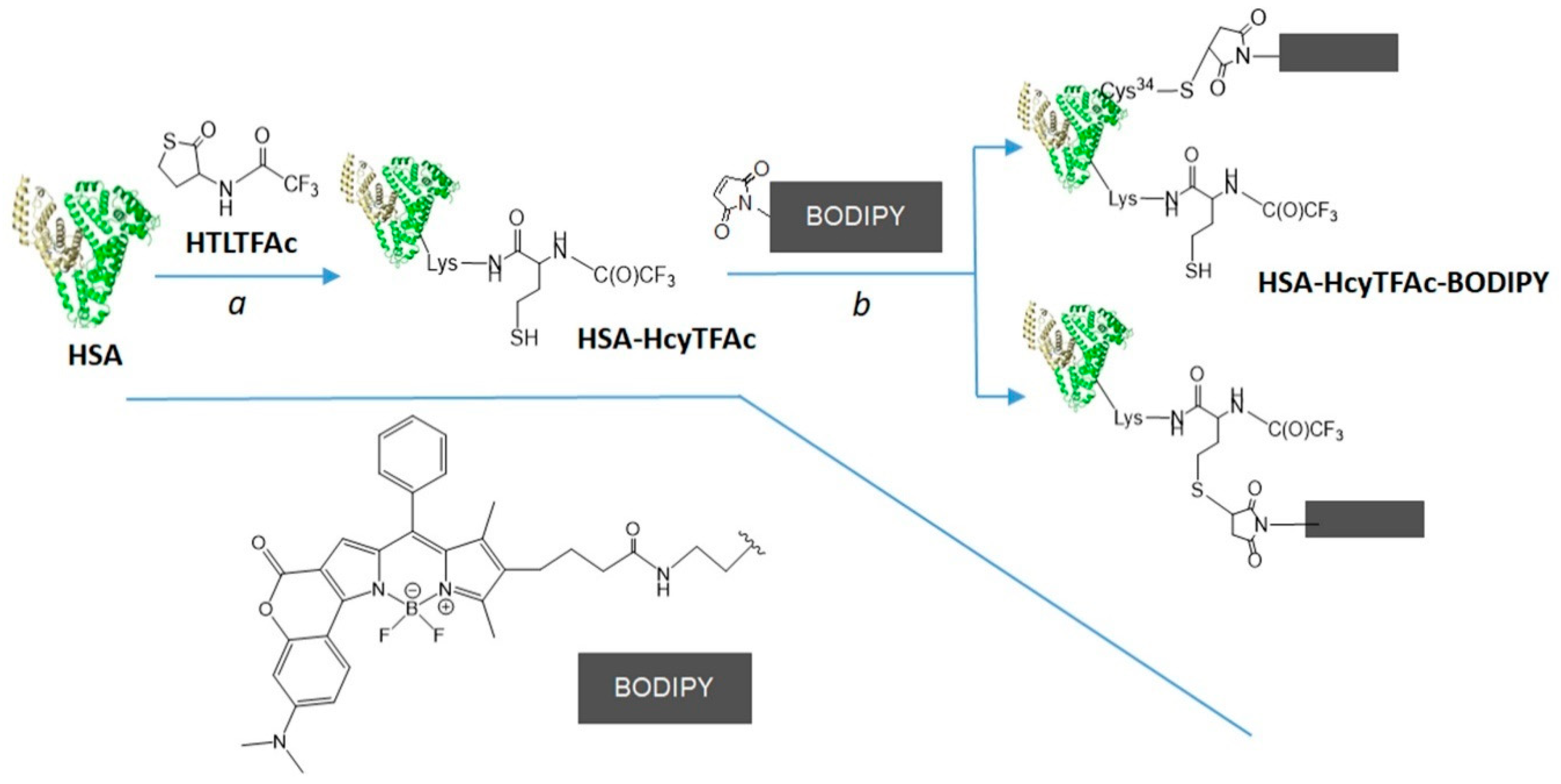
2.2. Bioconjugation
Synthesis of HSA-HcyTFAc-BODIPY Conjugate
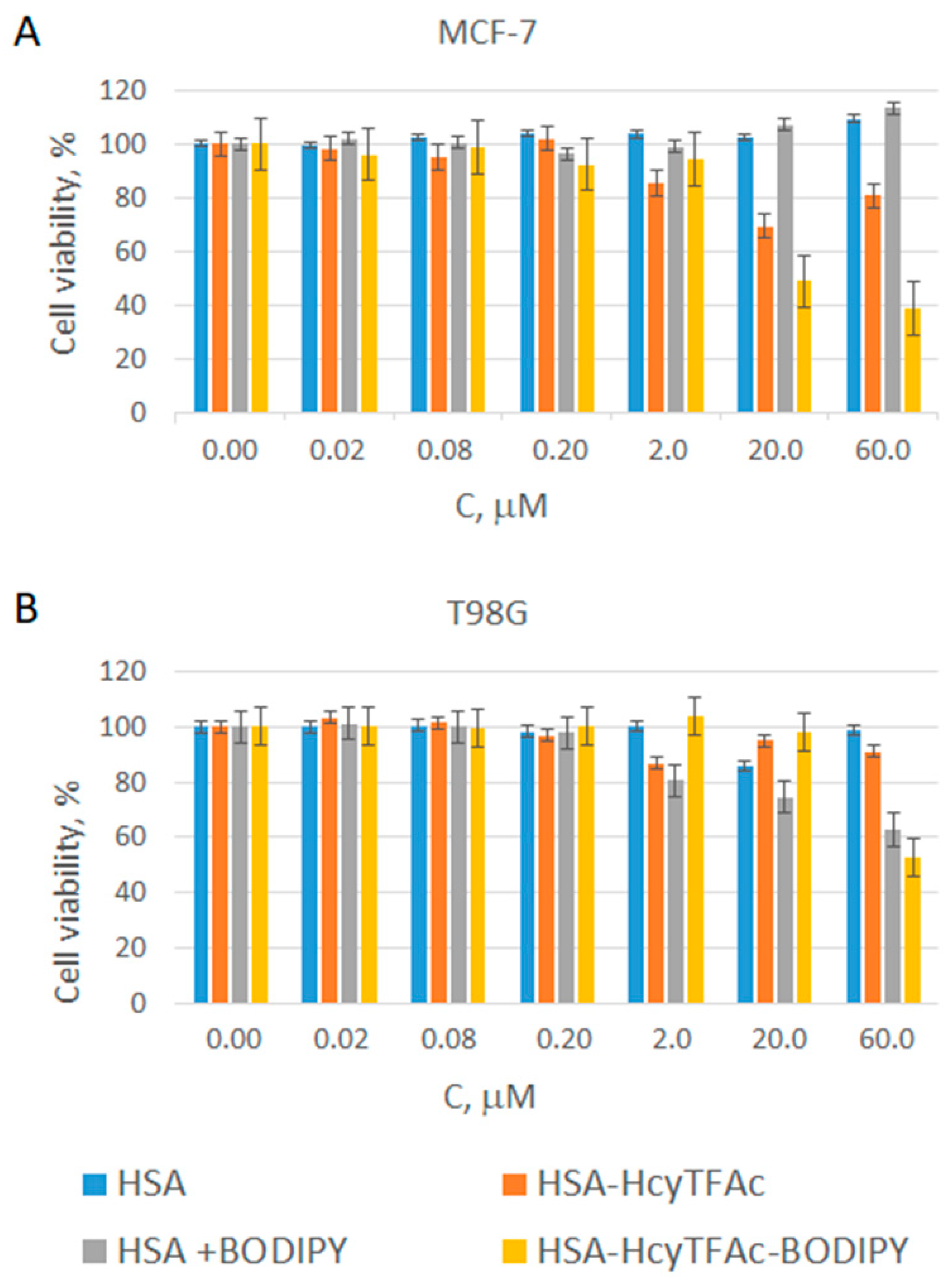
2.3. In Vitro Cytotoxicity Assay
3. Materials and Methods
3.1. BODIPY Compound 8
3.2. BODIPY-COOH 1
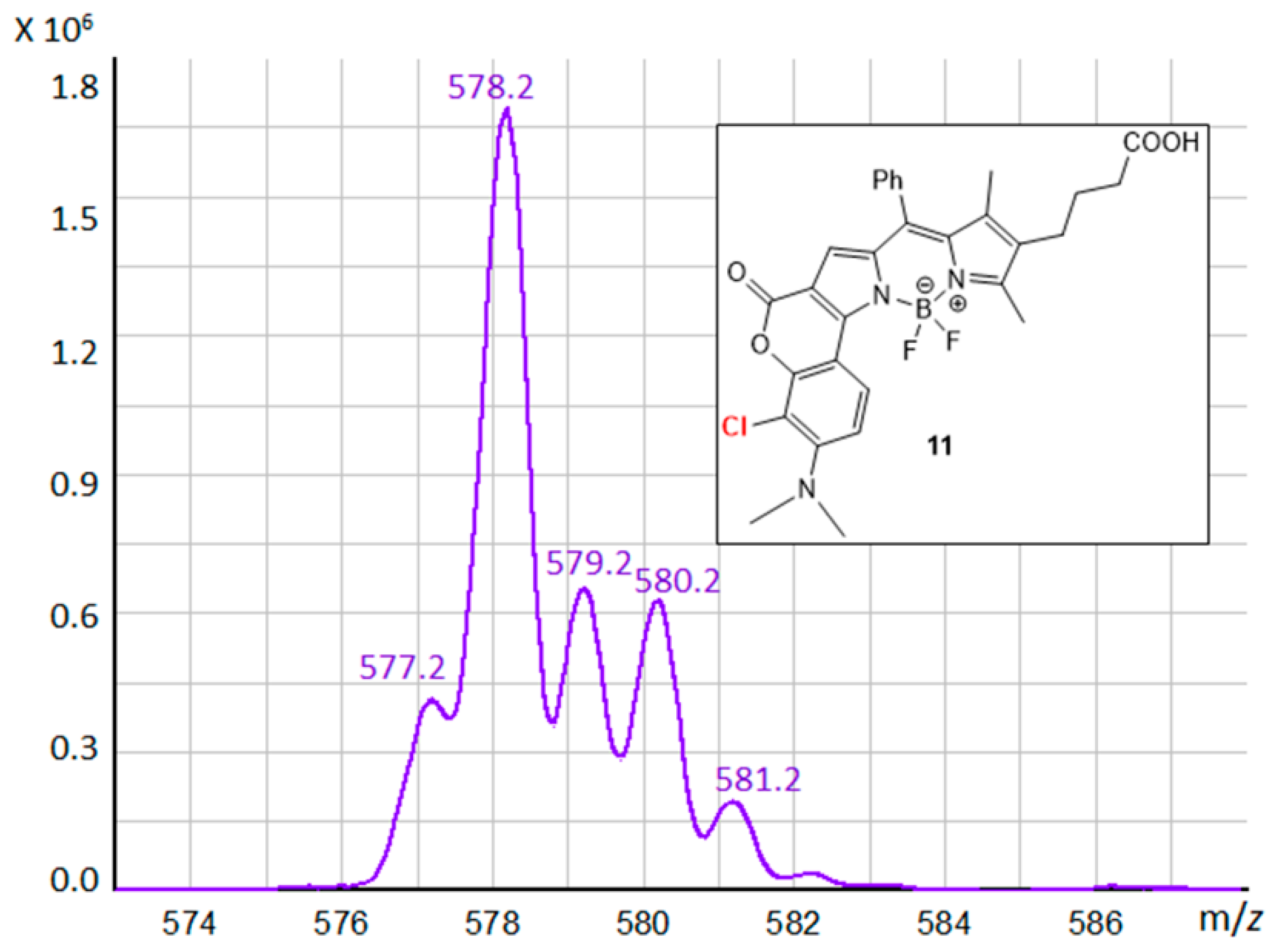
3.3. BODIPY Compound 11
3.4. BODIPY-Maleimide 12
3.5. HSA-HcyTFAc
3.6. HSA-HcyTFAc-BODIPY
3.7. In Vitro Cytotoxicity Assay
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Coderre, J.A.; Morris, G.M. The radiation biology of boron neutron capture therapy. Radiat. Res. 1999, 151, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Soloway, A.H.; Hatanaka, H.; Davis, M.A. Penetration of brain and brain tumor. VII. Tumor-binding sulfhydryl Boron compounds. J. Med. Chem. 1967, 10, 714–717. [Google Scholar] [CrossRef] [PubMed]
- Hatanaka, H.A. A revised boron-neutron capture therapy for malignant brain tumors. J. Neurol. 1975, 209, 81–94. [Google Scholar] [CrossRef] [PubMed]
- Mishima, Y.; Honda, C.; Ichikawa, M. Treatment of malignant melanoma by single thermal neutron capture therapy with melanoma-seeking 10B-compound. Lancet 1989, 334, 388–389. [Google Scholar] [CrossRef]
- Barth, R.F.; Mi, P.; Yang, W. Boron delivery agents for neutron capture therapy of cancer. Cancer Commun. 2018, 38, 35. [Google Scholar] [CrossRef] [PubMed]
- Ali, F.; Hosmane, N.S.; Zhu, Y. Boron chemistry for medical applications. Molecules 2020, 25, 828. [Google Scholar] [CrossRef] [PubMed]
- Rabbani, G.; Ahn, S.N. Structure, enzymatic activities, glycation and therapeutic potential of human serum albumin: A natural cargo. Int. J. Biol. Macromol. 2019, 123, 979–990. [Google Scholar] [CrossRef]
- Zia, M.K.; Siddiqui, T.; Ali, S.S.; Rehman, A.A.; Ahsan, H.; Khan, F.H. Chemotherapeutic drugs and plasma proteins: Exploring new dimensions. Curr. Protein Pept. Sci. 2018, 1, 937–947. [Google Scholar] [CrossRef]
- Amly, W.; Karaman, R. Recent updates in utilizing prodrugs in drug delivery (2013-2015). Expert Opin. Drug Deliv. 2016, 13, 571–591. [Google Scholar] [CrossRef]
- Gou, Y.; Yang, F.; Liang, H. Designing prodrugs based on special residues of human serum albumin. Curr. Top. Med. Chem. 2016, 16, 996–1008. [Google Scholar] [CrossRef]
- Popova, T.V.; Krumkacheva, O.A.; Burmakova, A.S.; Spitsyna, A.S.; Zakharova, O.D.; Lisitskiy, V.A.; Kirilyuk, I.A.; Silnikov, V.N.; Bowman, M.K.; Bagryanskaya, E.G.; et al. Protein modification by thiolactone homocysteine chemistry: A multifunctionalized human serum albumin theranostic. RSC Med. Chem. 2020, 1314–1325. [Google Scholar] [CrossRef]
- Popova, T.V.; Khan, H.; Chubarov, A.S.; Lisitskiy, V.A.; Antonova, N.M.; Akulov, A.E.; Shevelev, O.B.; Zavjalov, E.L.; Silnikov, V.N.; Ahmad, S.; et al. Biotin-decorated anti-cancer nucleotide theranostic conjugate of human serum albumin: Where the seed meets the soil? Bioorg. Med. Chem. Lett. 2018, 28, 260–264. [Google Scholar] [CrossRef]
- Lisitskiy, V.A.; Khan, H.; Popova, T.V.; Chubarov, A.S.; Zakharova, O.D.; Akulov, A.E.; Shevelev, O.B.; Zavjalov, E.L.; Koptyug, I.V.; Moshkin, M.P.; et al. Multifunctional human serum albumin-therapeutic nucleotide conjugate with redox and pH-sensitive drug release mechanism for cancer theranostics. Bioorg. Med. Chem. Lett. 2017, 27, 3925–3930. [Google Scholar] [CrossRef]
- Chubarov, A.S.; Zakharova, O.D.; Koval, O.A.; Romaschenko, A.V.; Akulov, A.E.; Zavjalov, E.L.; Razumov, I.A.; Koptyug, I.V.; Knorre, D.G.; Godovikova, T.S. Design of protein homocystamides with enhanced tumor uptake properties for (19)F magnetic resonance imaging. Bioorg. Med. Chem. 2015, 23, 6943–6954. [Google Scholar] [CrossRef]
- Popova, T.V.; Pyshnaya, I.A.; Zakharova, O.D.; Akulov, A.E.; Shevelev, O.B.; Poletaeva, J.; Zavjalov, E.L.; Silnikov, V.N.; Ryabchikova, E.I.; Godovikova, T.S. Rational design of albumin theranostic conjugates for gold nanoparticles anticancer drugs: Where the seed meets the soil? Biomedicines 2021, 9, 74. [Google Scholar] [CrossRef]
- Sato, S.; Ishii, S.; Nakamura, H. Development of albumin-closo-dodecaborate conjugates as Boron carriers for neutron-capture therapy by Ru(bpy)3-photocatalyzed modification of tyrosine. Eur. J. Inorg. Chem. 2017, 2017, 4406–4410. [Google Scholar] [CrossRef]
- Goszczyński, T.M.; Fink, K.; Kowalski, K.; Leśnikowski, Z.J.; Boratyński, J. Interactions of Boron clusters and their derivatives with serum albumin. Sci. Rep. 2017, 7, 9800. [Google Scholar] [CrossRef]
- Kawai, K.; Nishimura, K.; Okada, S.; Sato, S.; Suzuki, M.; Takata, T.; Nakamura, H. Cyclic RGD-functionalized closo-dodecaborate albumin conjugates as integrin targeting boron carriers for neutron capture therapy. Mol. Pharmaceutics 2020, 17, 3740–3747. [Google Scholar] [CrossRef]
- Takeuchi, I.; Nomura, K.; Makino, K. Hydrophobic boron compound-loaded poly(l-lactide-co-glycolide) nanoparticles for boron neutron capture therapy. Colloids Surf. B 2017, 159, 360–365. [Google Scholar] [CrossRef]
- Hoogenboezem, E.N.; Duvall, C.L. Harnessing albumin as a carrier for cancer therapies. Adv. Drug Delivery Rev. 2018, 130, 73–89. [Google Scholar]
- Loudet, A.; Burgess, K. BODIPY dyes and their derivatives: Syntheses and spectroscopic properties. Chem. Rev. 2007, 107, 4891–4932. [Google Scholar] [CrossRef]
- Liu, Y.; Yang, L.; Ma, C.; Tang, A. Quinoline-fused BODIPY with large Stokes shift as near-infrared dye for cell imaging. Dyes Pigm. 2020, 173, 107981. [Google Scholar] [CrossRef]
- Jiao, C.; Huang, K.-W.; Wu, J. Perylene-fused BODIPY dye with near-IR absorption/emission and high photostability. Org. Lett. 2011, 13, 632–635. [Google Scholar] [CrossRef]
- Sun, Z.-B.; Guo, M.; Zhao, C.-H. Synthesis and properties of benzothieno[b]-fused BODIPY dyes. J. Org. Chem. 2016, 81, 229–237. [Google Scholar]
- Bochkov, A.Y.; Akchurin, I.O.; Dyachenko, O.A.; Traven, V.F. NIR-fluorescent coumarin-fused BODIPY dyes with large Stokes shifts. Chem. Commun. 2013, 49, 11653–11655. [Google Scholar] [CrossRef]
- Urano, Y.; Asanuma, D.; Hama, Y.; Koyama, Y.; Barrett, T.; Kamiya, M.; Nagano, T.; Watanabe, T.; Hasegawa, A.; Choyke, P.L.; et al. Selective molecular imaging of viable cancer cells with pH activatable fluorescence probes. Nat. Med. 2009, 15, 104–109. [Google Scholar] [CrossRef]
- Zhang, W.; Lin, W.; Wang, X.; Li, C.; Liu, S.; Xie, Z. Hybrid nanomaterials of conjugated polymers and albumin for precise photothermal therapy. ACS Appl. Mater. Interfaces 2019, 11, 278–287. [Google Scholar]
- Han, J.; Loudet, A.; Barhoumi, R.; Burghardt, R.C.; Burgess, K. A Ratiometric pH reporter for imaging protein-dye conjugates in living cells. J. Am. Chem. Soc. 2009, 131, 1642–1643. [Google Scholar] [CrossRef]
- Chen, Y.; Liu, J.; Song, M.; Jiang, L.; Liu, L.; Liu, Y.; Fu, G.; Xue, J.; Liu, J.Y.; Huang, M.; et al. Insights into the binding mechanism of BODIPY-based photosensitizers to human serum albumin: A combined experimental and computational study. Spectrochim. Acta A. Mol. Biomol. Spectrosc. 2018, 203, 158–165. [Google Scholar] [CrossRef] [PubMed]
- Boens, N.; Verbelen, B.; Ortiz, M.J.; Jiao, L.; Dehaen, W. Synthesis of BODIPY dyes through postfunctionalization of the boron dipyrromethene core. Coord. Chem. Rev. 2019, 399, 213024. [Google Scholar] [CrossRef]
- Pliquett, J.; Dubois, A.; Racoeur, C.; Mabrouk, N.; Amor, S.; Lescure, R.; Bettaïeb, A.; Collin, B.; Bernhard, C.; Denat, F.; et al. Promising family of fluorescent water-soluble aza-BODIPY dyes for in vivo molecular imaging. Bioconjugate Chem. 2019, 30, 1061–1066. [Google Scholar] [CrossRef] [PubMed]
- Kalot, G.; Godard, A.; Busser, B.; Pliquett, J.; Broekgaarden, M.; Motto-Ros, V.; Wegner, K.D.; Resch-Genger, U.; Köster, U.; Denat, F.; et al. Aza-BODIPY: A new vector for enhanced theranostic Boron Neutron Capture Therapy Applications. Cells 2020, 9, 1953. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Fan, J.; Gao, X.; Wang, B.; Sun, S.; Peng, X. Carboxyl BODIPY Dyes from Bicarboxylic Anhydrides: One-Pot Preparation, Spectral Proper-ties, Photostability, and Biolabeling. J. Org. Chem. 2009, 74, 7675–7683. [Google Scholar] [CrossRef] [PubMed]
- Amorim, V.G.; Melo, S.M.G.; Leite, R.F.; Coutinho, P.A.; da Silva, S.M.P.; Silva, A.R.; Amorim, F.G.; Pires, R.G.W.; Coitinho, J.B.; Emery, F.S.; et al. Synthesis and characterization of two novel red-shifted isothiocyanate BODIPYs and their application in protein conjugation. Dyes Pigm. 2020, 182, 108646–108652. [Google Scholar] [CrossRef]
- Michnik, A.; Drzazga, Z. Effect of ethanol on the thermal stability of human serum albumin. J. Therm. Anal. Calorim. 2007, 88, 449–454. [Google Scholar] [CrossRef]
- Grigoryan, K.R. Preferential solvatation of human serum albumin in dimethylsulfoxide-H2O binary solution. Russ. J. Phys. Chem. A 2009, 83, 2368–2370. [Google Scholar] [CrossRef]
- Cheng, M.H.Y.; Savoie, H.; Bryden, F.; Boyle, R.W. A convenient method for multicolour labelling of proteins with BODIPY fluorophores via tyrosine residues. Photochem. Photobiol. Sci. 2017, 16, 1260–1267. [Google Scholar] [CrossRef]
- Kim, D.; Ma, D.; Kim, M.; Jung, Y.; Kim, N.H.; Lee, C.; Cho, S.W.; Park, S.; Huh, Y.; Jung, J.; et al. Fluorescent labeling of protein using blue-emitting 8-amino-BODIPY derivatives. J. Fluoresc. 2017, 27, 2231–2238. [Google Scholar] [CrossRef]
- Era, H.; Terada, S.; Minami, T.; Takahashi, T.; Arikawa, T. Heterogenity of commercially available human serum albumin products: Thiol oxidation and protein parbonylation. In Proceedings of the 37th Congress of IUPS, Birmingham, UK, 21–26 July 2013. [Google Scholar]
- Miyamura, S.; Imafuku, T.; Anraku, M.; Taguchi, K.; Yamasaki, K.; Tominaga, Y.; Maeda, H.; Ishima, Y.; Watanabe, H.; Otagiri, M.; et al. Comparison of posttranslational modification and the functional impairment of human serum albumin in commercial preparations. J. Pharm. Sci. 2016, 105, 1043–1049. [Google Scholar] [CrossRef]
- Chen, Y.-S.; Kuo, P.-Y.; Shie, T.-L.; Yang, D.-Y. Structure, reactivity, and application of some triketone derivaties. Tetrahedron 2006, 62, 9410–9416. [Google Scholar]
- Alexandrova, L.A.; Jasko, M.V.; Belobritskaya, E.E.; Chudinov, A.V.; Mityaeva, O.N.; Nasedkina, T.V.; Kukhanova, M.K. New triphosphate conjugates bearing reporter groups: Labeling of DNA fragments for microarray analysis. Bioconjugate Chem. 2007, 18, 886–893. [Google Scholar] [CrossRef]
- Puzicha, G.; Shrout, D.P.; Lightner, D.A. Synthesis and properties of homomologated and contracted dipyrrinone analogs of xanthobilirubic acid. J. Heterocycl. Chem. 1990, 27, 2117–2123. [Google Scholar] [CrossRef]
- Shrout, D.P.; Lightner, D.A. An efficient route to dipyrrinones: Synthesis of xanthobilirubic acid methyl ester. Synthesis 1990, 11, 1062–1065. [Google Scholar] [CrossRef]
- Lash, T.D.; Lamm, T.R.; Schaber, J.A.; Chung, W.; Johnson, E.K.; Jones, M.A. Normal and abnormal heme biosynthesis. Part 7. Synthesis and metabolism of coproporphyrinogen-III analogues with acetate or butyrate side chains on rings C and D. Development of a modified model for the active site of coproporphyrinogen oxidase. Bioorg. Med. Chem. 2011, 19, 1492–1504. [Google Scholar] [CrossRef]
- De Wael, E.V.; Pardoen, J.A.; van Koeveringe, J.A.; Lugtenburg, J. Pyrromethene-BF2 complexes (4,4′-difluoro-4-bora-3a,4a-diaza-s-indacenes). Synthesis and luminescence properties. Recl. Trav. Chim. Pays-Bas 2010, 96, 306–309. [Google Scholar] [CrossRef]
- Boens, N.; Leen, V.; Dehaen, W. Fluorescent indicators based on BODIPY. Chem. Soc. Rev. 2012, 41, 1130–1172. [Google Scholar] [CrossRef]
- Meltola, N.J.; Wahlroos, R.; Soini, A.E. Hydrophilic labeling reagents of dipyrrylmethene-BF2 Dyes for two-photon excited fluorometry: Syntheses and photophysical characterization. J. Fluoresc. 2004, 14, 635–647. [Google Scholar] [CrossRef]
- Gießler, K.; Griesser, H.; Göhringer, D.; Sabirov, T.; Richert, C. Synthesis of 3-BODIPY-labeled active esters of nucleotides and a chemical primer extension assay on beads. Eur. J. Org. Chem. 2010, 3611–3620. [Google Scholar] [CrossRef]
- Mukhopadhyay, S.; Chandalia, S.B. Oxidative Chlorination, desulphonation, or decarboxylation to synthesize pharmaceutical intermediates: 2,6-Dichlorotoluene, 2,6-dichloroaniline, and 2,6-dichlorophenol. Org. Process Res. Dev. 1999, 3, 10–16. [Google Scholar] [CrossRef]
- Xin, H.; Yang, S.; An, B.; An, Z. Selective water-based oxychlorination of phenol with hydrogen peroxide catalyzed by manganous sulfate. RSC Adv. 2017, 7, 13467–13472. [Google Scholar] [CrossRef]
- Sharma, S.K.; Agarwal, D.D. Oxidative chlorination of aromatic compounds in aqueous media. IJSRP 2014, 4. [Google Scholar]
- Bratulescu, G. A quick and advantageous synthesis of 2H-1-benzopyran-2-ones unsubstituted on the pyranic nucleus. Synthesis 2008, 2871–2873. [Google Scholar] [CrossRef]
- Choi, H.; Kim, J.; Lee, K. Metal-free, Bronsted acid-mediated synthesis of coumarin derivatives from phenols and propiolic acids. Tetrahedron Lett. 2016, 57, 3600–3603. [Google Scholar] [CrossRef]
- Bizzarri, B.M.; Botta, L.; Capecchi, E.; Celestino, E.; Checconi, P.; Palamara, A.T.; Nencioni, L.; Saladino, R. Regioselective IBX-mediated synthesis of coumarin derivatives with antioxidant andaAnti-influenza activities. J. Nat. Prod. 2017, 80, 3247–3254. [Google Scholar] [CrossRef]
- Masrani, K.V.; Rama, H.S.; Bafna, S.L. Ultraviolet absorption apectra: Some aubstituted coumarins. J. Appl. Chem. Biotechnol. 1974, 24, 331–341. [Google Scholar] [CrossRef]
- Zhao, N.; Xuan, S.; Fronczek, F.R.; Smith, K.M.; Vicente, M.G.H. Enhanced hypsochromic shifts, quantum yield, and π−π interactions in a meso,β-heteroaryl-fused BODIPY. J. Org. Chem. 2017, 82, 3880–3885. [Google Scholar] [CrossRef]
- Ravasco, J.M.; Faustino, H.; Trindade, A.; Gois, P.M.P. Bioconjugation with maleimides: A useful tool for chemical biology. Chem. Eur. J. 2019, 25, 43–59. [Google Scholar] [CrossRef]
- Horstmann, B.; Korbus, M.; Friedmann, T.; Wolff, C.; Thiele, C.M.; Meyer-Almes, F.-J. Synthesis of azobenzenealkylmaleimide probes to photocontrol the enzyme activity of a bacterial histone deacetylase-like amidohydrolase. Bioorg. Chem. 2014, 57, 155–161. [Google Scholar]
- Pisanti, F.A.; Frascatore, S.; Vuttariello, E.; Grillo, A. Influence of acetyl homocysteine thiolactone on erythrocyte superoxide dismutase activity. Biochem. Med. Metab. Biol. 1987, 37, 265–267. [Google Scholar] [CrossRef]
- Papaccio, G.; Pisanti, F.A.; Frascatore, S. Acetyl-homocysteine-thiolactone-induced increase of superoxide dismutase counteracts the effect of subdiabetogenic doses of streptozocin. Diabetes 1986, 35, 470–474. [Google Scholar] [CrossRef]
- Linkova, M.G.; Kuleshova, N.D.; Knunyants, I.L. Thiolactones. Russ. Chem. Rev. 1964, 33, 493–507. [Google Scholar] [CrossRef]
- du Vigneaud, V.; Patterson, W.I.; Hunt, M. Opening of the ring of the thiolactone of homocysteine. J. Biol. Chem. 1938, 126, 217–231. [Google Scholar] [CrossRef]
- Benesch, R.; Benesch, R.E. Formation of peptide bonds by aminolysis of homocysteine thiolactones. J. Am. Chem. Soc. 1956, 78, 1597–1599. [Google Scholar] [CrossRef]
- Peters, T. All About Albumin: Biochemistry, Genetics and Medical Applications; Academic Press: San Diego, CA, USA, 1996; p. 432. [Google Scholar]
- Watanabe, H.; Imafuku, T.; Otagiri, M.; Maruyama, T. Clinical implications associated with the posttranslational modification-induced functional impairment of albumin in oxidative stress-related diseases. J. Pharm. Sci. 2017, 106, 2195–2203. [Google Scholar] [CrossRef] [PubMed]
- Oettl, K.; Marsche, G. Redox state of human serum albumin in terms of cysteine-34 in health and disease. Methods Enzymol. 2010, 474, 181–195. [Google Scholar]
- Jakubowski, H. Homocysteine in Protein Structure/Function and Human Disease: Chemical Biology of Homocysteine-Containing Proteins; Springer: Wien, Austria, 2013; pp. 1–166. [Google Scholar]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Lopes, S.; Nunes, C.M.; Gomes-Zavaglia, A.; Pinho E Melo, T.M.V.D.; Fausto, R. Structure and photochemical behaviour of 3-azido-acrylophenones: A matrix isolation infrared spectroscopy study. Tetrahedron 2011, 67, 7794–7804. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
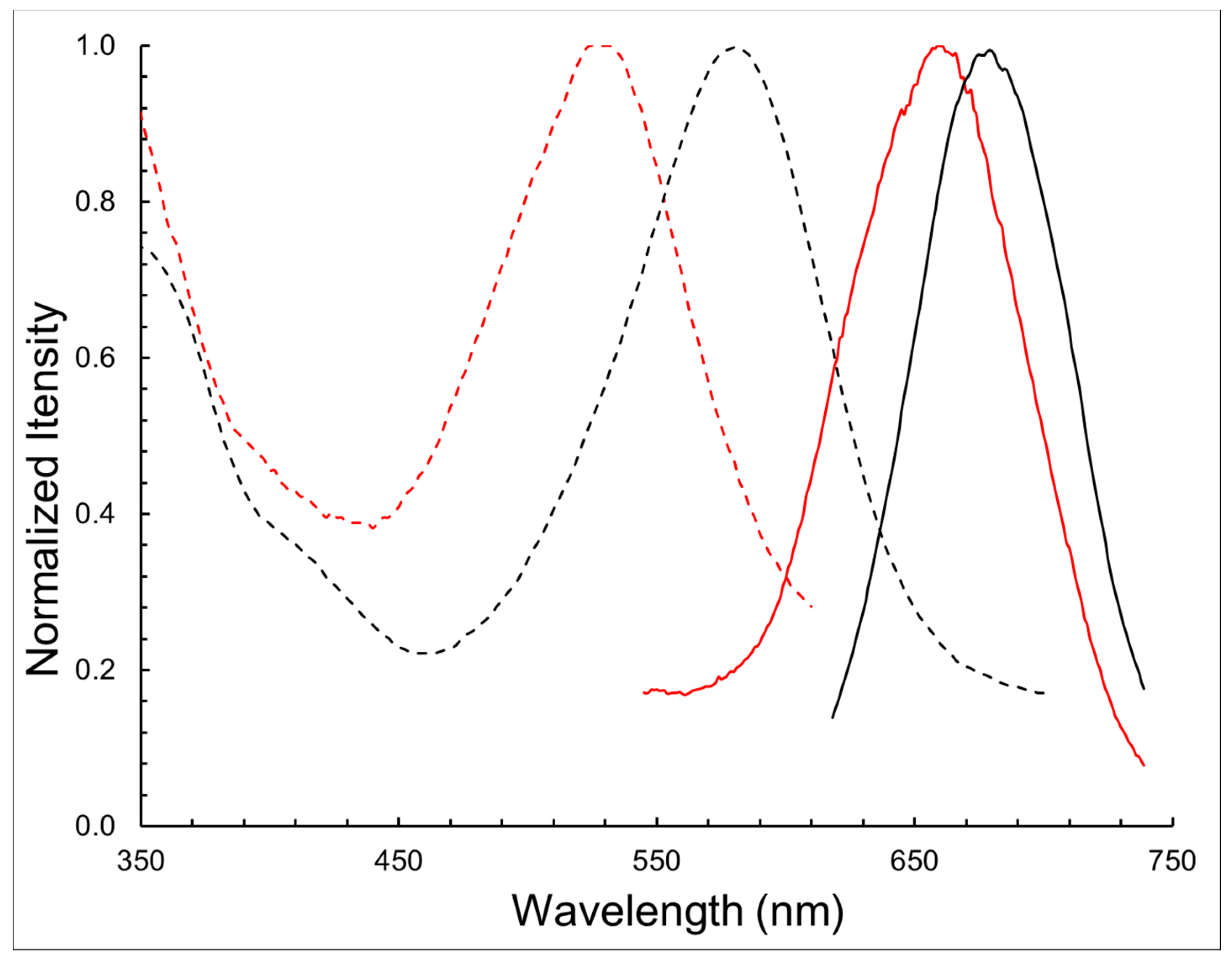
| Compound | λabs (nm) | λem (nm) | log ε | ∆λ (nm) |
|---|---|---|---|---|
| BODIPY 1 | 580 | 678 | 4.54 | 98 |
| BODIPY 11 | 528 | 662 | 4.32 | 134 |
| Conditions | HSA Type | Higher Aggregates | Monomer~66.5 kDa |
|---|---|---|---|
| HSA | 23.5 | 76.5 | |
| Without DTT | HSA-HcyTFAc | 57.2 | 42.8 |
| HSA-HcyTFAc-BODIPY | 66.7 | 33.3 | |
| With DTT (data not shown) | HSA | 16.5 | 83.5 |
| HSA-HcyTFAc | 15.4 | 84.6 | |
| HSA-HcyTFAc-BODIPY | 14.8 | 85.2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Raskolupova, V.I.; Popova, T.V.; Zakharova, O.D.; Nikotina, A.E.; Abramova, T.V.; Silnikov, V.N. Human Serum Albumin Labelling with a New BODIPY Dye Having a Large Stokes Shift. Molecules 2021, 26, 2679. https://doi.org/10.3390/molecules26092679
Raskolupova VI, Popova TV, Zakharova OD, Nikotina AE, Abramova TV, Silnikov VN. Human Serum Albumin Labelling with a New BODIPY Dye Having a Large Stokes Shift. Molecules. 2021; 26(9):2679. https://doi.org/10.3390/molecules26092679
Chicago/Turabian StyleRaskolupova, Valeria I., Tatyana V. Popova, Olga D. Zakharova, Anastasia E. Nikotina, Tatyana V. Abramova, and Vladimir N. Silnikov. 2021. "Human Serum Albumin Labelling with a New BODIPY Dye Having a Large Stokes Shift" Molecules 26, no. 9: 2679. https://doi.org/10.3390/molecules26092679
APA StyleRaskolupova, V. I., Popova, T. V., Zakharova, O. D., Nikotina, A. E., Abramova, T. V., & Silnikov, V. N. (2021). Human Serum Albumin Labelling with a New BODIPY Dye Having a Large Stokes Shift. Molecules, 26(9), 2679. https://doi.org/10.3390/molecules26092679