
Metformin Inhibits the Urea Cycle and Reduces Putrescine Generation in Colorectal Cancer Cell Lines

 , ,
, ,
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. Metformin Reduced the Expression of ODC and Inhibited the Growth of Colorectal Cancer Cells In Vivo
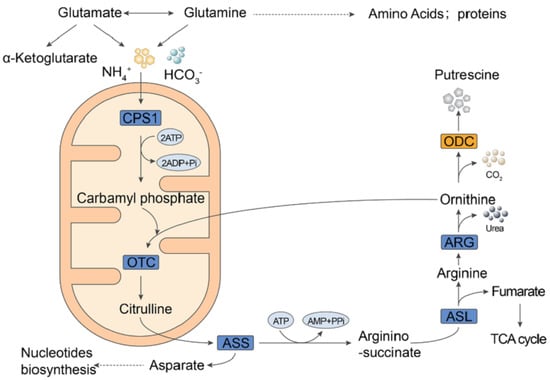
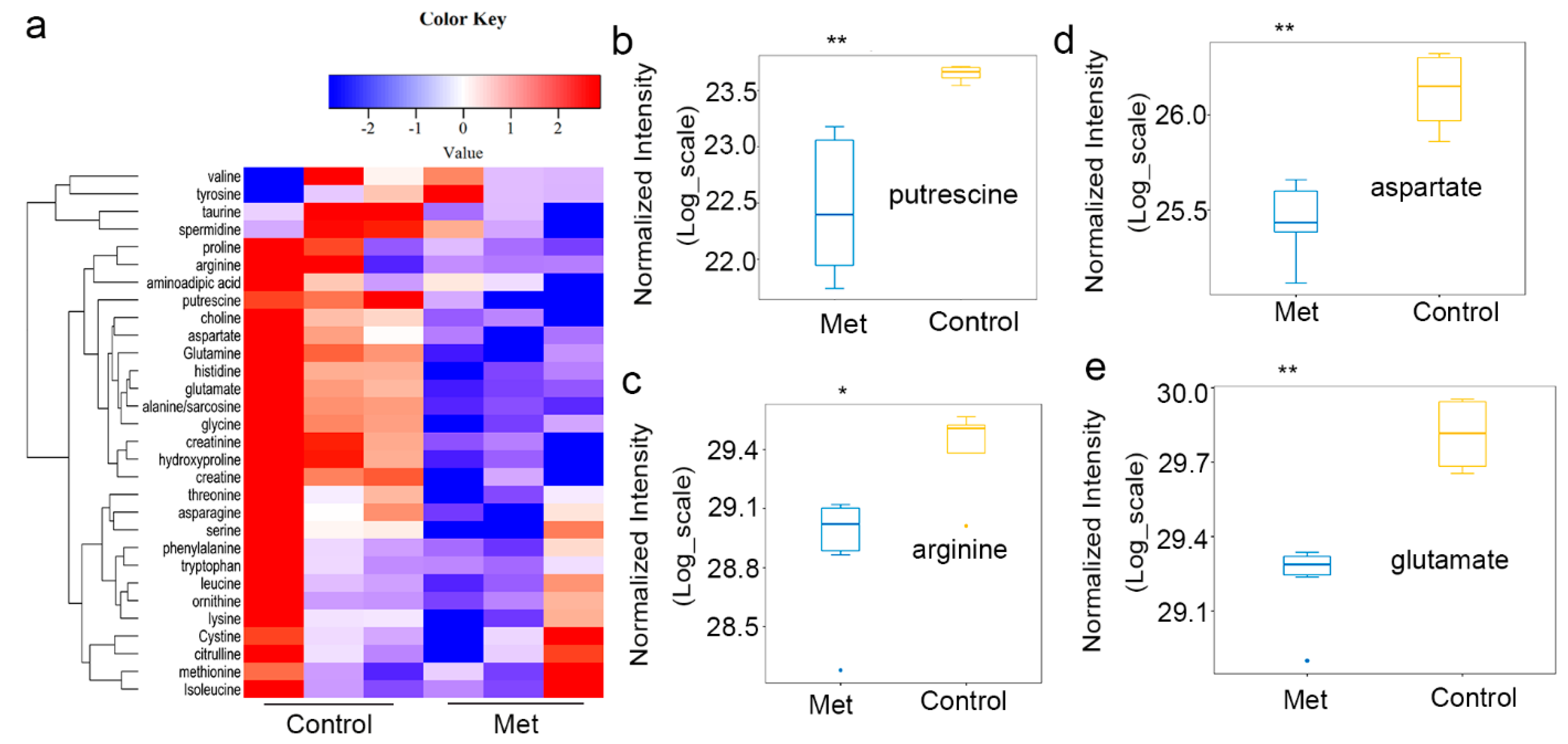
2.2. Metformin Reduced Putrescine Levels and Urea Cycle Intermediates in HCT116 Xenografts
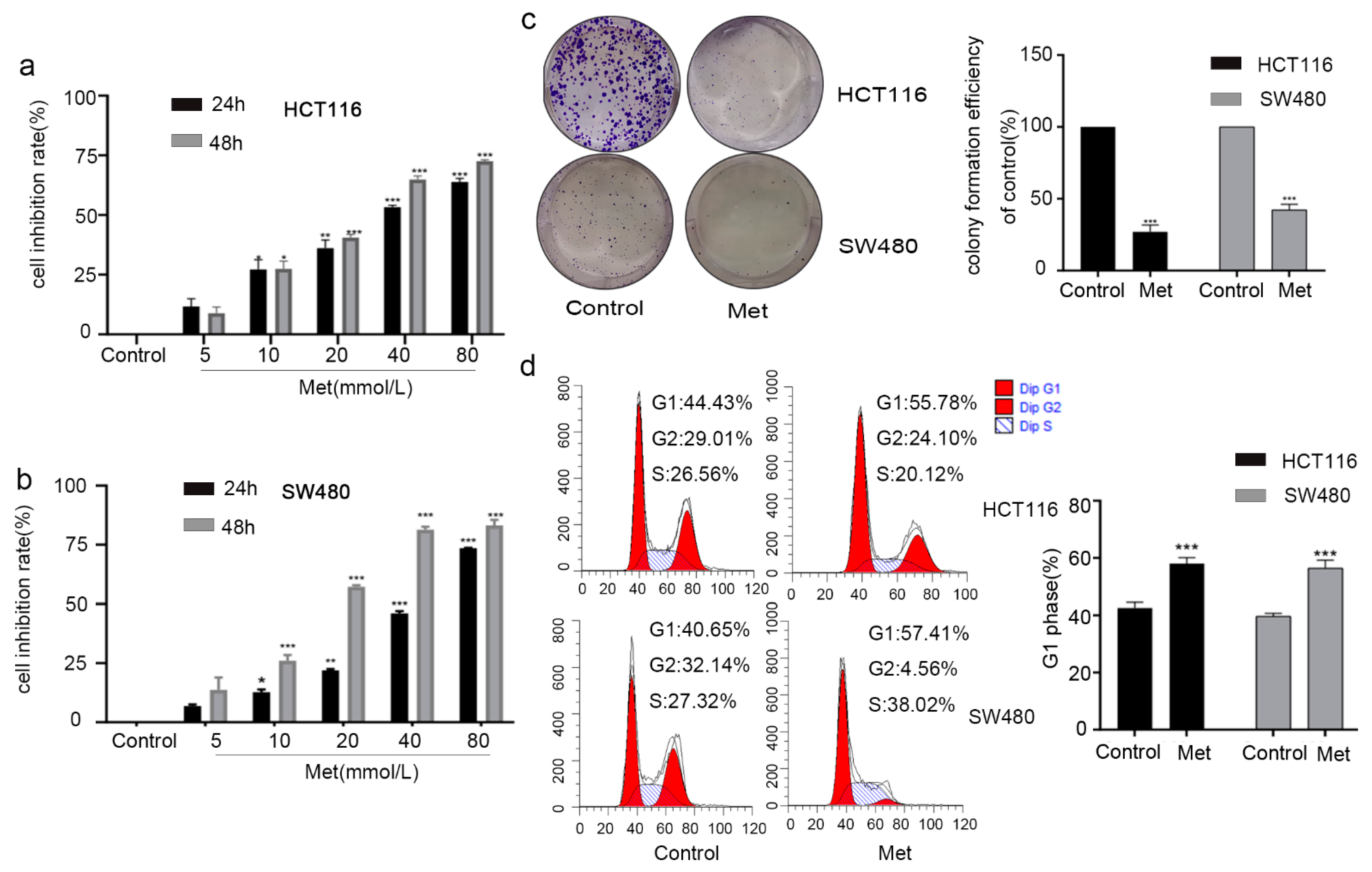
2.3. Metformin Inhibited Cell Viability by Inducing G1 Phase Cell Cycle Arrest in Colorectal Cancer Cells
2.4. Metformin Inhibited the Proliferation of HCT116 Cells via Decreasing the Level of Putrescine and Increasing the Levels of Branched-Chain Amino Acids (BCAAs: Val, Ile, and Leu)
2.5. Metformin Inhibited Proliferation of Colorectal Cancer Cells by Changing Expression of AMPK and Urea Cycle Enzymes
2.6. Metformin Inhibited Colorectal Cancer HCT116 Cell Proliferation by Activating p53
2.7. NH4Cl Suppressed the Proliferation of Colorectal Cancer Cells and Expression of ODC In Vitro
3. Discussion
4. Materials and Methods
4.1. Reagents and Antibodies
4.2. Cell Culture and Culture Conditions
4.3. Tumor Xenograft Models in Nude Mice
4.4. Cell Viability Assay
4.5. Cell cycle Analysis
4.6. Colony-Formation Assay
4.7. Immunoblot Assay
4.8. EdU Proliferation Assay
4.9. LC-MS Analysis for Metabolites
4.10. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Mathers, C.; Parkin, D.M.; Pineros, M.; Znaor, A.; Bray, F. Estimating the global cancer incidence and mortality in 2018: GLOBOCAN sources and methods. Int. J. Cancer 2019, 144, 1941–1953. [Google Scholar] [CrossRef]
- Obrand, D.I.; Gordon, P.H. Incidence and patterns of recurrence following curative resection for colorectal carcinoma. Dis. Colon Rectum 1997, 40, 15–24. [Google Scholar] [CrossRef]
- O’Connell, M.J.; Campbell, M.E.; Goldberg, R.M.; Grothey, A.; Seitz, J.-F.; Benedetti, J.K.; André, T.; Haller, D.G.; Sargent, D.J. Survival Following Recurrence in Stage II and III Colon Cancer: Findings From the ACCENT Data Set. J. Clin. Oncol. 2008, 26, 2336–2341. [Google Scholar] [CrossRef] [PubMed]
- André, T.; De Gramont, A.A.; Vernerey, D.; Chibaudel, B.B.; Bonnetain, F.; Tijeras-Raballand, A.A.; Scriva, A.A.; Hickish, T.T.; Tabernero, J.; Van Laethem, J.L.; et al. Adjuvant Fluorouracil, Leucovorin, and Oxaliplatin in Stage II to III Colon Cancer: Updated 10-Year Survival and Outcomes According to BRAF Mutation and Mismatch Repair Status of the MOSAIC Study. J. Clin. Oncol. 2015, 33, 4176–4187. [Google Scholar] [CrossRef] [PubMed]
- Katona, B.W.; Weiss, J.M. Chemoprevention of Colorectal Cancer. Gastroenterology 2020, 158, 368–388. [Google Scholar] [CrossRef] [PubMed]
- Dupont, A.W.; Arguedas, M.R.; Wilcox, C.M. Aspirin chemoprevention in patients with increased risk for colorectal cancer: A cost-effectiveness analysis. Aliment. Pharmacol. Ther. 2007, 26, 431–441. [Google Scholar] [CrossRef] [PubMed]
- Ward, P.S.; Thompson, C.B. Metabolic Reprogramming: A Cancer Hallmark Even Warburg Did Not Anticipate. Cancer Cell 2012, 21, 297–308. [Google Scholar] [CrossRef] [PubMed]
- Holmes, F.L. Hans Krebs and the discovery of the ornithine cycle. Fed. Proc. 1980, 39, 216–225. [Google Scholar]
- Krebs, H.A. The History of the Tricarboxylic Acid Cycle. Perspect. Biol. Med. 1970, 14, 154–172. [Google Scholar] [CrossRef]
- Lanpher, B.; Brunetti-Pierri, N.; Lee, B. Inborn errors of metabolism: The flux from Mendelian to complex diseases. Nat. Rev. Genet. 2006, 7, 449–459. [Google Scholar] [CrossRef]
- Wang, H.; Huang, B.; Wang, W.; Li, J.; Chen, Y.; Flynn, T.; Zhao, M.; Zhou, Z.; Lin, X.; Zhang, Y.; et al. High urea induces depression and LTP impairment through mTOR signalling suppression caused by carbamylation. EBioMedicine 2019, 48, 478–490. [Google Scholar] [CrossRef]
- Kim, J.; Hu, Z.; Cai, L.; Li, K.; Choi, E.; Faubert, B.; Bezwada, D.; Rodriguez-Canales, J.; Villalobos, P.; Lin, Y.-F.; et al. CPS1 maintains pyrimidine pools and DNA synthesis in KRAS/LKB1-mutant lung cancer cells. Nat. Cell Biol. 2017, 546, 168–172. [Google Scholar] [CrossRef]
- Dillon, B.J.; Prieto, V.G.; Curley, S.A.; Ensor, C.M.; Holtsberg, F.W.; Bomalaski, J.S.; Clark, M.A. Incidence and distribution of argininosuccinate synthetase deficiency in human cancers. Cancer 2004, 100, 826–833. [Google Scholar] [CrossRef]
- Gao, Y.; Wang, X.; Sang, Z.; Li, Z.; Liu, F.; Mao, J.; Yan, D.; Zhao, Y.; Wang, H.; Li, P.; et al. Quantitative proteomics by SWATH-MS reveals sophisticated metabolic reprogramming in hepatocellular carcinoma tissues. Sci. Rep. 2017, 7, 45913. [Google Scholar] [CrossRef]
- Huang, H.-L.; Chen, W.-C.; Hsu, H.-P.; Cho, C.-Y.; Hung, Y.-H.; Wang, C.-Y.; Lai, M.-D. Silencing of argininosuccinate lyase inhibits colorectal cancer formation. Oncol. Rep. 2016, 37, 163–170. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.-L.; Chen, W.-C.; Hsu, H.-P.; Cho, C.-Y.; Hung, Y.-H.; Wang, C.-Y.; Lai, M.-D. Argininosuccinate lyase is a potential therapeutic target in breast cancer. Oncol. Rep. 2015, 34, 3131–3139. [Google Scholar] [CrossRef]
- Cervelli, M.; Pietropaoli, S.; Signore, F.; Amendola, R.; Mariottini, P. Polyamines metabolism and breast cancer: State of the art and perspectives. Breast Cancer Res. Treat. 2014, 148, 233–248. [Google Scholar] [CrossRef] [PubMed]
- Bachrach, U. Polyamines and cancer: Minireview article. Amino Acids 2004, 26, 307–309. [Google Scholar] [CrossRef] [PubMed]
- Linsalata, M.; Caruso, M.G.; Leo, S.; Guerra, V.; D’Attoma, B.; Di Leo, A. Prognostic value of tissue polyamine levels in human colorectal carcinoma. Anticancer Res. 2002, 22, 2465–2469. [Google Scholar] [PubMed]
- Linsalata, M.; Giannini, R.; Notarnicola, M.; Cavallini, A. Peroxisome proliferator-activated receptor gamma and spermidine/spermine N1-acetyltransferase gene expressions are significantly correlated in human colorectal cancer. BMC Cancer 2006, 6, 191. [Google Scholar] [CrossRef]
- Babbar, N.; Gerner, E.W. Targeting Polyamines and Inflammation for Cancer Prevention. Methods Mol. Biol. 2010, 188, 49–64. [Google Scholar]
- Rena, G.; Hardie, D.G.; Pearson, E.R. The mechanisms of action of metformin. Diabetology 2017, 60, 1577–1585. [Google Scholar] [CrossRef] [PubMed]
- Cardel, M.; Jensen, S.M.; Pottegård, A.; Jørgensen, T.L.; Hallas, J. Long-term use of metformin and colorectal cancer risk in type II diabetics: A population-based case–control study. Cancer Med. 2014, 3, 1458–1466. [Google Scholar] [CrossRef]
- Zhou, G.; Myers, R.; Li, Y.; Chen, Y.; Shen, X.; Fenyk-Melody, J.; Wu, M.; Ventre, J.; Doebber, T.; Fujii, N.; et al. Role of AMP-activated protein kinase in mechanism of metformin action. J. Clin. Investig. 2001, 108, 1167–1174. [Google Scholar] [CrossRef]
- Jalving, M.; Gietema, J.A.; Lefrandt, J.D.; De Jong, S.; Reyners, A.K.; Gans, R.O.; De Vries, E.G. Metformin: Taking away the candy for cancer? Eur. J. Cancer 2010, 46, 2369–2380. [Google Scholar] [CrossRef]
- Saraei, P.; Asadi, I.; Kakar, M.A.; Moradi-Kor, N. The beneficial effects of metformin on cancer prevention and therapy: A comprehensive review of recent advances. Cancer Manag. Res. 2019, ume 11, 3295–3313. [Google Scholar] [CrossRef]
- Bieging, K.T.; Mello, S.S.; Attardi, L.D. Unravelling mechanisms of p53-mediated tumour suppression. Nat. Rev. Cancer 2014, 14, 359–370. [Google Scholar] [CrossRef]
- Berkers, C.R.; Maddocks, O.D.; Cheung, E.C.; Mor, I.; Vousden, K.H. Metabolic Regulation by p53 Family Members. Cell Metab. 2013, 18, 617–633. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Kon, N.; Li, T.; Wang, S.-J.; Su, T.; Hibshoosh, H.; Baer, R.; Gu, W. Ferroptosis as a p53-mediated activity during tumour suppression. Nat. Cell Biol. 2015, 520, 57–62. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Mao, Y.; Zhao, L.; Li, L.; Wu, J.; Zhao, M.; Du, W.; Yu, L.; Jiang, P. p53 regulation of ammonia metabolism through urea cycle controls polyamine biosynthesis. Nat. Cell Biol. 2019, 567, 253–256. [Google Scholar] [CrossRef]
- Meyskens, F.L.; Emerson, S.S.; Pelot, D.; Meshkinpour, H.; Shassetz, L.R.; Einspahr, J.G.; Alberts, D.S.; Gerner, E.W. Dose De-escalation Chemoprevention Trial of -Difluoromethylornithine in Patients With Colon Polyps. J. Natl. Cancer Inst. 1994, 86, 1122–1130. [Google Scholar] [CrossRef]
- Glass, J.R.; Gerner, E.W.; Shantz, L.M.; Porter, C.W.; Berger, F.G.; Ganis, B.; Bergeron, R.J.; Feith, D.J.; Pegg, A.E.; Madhubala, R.; et al. Polyamine-mediated turnover of ornithine decarboxylase in Chinese-hamster ovary cells. Biochem. J. 1986, 236, 351–357. [Google Scholar] [CrossRef]
- Klimberg, V.S.; McClellan, J.L. Glutamine, cancer, and its therapy. Am. J. Surg. 1996, 172, 418–424. [Google Scholar] [CrossRef]
- Chen, M.K.; Espat, N.J.; Bland, K.I.; Copeland, E.M.; Souba, W.W. Influence of Progressive Tumor Growth on Glutamine Metabolism in Skeletal Muscle and Kidney. Ann. Surg. 1993, 217, 655–667. [Google Scholar] [CrossRef]
- Ma, Z.; Lian, J.; Yang, M.; Wuyang, J.; Zhao, C.; Chen, W.; Liu, C.; Zhao, Q.; Lou, C.; Han, J.; et al. Overexpression of Arginase-1 is an indicator of poor prognosis in patients with colorectal cancer. Pathol. Res. Pr. 2019, 215, 152383. [Google Scholar] [CrossRef]
- Cheong, H.; Lindsten, T.; Wu, J.; Lu, C.; Thompson, C. Ammonia-induced autophagy is independent of ULK1/ULK2 kinases. Proc. Natl. Acad. Sci. USA 2011, 108, 11121–11126. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Xu, W.; Yan, Z.; Zhao, W.; Mi, J.; Li, J.; Yan, H. Metformin induces autophagy and G0/G1 phase cell cycle arrest in myeloma by targeting the AMPK/mTORC1 and mTORC2 pathways. J. Exp. Clin. Cancer Res. 2018, 37, 1–12. [Google Scholar] [CrossRef]
- Lin, O.S.; Kozarek, R.A.; Cha, J.M. Impact of Sigmoidoscopy and Colonoscopy on Colorectal Cancer Incidence and Mortality: An Evidence-Based Review of Published Prospective and Retrospective Studies. Intest. Res. 2014, 12, 268–274. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ng, S.C.; Wong, S.H. Colorectal cancer screening in Asia. Br. Med. Bull. 2013, 105, 29–42. [Google Scholar] [CrossRef]
- Arruabarrena-Aristorena, A.; Zabala-Letona, A.; Carracedo, A. Oil for the cancer engine: The cross-talk between oncogenic signaling and polyamine metabolism. Sci. Adv. 2018, 4, eaar2606. [Google Scholar] [CrossRef] [PubMed]
- Casero, R.A., Jr.; Marton, L.J. Targeting polyamine metabolism and function in cancer and other hyperproliferative diseases. Nat. Rev. Drug Discov. 2007, 6, 373–390. [Google Scholar] [CrossRef] [PubMed]
- Manni, A.; Mauger, D.; Gimotty, P.; Badger, B. Prognostic influence on survival of increased ornithine decarboxylase activity in human breast cancer. Clin. Cancer Res. 1996, 2, 1901–1906. [Google Scholar] [PubMed]
- Gupta, S.; Ahmad, N.; Marengo, S.R.; MacLennan, G.T.; Greenberg, N.M.; Mukhtar, H. Chemoprevention of prostate carcinogenesis by alpha-difluoromethylornithine in TRAMP mice. Cancer Res. 2000, 60, 5125–5133. [Google Scholar]
- Agostinelli, E. Polyamines in biological systems. Amino Acids 2009, 38, 351–352. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Agostinelli, E.; Marques, M.P.M.; Calheiros, R.; Gil, F.P.S.C.; Tempera, G.; Viceconte, N.; Battaglia, V.; Grancara, S.; Toninello, A. Polyamines: Fundamental characters in chemistry and biology. Amino Acids 2009, 38, 393–403. [Google Scholar] [CrossRef] [PubMed]
- Seiler, N.; Atanassov, C.L.; Raul, F. Polyamine metabolism as target for cancer chemoprevention (review). Int. J. Oncol. 1998, 13, 13. [Google Scholar] [CrossRef] [PubMed]
- Meyskens, F.L.; Gerner, E.W.; Emerson, S.; Pelot, D.; Durbin, T.; Doyle, K.; Lagerberg, W. Effect of α-Difluoromethylornithine on Rectal Mucosal Levels of Polyamines in a Randomized, Double-Blinded Trial for Colon Cancer Prevention. J. Natl. Cancer Inst. 1998, 90, 1212–1218. [Google Scholar] [CrossRef]
- Meyskens, F.L.; McLaren, C.E.; Pelot, D.; Fujikawa-Brooks, S.; Carpenter, P.M.; Hawk, E.; Kelloff, G.; Lawson, M.J.; Kidao, J.; McCracken, J.; et al. Difluoromethylornithine Plus Sulindac for the Prevention of Sporadic Colorectal Adenomas: A Randomized Placebo-Controlled, Double-Blind Trial. Cancer Prev. Res. 2008, 1, 32–38. [Google Scholar] [CrossRef]
- Kourelis, T.V.; Siegel, R.D. Metformin and cancer: New applications for an old drug. Med. Oncol. 2012, 29, 1314–1327. [Google Scholar] [CrossRef] [PubMed]
- Nangia-Makker, P.; Yu, Y.; Vasudevan, A.; Farhana, L.; Rajendra, S.G.; Levi, E.; Majumdar, A.P.N. Metformin: A Potential Therapeutic Agent for Recurrent Colon Cancer. PLoS ONE 2014, 9, e84369. [Google Scholar] [CrossRef]
- Okoshi, R.; Ozaki, T.; Yamamoto, H.; Ando, K.; Koida, N.; Ono, S.; Koda, T.; Kamijo, T.; Nakagawara, A.; Kizaki, H. Activation of AMP-activated Protein Kinase Induces p53-dependent Apoptotic Cell Death in Response to Energetic Stress. J. Biol. Chem. 2008, 283, 3979–3987. [Google Scholar] [CrossRef] [PubMed]
- Jones, R.G.; Plas, D.R.; Kubek, S.; Buzzai, M.; Mu, J.; Xu, Y.; Birnbaum, M.J.; Thompson, C.B. AMP-Activated Protein Kinase Induces a p53-Dependent Metabolic Checkpoint. Mol. Cell 2005, 18, 283–293. [Google Scholar] [CrossRef] [PubMed]
- Feng, Z.; Hu, W.; De Stanchina, E.; Teresky, A.K.; Jin, S.; Lowe, S.W.; Levine, A.J. The Regulation of AMPK β1, TSC2, and PTEN Expression by p53: Stress, Cell and Tissue Specificity, and the Role of These Gene Products in Modulating the IGF-1-AKT-mTOR Pathways. Cancer Res. 2007, 67, 3043–3053. [Google Scholar] [CrossRef] [PubMed]
- Shaw, R.J. Glucose metabolism and cancer. Curr. Opin. Cell Biol. 2006, 18, 598–608. [Google Scholar] [CrossRef] [PubMed]
- Prives, C.; Hall, P.A. The p53 pathway. J. Pathol. 1999, 187, 112–126. [Google Scholar] [CrossRef]
- Sionov, R.V.; Haupt, Y. The cellular response to p53: The decision between life and death. Oncogene 1999, 18, 6145–6157. [Google Scholar] [CrossRef] [PubMed]
- Vousden, K.H.; Lu, X. Live or let die: The cell’s response to p53. Nat. Rev. Cancer 2002, 2, 594–604. [Google Scholar] [CrossRef]
- Ben Sahra, I.; Laurent, K.; Loubat, A.; Peraldi, S.G.; Colosetti, P.; Auberger, P.; Tanti, J.-F.; Brustel, Y.L.M.; Bost, F. The antidiabetic drug metformin exerts an antitumoral effect in vitro and in vivo through a decrease of cyclin D1 level. Oncogene 2008, 27, 3576–3586. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, Q.; Zeng, S.X.; Zhang, Y.; Mayo, L.D.; Lu, H. Inauhzin and Nutlin3 synergistically activate p53 and suppress tumor growth. Cancer Biol. Ther. 2012, 13, 915–924. [Google Scholar] [CrossRef]
- Vogelstein, B.; Lane, D.; Levine, A.J. Surfing the p53 network. Nat. Cell Biol. 2000, 408, 307–310. [Google Scholar] [CrossRef]
- Vousden, K.H. Outcomes of p53 activation—Spoilt for choice. J. Cell Sci. 2006, 119, 5015–5020. [Google Scholar] [CrossRef] [PubMed]
- Bensaad, K.; Vousden, K.H. p53: New roles in metabolism. Trends Cell Biol. 2007, 17, 286–291. [Google Scholar] [CrossRef] [PubMed]
- Lacroix, M.; Riscal, R.; Arena, G.; Linares, L.K.; Le Cam, L. Metabolic functions of the tumor suppressor p53: Implications in normal physiology, metabolic disorders, and cancer. Mol. Metab. 2019. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.-Y.; Li, C.-F.; Lin, C.-Y.; Lee, S.-W.; Sheu, M.-J.; Lin, L.-C.; Chen, T.-J.; Wu, T.-F.; Hsing, C.-H. Overexpression of CPS1 is an independent negative prognosticator in rectal cancers receiving concurrent chemoradiotherapy. Tumor Biol. 2014, 35, 11097–11105. [Google Scholar] [CrossRef] [PubMed]
- Palaniappan, A.; Ramar, K.; Ramalingam, S. Computational Identification of Novel Stage-Specific Biomarkers in Colorectal Cancer Progression. PLoS ONE 2016, 11, e0156665. [Google Scholar] [CrossRef]
- Liu, H.; Dong, H.; Robertson, K.; Liu, C. DNA Methylation Suppresses Expression of the Urea Cycle Enzyme Carbamoyl Phosphate Synthetase 1 (CPS1) in Human Hepatocellular Carcinoma. Am. J. Pathol. 2011, 178, 652–661. [Google Scholar] [CrossRef]
- Vardon, A.; Dandapani, M.; Cheng, D.; Cheng, P.; De Santo, C.; Mussai, F. Arginine auxotrophic gene signature in paediatric sarcomas and brain tumours provides a viable target for arginine depletion therapies. Oncotarget 2017, 8, 63506–63517. [Google Scholar] [CrossRef]
- De Santo, C.; Booth, S.; Vardon, A.; Cousins, A.; Tubb, V.; Perry, T.; Noyvert, B.; Beggs, A.; Ng, M.; Halsey, C.; et al. The arginine metabolome in acute lymphoblastic leukemia can be targeted by the pegylated-recombinant arginase I BCT-100. Int. J. Cancer 2017, 142, 1490–1502. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.S.; Adler, L.; Karathia, H.; Carmel, N.; Rabinovich, S.; Auslander, N.; Keshet, R.; Stettner, N.; Silberman, A.; Agemy, L.; et al. Urea Cycle Dysregulation Generates Clinically Relevant Genomic and Biochemical Signatures. Cell 2018, 174, 1559–1570.e22. [Google Scholar] [CrossRef] [PubMed]
- Kodama, M.; Oshikawa, K.; Shimizu, H.; Yoshioka, S.; Takahashi, M.; Izumi, Y.; Bamba, T.; Tateishi, C.; Tomonaga, T.; Matsumoto, M.; et al. A shift in glutamine nitrogen metabolism contributes to the malignant progression of cancer. Nat. Commun. 2020, 11, 1–16. [Google Scholar] [CrossRef]
- Spinelli, J.B.; Yoon, H.; Ringel, A.E.; Jeanfavre, S.; Clish, C.B.; Haigis, M.C. Metabolic recycling of ammonia via glutamate dehydrogenase supports breast cancer biomass. Science 2017, 358, 941–946. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Z.; Wang, W.; Wu, C.; Zou, X.; Fang, L.; Su, W.; Wang, P. Novel Pyrrole–Imidazole Polyamide Hoechst Conjugate Suppresses Epstein–Barr Virus Replication and Virus-Positive Tumor Growth. J. Med. Chem. 2018, 61, 6674–6684. [Google Scholar] [CrossRef]
- Guo, X.-X.; Li, X.-P.; Zhou, P.; Li, D.-Y.; Lyu, X.-T.; Chen, Y.; Lyu, Y.-W.; Tian, K.; Yuan, D.-Z.; Ran, J.-H.; et al. Evodiamine Induces Apoptosis in SMMC-7721 and HepG2 Cells by Suppressing NOD1 Signal Pathway. Int. J. Mol. Sci. 2018, 19, 3419. [Google Scholar] [CrossRef] [PubMed]
- Hu, L.; Tian, K.; Zhang, T.; Fan, C.-H.; Zhou, P.; Zeng, D.; Zhao, S.; Li, L.-S.; Smith, H.S.; Li, J.; et al. Cyanate Induces Oxidative Stress Injury and Abnormal Lipid Metabolism in Liver through Nrf2/HO-1. Molecules 2019, 24, 3231. [Google Scholar] [CrossRef] [PubMed]
- Lin, R.; Mo, Y.; Zha, H.; Qu, Z.; Xie, P.; Zhu, Z.-J.; Xu, Y.; Xiong, Y.; Guan, K.-L. CLOCK Acetylates ASS1 to Drive Circadian Rhythm of Ureagenesis. Mol. Cell 2017, 68, 198–209.e6. [Google Scholar] [CrossRef]
- Bai, Z.; Qi, T.; Liu, Y.; Wu, Z.; Ma, L.; Liu, W.; Cao, Y.; Bao, Y.; Fu, C. Alteration of S-adenosylhomocysteine levels affects lignin biosynthesis in switchgrass. Plant. Biotechnol. J. 2018, 16, 2016–2026. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, T.; Hu, L.; Tang, J.-F.; Xu, H.; Tian, K.; Wu, M.-N.; Huang, S.-Y.; Du, Y.-M.; Zhou, P.; Lu, R.-J.; et al. Metformin Inhibits the Urea Cycle and Reduces Putrescine Generation in Colorectal Cancer Cell Lines. Molecules 2021, 26, 1990. https://doi.org/10.3390/molecules26071990
Zhang T, Hu L, Tang J-F, Xu H, Tian K, Wu M-N, Huang S-Y, Du Y-M, Zhou P, Lu R-J, et al. Metformin Inhibits the Urea Cycle and Reduces Putrescine Generation in Colorectal Cancer Cell Lines. Molecules. 2021; 26(7):1990. https://doi.org/10.3390/molecules26071990
Chicago/Turabian StyleZhang, Tao, Ling Hu, Jia-Feng Tang, Hang Xu, Kuan Tian, Meng-Na Wu, Shi-Ying Huang, Yu-Mei Du, Peng Zhou, Rui-Jin Lu, and et al. 2021. "Metformin Inhibits the Urea Cycle and Reduces Putrescine Generation in Colorectal Cancer Cell Lines" Molecules 26, no. 7: 1990. https://doi.org/10.3390/molecules26071990
APA StyleZhang, T., Hu, L., Tang, J.-F., Xu, H., Tian, K., Wu, M.-N., Huang, S.-Y., Du, Y.-M., Zhou, P., Lu, R.-J., He, S., Xu, J.-M., Si, J.-J., Li, J., Chen, D.-L., & Ran, J.-H. (2021). Metformin Inhibits the Urea Cycle and Reduces Putrescine Generation in Colorectal Cancer Cell Lines. Molecules, 26(7), 1990. https://doi.org/10.3390/molecules26071990