
Design and Synthesis of Non-Covalent Imidazo[1,2-a]quinoxaline-Based Inhibitors of EGFR and Their Anti-Cancer Assessment

 , , ,
, , ,
Abstract
1. Introduction
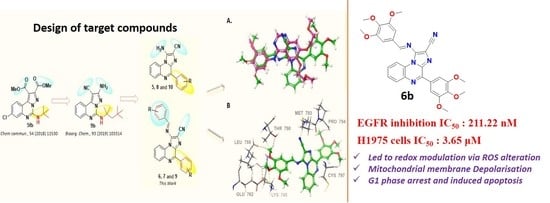
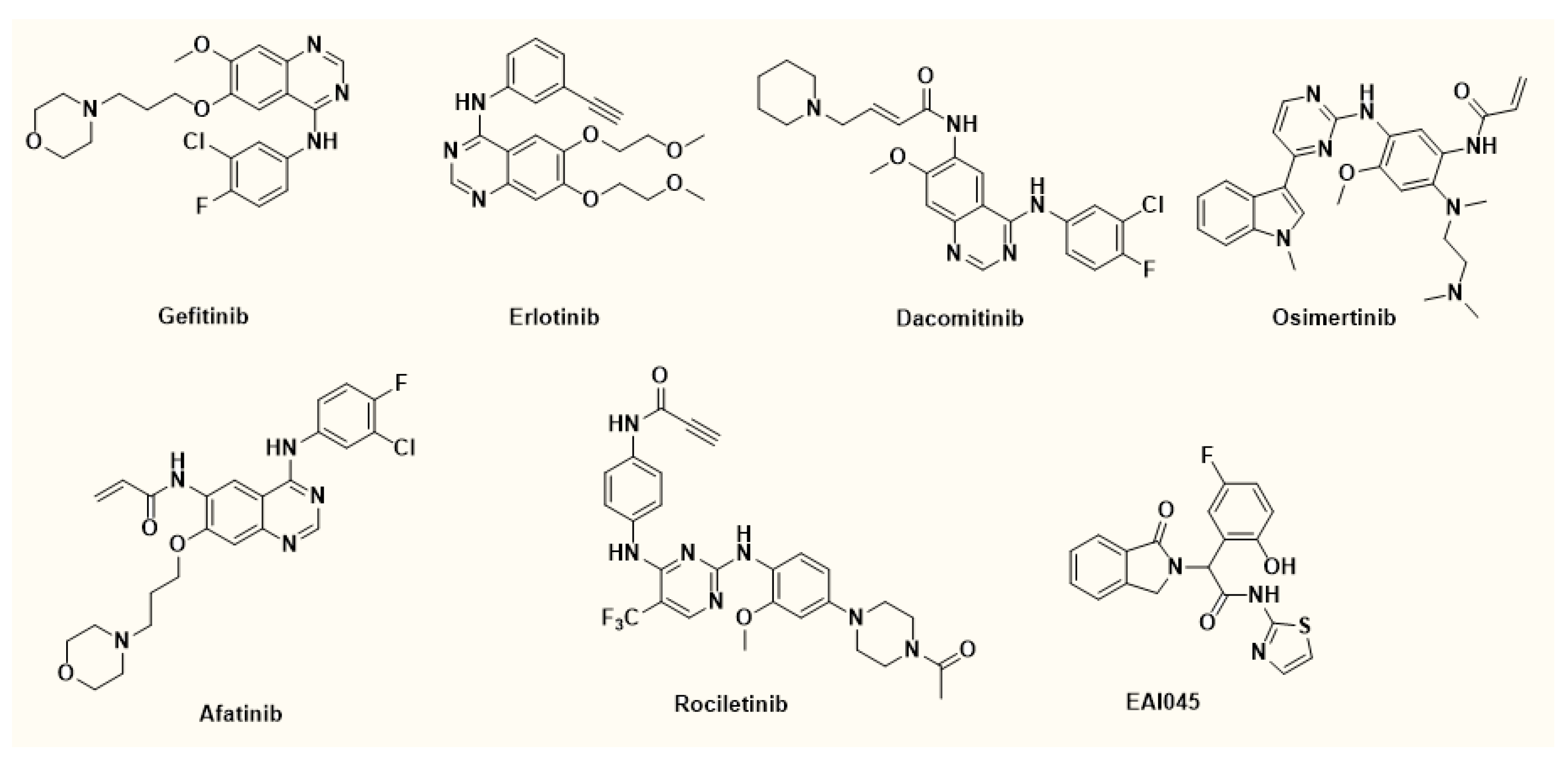
2. Results and Discussion
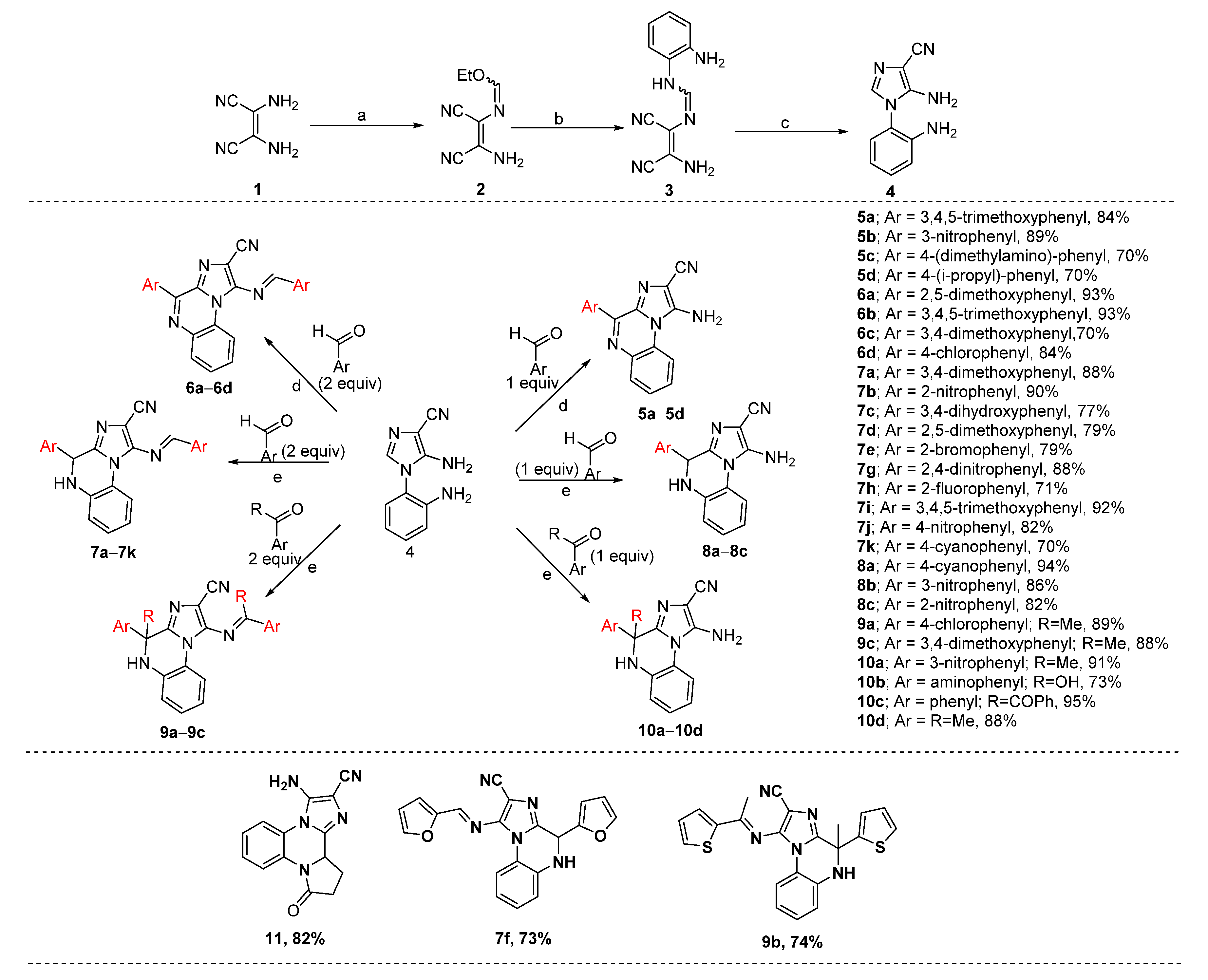
2.1. Chemistry
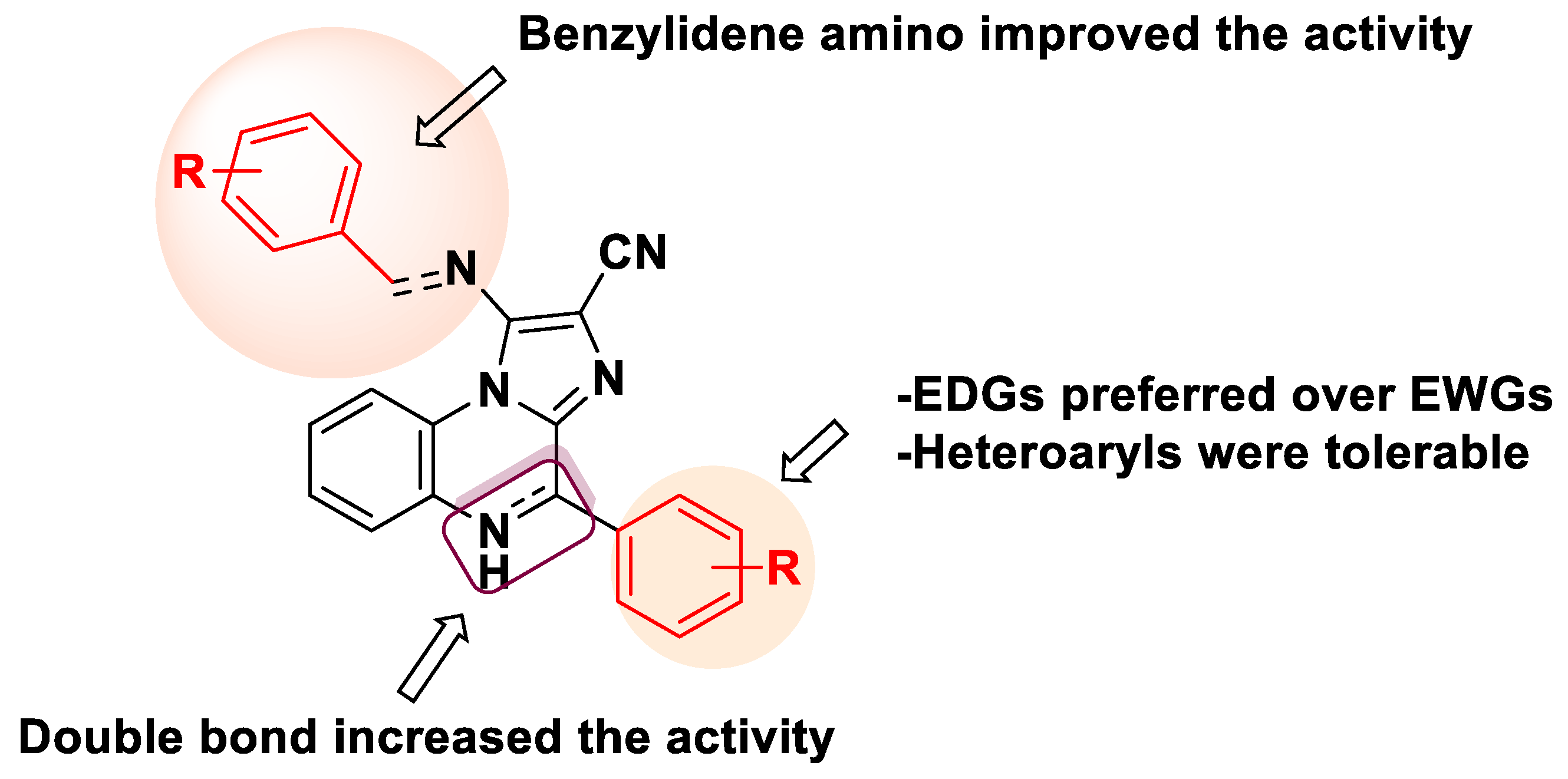
2.2. Biological Evaluation
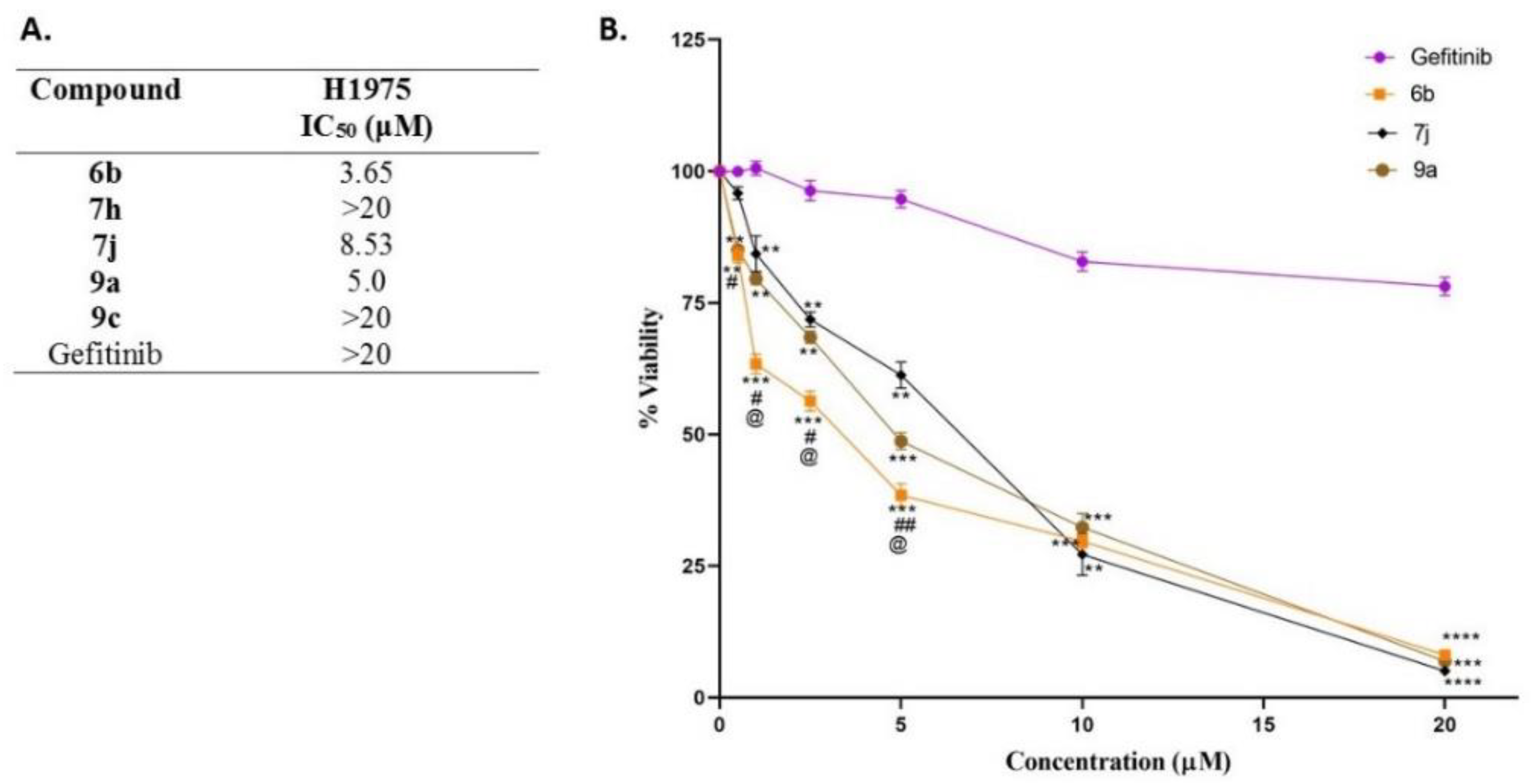
In Vitro EGFR Kinase Inhibitory Activity and Antiproliferative Assay
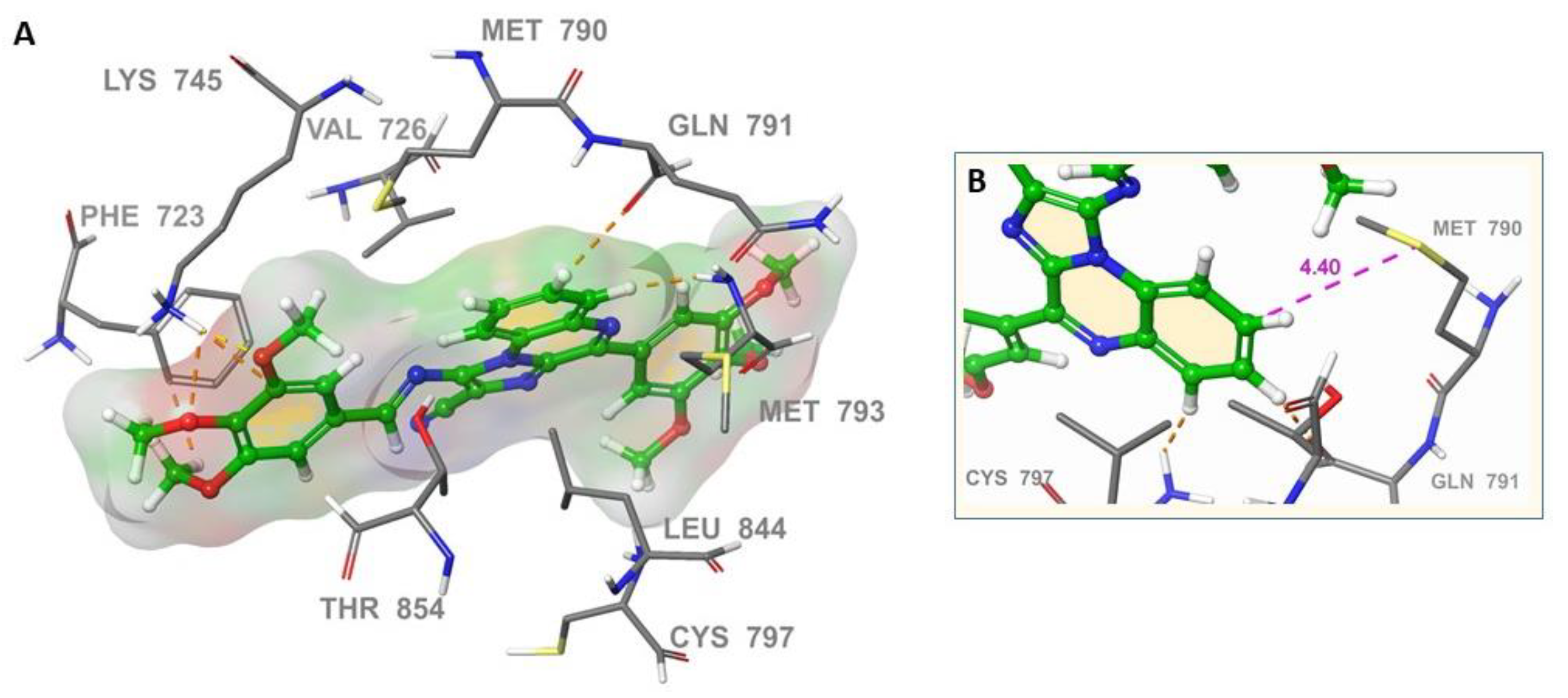
2.3. Molecular Docking
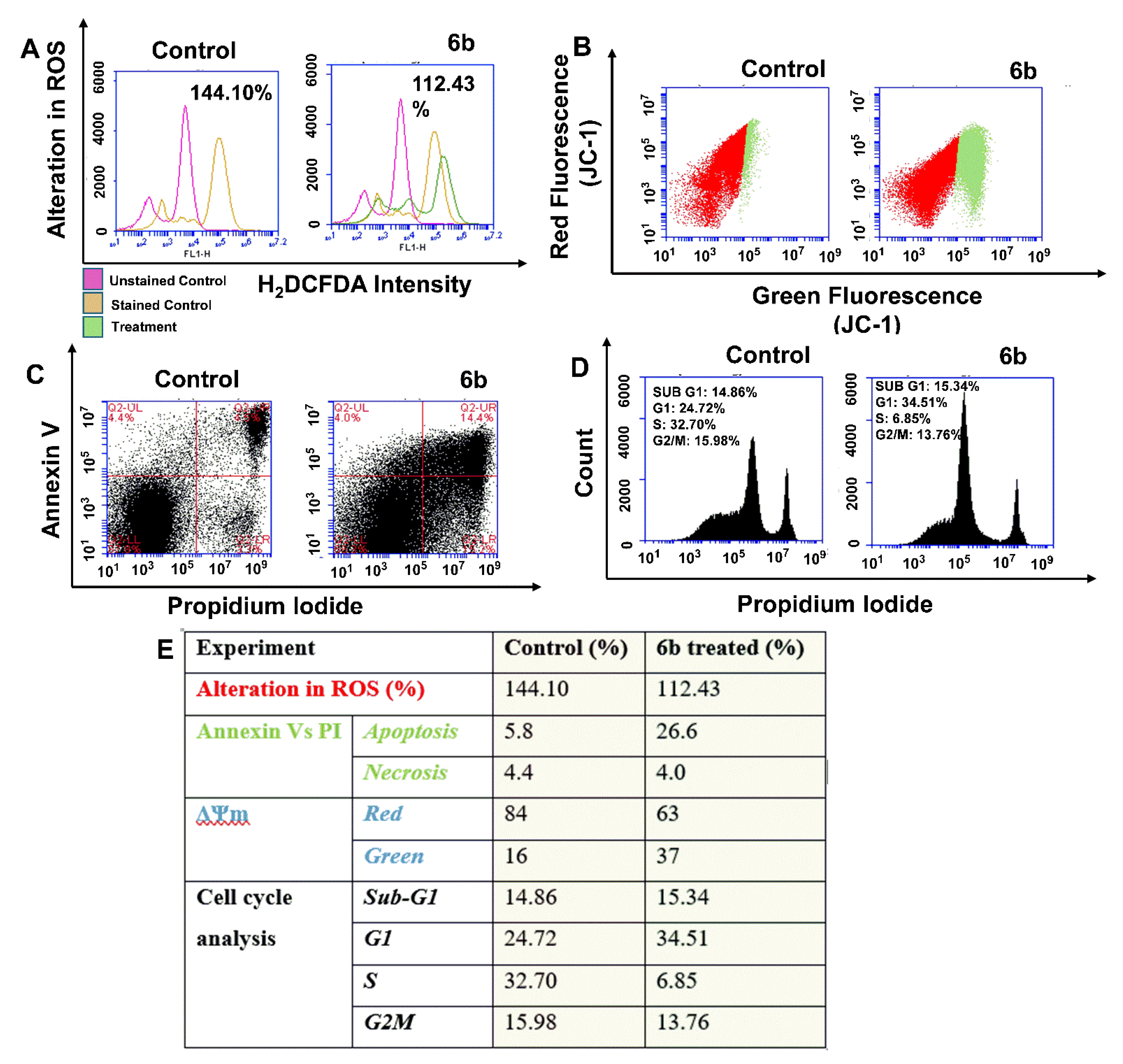
2.4. Flow Cytometric Analysis of Reactive Oxygen Species (ROS), Mitochondrial Membrane Potential and Apoptosis
3. Experimental
3.1. Synthesis of Intermediates (2–4)
3.1.1. N-(2-Amino-1, 2-dicyanovinyl)-formimydic acid ester (2)
3.1.2. N-(2-Amino-1,2-dicyanovinyl)-N’-(2-amino-phenyl)formimidine (3)
3.1.3. 5-Amino-1-(2-amino-phenyl)-1H-imidazole-4-carbonitrile (4)
3.2. Synthesis of Target Compounds (5–10)
3.2.1. Representative Procedure for the Synthesis of (E)-1-((3,4,5-Trimethoxybenzylidene)amino)-4-(3,4,5-trimethoxyphenyl)imidazo[1,2-a]quinoxaline-2-carbonitrile (6b)
3.2.2. 1-Amino-4-(4-(dimethylamino)phenyl)imidazo[1,2-a]quinoxaline-2-carbonitrile (5c)
3.2.3. 1-Amino-4-(4-isopropylphenyl)imidazo[1,2-a]quinoxaline-2-carbonitrile (5d)
3.2.4. (E)-1-((3,4-Dimethoxybenzylidene)amino)-4-(3,4-dimethoxy-phenyl)imidazo[1,2-a]quinoxaline-2-carbonitrile (6c)
3.2.5. Representative Procedure for the Synthesis of (E)-1-((4-Cyanobenzylidene)amino)-4-(4-cyanophenyl)-4,5-dihydroimidazo[1,2-a]quinoxaline-2-carbonitrile (7k)
3.2.6. 1-Amino-4-(2-nitrophenyl)-4,5-dihydroimidazo[1,2-a]quinoxaline-2-carbonitrile (8c)
3.3. Biology
3.3.1. EGFR Inhibitory Assay
Calculations
3.3.2. Cell Culture and Maintenance
3.3.3. 3-(4,5-Dimethythiazol-2-yl)-2,5-diphenyl Tetrazolium Bromide-Based MTT Assay
3.3.4. Molecular Docking
3.3.5. Flow Cytometric-Based Analysis
3.4. Statistical Treatment
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Wee, P.; Wang, Z. Epidermal growth factor receptor cell proliferation signaling pathways. Cancers 2017, 9, 52. [Google Scholar]
- Kumar, M.; Joshi, G.; Chatterjee, J.; Kumar, R. Epidermal Growth Factor Receptor and its Trafficking Regulation by Acetylation: Implication in Resistance and Exploring the Newer Therapeutic Avenues in Cancer. Curr. Top. Med. Chem. 2020, 20, 1105–1123. [Google Scholar] [CrossRef]
- Shepherd, F.A.; Rodrigues Pereira, J.; Ciuleanu, T.; Tan, E.H.; Hirsh, V.; Thongprasert, S.; Campos, D.; Maoleekoonpiroj, S.; Smylie, M.; Martins, R. Erlotinib in previously treated non–small-cell lung cancer. N. Engl. J. Med. 2005, 353, 123–132. [Google Scholar] [CrossRef] [PubMed]
- Paez, J.G.; Jänne, P.A.; Lee, J.C.; Tracy, S.; Greulich, H.; Gabriel, S.; Herman, P.; Kaye, F.J.; Lindeman, N.; Boggon, T.J. EGFR mutations in lung cancer: Correlation with clinical response to gefitinib therapy. Science 2004, 304, 1497–1500. [Google Scholar] [CrossRef]
- Cheng, H.; Nair, S.K.; Murray, B.W. Recent progress on third generation covalent EGFR inhibitors. Bioorg. Med. Chem. Lett. 2016, 26, 1861–1868. [Google Scholar] [CrossRef] [PubMed]
- Grabe, T.; Lategahn, J.; Rauh, D. C797S Resistance: The undruggable EGFR mutation in non-small cell lung cancer? ACS Med. Chem. Lett. 2018, 9, 779–782. [Google Scholar] [CrossRef]
- Lacouture, M.E. Mechanisms of cutaneous toxicities to EGFR inhibitors. Nat. Rev. Cancer 2006, 6, 803–812. [Google Scholar] [CrossRef]
- Gray, N.; Jaebong, J.; De Clercq, D.; Eck, M.; Janne, P. Bifunctional Molecules for Degradation of EGFR and Methods of Use. WO2017185036A1, 26 October 2017. [Google Scholar]
- Niederst, M.J.; Hu, H.; Mulvey, H.E.; Lockerman, E.L.; Garcia, A.R.; Piotrowska, Z.; Sequist, L.V.; Engelman, J.A. The allelic context of the C797S mutation acquired upon treatment with third-generation EGFR inhibitors impacts sensitivity to subsequent treatment strategies. Clin. Cancer Res. 2015, 21, 3924–3933. [Google Scholar] [CrossRef]
- Li, Q.; Zhang, T.; Li, S.; Tong, L.; Li, J.; Su, Z.; Feng, F.; Sun, D.; Tong, Y.; Wang, X. Discovery of potent and non-covalent reversible EGFR kinase inhibitors of EGFRL858R/T790M/C797S. ACS Med. Chem. Lett. 2019, 10, 869–873. [Google Scholar] [CrossRef] [PubMed]
- Kalra, S.; Joshi, G.; Kumar, M.; Arora, S.; Kaur, H.; Singh, S.; Munshi, A.; Kumar, R. Anticancer potential of some imidazole and fused imidazole derivatives: Exploring the mechanism via epidermal growth factor receptor (EGFR) inhibition. RSC Med. Chem. 2020, 11, 923–939. [Google Scholar] [CrossRef]
- Garg, M.; Chauhan, M.; Singh, P.K.; Alex, J.M.; Kumar, R. Pyrazoloquinazolines: Synthetic strategies and bioactivities. Eur. J. Med. Chem. 2015, 97, 444–461. [Google Scholar] [CrossRef] [PubMed]
- Kaur, G.; Cholia, R.P.; Joshi, G.; Amrutkar, S.M.; Kalra, S.; Mantha, A.K.; Banerjee, U.C.; Kumar, R. Anti-cancer activity of dihydropyrazolo [1, 5-c] quinazolines against rat C6 glioma cells via inhibition of topoisomerase II. Arch. Pharm. 2018, 351, 1800023. [Google Scholar] [CrossRef] [PubMed]
- Kumar, D.; Kaur, G.; Negi, A.; Kumar, S.; Singh, S.; Kumar, R. Synthesis and xanthine oxidase inhibitory activity of 5, 6-dihydropyrazolo/pyrazolo [1, 5-c] quinazoline derivatives. Bioorg. Chem. 2014, 57, 57–64. [Google Scholar] [CrossRef]
- Kumar, D.; Kumar, R. Microwave-assisted synthesis of pyrazolo [1, 5-c] quinazolines and their derivatives. Tetrahedron Lett. 2014, 55, 2679–2683. [Google Scholar] [CrossRef]
- Sawant, D.M.; Sharma, S.; Pathare, R.S.; Joshi, G.; Kalra, S.; Sukanya, S.; Maurya, A.K.; Metre, R.K.; Agnihotri, V.K.; Khan, S. Relay tricyclic Pd (ii)/Ag (i) catalysis: Design of a four-component reaction driven by nitrene-transfer on isocyanide yields inhibitors of EGFR. Chem. Commun. 2018, 54, 11530–11533. [Google Scholar] [CrossRef]
- Ansari, A.J.; Joshi, G.; Yadav, U.P.; Maurya, A.K.; Agnihotri, V.K.; Kalra, S.; Kumar, R.; Singh, S.; Sawant, D.M. Exploration of Pd-catalysed four-component tandem reaction for one-pot assembly of pyrazolo [1, 5-c] quinazolines as potential EGFR inhibitors. Bioorg. Chem. 2019, 93, 103314. [Google Scholar] [CrossRef]
- Negi, A.; Alex, J.M.; Amrutkar, S.M.; Baviskar, A.T.; Joshi, G.; Singh, S.; Banerjee, U.C.; Kumar, R. Imine/amide–imidazole conjugates derived from 5-amino-4-cyano-N1-substituted benzyl imidazole: Microwave-assisted synthesis and anti-cancer activity via selective topoisomerase-II-α inhibition. Bioorg. Med. Chem. 2015, 23, 5654–5661. [Google Scholar] [CrossRef] [PubMed]
- El Newahie, A.M.; Ismail, N.S.; Abou El Ella, D.A.; Abouzid, K.A. Quinoxaline-Based Scaffolds Targeting Tyrosine Kinases and Their Potential Anti-cancer Activity. Arch. Pharm. 2016, 349, 309–326. [Google Scholar] [CrossRef] [PubMed]
- Chouchou, A.; Patinote, C.; Cuq, P.; Bonnet, P.-A.; Deleuze-Masquéfa, C. Imidazo [1, 2-a] quinoxalines derivatives grafted with amino acids: Synthesis and evaluation on A375 melanoma cells. Molecules 2018, 23, 2987. [Google Scholar] [CrossRef]
- Moarbess, G.; Deleuze-Masquefa, C.; Bonnard, V.; Gayraud-Paniagua, S.; Vidal, J.-R.; Bressolle, F.; Pinguet, F.; Bonnet, P.-A. In vitro and in vivo anti-tumoral activities of imidazo [1, 2-a] quinoxaline, imidazo [1, 5-a] quinoxaline, and pyrazolo [1, 5-a] quinoxaline derivatives. Bioorg. Med. Chem. 2008, 16, 6601–6610. [Google Scholar] [CrossRef]
- Moarbess, G.; Guichou, J.-F.; Paniagua-Gayraud, S.; Chouchou, A.; Marcadet, O.; Leroy, F.; Ruédas, R.; Cuq, P.; Deleuze-Masquéfa, C.; Bonnet, P.-A. New IKK inhibitors: Synthesis of new imidazo [1, 2-a] quinoxaline derivatives using microwave assistance and biological evaluation as IKK inhibitors. Eur. J. Med. Chem. 2016, 115, 268–274. [Google Scholar] [CrossRef]
- Chen, P.; Iwanowicz, E.J.; Norris, D.; Gu, H.H.; Lin, J.; Moquin, R.V.; Das, J.; Wityak, J.; Spergel, S.H.; de Fex, H. Synthesis and SAR of novel imidazoquinoxaline-based Lck inhibitors: Improvement of cell potency. Bioorg. Med. Chem. Lett. 2002, 12, 3153–3156. [Google Scholar] [CrossRef]
- Sharma, S.; Joshi, G.; Kalra, S.; Singh, S.; Kumar, R. Synthetic versus enzymatic pictet-spengler reaction: An overview. Curr. Org. Synth. 2018, 15, 924–939. [Google Scholar] [CrossRef]
- Joshi, G.; Chauhan, M.; Kumar, R.; Thakur, A.; Sharma, S.; Singh, R.; Wani, A.A.; Sharon, A.; Bharatam, P.V.; Kumar, R. Cyclocondensation reactions of an electron deactivated 2-aminophenyl tethered imidazole with mono/1, 2-biselectrophiles: Synthesis and DFT studies on the rationalization of imidazo [1, 2-a] quinoxaline versus benzo [f] imidazo [1, 5-a][1, 3, 5] triazepine selectivity switches. Org. Chem. Front. 2018, 5, 3526–3533. [Google Scholar]
- Kumar, R.; Ujjinamatada, R.K.; Hosmane, R.S. The first synthesis of a novel 5: 7: 5-fused diimidazodiazepine ring system and some of its chemical properties. Org. Lett. 2008, 10, 4681–4684. [Google Scholar] [CrossRef]
- Bissantz, C.; Kuhn, B.; Stahl, M. A medicinal chemist’s guide to molecular interactions. J. Med. Chem. 2010, 53, 5061–5084. [Google Scholar] [CrossRef]
- Heald, R.; Bowman, K.K.; Bryan, M.C.; Burdick, D.; Chan, B.; Chan, E.; Chen, Y.; Clausen, S.; Dominguez-Fernandez, B.; Eigenbrot, C. Non-covalent mutant selective epidermal growth factor receptor inhibitors: A lead optimization case study. J. Med. Chem. 2015, 58, 8877–8895. [Google Scholar] [CrossRef]
- Orphanos, G.S.; Ioannidis, G.N.; Ardavanis, A.G. Cardiotoxicity induced by tyrosine kinase inhibitors. Acta Oncol. 2009, 48, 964–970. [Google Scholar] [CrossRef] [PubMed]
- Force, T.; Kolaja, K.L. Cardiotoxicity of kinase inhibitors: The prediction and translation of preclinical models to clinical outcomes. Nat. Rev. Drug Discov. 2011, 10, 111–126. [Google Scholar] [CrossRef]
- Joshi, G.; Kalra, S.; Yadav, U.P.; Sharma, P.; Singh, P.K.; Amrutkar, S.; Ansari, A.J.; Kumar, S.; Sharon, A.; Sharma, S. E-pharmacophore guided discovery of pyrazolo [1, 5-c] quinazolines as dual inhibitors of topoisomerase-I and histone deacetylase. Bioorg. Chem. 2020, 94, 103409. [Google Scholar] [CrossRef]
- Chauhan, M.; Joshi, G.; Kler, H.; Kashyap, A.; Amrutkar, S.M.; Sharma, P.; Bhilare, K.D.; Banerjee, U.C.; Singh, S.; Kumar, R. Dual inhibitors of epidermal growth factor receptor and topoisomerase IIα derived from a quinoline scaffold. RSC Adv. 2016, 6, 77717–77734. [Google Scholar] [CrossRef]
- Joshi, G.; Nayyar, H.; Kalra, S.; Sharma, P.; Munshi, A.; Singh, S.; Kumar, R. Pyrimidine containing epidermal growth factor receptor kinase inhibitors: Synthesis and biological evaluation. Chem. Biol. Drug Des. 2017, 90, 995–1006. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | EGFR Inhibition IC50 (nM) ± SEM a,b | Compound | EGFR Inhibition IC50 (nM) a ± SEM a,b |
|---|---|---|---|
| 5a | >720 | 7i | 613.37 ± 0.024 |
| 5b | 244.45 ± 0.032 | 7j | 193.18 ± 0.021 |
| 5c | >720 | 7k | >720 |
| 5d | >720 | 8a | >720 |
| 6a | 429.81 ± 0.018 | 8b | >720 |
| 6b | 211.22 ± 0.027 | 8c | >720 |
| 6c | >720 | 9a | 223.32 ± 0.031 |
| 6d | >720 | 9b | 244.51 ± 0.021 |
| 7a | >720 | 9c | 221.53 ± 0.028 |
| 7b | >720 | 10a | 247.32 ± 0.021 |
| 7c | 620.17 ± 0.019 | 10b | >720 |
| 7d | 652.56 ± 0.024 | 10c | 716.32 ± 0.026 |
| 7e | 665.89 ± 0.029 | 10d | 624.57 ± 0.024 |
| 7f | 232.40 ± 0.031 | 11 | 669.39 ± 0.019 |
| 7g | 476.87 ± 0.022 | Erlotinib | 221.03 ± 0.028 |
| 7h | 222.21 ± 0.028 |
| Code | A549 (Lung Cancer) | HCT-116 WT (Colon Cancer) | MDA-MB-231 (Breast Cancer) |
|---|---|---|---|
| IC50 (μM) ± SEM a,b | |||
| 6b | 2.7 ± 0.032 | 5.1 ± 0.029 | 4.1 ± 0.031 |
| 7h | 4.09 ± 0.024 | <1 | 11.2 ± 0.022 |
| 7j | 8.75 ± 0.028 | <1 | 2.2 ± 0.026 |
| 9a | 6.63 ± 0.031 | >25 | 14.1 ± 0.021 |
| 9c | 12.06 ± 0.021 | 7.90 ± 0.027 | 13.6 ± 0.028 |
| Erlotinib | 4.56 ± 0.019 | 2.98 ± 0.023 | 3.33 ± 0.018 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kumar, M.; Joshi, G.; Arora, S.; Singh, T.; Biswas, S.; Sharma, N.; Bhat, Z.R.; Tikoo, K.; Singh, S.; Kumar, R. Design and Synthesis of Non-Covalent Imidazo[1,2-a]quinoxaline-Based Inhibitors of EGFR and Their Anti-Cancer Assessment. Molecules 2021, 26, 1490. https://doi.org/10.3390/molecules26051490
Kumar M, Joshi G, Arora S, Singh T, Biswas S, Sharma N, Bhat ZR, Tikoo K, Singh S, Kumar R. Design and Synthesis of Non-Covalent Imidazo[1,2-a]quinoxaline-Based Inhibitors of EGFR and Their Anti-Cancer Assessment. Molecules. 2021; 26(5):1490. https://doi.org/10.3390/molecules26051490
Chicago/Turabian StyleKumar, Manvendra, Gaurav Joshi, Sahil Arora, Tashvinder Singh, Sajal Biswas, Nisha Sharma, Zahid Rafiq Bhat, Kulbhushan Tikoo, Sandeep Singh, and Raj Kumar. 2021. "Design and Synthesis of Non-Covalent Imidazo[1,2-a]quinoxaline-Based Inhibitors of EGFR and Their Anti-Cancer Assessment" Molecules 26, no. 5: 1490. https://doi.org/10.3390/molecules26051490
APA StyleKumar, M., Joshi, G., Arora, S., Singh, T., Biswas, S., Sharma, N., Bhat, Z. R., Tikoo, K., Singh, S., & Kumar, R. (2021). Design and Synthesis of Non-Covalent Imidazo[1,2-a]quinoxaline-Based Inhibitors of EGFR and Their Anti-Cancer Assessment. Molecules, 26(5), 1490. https://doi.org/10.3390/molecules26051490