
Genetically Encodable Scaffolds for Optimizing Enzyme Function


Abstract
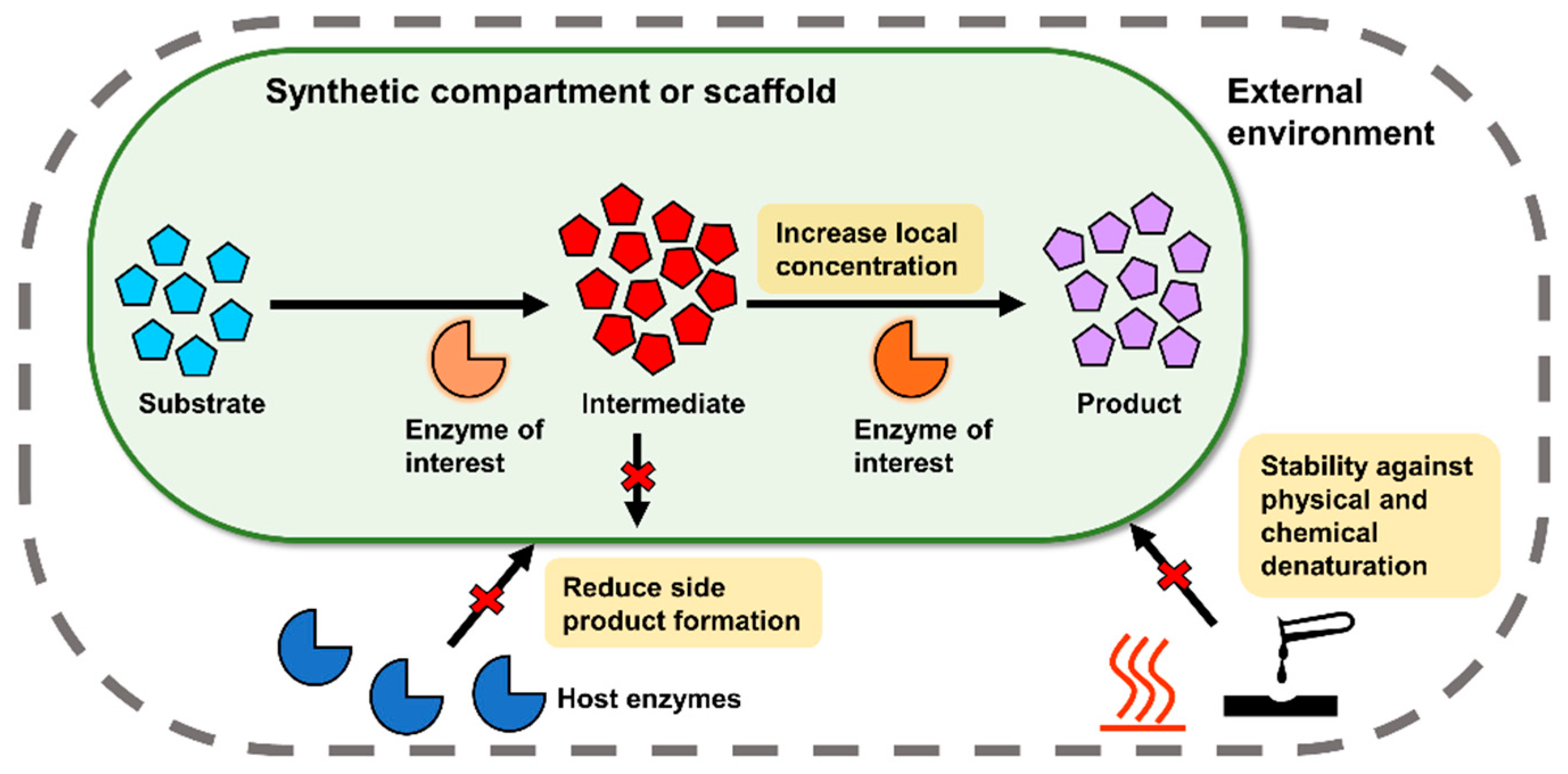
1. Introduction
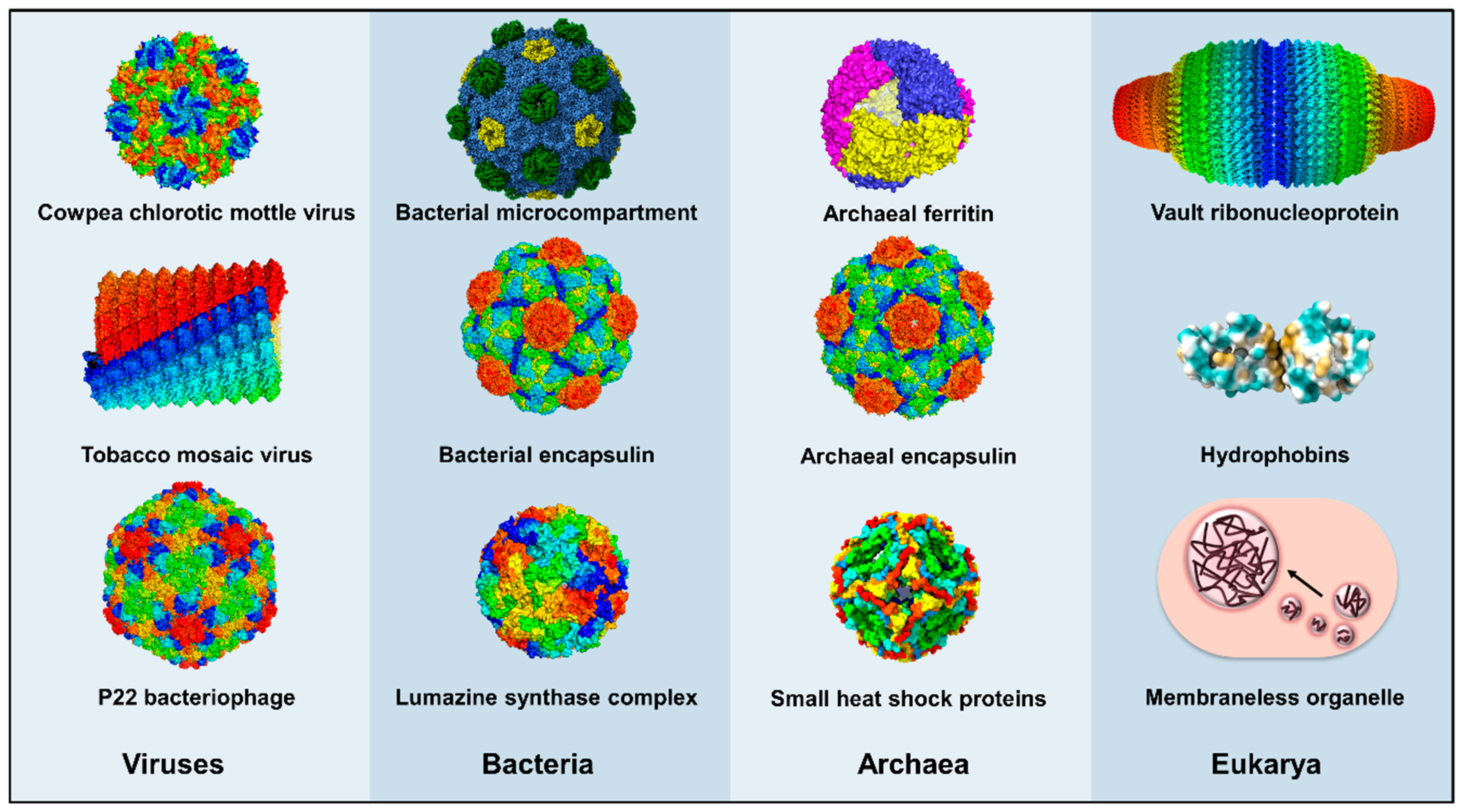
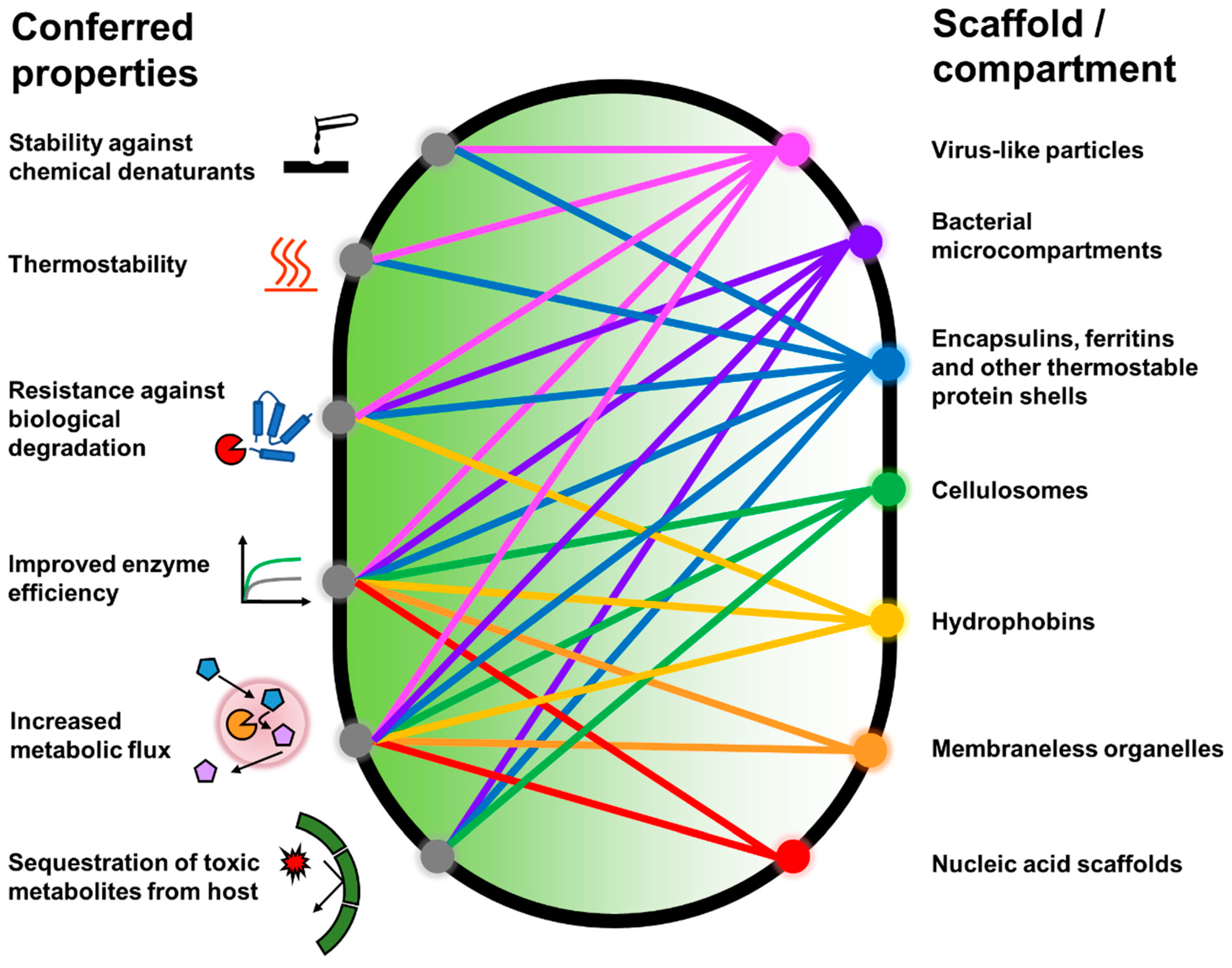
2. Protein Shells and Scaffolds
2.1. Virus-Derived Protein Shells

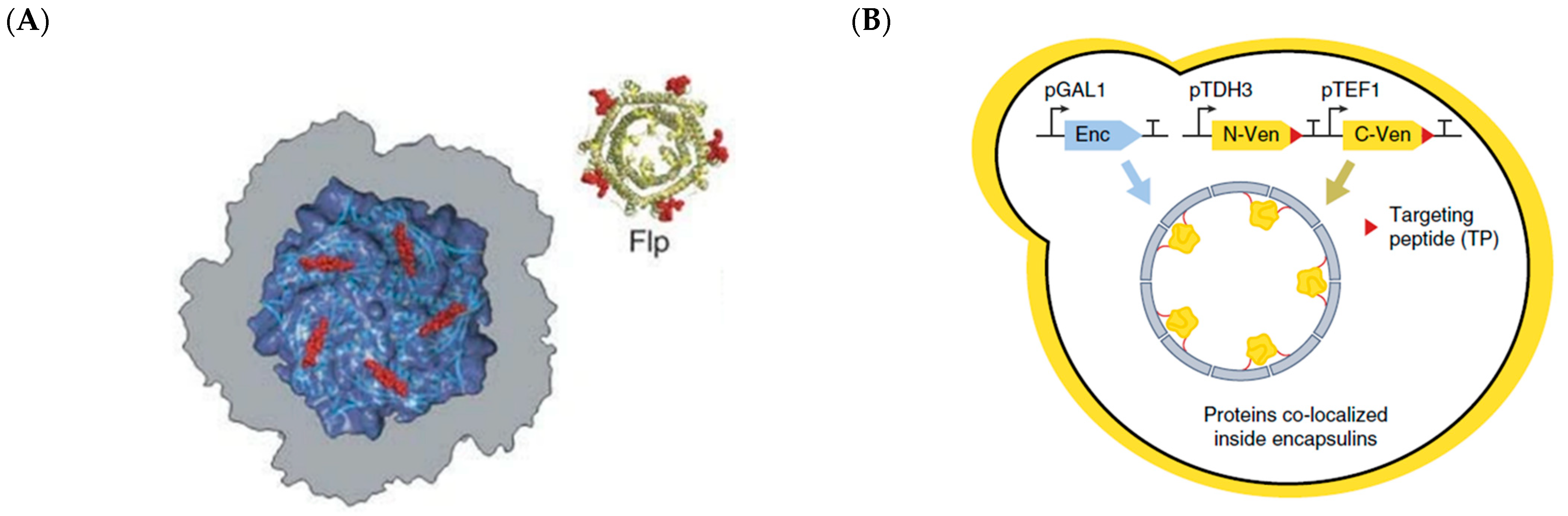
2.2. Encapsulins, Ferritins, and Small Heat Shock Proteins
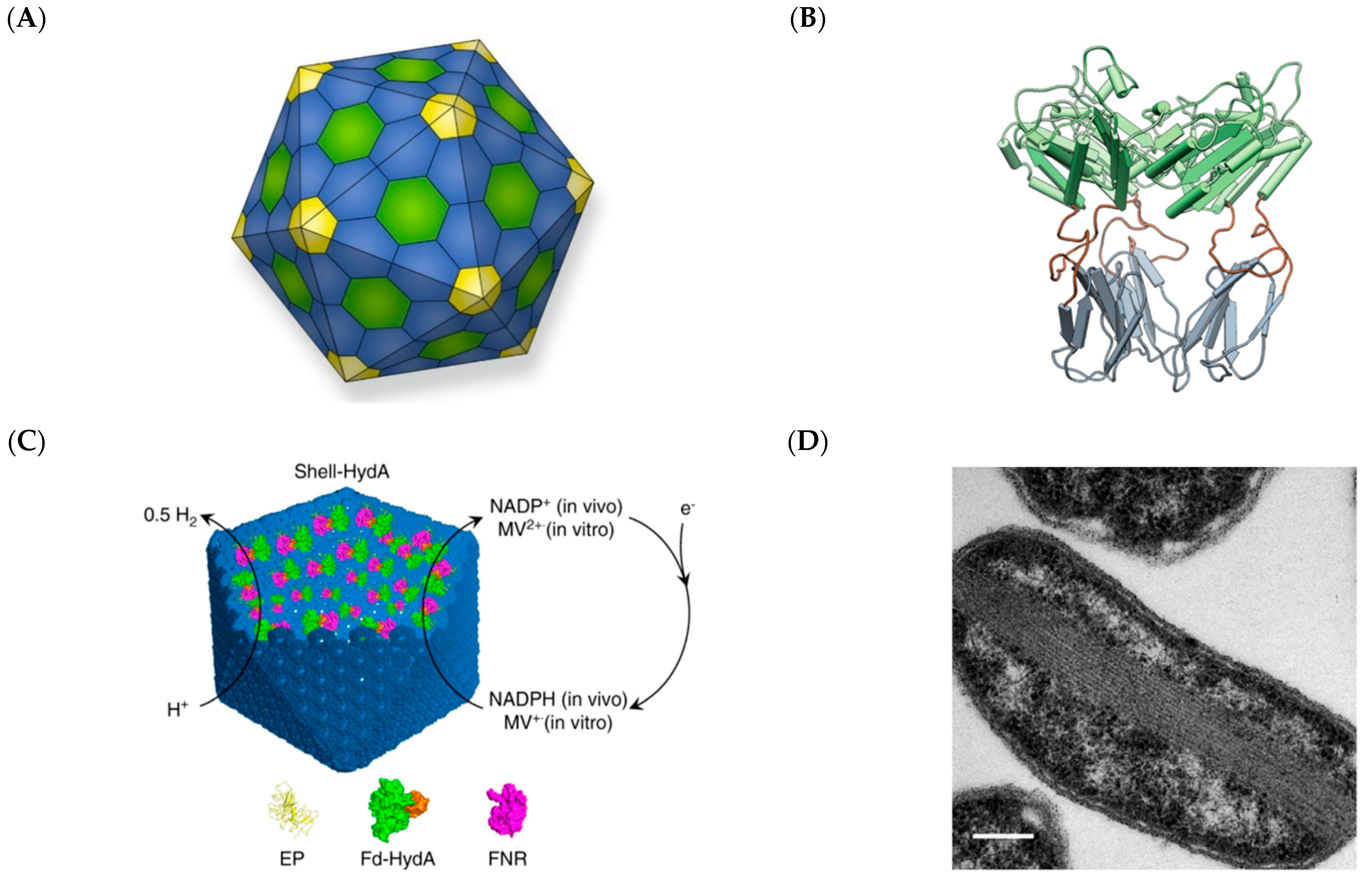
2.3. Bacterial Microcompartments

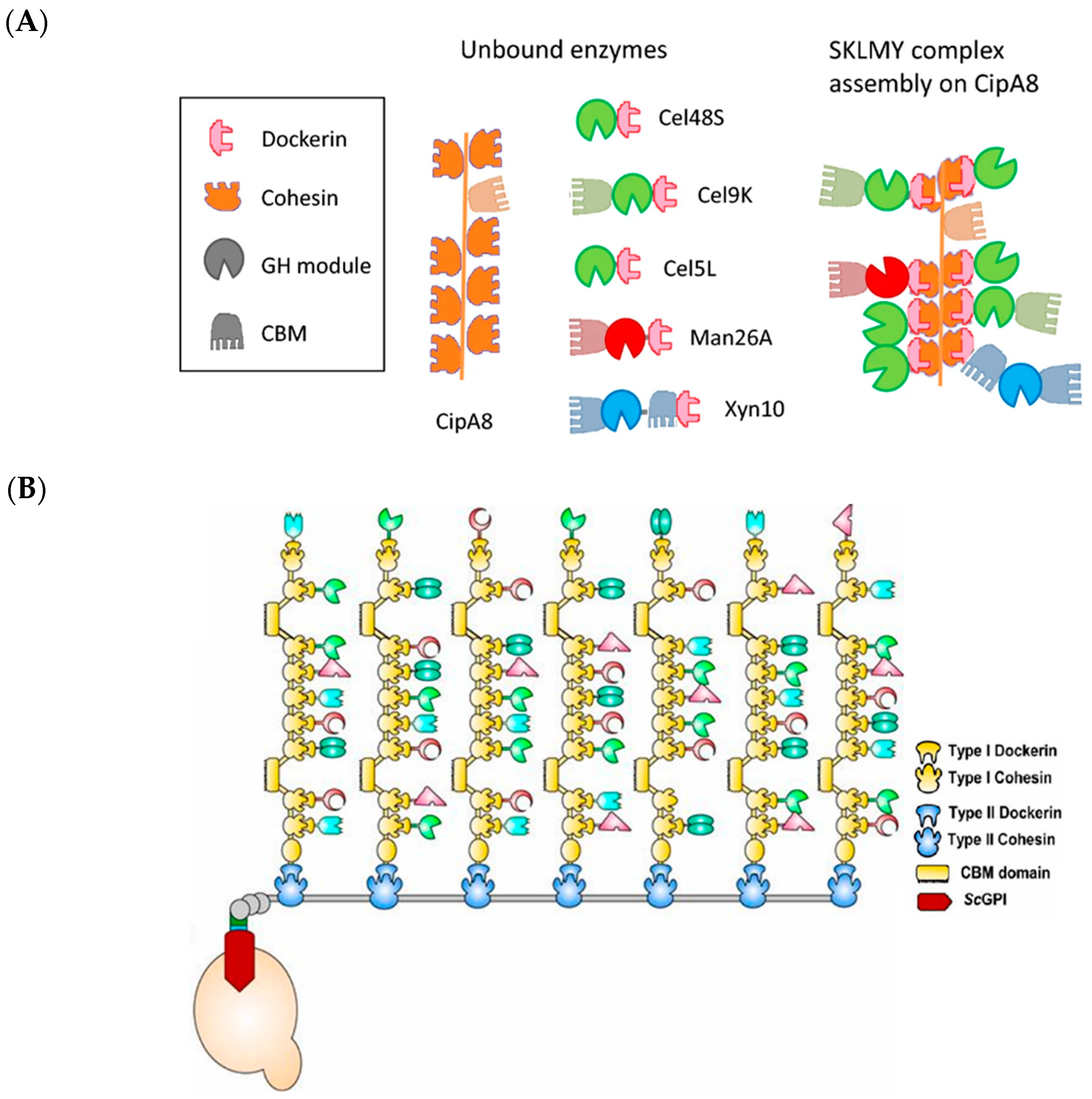
2.4. Cellulosomes
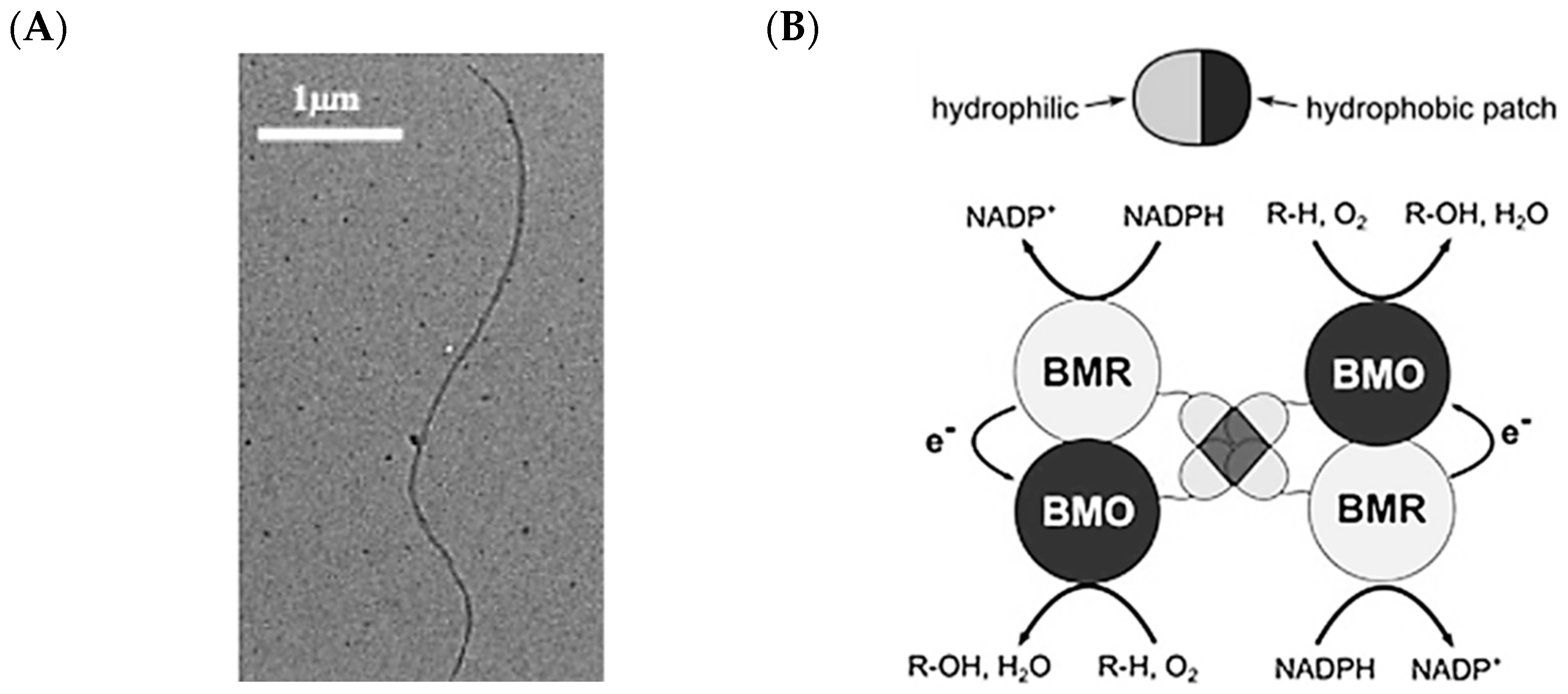
2.5. Hydrophobins

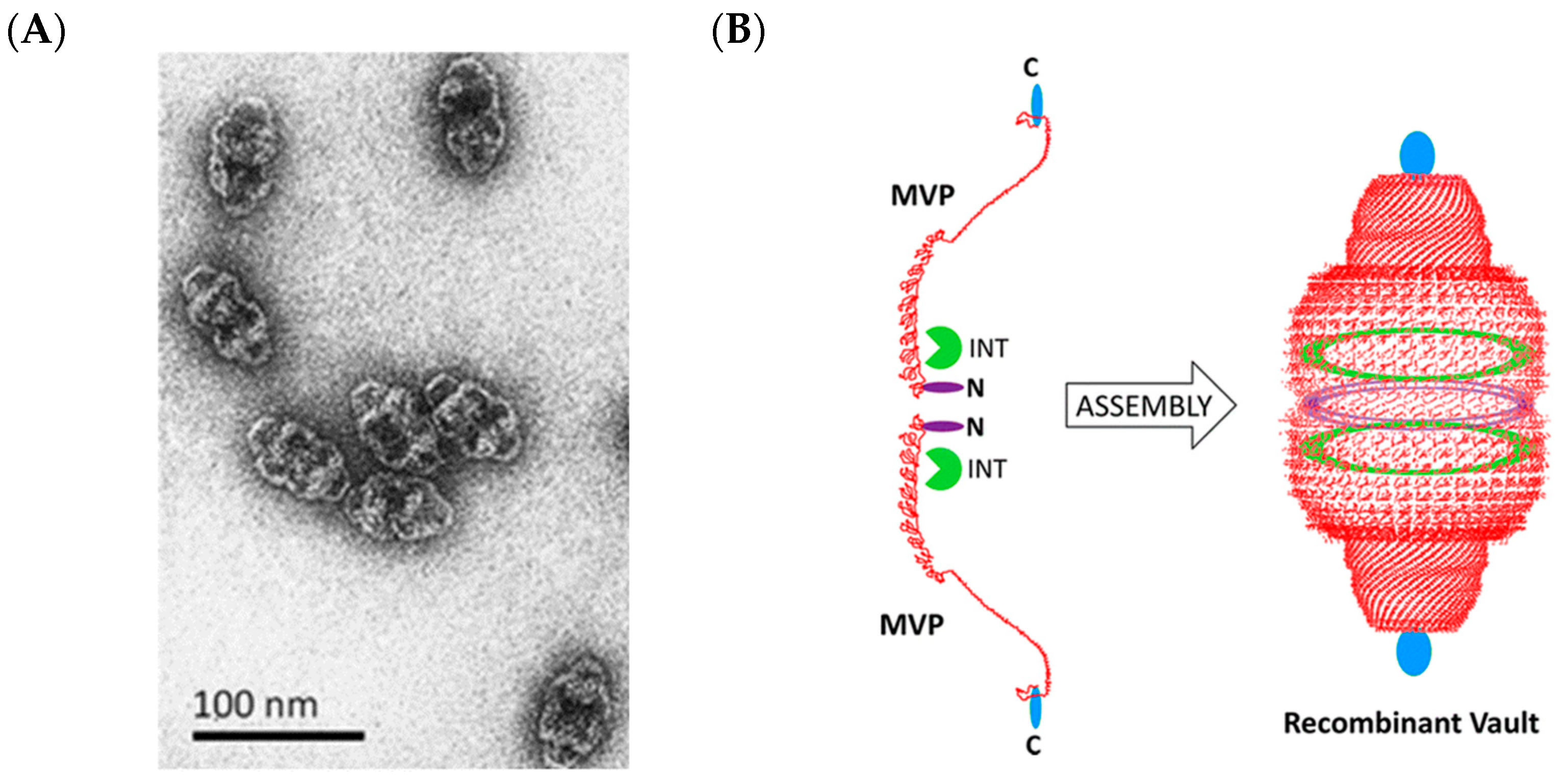
2.6. Vault Ribonucleoprotein Complexes

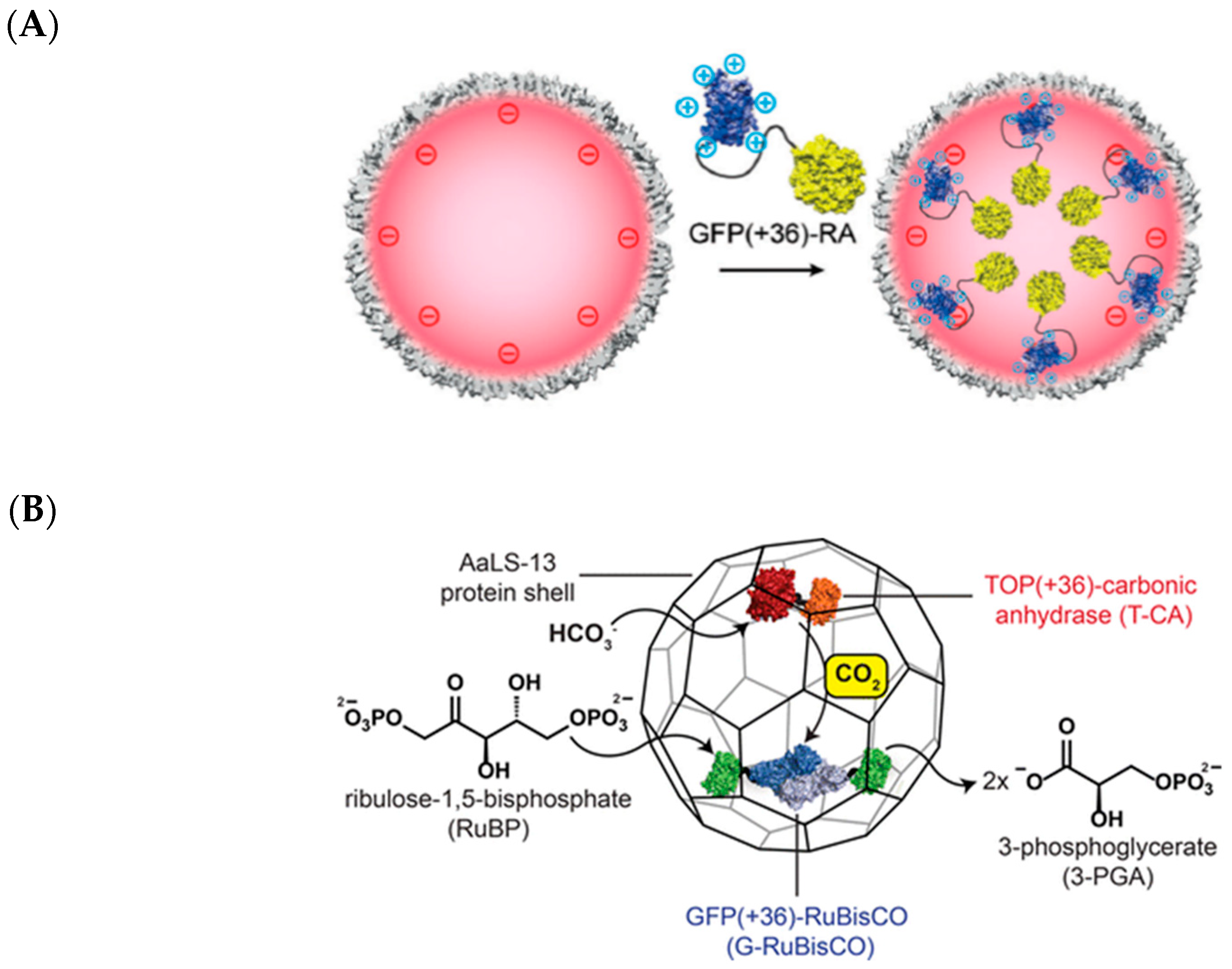
2.7. Lumazine Synthase Assemblies

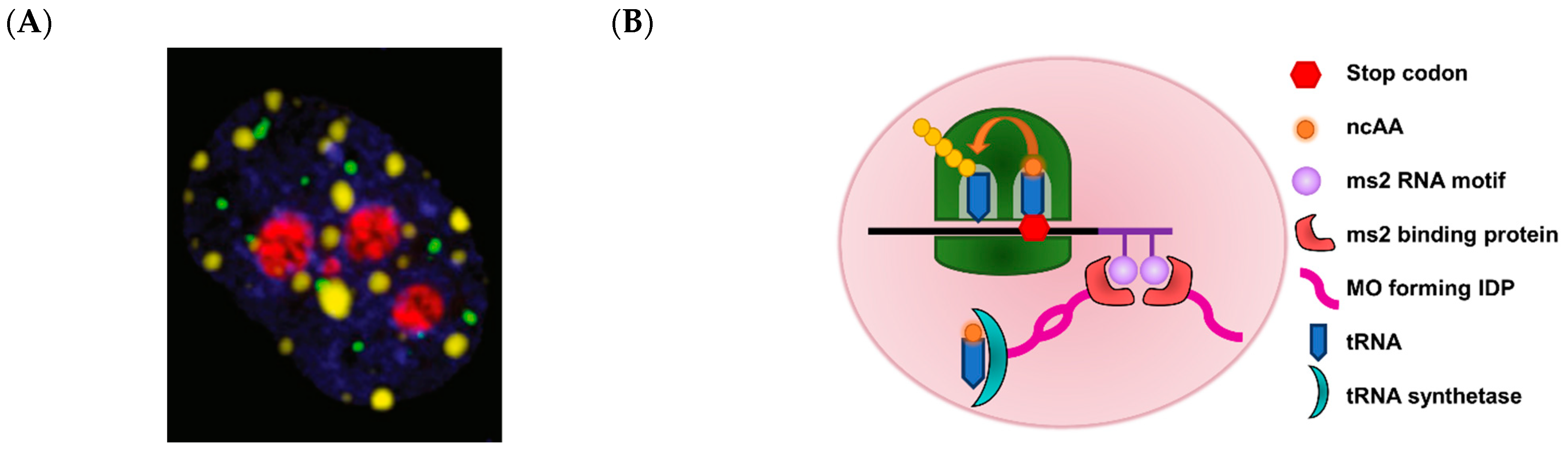
2.8. Membraneless Organelles
3. Nucleic Acid Scaffolds
3.1. DNA Scaffolds

3.2. RNA Scaffolds
4. Molecular Tools for Constructing Enzymatic Scaffolds
4.1. Multipartite DNA Assembly Enabled by Synthetic Biology
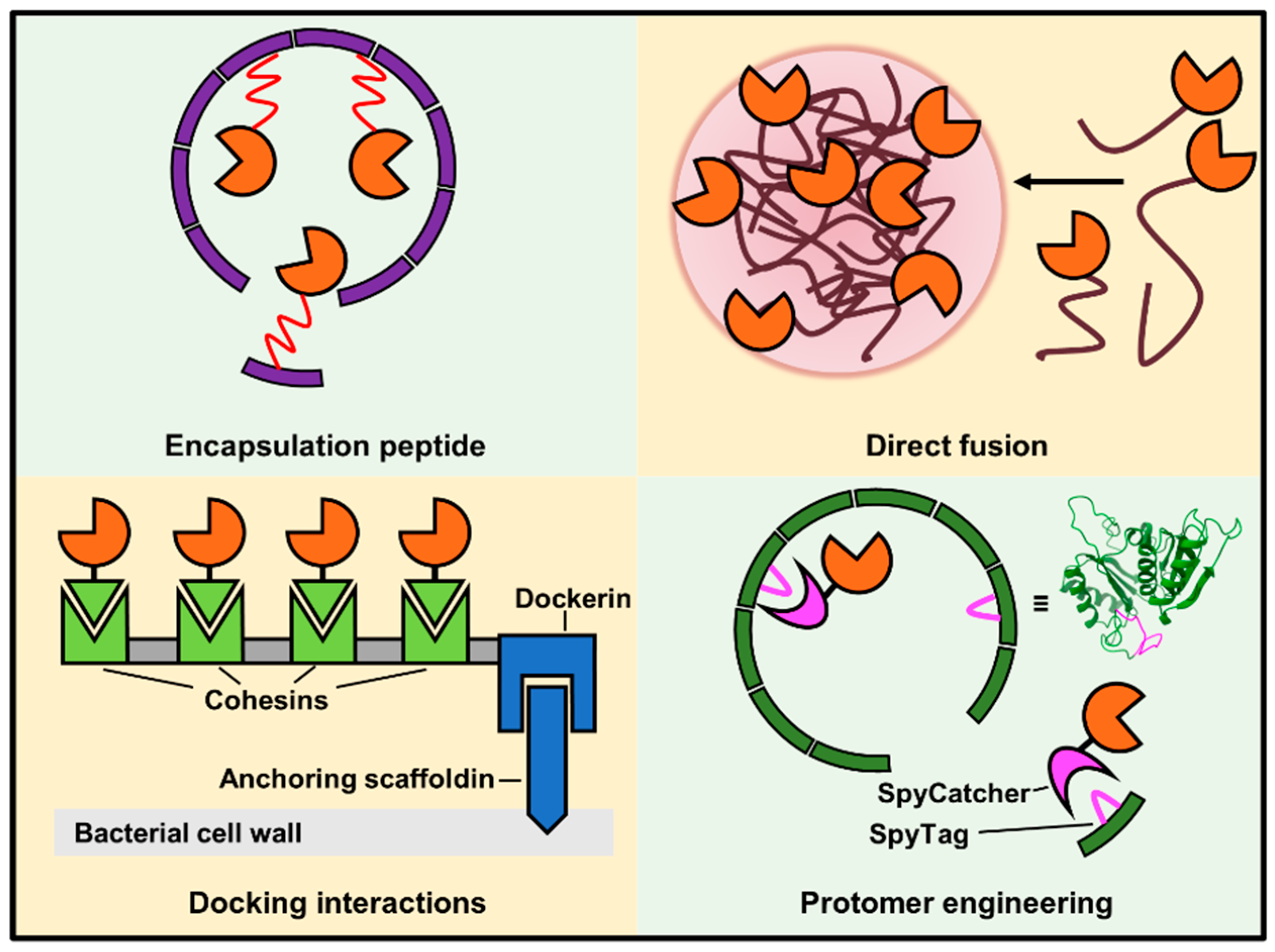
4.2. Strategies for Installing Enzymes on Scaffolds
5. Conclusions and Outlook
Author Contributions
Funding
Institutional Review Board Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AAR | Acyl-ACP reductase |
| ADH | Alcohol dehydrogenase |
| ADO | Aldehyde deformylating oxygenase |
| AuNP | Gold nanoparticle |
| BIA | Benzylisoquinoline alkaloid |
| BMC | Bacterial microcompartment |
| CBM | Carbohydrate-binding module |
| CPMV | Cowpea mosaic virus |
| CYP450 | Cytochrome P450 |
| EP | Encapsulation peptide |
| EPSPS | 5-enolpyruvylshikimate-3-phosphate |
| G6pDH | Glucose-6-phosphate dehydrogenase |
| GCK | Gluconokinase |
| GFP | Green fluorescent protein |
| GH | Glycoside hydrolase |
| GOX | Glucose oxidase |
| GRAS | Generally regarded as safe |
| GSH | Glutathione |
| GST | Glutathione S-transferase |
| HRP | Horseradish peroxidase |
| IMAC | Immobilized metal affinity chromatography |
| INT | MVP interaction domain |
| LDH | Lactate dehydrogenase |
| LLPS | Liquid-liquid phase separation |
| MDH | Malate dehydrogenase |
| MnP | Manganese peroxidase |
| MO | Membraneless organelle |
| MOFS | Membraneless organelle formation sequence |
| MoClo | Modular cloning |
| MOFS | Membraneless organelle formation sequence |
| MVP | Major vault protein |
| ncAA | Non-canonical amino acid |
| ncNT | Non-canonical nucleotide |
| NHS | N-hydroxysuccinimide |
| NTA | Nitrilotriacetate |
| ORF | Open reading frame |
| PDC | Pyruvate decarboxylase |
| Pyl | Pyrrolysine |
| RBS | Ribosomal binding site |
| RE | Restriction enzyme |
| ROS | Reactive oxygen species |
| SDS | Sodium dodecylsulfate |
| sHSP | Small heat shock protein |
| TEM | Transmission electron microscopy |
| TMV | Tobacco mosaic virus |
| TP | Targeting peptide |
| VLP | Virus-like particle |
| YFP | Yellow fluorescent protein |
References
- Nielsen, J.; Keasling, J.D. Engineering cellular metabolism. Cell 2016, 164, 1185–1197. [Google Scholar] [CrossRef] [PubMed]
- Galanie, S.; Thodey, K.; Trenchard, I.J.; Filsinger Interrante, M.; Smolke, C.D. Complete biosynthesis of opioids in yeast. Science 2015, 349, 1095–1100. [Google Scholar] [CrossRef]
- Luo, X.; Reiter, M.A.; d’Espaux, L.; Wong, J.; Denby, C.M.; Lechner, A.; Zhang, Y.; Grzybowski, A.T.; Harth, S.; Lin, W.; et al. Complete biosynthesis of cannabinoids and their unnatural analogues in yeast. Nature 2019, 567, 123–126. [Google Scholar] [CrossRef] [PubMed]
- Zhou, K.; Qiao, K.; Edgar, S.; Stephanopoulos, G. Distributing a metabolic pathway among a microbial consortium enhances production of natural products. Nat. Biotech. 2015, 33, 377–383. [Google Scholar] [CrossRef] [PubMed]
- Tournier, V.; Topham, C.M.; Gilles, A.; David, B.; Folgoas, C.; Moya-Leclair, E.; Kamionka, E.; Desrousseaux, M.L.; Texier, H.; Gavalda, S.; et al. An engineered pet depolymerase to break down and recycle plastic bottles. Nature 2020, 580, 216–219. [Google Scholar] [CrossRef]
- Ru, J.; Huo, Y.; Yang, Y. Microbial degradation and valorization of plastic wastes. Front. Microbiol. 2020, 11. [Google Scholar] [CrossRef]
- Go, M.K.; Wongsantichon, J.; Cheung, V.W.N.; Chow, J.Y.; Robinson, R.C.; Yew, W.S. Synthetic polyketide enzymology: Platform for biosynthesis of antimicrobial polyketides. ACS Catal. 2015, 5, 4033–4042. [Google Scholar] [CrossRef]
- Awan, A.R.; Blount, B.A.; Bell, D.J.; Shaw, W.M.; Ho, J.C.H.; McKiernan, R.M.; Ellis, T. Biosynthesis of the antibiotic nonribosomal peptide penicillin in baker’s yeast. Nat. Commun. 2017, 8, 15202. [Google Scholar] [CrossRef]
- Wang, J. Electrochemical glucose biosensors. Chem. Rev. 2008, 108, 814–825. [Google Scholar] [CrossRef] [PubMed]
- Gerlt, J.A. Genomic enzymology: Web tools for leveraging protein family sequence–function space and genome context to discover novel functions. Biochemistry 2017, 56, 4293–4308. [Google Scholar] [CrossRef]
- Li, C.; Zhang, R.; Wang, J.; Wilson, L.M.; Yan, Y. Protein engineering for improving and diversifying natural product biosynthesis. Trends Biotechnol. 2020, 38, 729–744. [Google Scholar] [CrossRef] [PubMed]
- Jäckel, C.; Kast, P.; Hilvert, D. Protein design by directed evolution. Annu. Rev. Biophys. 2008, 37, 153–173. [Google Scholar] [CrossRef]
- Currin, A.; Swainston, N.; Day, P.J.; Kell, D.B. Synthetic biology for the directed evolution of protein biocatalysts: Navigating sequence space intelligently. Chem. Soc. Rev. 2015, 44, 1172–1239. [Google Scholar] [CrossRef]
- Agapakis, C.M.; Boyle, P.M.; Silver, P.A. Natural strategies for the spatial optimization of metabolism in synthetic biology. Nat. Chem. Biol. 2012, 8, 527–535. [Google Scholar] [CrossRef]
- Deshpande, S.; Masurkar, N.D.; Girish, V.M.; Desai, M.; Chakraborty, G.; Chan, J.M.; Drum, C.L. Thermostable exoshells fold and stabilize recombinant proteins. Nat. Commun. 2017, 8, 1442. [Google Scholar] [CrossRef]
- Bari, N.K.; Kumar, G.; Hazra, J.P.; Kaur, S.; Sinha, S. Functional protein shells fabricated from the self-assembling protein sheets of prokaryotic organelles. J. Mater. Chem. B 2020, 8, 523–533. [Google Scholar] [CrossRef] [PubMed]
- Rahmanpour, R.; Bugg, T.D.H. Assembly in vitro of rhodococcus jostii rha1 encapsulin and peroxidase dypb to form a nanocompartment. FEBS J. 2013, 280, 2097–2104. [Google Scholar] [CrossRef] [PubMed]
- Lončar, N.; Rozeboom, H.J.; Franken, L.E.; Stuart, M.C.A.; Fraaije, M.W. Structure of a robust bacterial protein cage and its application as a versatile biocatalytic platform through enzyme encapsulation. Biochem. Biophys. Res. Commun. 2020, 529, 548–553. [Google Scholar] [CrossRef]
- Avalos, J.L.; Fink, G.R.; Stephanopoulos, G. Compartmentalization of metabolic pathways in yeast mitochondria improves production of branched chain alcohols. Nat. Biotechnol. 2013, 31, 335–341. [Google Scholar] [CrossRef]
- Hammer, S.K.; Avalos, J.L. Harnessing yeast organelles for metabolic engineering. Nat. Chem. Biol. 2017, 13, 823. [Google Scholar] [CrossRef] [PubMed]
- Engels, B.; Dahm, P.; Jennewein, S. Metabolic engineering of taxadiene biosynthesis in yeast as a first step towards taxol (paclitaxel) production. Metab. Eng. 2008, 10, 201–206. [Google Scholar] [CrossRef] [PubMed]
- Delebecque, C.J.; Lindner, A.B.; Silver, P.A.; Aldaye, F.A. Organization of intracellular reactions with rationally designed rna assemblies. Science 2011, 333, 470–474. [Google Scholar] [CrossRef]
- Spillmann, C.M.; Medintz, I.L. Use of biomolecular scaffolds for assembling multistep light harvesting and energy transfer devices. J. Photochem. Photobiol. C Photochem. Rev. 2015, 23, 1–24. [Google Scholar] [CrossRef]
- Hartl, F.U.; Hayer-Hartl, M. Converging concepts of protein folding in vitro and in vivo. Nat. Struct. Mol. Biol. 2009, 16, 574–581. [Google Scholar] [CrossRef] [PubMed]
- Ellis, G.A.; Klein, W.P.; Lasarte-Aragonés, G.; Thakur, M.; Walper, S.A.; Medintz, I.L. Artificial multienzyme scaffolds: Pursuing in vitro substrate channeling with an overview of current progress. ACS Catal. 2019, 9, 10812–10869. [Google Scholar] [CrossRef]
- Medema, M.H.; Breitling, R.; Bovenberg, R.; Takano, E. Exploiting plug-and-play synthetic biology for drug discovery and production in microorganisms. Nat. Rev. Microbiol. 2011, 9, 131–137. [Google Scholar] [CrossRef]
- Halperin, L.; Jung, J.; Michalak, M. The many functions of the endoplasmic reticulum chaperones and folding enzymes. IUBMB Life 2014, 66, 318–326. [Google Scholar] [CrossRef]
- del Río, L.A.; Corpas, F.J.; Sandalio, L.M.; Palma, J.M.; Gómez, M.; Barroso, J.B. Reactive oxygen species, antioxidant systems and nitric oxide in peroxisomes. J. Exp. Bot. 2002, 53, 1255–1272. [Google Scholar] [CrossRef]
- Boeynaems, S.; Alberti, S.; Fawzi, N.L.; Mittag, T.; Polymenidou, M.; Rousseau, F.; Schymkowitz, J.; Shorter, J.; Wolozin, B.; Van Den Bosch, L.; et al. Protein phase separation: A new phase in cell biology. Trends Cell Biol. 2018, 28, 420–435. [Google Scholar] [CrossRef] [PubMed]
- Kerfeld, C.A.; Aussignargues, C.; Zarzycki, J.; Cai, F.; Sutter, M. Bacterial microcompartments. Nat. Rev. Microbiol. 2018, 16, 277. [Google Scholar] [CrossRef]
- Nichols, R.J.; Cassidy-Amstutz, C.; Chaijarasphong, T.; Savage, D.F. Encapsulins: Molecular biology of the shell. Crit. Rev. Biochem. Mol. Biol. 2017, 52, 583–594. [Google Scholar] [CrossRef]
- Wong, J.X.; Ogura, K.; Chen, S.; Rehm, B.H.A. Bioengineered polyhydroxyalkanoates as immobilized enzyme scaffolds for industrial applications. Front. Bioeng. Biotechnol. 2020, 8, 156. [Google Scholar] [CrossRef] [PubMed]
- Sharmeen, S.; Rahman, M.S.; Islam, M.M.; Islam, M.S.; Shahruzzaman, M.; Mallik, A.K.; Haque, P.; Rahman, M.M. Applications of polysaccharides in enzyme immobilization. In Functional Polysaccharides for Biomedical Applications; Maiti, S., Jana, S., Eds.; Woodhead Publishing: Cambridge, UK, 2019; pp. 357–395. [Google Scholar]
- Yoshimoto, M. Stabilization of enzymes through encapsulation in liposomes. Methods Mol. Biol. 2017, 1504, 9–18. [Google Scholar] [PubMed]
- Long, B.M.; Hee, W.Y.; Sharwood, R.E.; Rae, B.D.; Kaines, S.; Lim, Y.-L.; Nguyen, N.D.; Massey, B.; Bala, S.; von Caemmerer, S.; et al. Carboxysome encapsulation of the co2-fixing enzyme rubisco in tobacco chloroplasts. Nat. Commun. 2018, 9, 3570. [Google Scholar] [CrossRef] [PubMed]
- Wei, S.-P.; Qian, Z.-G.; Hu, C.-F.; Pan, F.; Chen, M.-T.; Lee, S.Y.; Xia, X.-X. Formation and functionalization of membraneless compartments in escherichia coli. Nat. Chem. Biol. 2020, 16, 1143–1148. [Google Scholar] [CrossRef]
- Subramaniam, S.; Earl, L.A.; Falconieri, V.; Milne, J.L.; Egelman, E.H. Resolution advances in cryo-em enable application to drug discovery. Curr. Opin. Struct. Biol. 2016, 41, 194–202. [Google Scholar] [CrossRef]
- Speir, J.A.; Bothner, B.; Qu, C.; Willits, D.A.; Young, M.J.; Johnson, J.E. Enhanced local symmetry interactions globally stabilize a mutant virus capsid that maintains infectivity and capsid dynamics. J. Virol. 2006, 80, 3582–3591. [Google Scholar] [CrossRef]
- Schmidli, C.; Albiez, S.; Rima, L.; Righetto, R.; Mohammed, I.; Oliva, P.; Kovacik, L.; Stahlberg, H.; Braun, T. Microfluidic protein isolation and sample preparation for high-resolution cryo-em. Proc. Natl. Acad. Sci. USA 2019, 116, 15007–15012. [Google Scholar] [CrossRef] [PubMed]
- Hryc, C.F.; Chen, D.H.; Afonine, P.V.; Jakana, J.; Wang, Z.; Haase-Pettingell, C.; Jiang, W.; Adams, P.D.; King, J.A.; Schmid, M.F.; et al. Accurate model annotation of a near-atomic resolution cryo-em map. Proc. Natl. Acad. Sci. USA 2017, 114, 3103–3108. [Google Scholar] [CrossRef] [PubMed]
- Sutter, M.; Greber, B.; Aussignargues, C.; Kerfeld, C.A. Assembly principles and structure of a 6.5-mda bacterial microcompartment shell. Science 2017, 356, 1293–1297. [Google Scholar] [CrossRef]
- McHugh, C.A.; Fontana, J.; Nemecek, D.; Cheng, N.; Aksyuk, A.A.; Heymann, J.B.; Winkler, D.C.; Lam, A.S.; Wall, J.S.; Steven, A.C.; et al. A virus capsid-like nanocompartment that stores iron and protects bacteria from oxidative stress. EMBO J. 2014, 33, 1896–1911. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Meining, W.; Fischer, M.; Bacher, A.; Ladenstein, R. X-ray structure analysis and crystallographic refinement of lumazine synthase from the hyperthermophile aquifex aeolicus at 1.6 a resolution: Determinants of thermostability revealed from structural comparisons. J. Mol. Biol. 2001, 306, 1099–1114. [Google Scholar] [CrossRef] [PubMed]
- Johnson, E.; Cascio, D.; Sawaya, M.R.; Gingery, M.; Schröder, I. Crystal structures of a tetrahedral open pore ferritin from the hyperthermophilic archaeon archaeoglobus fulgidus. Structure 2005, 13, 637–648. [Google Scholar] [CrossRef] [PubMed]
- Akita, F.; Chong, K.T.; Tanaka, H.; Yamashita, E.; Miyazaki, N.; Nakaishi, Y.; Suzuki, M.; Namba, K.; Ono, Y.; Tsukihara, T.; et al. The crystal structure of a virus-like particle from the hyperthermophilic archaeon pyrococcus furiosus provides insight into the evolution of viruses. J. Mol. Biol. 2007, 368, 1469–1483. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.K.; Kim, R.; Kim, S.H. Crystal structure of a small heat-shock protein. Nature 1998, 394, 595–599. [Google Scholar] [CrossRef]
- Tanaka, H.; Kato, K.; Yamashita, E.; Sumizawa, T.; Zhou, Y.; Yao, M.; Iwasaki, K.; Yoshimura, M.; Tsukihara, T. The structure of rat liver vault at 3.5 angstrom resolution. Science 2009, 323, 384–388. [Google Scholar] [CrossRef]
- Hakanpää, J.; Paananen, A.; Askolin, S.; Nakari-Setälä, T.; Parkkinen, T.; Penttilä, M.; Linder, M.B.; Rouvinen, J. Atomic resolution structure of the hfbii hydrophobin, a self-assembling amphiphile. J. Biol. Chem. 2004, 279, 534–539. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. Ucsf chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Schrodinger-LLC. The Pymol Molecular Graphics System. 2010. Available online: https://pymol.org/2/ (accessed on 4 March 2021).
- Lau, Y.H.; Giessen, T.W.; Altenburg, W.J.; Silver, P.A. Prokaryotic nanocompartments form synthetic organelles in a eukaryote. Nat. Commun. 2018, 9, 1311. [Google Scholar] [CrossRef]
- Lawrence, A.D.; Frank, S.; Newnham, S.; Lee, M.J.; Brown, I.R.; Xue, W.-F.; Rowe, M.L.; Mulvihill, D.P.; Prentice, M.B.; Howard, M.J.; et al. Solution structure of a bacterial microcompartment targeting peptide and its application in the construction of an ethanol bioreactor. ACS Synth. Biol. 2014, 3, 454–465. [Google Scholar] [CrossRef]
- Anandharaj, M.; Lin, Y.-J.; Rani, R.P.; Nadendla, E.K.; Ho, M.-C.; Huang, C.-C.; Cheng, J.-F.; Chang, J.-J.; Li, W.-H. Constructing a yeast to express the largest cellulosome complex on the cell surface. Proc. Nat. Acad. Sci. USA 2020, 117, 2385–2394. [Google Scholar] [CrossRef] [PubMed]
- Hagen, A.; Sutter, M.; Sloan, N.; Kerfeld, C.A. Programmed loading and rapid purification of engineered bacterial microcompartment shells. Nat. Commun. 2018, 9, 2881. [Google Scholar] [CrossRef]
- Schwarz, B.; Uchida, M.; Douglas, T. Chapter one-biomedical and catalytic opportunities of virus-like particles in nanotechnology. In Advances in Virus Research; Kielian, M., Mettenleiter, T.C., Roossinck, M.J., Eds.; Academic Press: Cambridge, MA, USA, 2017; Volume 97, pp. 1–60. [Google Scholar]
- Brasch, M.; Putri, R.M.; de Ruiter, M.V.; Luque, D.; Koay, M.S.T.; Castón, J.R.; Cornelissen, J.J.L.M. Assembling enzymatic cascade pathways inside virus-based nanocages using dual-tasking nucleic acid tags. J. Am. Chem. Soc. 2017, 139, 1512–1519. [Google Scholar] [CrossRef]
- Rurup, W.F.; Verbij, F.; Koay, M.S.T.; Blum, C.; Subramaniam, V.; Cornelissen, J.J.L.M. Predicting the loading of virus-like particles with fluorescent proteins. Biomacromolecules 2014, 15, 558–563. [Google Scholar] [CrossRef] [PubMed]
- Comellas-Aragonès, M.; Engelkamp, H.; Claessen, V.I.; Sommerdijk, N.A.J.M.; Rowan, A.E.; Christianen, P.C.M.; Maan, J.C.; Verduin, B.J.M.; Cornelissen, J.J.L.M.; Nolte, R.J.M.; et al. A virus-based single-enzyme nanoreactor. Nat. Nanotechnol. 2007, 2, 635–639. [Google Scholar] [CrossRef] [PubMed]
- Lemire, S.; Yehl, K.M.; Lu, T.K. Phage-based applications in synthetic biology. Annu. Rev. Virol. 2018, 5, 453–476. [Google Scholar] [CrossRef] [PubMed]
- Patterson, D.P.; Prevelige, P.E.; Douglas, T. Nanoreactors by programmed enzyme encapsulation inside the capsid of the bacteriophage p22. ACS Nano 2012, 6, 5000–5009. [Google Scholar] [CrossRef] [PubMed]
- O’Neil, A.; Reichhardt, C.; Johnson, B.; Prevelige, P.E.; Douglas, T. Genetically programmed in vivo packaging of protein cargo and its controlled release from bacteriophage p22. Angew. Chem. 2011, 50, 7425–7428. [Google Scholar] [CrossRef] [PubMed]
- Patterson, D.P.; Schwarz, B.; El-Boubbou, K.; van der Oost, J.; Prevelige, P.E.; Douglas, T. Virus-like particle nanoreactors: Programmed encapsulation of the thermostable celb glycosidase inside the p22 capsid. Soft Matter 2012, 8, 10158–10166. [Google Scholar] [CrossRef]
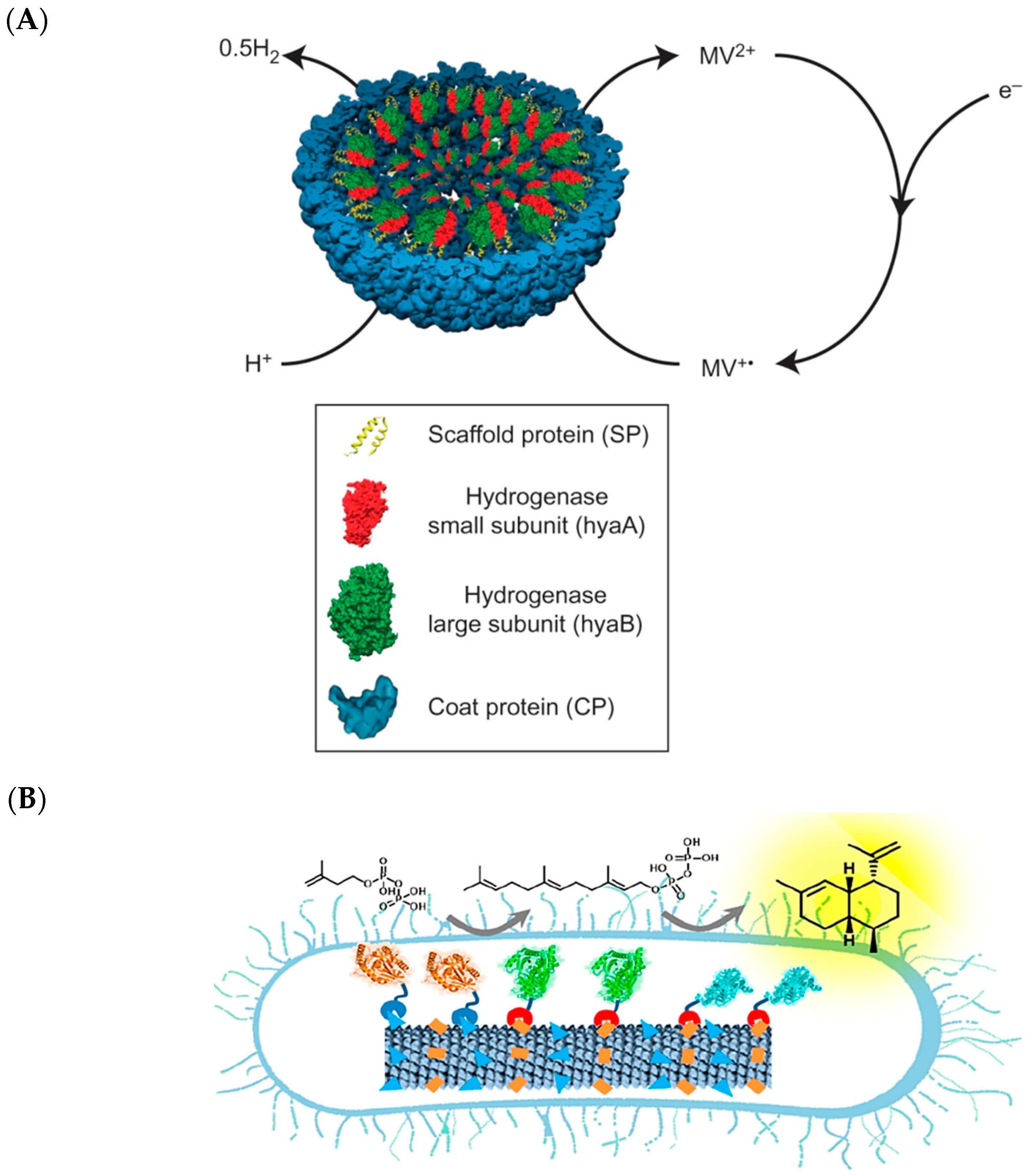
- Jordan, P.C.; Patterson, D.P.; Saboda, K.N.; Edwards, E.J.; Miettinen, H.M.; Basu, G.; Thielges, M.C.; Douglas, T. Self-assembling biomolecular catalysts for hydrogen production. Nat. Chem. 2016, 8, 179–185. [Google Scholar] [CrossRef]
- Kalms, J.; Schmidt, A.; Frielingsdorf, S.; Utesch, T.; Gotthard, G.; von Stetten, D.; van der Linden, P.; Royant, A.; Mroginski, M.A.; Carpentier, P.; et al. Tracking the route of molecular oxygen in o2-tolerant membrane-bound [NiFe] hydrogenase. Proc. Nat. Acad. Sci. USA 2018, 115, E2229–E2237. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Uchida, M.; Waghwani, H.K.; Douglas, T. Synthetic virus-like particles for glutathione biosynthesis. ACS Synth. Biol. 2020, 9, 3298–3310. [Google Scholar] [CrossRef]
- Jørgensen, K.; Rasmussen, A.V.; Morant, M.; Nielsen, A.H.; Bjarnholt, N.; Zagrobelny, M.; Bak, S.; Møller, B.L. Metabolon formation and metabolic channeling in the biosynthesis of plant natural products. Curr. Opin. Plant Biol. 2005, 8, 280–291. [Google Scholar] [CrossRef]
- Wei, Q.; He, S.; Qu, J.; Xia, J. Synthetic multienzyme complexes assembled on virus-like particles for cascade biosynthesis in cellulo. Bioconjugate Chem. 2020, 31, 2413–2420. [Google Scholar] [CrossRef] [PubMed]
- Giessen, T.W.; Silver, P.A. Widespread distribution of encapsulin nanocompartments reveals functional diversity. Nat. Microbiol. 2017, 2, 17029. [Google Scholar] [CrossRef]
- Sutter, M.; Boehringer, D.; Gutmann, S.; Günther, S.; Prangishvili, D.; Loessner, M.J.; Stetter, K.O.; Weber-Ban, E.; Ban, N. Structural basis of enzyme encapsulation into a bacterial nanocompartment. Nat. Struct. Mol. Biol. 2008, 15, 939–947. [Google Scholar] [CrossRef] [PubMed]
- Grewal, P.S.; Samson, J.A.; Baker, J.J.; Choi, B.; Dueber, J.E. Peroxisome compartmentalization of a toxic enzyme improves alkaloid production. Nat. Chem. Biol. 2021, 17, 96–103. [Google Scholar] [CrossRef]
- Sigmund, F.; Massner, C.; Erdmann, P.; Stelzl, A.; Rolbieski, H.; Desai, M.; Bricault, S.; Wörner, T.P.; Snijder, J.; Geerlof, A.; et al. Bacterial encapsulins as orthogonal compartments for mammalian cell engineering. Nat. Commun. 2018, 9, 1990. [Google Scholar] [CrossRef]
- Jathoul, A.P.; Laufer, J.; Ogunlade, O.; Treeby, B.; Cox, B.; Zhang, E.; Johnson, P.; Pizzey, A.R.; Philip, B.; Marafioti, T.; et al. Deep in vivo photoacoustic imaging of mammalian tissues using a tyrosinase-based genetic reporter. Nat. Photonics 2015, 9, 239–246. [Google Scholar] [CrossRef]
- Lam, S.S.; Martell, J.D.; Kamer, K.J.; Deerinck, T.J.; Ellisman, M.H.; Mootha, V.K.; Ting, A.Y. Directed evolution of apex2 for electron microscopy and proximity labeling. Nat. Methods 2015, 12, 51–54. [Google Scholar] [CrossRef]
- Dunleavy, R.; Lu, L.; Kiely, C.J.; McIntosh, S.; Berger, B.W. Single-enzyme biomineralization of cadmium sulfide nanocrystals with controlled optical properties. Proc. Nat. Acad. Sci. USA 2016, 113, 5275–5280. [Google Scholar] [CrossRef]
- Demchuk, A.M.; Patel, T.R. The biomedical and bioengineering potential of protein nanocompartments. Biotechnol. Adv. 2020, 41, 107547. [Google Scholar] [CrossRef]
- Jiang, B.; Fang, L.; Wu, K.; Yan, X.; Fan, K. Ferritins as natural and artificial nanozymes for theranostics. Theranostics 2020, 10, 687–706. [Google Scholar] [CrossRef]
- Hagen, W.R.; Hagedoorn, P.L.; Honarmand Ebrahimi, K. The workings of ferritin: A crossroad of opinions. Metallomics Integr. Biometal Sci. 2017, 9, 595–605. [Google Scholar] [CrossRef] [PubMed]
- Tetter, S.; Hilvert, D. Enzyme encapsulation by a ferritin cage. Angew. Chem. Int. Ed. 2017, 56, 14933–14936. [Google Scholar] [CrossRef]
- Jutz, G.; van Rijn, P.; Santos Miranda, B.; Böker, A. Ferritin: A versatile building block for bionanotechnology. Chem. Rev. 2015, 115, 1653–1701. [Google Scholar] [CrossRef]
- Chakraborti, S.; Korpi, A.; Kumar, M.; Stępień, P.; Kostiainen, M.A.; Heddle, J.G. Three-dimensional protein cage array capable of active enzyme capture and artificial chaperone activity. Nano Lett. 2019, 19, 3918–3924. [Google Scholar] [CrossRef] [PubMed]
- Han, M.-J.; Yun, H.; Lee, S.Y. Microbial small heat shock proteins and their use in biotechnology. Biotechnol. Adv. 2008, 26, 591–609. [Google Scholar] [CrossRef] [PubMed]
- Flenniken, M.L.; Willits, D.A.; Brumfield, S.; Young, M.J.; Douglas, T. The small heat shock protein cage from methanococcus jannaschii is a versatile nanoscale platform for genetic and chemical modification. Nano Lett. 2003, 3, 1573–1576. [Google Scholar] [CrossRef]
- Kim, R.; Kim, K.K.; Yokota, H.; Kim, S.-H. Small heat shock protein of methanococcus jannaschii, a hyperthermophile. Proc. Nat. Acad. Sci. USA 1998, 95, 9129–9133. [Google Scholar] [CrossRef] [PubMed]
- Yeates, T.O.; Thompson, M.C.; Bobik, T.A. The protein shells of bacterial microcompartment organelles. Curr. Opin. Struct. Biol. 2011, 21, 223–231. [Google Scholar] [CrossRef]
- Thompson, M.C.; Cascio, D.; Leibly, D.J.; Yeates, T.O. An allosteric model for control of pore opening by substrate binding in the eutl microcompartment shell protein. Protein Sci. Publ. Protein Soc. 2015, 24, 956–975. [Google Scholar] [CrossRef]
- Parsons, J.B.; Frank, S.; Bhella, D.; Liang, M.; Prentice, M.B.; Mulvihill, D.P.; Warren, M.J. Synthesis of empty bacterial microcompartments, directed organelle protein incorporation, and evidence of filament-associated organelle movement. Mol. Cell 2010, 38, 305–315. [Google Scholar] [CrossRef]
- Bonacci, W.; Teng, P.K.; Afonso, B.; Niederholtmeyer, H.; Grob, P.; Silver, P.A.; Savage, D.F. Modularity of a carbon-fixing protein organelle. Proc. Nat. Acad. Sci. USA 2012, 109, 478–483. [Google Scholar] [CrossRef] [PubMed]
- Cai, F.; Bernstein, S.L.; Wilson, S.C.; Kerfeld, C.A. Production and characterization of synthetic carboxysome shells with incorporated luminal proteins. Plant Physiol. 2016, 170, 1868–1877. [Google Scholar]
- Lee, M.J.; Brown, I.R.; Juodeikis, R.; Frank, S.; Warren, M.J. Employing bacterial microcompartment technology to engineer a shell-free enzyme-aggregate for enhanced 1,2-propanediol production in escherichia coli. Metab. Eng. 2016, 36, 48–56. [Google Scholar] [CrossRef] [PubMed]
- Dou, Z.; Heinhorst, S.; Williams, E.B.; Murin, C.D.; Shively, J.M.; Cannon, G.C. Co2 fixation kinetics of halothiobacillus neapolitanus mutant carboxysomes lacking carbonic anhydrase suggest the shell acts as a diffusional barrier for co2. J. Biol. Chem. 2008, 283, 10377–10384. [Google Scholar] [CrossRef]
- Oltrogge, L.M.; Chaijarasphong, T.; Chen, A.W.; Bolin, E.R.; Marqusee, S.; Savage, D.F. Multivalent interactions between csos2 and rubisco mediate α-carboxysome formation. Nat. Struct. Mol. Biol. 2020, 27, 281–287. [Google Scholar] [CrossRef]
- Ferlez, B.; Sutter, M.; Kerfeld, C.A. A designed bacterial microcompartment shell with tunable composition and precision cargo loading. Metab. Eng. 2019, 54, 286–291. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Jiang, Q.; Huang, J.; Aitchison, C.M.; Huang, F.; Yang, M.; Dykes, G.F.; He, H.-L.; Wang, Q.; Sprick, R.S.; et al. Reprogramming bacterial protein organelles as a nanoreactor for hydrogen production. Nat. Commun. 2020, 11, 5448. [Google Scholar] [CrossRef] [PubMed]
- Esselborn, J.; Kertess, L.; Apfel, U.-P.; Hofmann, E.; Happe, T. Loss of specific active-site iron atoms in oxygen-exposed [FeFe]-hydrogenase determined by detailed x-ray structure analyses. J. Am. Chem. Soc. 2019, 141, 17721–17728. [Google Scholar] [CrossRef]
- Lee, M.J.; Mantell, J.; Hodgson, L.; Alibhai, D.; Fletcher, J.M.; Brown, I.R.; Frank, S.; Xue, W.-F.; Verkade, P.; Woolfson, D.N.; et al. Engineered synthetic scaffolds for organizing proteins within the bacterial cytoplasm. Nat. Chem. Biol. 2017, 14, 142. [Google Scholar] [CrossRef]
- Zhang, G.; Johnston, T.; Quin, M.B.; Schmidt-Dannert, C. Developing a protein scaffolding system for rapid enzyme immobilization and optimization of enzyme functions for biocatalysis. ACS Synth. Biol. 2019, 8, 1867–1876. [Google Scholar] [CrossRef] [PubMed]
- Kirst, H.; Kerfeld, C.A. Bacterial microcompartments: Catalysis-enhancing metabolic modules for next generation metabolic and biomedical engineering. BMC Biol. 2019, 17, 79. [Google Scholar] [CrossRef] [PubMed]
- Artzi, L.; Bayer, E.A.; Moraïs, S. Cellulosomes: Bacterial nanomachines for dismantling plant polysaccharides. Nat. Rev. Microbiol. 2017, 15, 83–95. [Google Scholar] [CrossRef] [PubMed]
- Leis, B.; Held, C.; Andreeßen, B.; Liebl, W.; Graubner, S.; Schulte, L.-P.; Schwarz, W.H.; Zverlov, V.V. Optimizing the composition of a synthetic cellulosome complex for the hydrolysis of softwood pulp: Identification of the enzymatic core functions and biochemical complex characterization. Biotechnol. Biofuels 2018, 11, 220. [Google Scholar] [CrossRef] [PubMed]
- Zverlov, V.V.; Klupp, M.; Krauss, J.; Schwarz, W.H. Mutations in the scaffoldin gene, cipa, of clostridium thermocellum with impaired cellulosome formation and cellulose hydrolysis: Insertions of a new transposable element, IS1447, and implications for cellulase synergism on crystalline cellulose. J. Bacteriol. 2008, 190, 4321. [Google Scholar] [CrossRef]
- Cherf, G.M.; Cochran, J.R. Applications of yeast surface display for protein engineering. Methods Mol. Biol. 2015, 1319, 155–175. [Google Scholar] [PubMed]
- Tanaka, T.; Kondo, A. Cell-surface display of enzymes by the yeast saccharomyces cerevisiae for synthetic biology. FEMS Yeast Res. 2015, 15, 1–9. [Google Scholar]
- Liu, F.; Banta, S.; Chen, W. Functional assembly of a multi-enzyme methanol oxidation cascade on a surface-displayed trifunctional scaffold for enhanced nadh production. Chem. Commun. 2013, 49, 3766–3768. [Google Scholar] [CrossRef]
- Dong, C.; Qiao, J.; Wang, X.; Sun, W.; Chen, L.; Li, S.; Wu, K.; Ma, L.; Liu, Y. Engineering pichia pastoris with surface-display minicellulosomes for carboxymethyl cellulose hydrolysis and ethanol production. Biotechnol. Biofuels 2020, 13, 108. [Google Scholar] [CrossRef] [PubMed]
- Macindoe, I.; Kwan, A.H.; Ren, Q.; Morris, V.K.; Yang, W.; Mackay, J.P.; Sunde, M. Self-assembly of functional, amphipathic amyloid monolayers by the fungal hydrophobin eas. Proc. Nat. Acad. Sci. USA 2012, 109, E804–E811. [Google Scholar] [CrossRef]
- Winandy, L.; Hilpert, F.; Schlebusch, O.; Fischer, R. Comparative analysis of surface coating properties of five hydrophobins from aspergillus nidulans and trichoderma reseei. Sci. Rep. 2018, 8, 12033. [Google Scholar] [CrossRef] [PubMed]
- Longobardi, S.; Picone, D.; Ercole, C.; Spadaccini, R.; Stefano, L.D.; Rea, I.; Giardina, P. Environmental conditions modulate the switch among different states of the hydrophobin vmh2 from pleurotus ostreatus. Biomacromolecules 2012, 13, 743–750. [Google Scholar] [CrossRef] [PubMed]
- Piscitelli, A.; Pennacchio, A.; Longobardi, S.; Velotta, R.; Giardina, P. Vmh2 hydrophobin as a tool for the development of “self-immobilizing” enzymes for biosensing. Biotechnol. Bioeng. 2017, 114, 46–52. [Google Scholar] [CrossRef]
- Döring, J.; Rettke, D.; Rödel, G.; Pompe, T.; Ostermann, K. Surface functionalization by hydrophobin-epsps fusion protein allows for the fast and simple detection of glyphosate. Biosensors 2019, 9, 104. [Google Scholar] [CrossRef]
- Schulz, S.; Schumacher, D.; Raszkowski, D.; Girhard, M.; Urlacher, V.B. Fusion to hydrophobin hfbi improves the catalytic performance of a cytochrome p450 system. Front. Bioeng. Biotechnol. 2016, 4, 57. [Google Scholar] [CrossRef] [PubMed]
- Hlavica, P. Assembly of non-natural electron transfer conduits in the cytochrome p450 system: A critical assessment and update of artificial redox constructs amenable to exploitation in biotechnological areas. Biotechnol. Adv. 2009, 27, 103–121. [Google Scholar] [CrossRef]
- Piscitelli, A.; Pennacchio, A.; Cicatiello, P.; Politi, J.; De Stefano, L.; Giardina, P. Rapid and ultrasensitive detection of active thrombin based on the vmh2 hydrophobin fused to a green fluorescent protein. Biosens. Bioelectron. 2017, 87, 816–822. [Google Scholar] [CrossRef] [PubMed]
- Kedersha, N.L.; Miquel, M.C.; Bittner, D.; Rome, L.H. Vaults. Ii. Ribonucleoprotein structures are highly conserved among higher and lower eukaryotes. J. Cell Biol. 1990, 110, 895–901. [Google Scholar] [CrossRef]
- Berger, W.; Steiner, E.; Grusch, M.; Elbling, L.; Micksche, M. Vaults and the major vault protein: Novel roles in signal pathway regulation and immunity. Cell. Mol. Life Sci. 2008, 66, 43. [Google Scholar] [CrossRef] [PubMed]
- Goldsmith, L.E.; Pupols, M.; Kickhoefer, V.A.; Rome, L.H.; Monbouquette, H.G. Utilization of a protein “shuttle” to load vault nanocapsules with gold probes and proteins. ACS Nano 2009, 3, 3175–3183. [Google Scholar] [CrossRef] [PubMed]
- Kickhoefer, V.A.; Garcia, Y.; Mikyas, Y.; Johansson, E.; Zhou, J.C.; Raval-Fernandes, S.; Minoofar, P.; Zink, J.I.; Dunn, B.; Stewart, P.L.; et al. Engineering of vault nanocapsules with enzymatic and fluorescent properties. Proc. Nat. Acad. Sci USA 2005, 102, 4348–4352. [Google Scholar] [CrossRef]
- Hammel, K.E.; Kalyanaraman, B.; Kirk, T.K. Substrate free radicals are intermediates in ligninase catalysis. Proc. Natl. Acad. Sci. USA 1986, 83, 3708–3712. [Google Scholar] [CrossRef]
- Heinfling, A.; Ruiz-Dueñas, F.J.; Martínez, M.a.J.; Bergbauer, M.; Szewzyk, U.; Martínez, A.T. A study on reducing substrates of manganese-oxidizing peroxidases from pleurotus eryngii and bjerkandera adusta. FEBS Lett. 1998, 428, 141–146. [Google Scholar] [CrossRef]
- Rome, L.H.; Kickhoefer, V.A. Development of the vault particle as a platform technology. ACS Nano 2013, 7, 889–902. [Google Scholar] [CrossRef] [PubMed]
- Ladenstein, R.; Fischer, M.; Bacher, A. The lumazine synthase/riboflavin synthase complex: Shapes and functions of a highly variable enzyme system. FEBS J. 2013, 280, 2537–2563. [Google Scholar] [CrossRef] [PubMed]
- Azuma, Y.; Edwardson, T.G.W.; Hilvert, D. Tailoring lumazine synthase assemblies for bionanotechnology. Chem. Soc. Rev. 2018, 47, 3543–3557. [Google Scholar] [CrossRef]
- Wörsdörfer, B.; Woycechowsky, K.J.; Hilvert, D. Directed evolution of a protein container. Science 2011, 331, 589–592. [Google Scholar] [CrossRef]
- Azuma, Y.; Zschoche, R.; Tinzl, M.; Hilvert, D. Quantitative packaging of active enzymes into a protein cage. Angew. Chem. Int. Ed. 2016, 55, 1531–1534. [Google Scholar] [CrossRef] [PubMed]
- Frey, R.; Hayashi, T.; Hilvert, D. Enzyme-mediated polymerization inside engineered protein cages. Chem. Commun. 2016, 52, 10423–10426. [Google Scholar] [CrossRef] [PubMed]
- Frey, R.; Mantri, S.; Rocca, M.; Hilvert, D. Bottom-up construction of a primordial carboxysome mimic. J. Am. Chem. Soc. 2016, 138, 10072–10075. [Google Scholar] [CrossRef]
- Nott, T.J.; Petsalaki, E.; Farber, P.; Jervis, D.; Fussner, E.; Plochowietz, A.; Craggs, T.D.; Bazett-Jones, D.; Pawson, T.; Forman-Kay, J.; et al. Phase transition of a disordered nuage protein generates environmentally responsive membraneless organelles. Mol. Cell 2015, 57, 936–947. [Google Scholar] [CrossRef] [PubMed]
- Reinkemeier, C.D.; Girona, G.E.; Lemke, E.A. Designer membraneless organelles enable codon reassignment of selected mrnas in eukaryotes. Science 2019, 363, eaaw2644. [Google Scholar] [CrossRef] [PubMed]
- Kato, Y. Translational control using an expanded genetic code. Int. J. Mol. Sci. 2019, 20, 887. [Google Scholar] [CrossRef]
- Liu, M.; He, S.; Cheng, L.; Qu, J.; Xia, J. Phase-separated multienzyme biosynthesis. Biomacromolecules 2020, 21, 2391–2399. [Google Scholar] [CrossRef]
- Nott, T.J.; Craggs, T.D.; Baldwin, A.J. Membraneless organelles can melt nucleic acid duplexes and act as biomolecular filters. Nat. Chem. 2016, 8, 569. [Google Scholar] [CrossRef]
- Pinheiro, A.V.; Han, D.; Shih, W.M.; Yan, H. Challenges and opportunities for structural DNA nanotechnology. Nat. Nanotechnol. 2011, 6, 763–772. [Google Scholar] [CrossRef]
- Zhang, Y.; Tsitkov, S.; Hess, H. Proximity does not contribute to activity enhancement in the glucose oxidase–horseradish peroxidase cascade. Nat. Commun. 2016, 7, 13982. [Google Scholar] [CrossRef] [PubMed]
- Seeman, N.C.; Sleiman, H.F. DNA nanotechnology. Nat. Rev. Mater. 2017, 3, 17068. [Google Scholar] [CrossRef]
- Kosuri, S.; Church, G.M. Large-scale de novo DNA synthesis: Technologies and applications. Nat. Methods 2014, 11, 499–507. [Google Scholar] [CrossRef]
- Rothemund, P.W.K. Folding DNA to create nanoscale shapes and patterns. Nature 2006, 440, 297–302. [Google Scholar] [CrossRef] [PubMed]
- Linko, V.; Eerikäinen, M.; Kostiainen, M.A. A modular DNA origami-based enzyme cascade nanoreactor. Chem. Commun. 2015, 51, 5351–5354. [Google Scholar] [CrossRef]
- Shimada, J.; Maruyama, T.; Hosogi, T.; Tominaga, J.; Kamiya, N.; Goto, M. Conjugation of DNA with protein using his-tag chemistry and its application to the aptamer-based detection system. Biotechnol. Lett. 2008, 30, 2001–2006. [Google Scholar] [CrossRef]
- Rosen, C.B.; Kodal, A.L.B.; Nielsen, J.S.; Schaffert, D.H.; Scavenius, C.; Okholm, A.H.; Voigt, N.V.; Enghild, J.J.; Kjems, J.; Tørring, T.; et al. Template-directed covalent conjugation of DNA to native antibodies, transferrin and other metal-binding proteins. Nat. Chem. 2014, 6, 804–809. [Google Scholar] [CrossRef]
- Fu, J.; Yang, Y.R.; Johnson-Buck, A.; Liu, M.; Liu, Y.; Walter, N.G.; Woodbury, N.W.; Yan, H. Multi-enzyme complexes on DNA scaffolds capable of substrate channelling with an artificial swinging arm. Nat. Nanotechnol. 2014, 9, 531–536. [Google Scholar] [CrossRef]
- Perham, R.N. Swinging arms and swinging domains in multifunctional enzymes: Catalytic machines for multistep reactions. Annu. Rev. Biochem. 2000, 69, 961–1004. [Google Scholar] [CrossRef]
- Lim, Y.P.; Go, M.K.; Yew, W.S. Exploiting the biosynthetic potential of type iii polyketide synthases. Molecules 2016, 21, 806. [Google Scholar] [CrossRef]
- Fu, T.J.; Seeman, N.C. DNA double-crossover molecules. Biochemistry 1993, 32, 3211–3220. [Google Scholar] [CrossRef] [PubMed]
- Grossi, G.; Dalgaard Ebbesen Jepsen, M.; Kjems, J.; Andersen, E.S. Control of enzyme reactions by a reconfigurable DNA nanovault. Nat. Commun. 2017, 8, 992. [Google Scholar] [CrossRef]
- Kim, C.H.; Axup, J.Y.; Schultz, P.G. Protein conjugation with genetically encoded unnatural amino acids. Curr. Opin. Chem. Biol. 2013, 17, 412–419. [Google Scholar] [CrossRef]
- Siu, K.-H.; Chen, R.P.; Sun, Q.; Chen, L.; Tsai, S.-L.; Chen, W. Synthetic scaffolds for pathway enhancement. Curr. Opin. Biotechnol. 2015, 36, 98–106. [Google Scholar] [CrossRef]
- Kushner, S.R. Mrna decay in escherichia coli comes of age. J. Bacteriol. 2002, 184, 4658–4665. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ma, Q.; Lee, D.; Tan, Y.Q.; Wong, G.; Gao, Z. Synthetic genetic polymers: Advances and applications. Polym. Chem. 2016, 7, 5199–5216. [Google Scholar] [CrossRef]
- Sachdeva, G.; Garg, A.; Godding, D.; Way, J.C.; Silver, P.A. In vivo co-localization of enzymes on rna scaffolds increases metabolic production in a geometrically dependent manner. Nucleic Acids Res. 2014, 42, 9493–9503. [Google Scholar] [CrossRef]
- Schirmer, A.; Rude, M.A.; Li, X.; Popova, E.; del Cardayre, S.B. Microbial biosynthesis of alkanes. Science 2010, 329, 559–562. [Google Scholar] [CrossRef]
- Ellis, T.; Adie, T.; Baldwin, G.S. DNA assembly for synthetic biology: From parts to pathways and beyond. Integr. Biol. Quant. Biosci. Nano Macro 2011, 3, 109–118. [Google Scholar] [CrossRef]
- Shetty, R.P.; Endy, D.; Knight, T.F., Jr. Engineering biobrick vectors from biobrick parts. J. Biol. Eng. 2008, 2, 5. [Google Scholar] [CrossRef] [PubMed]
- Engler, C.; Gruetzner, R.; Kandzia, R.; Marillonnet, S. Golden gate shuffling: A one-pot DNA shuffling method based on type iis restriction enzymes. PLoS ONE 2009, 4, e5553. [Google Scholar] [CrossRef] [PubMed]
- Wade, Y.; Daniel, R.A.; Leak, D.J. Heterologous microcompartment assembly in bacillaceae: Establishing the components necessary for scaffold formation. ACS Synth. Biol. 2019, 8, 1642–1654. [Google Scholar] [CrossRef]
- Slininger Lee, M.F.; Jakobson, C.M.; Tullman-Ercek, D. Evidence for improved encapsulated pathway behavior in a bacterial microcompartment through shell protein engineering. ACS Synth. Biol. 2017, 6, 1880–1891. [Google Scholar] [CrossRef] [PubMed]
- Gibson, D.G.; Young, L.; Chuang, R.-Y.; Venter, J.C.; Hutchison, C.A.; Smith, H.O. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods 2009, 6, 343–345. [Google Scholar] [CrossRef] [PubMed]
- Roth, T.L.; Milenkovic, L.; Scott, M.P. A rapid and simple method for DNA engineering using cycled ligation assembly. PLoS ONE 2014, 9, e107329. [Google Scholar] [CrossRef] [PubMed]
- Aljabali, A.A.A.; Barclay, J.E.; Steinmetz, N.F.; Lomonossoff, G.P.; Evans, D.J. Controlled immobilisation of active enzymes on the cowpea mosaic virus capsid. Nanoscale 2012, 4, 5640–5645. [Google Scholar] [CrossRef]
- Deiters, A.; Cropp, T.A.; Mukherji, M.; Chin, J.W.; Anderson, J.C.; Schultz, P.G. Adding amino acids with novel reactivity to the genetic code of saccharomyces cerevisiae. J. Am. Chem. Soc. 2003, 125, 11782–11783. [Google Scholar] [CrossRef]
- Loakes, D.; Holliger, P. Polymerase engineering: Towards the encoded synthesis of unnatural biopolymers. Chem. Commun. 2009, 31, 4619–4631. [Google Scholar] [CrossRef]
- Brune, K.D.; Leneghan, D.B.; Brian, I.J.; Ishizuka, A.S.; Bachmann, M.F.; Draper, S.J.; Biswas, S.; Howarth, M. Plug-and-display: Decoration of virus-like particles via isopeptide bonds for modular immunization. Sci. Rep. 2016, 6, 19234. [Google Scholar] [CrossRef]
- Theile, C.S.; Witte, M.D.; Blom, A.E.M.; Kundrat, L.; Ploegh, H.L.; Guimaraes, C.P. Site-specific n-terminal labeling of proteins using sortase-mediated reactions. Nat. Protoc. 2013, 8, 1800–1807. [Google Scholar] [CrossRef]
- Nguyen, G.K.T.; Wang, S.; Qiu, Y.; Hemu, X.; Lian, Y.; Tam, J.P. Butelase 1 is an asx-specific ligase enabling peptide macrocyclization and synthesis. Nat. Chem. Biol. 2014, 10, 732–738. [Google Scholar] [CrossRef]
- Zalatan, J.G.; Herschlag, D. The far reaches of enzymology. Nat. Chem. Biol. 2009, 5, 516–520. [Google Scholar] [CrossRef]
- Silpe, J.E.; Balskus, E.P. Deciphering human microbiota–host chemical interactions. ACS Cent. Sci. 2020, 7, 20–29. [Google Scholar] [CrossRef] [PubMed]
- Nakane, T.; Kotecha, A.; Sente, A.; McMullan, G.; Masiulis, S.; Brown, P.M.G.E.; Grigoras, I.T.; Malinauskaite, L.; Malinauskas, T.; Miehling, J.; et al. Single-particle cryo-em at atomic resolution. Nature 2020, 587, 152–156. [Google Scholar] [CrossRef] [PubMed]
- Chapman, J.; Ismail, A.E.; Dinu, C.Z. Industrial applications of enzymes: Recent advances, techniques, and outlooks. Catalysts 2018, 8, 238. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein Compartment/Scaffold | Origin | Cognate Function | Diameter/Size (nm) | Composition |
|---|---|---|---|---|
| Cowpea chlorotic mottle virus shell | Cowpea chlorotic mottle virus | Packaging of viral nucleic acids | 28 | Homomeric |
| Tobacco mosaic virus shell | Tobacco mosaic virus shell | Packaging of viral nucleic acids | 18 (exterior) 4 (interior) | Homomeric |
| P22 bacteriophage head capsid | P22 bacteriophage | Packaging of viral nucleic acids | 60 | Homomeric |
| Bacterial microcompartment—carboxysome | Cyanobacteria, chemoautotrophic bacteria | Inorganic carbon fixation | ~80–200 | Heteromeric |
| Bacterial microcompartment—metabolosome | Various enteric and soil bacteria species | Catabolism of short chain carbon sources | ~100–300 | Heteromeric |
| Cellulosome | Various anaerobic bacteria species | Lignocellulose degradation | Highly variable | Heteromeric |
| Encapsulin | Various bacterial and archaeal species | Iron and redox metabolism | ~25–30 | Homomeric |
| Lumazine synthase | Various bacterial and fungal species | Riboflavin biosynthesis | ~15–35 | Homomeric |
| Ferritin | Bacteria, archaea, eukarya | Iron storage | ~10 | Homomeric |
| Small heat shock proteins | Bacteria, archaea, eukarya | Aids in protein refolding | ~10 | Homomeric |
| Vault ribonucleoprotein | Eukarya | Nuclear pore assembly, but exact function unclear | 35 × 65 | Homomeric |
| Hydrophobins | Filamentous fungi | Surface attachment, spore dispersion | - | Homomeric |
| Membraneless organelles | Eukarya | RNA processing, stress response | Highly variable | Homomeric/heteromeric |
| Method | Examples | Advantages | Limitations |
|---|---|---|---|
| Chemical crosslinking | N-hydroxysuccinimide (NHS), sulfhydryl, carbodiimide | Wide variety of reagents commercially available Covalent conjugation | Modifications are often non-regiospecific and may inactivate enzymes Incompatible with in vivo applications |
| Non-canonical amino acids (ncAA)/nucleotides (ncNT) | Amino acids and nucleotides containing azido, alkynyl, and other crosslinking moieties | Regiospecific and bioorthogonal Conjugation is often covalent | Inefficient and challenging incorporation of ncAA/ncNT into target molecules in vivo ncAA and ncNT are expensive or have to be synthesized in-house |
| Affinity tags | Encapsulation/targeting peptides, supercharged proteins (e.g., GFP(+36)), avidin-biotin, aptamers | Regiospecific Tags are often short sequences that do not severely impact protein folding/function | Dissociable linkage Tags sometimes do significantly affect protein property |
| Covalent protein ligation | SpyCatcher/SpyTag, SnoopCatcher/SnoopTag, peptide ligases | Covalent conjugation Regiospecific | Some protein ligation domains (e.g., SpyCatcher, SnoopCatcher) are relatively bulky and may affect protein folding/function |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tan, Y.Q.; Xue, B.; Yew, W.S. Genetically Encodable Scaffolds for Optimizing Enzyme Function. Molecules 2021, 26, 1389. https://doi.org/10.3390/molecules26051389
Tan YQ, Xue B, Yew WS. Genetically Encodable Scaffolds for Optimizing Enzyme Function. Molecules. 2021; 26(5):1389. https://doi.org/10.3390/molecules26051389
Chicago/Turabian StyleTan, Yong Quan, Bo Xue, and Wen Shan Yew. 2021. "Genetically Encodable Scaffolds for Optimizing Enzyme Function" Molecules 26, no. 5: 1389. https://doi.org/10.3390/molecules26051389
APA StyleTan, Y. Q., Xue, B., & Yew, W. S. (2021). Genetically Encodable Scaffolds for Optimizing Enzyme Function. Molecules, 26(5), 1389. https://doi.org/10.3390/molecules26051389