
Boulton-Katritzky Rearrangement of 5-Substituted Phenyl-3-[2-(morpholin-1-yl)ethyl]-1,2,4-oxadiazoles as a Synthetic Path to Spiropyrazoline Benzoates and Chloride with Antitubercular Properties

 ,
,  , ,
, ,
Abstract

1. Introduction
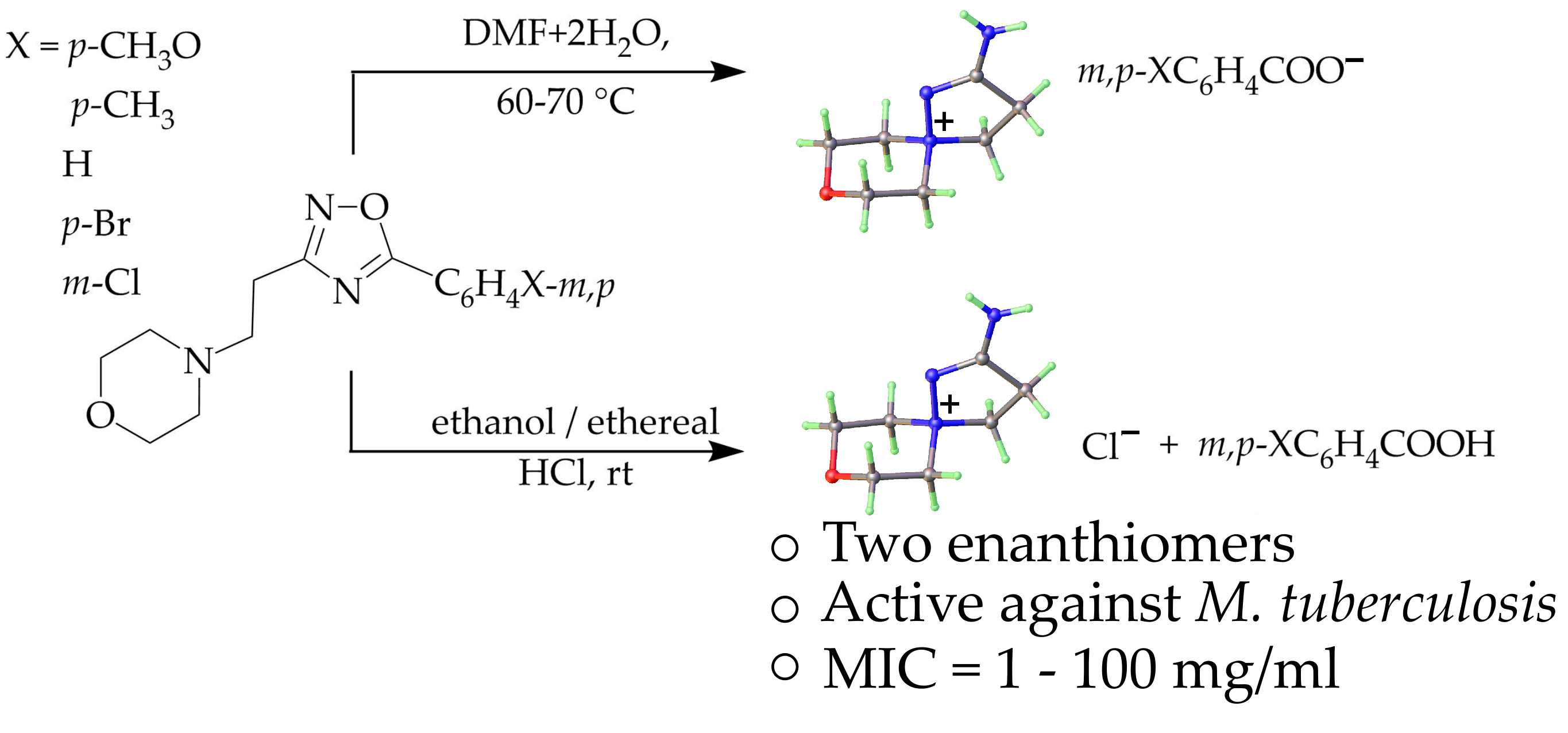
2. Results and Discussion
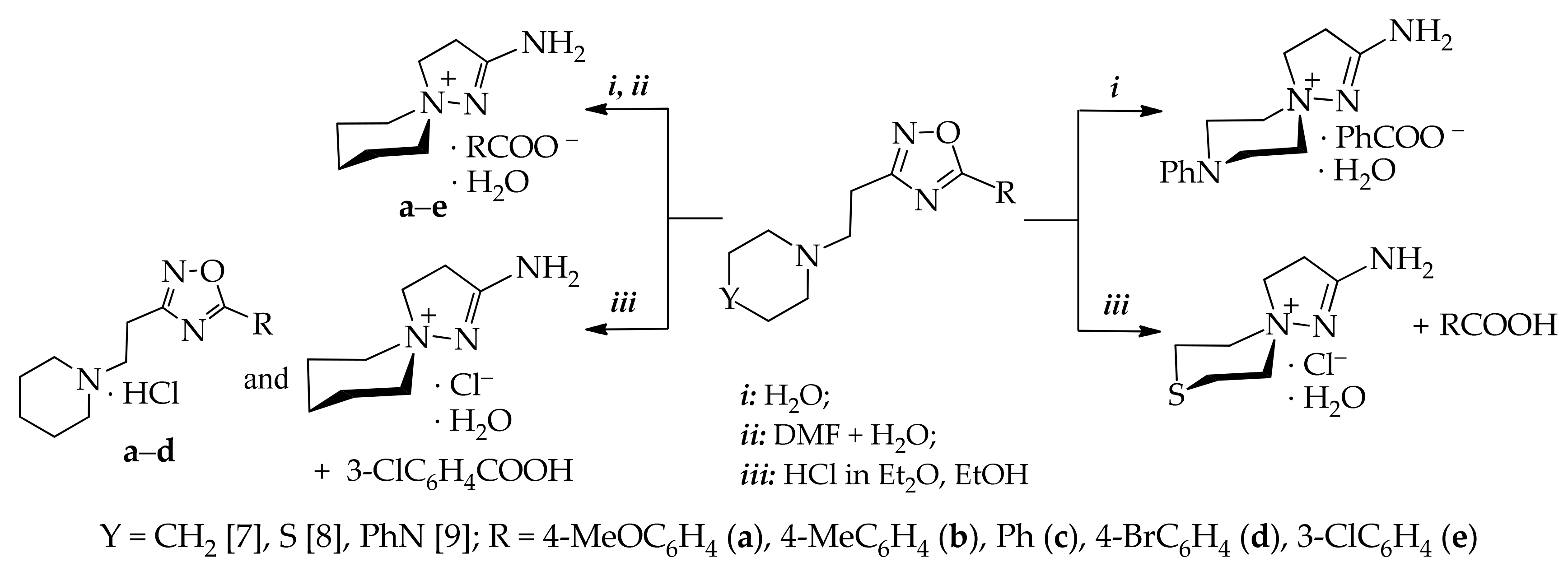
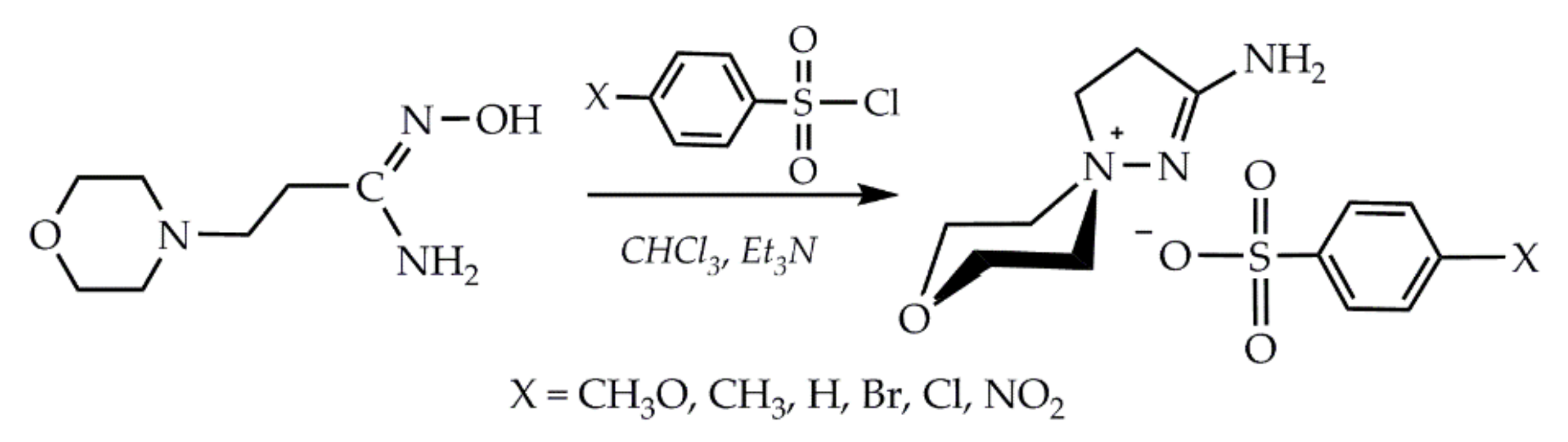
2.1. Synthesis and Spectra
2.2. In Vitro Antitubercular Screening of 2-amino-8-oxa-1,5-diazaspiro[4.5]dec-1-en-5-ium Benzoates and Chloride (5a–e, 6)
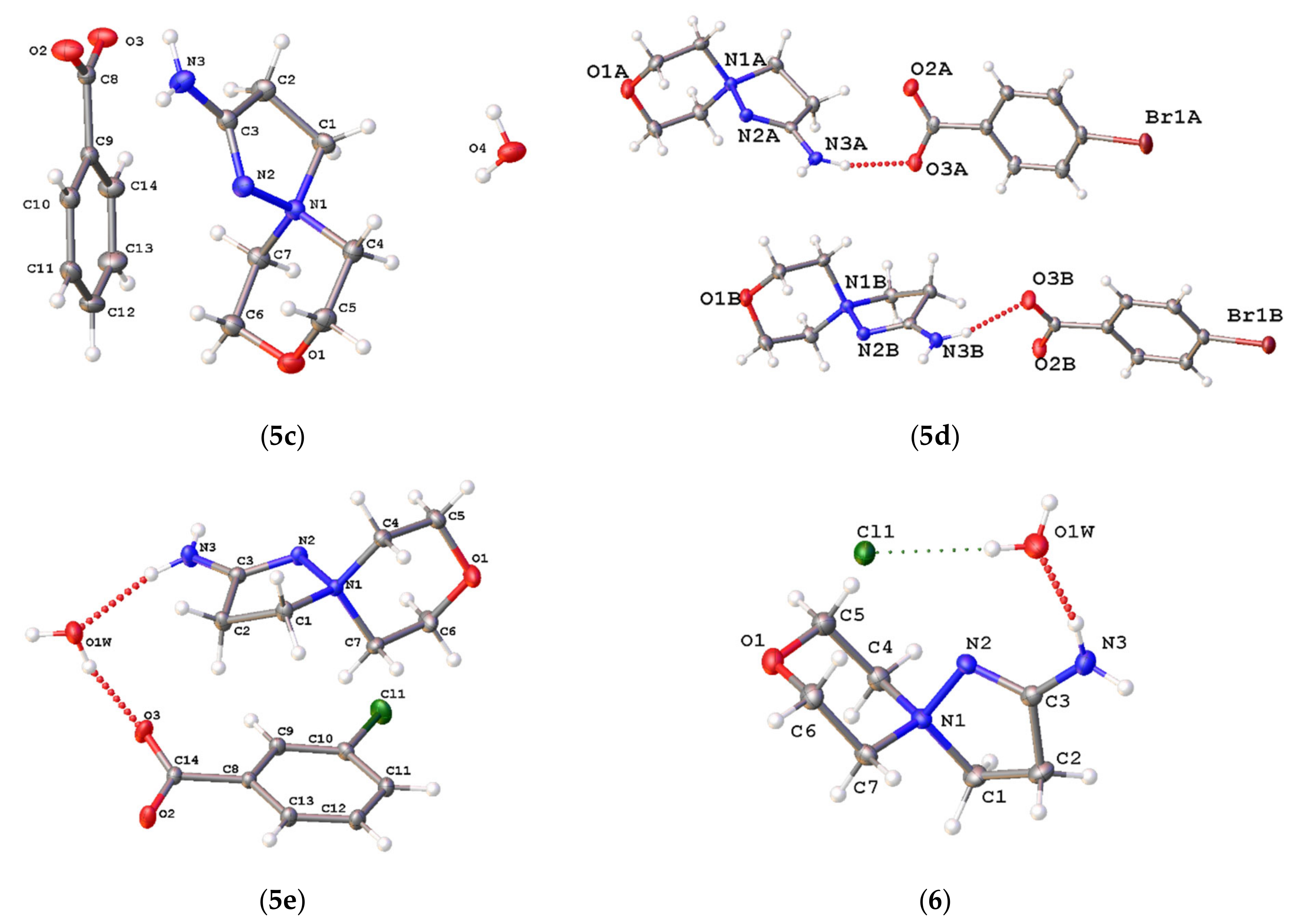
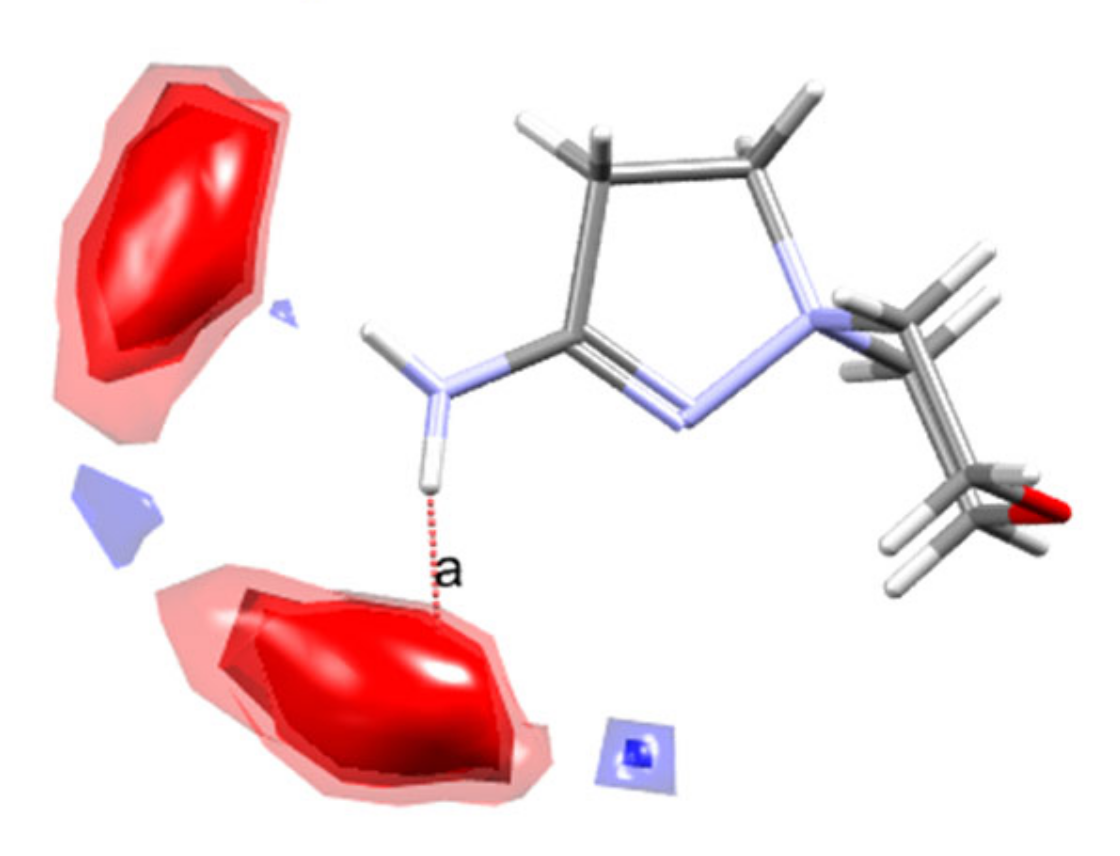
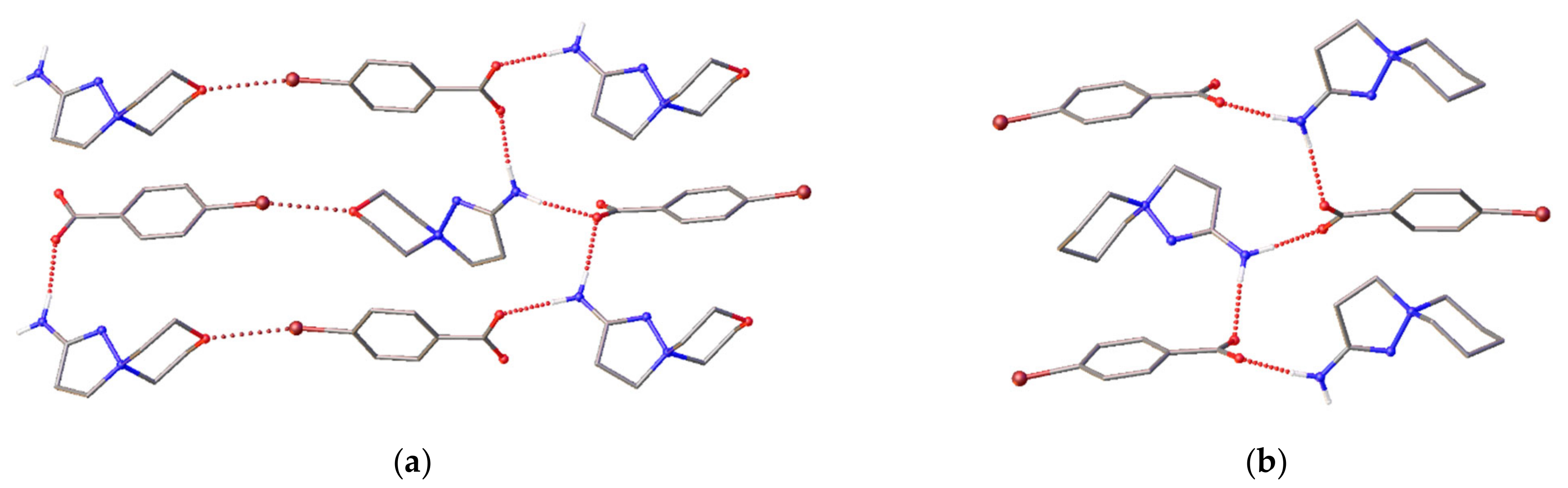
2.3. X-ray Diffraction
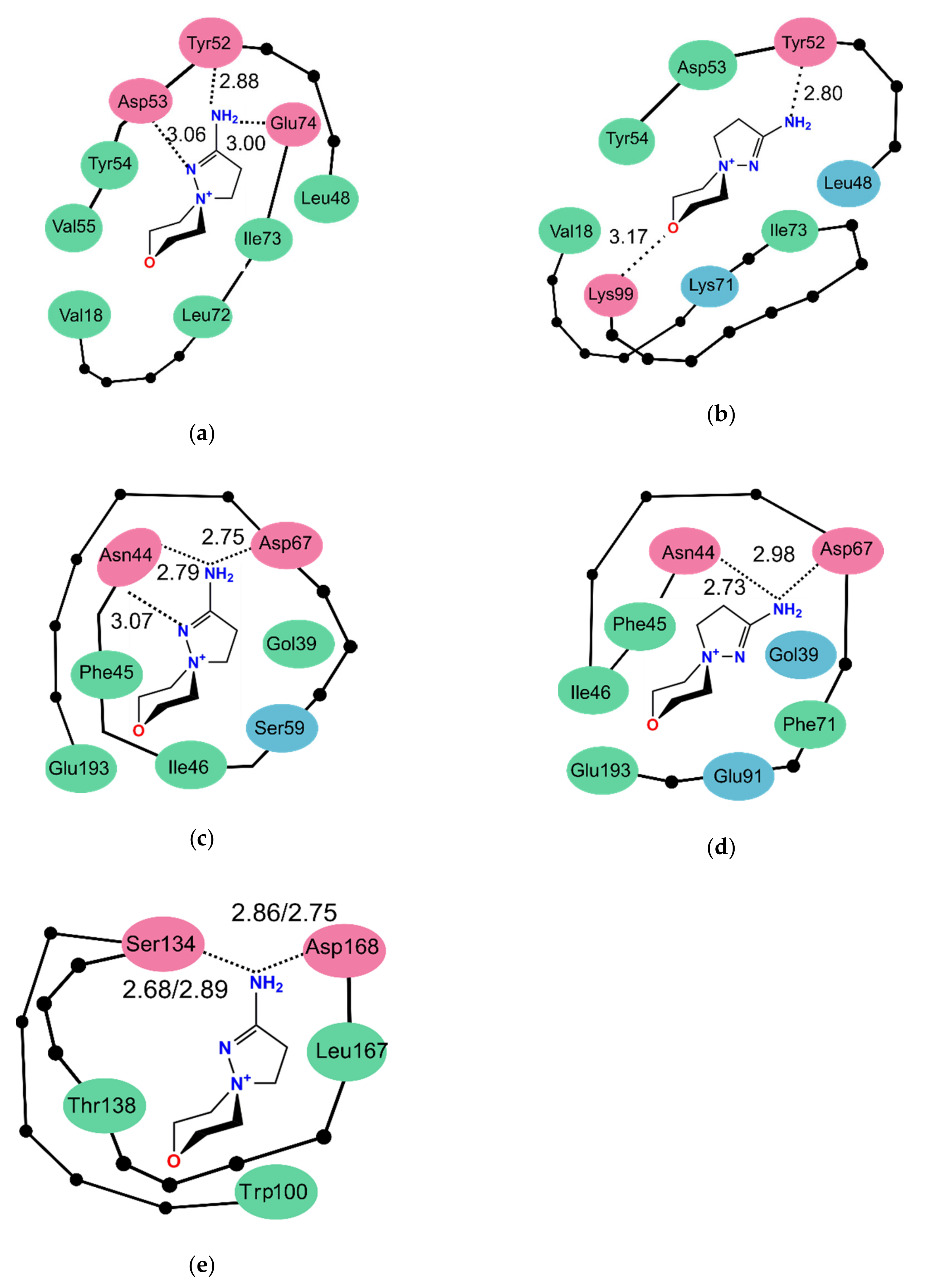
2.4. Molecular Docking Studies
3. Materials and Methods
3.1. Synthesis
3.1.1. A General Procedure for the Synthesis of 5-Aryl-3-[2-(morpholin-1-yl)ethyl]-1,2,4-oxadiazoles (4a–e)
3.1.2. A General Procedure of the Formation of 2-Amino-8-oxa-1,5-diazaspiro[4.5]dec-1-ene-5-ammonium benzoates (5a–e)
3.1.3. A General Method of the Formation of 2-Amino-8-oxa-1,5-diazaspiro[4.5]dec-1-ene-5-ammonium chloride (6) and Substituted Benzoic Acids
3.2. Single-Crystal X-ray Diffraction
3.3. Molecular Docking Studies
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- List of Drugs–Wikipedia. Available online: https://en.wikipedia.org/wiki/List_of_drugs (accessed on 12 December 2020).
- Fischer, E.; Knovenagel, O. Ueber die Verbindungen des Phenylhydrazinsmit Acroleïn, Mesityloxyd und Allylbromid. Ann. Chem. 1887, 239, 194–206. [Google Scholar] [CrossRef]
- Santos, C.M.M.; Silva, V.L.M.; Silva, A.M.S. Synthesis of Chromone-Related Pyrazole Compounds. Molecules 2017, 22, 1665. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Kaur, S.; Bansal, T.; Gaba, J. Review on Synthesis of Bioactive Pyrazoline Derivatives. Chem. Sci. Trans. 2014, 3, 861–875. [Google Scholar] [CrossRef]
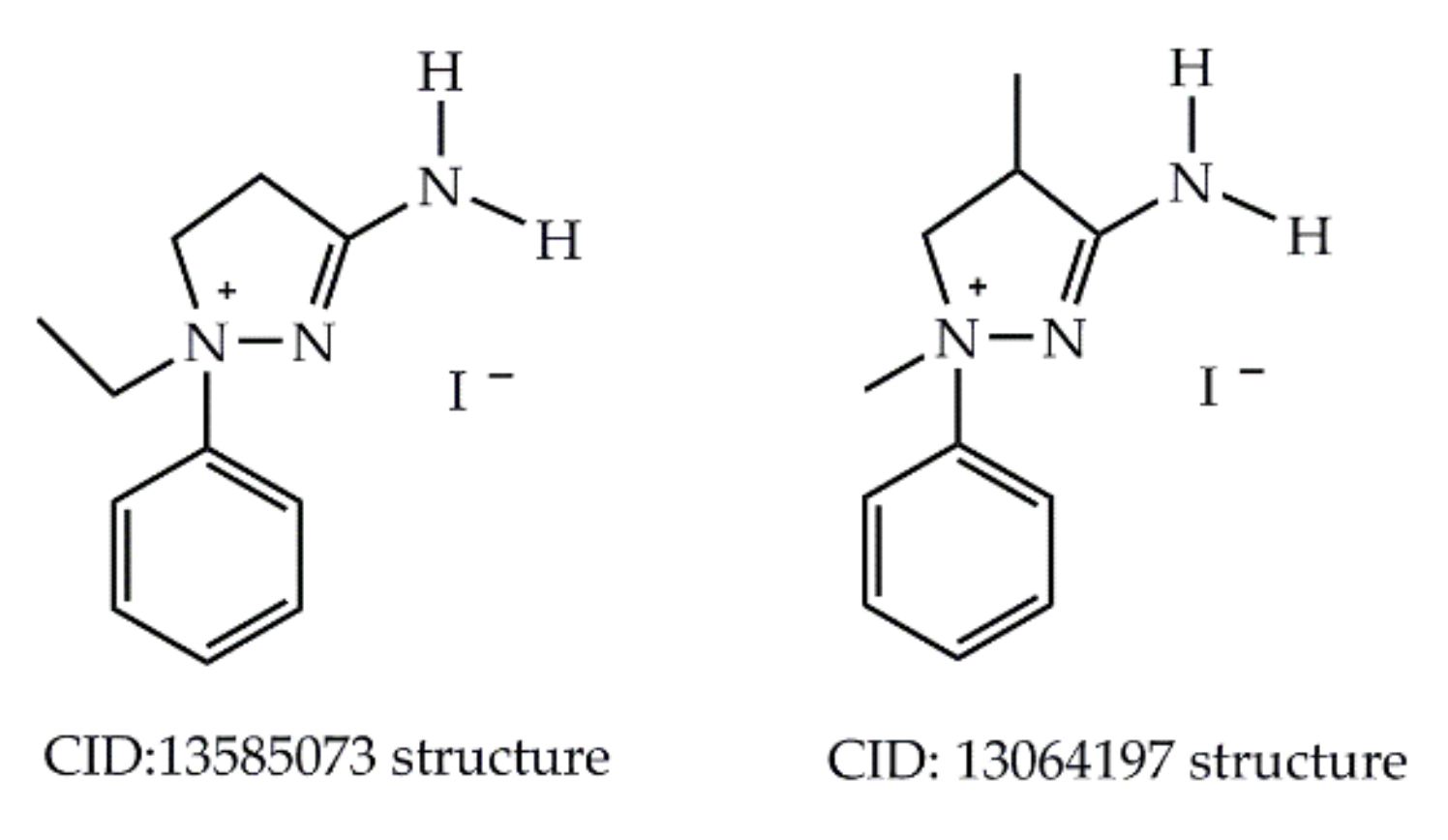
- 1-H Pyrazolium at National Library of Medicine. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/13585073 (accessed on 12 December 2020).
- 3-Amino-1,4-Dimethyl-1-Phenyl-2-Pyrazolinium Iodide at National Library of Medicine. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/13064197 (accessed on 12 December 2020).
- Kayukova, L.A.; Orazbaeva, M.A.; Gapparova, G.I.; Beketov, K.M.; Espenbetov, A.A.; Faskhutdinov, M.F.; Tashkhodjaev, B.T. Rapid acid hydrolysis of 5-aryl-3-(β-thiomorpholinoethyl)-1,2,4-oxadiazoles. Chem. Heterocycl. Compd. 2010, 46, 879–886. [Google Scholar] [CrossRef]
- Kayukova, L.A.; Beketov, K.M.; Baitursynova, G.P. 5-Aryl-3-(β-aminoethyl)-1,2,4-oxadiazoles in Boulton-Katritzky Rearrangement. In Proceedings of the International Conference Catalysis in Organic Synthesis ICCOS-2012 Book of Abstracts, Moscow, Russia, 15–20 September 2012; p. 214. Available online: http://www.spsl.nsc.ru/FullText/konfe/ICCOS2012.pdf (accessed on 26 January 2021).
- Kayukova, L.A.; Uzakova, A.B.; Vologzhanina, A.V.; Akatan, K.; Shaymardan, E.; Kabdrakhmanova, S.K. Rapid Boulton–Katritzky rearrangement of 5-aryl-3-[2-(piperidin-1-yl)ethyl]-1,2,4-oxadiazoles upon exposure to water and HCl. Chem. Heterocycl. Compd. 2018, 54, 643–649. [Google Scholar] [CrossRef]
- Li, J.J. Boulton–Katritzky rearrangement. In Name Reactions; Springer: Cham, Switzerland, 2014. [Google Scholar] [CrossRef]
- Korbonits, D.; Kanzel-Szvoboda, I.; Horváth, K. Ring transformation of 3-(2-aminoaryl)-1,2,4-oxadiazoles into 3-acylaminoindazoles; extension of the Boulton–Katritzky scheme. J. Chem. Soc. Perkin Trans. 1982, 1, 759–766. [Google Scholar] [CrossRef]
- Kayukova, L.A.; Praliyev, K.D.; Myrzabek, A.B.; Kainarbayeva, Z.N. Arylsulfochlorination of β-aminopropioamidoximes giving 2-aminospiropyrazolylammonium arylsulfonates. Rus. Chem. Bull. 2020, 69, 496–503. [Google Scholar] [CrossRef]
- Kayukova, L.A.; Praliev, K.D.; Akhelova, A.L.; Kemel’bekov, U.S.; Pichkhadze, G.M.; Mukhamedzhanova, G.S.; Kadyrova, D.M.; Nasyrova, S. Local anesthetic activity of new amidoxime derivatives. Pharm. Chem. J. 2011, 45, 468–471. [Google Scholar] [CrossRef]
- Kayukova, L.A.; Jussipbekov, U.; Praliyev, K. Amidoxime Derivatives with Local Anesthetic, Antitubercular, and Antidiabetic Activity. In Heterocycles-Synthesis and Biological Activities; Nandeshwarappa, B.P., Sadashiv, S.O., Eds.; IntechOpen: London, UK, 17 December 2019. [Google Scholar] [CrossRef]
- Kayukova, L.A.; Uzakova, A.B.; Baitursynova, G.P.; Dyusembaeva, G.T.; Shul’gau, Z.T.; Gulyaev, A.E.; Sergazy, S.D. Inhibition of α-Amylase and α-Glucosidase by New β-Aminopropionamidoxime Derivatives. Pharm. Chem. J. 2019, 53, 129–133. [Google Scholar] [CrossRef]
- The Use of a Derivative of Beta-Morpholinopropioamidoxime as an Antidiabetic Agent. Patent Owner: JSC «A.B. Institute of Chemical Sciences». Russia Patent No. 2684779, 15 April 2019.
- Ashenhurst, J. Homotopic, Enantiotopic, and Diastereotopic Groups: What Does It Mean? Last updated: October 16th, 2020. Available online: https://www.masterorganicchemistry.com/homotopic-enantiotopic-diastereotopic/ (accessed on 26 January 2021).
- Kayukova, L.A. Conditions for the Heterocyclization of O-Aroyl-β-morpholinopropioamide Oximes to 5-Aryl-3-(β-morpholino)ethyl-1,2,4-oxadiazoles. Chem. Heterocycl. Compd. 2003, 39, 223–227. [Google Scholar] [CrossRef]
- Usachev, S.V.; Nikiforov, G.A.; Lyssenko, K.A.; Nelyubina, Y.V.; Levkin, P.A.; Kostyanovsky, R.G. Sterically hindered and completely arrested nitrogen inversion in pyrazolidines. Tetrahedron Asymmetry 2007, 18, 1540–1547. [Google Scholar] [CrossRef]
- Yap, G.P.A.; Alkorta, I.; Elguero, J.; Jagerovic, N. The structure of 1,1,3-trimethyl-Δ2-pyrazolinium perchlorate: An X-ray crystallographic and GIAO/DFT multinuclear NMR study. Spectrosc. Int. J. 2004, 18, 605–611. [Google Scholar] [CrossRef]
- Wood, P.A.; Olsson, T.S.G.; Cole, J.C.; Cottrell, S.J.; Feeder, N.; Galek, P.T.A.; Groom, C.R.; Pidcock, E. Evaluation of molecular crystal structures using Full Interaction Maps. Cryst. Eng. Comm. 2013, 15, 65–72. [Google Scholar] [CrossRef]
- Vologzhanina, A.V. Intermolecular Interactions in Functional Crystalline Materials: From Data to Knowledge. Crystals 2019, 9, 478. [Google Scholar] [CrossRef]
- Macrae, C.F.; Sovago, I.; Cottrell, S.J.; Galek, P.T.A.; McCabe, P.; Pidcock, E.; Platings, M.; Shields, G.P.; Stevens, J.S.; Towler, M.; et al. Mercury 4.0: From visualization to analysis, design and prediction. J. Appl. Cryst. 2020, 53, 226–235. [Google Scholar] [CrossRef]
- French, F.A.; Freedlander, B.L. Derivatives of benzoic acid and simple phenols in the chemotherapy of tuberculosis. J. Am. Pharm. Assoc. 1949, 38, 343–346. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, H.; Sun, Z. Susceptibility of Mycobacterium tuberculosis to weak acids. J. Antimicrob. Chemother. 2003, 52, 56–60. [Google Scholar] [CrossRef]
- Maresca, A.; Vullo, D.; Scozzafava, A.; Manole, G.; Supuran, C.T. Inhibition of the β-class carbonic anhydrases from Mycobacterium tuberculosis with carboxylic acids. J. Enz. Inhib. Med. Chem. 2013, 28, 392–396. [Google Scholar] [CrossRef]
- Taylor, F.F.; Faloon, W.W. The role of potassium in the natriuretic response to a steroidal lactone (SC-9420). J. Clin. Endocrinol. Metab. 1959, 19, 1683–1687. [Google Scholar] [CrossRef]
- Brunner, H.R. The New Angiotensin II Receptor Antagonist, Irbesartan: Pharmacokinetic and Pharmacodynamic Considerations. Am. J. Hypertens. 1997, 10, 311S–317S. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Hanson, A.D.; Gregory, J.F., III. Folate Biosynthesis, Turnover, and Transport in Plants. Annu. Rev. Plant Biol. 2011, 62, 105–125. [Google Scholar] [CrossRef] [PubMed]
- Gavezzotti, A. Are crystal structures predictable? Acc. Chem. Res. 1994, 27, 309–314. [Google Scholar] [CrossRef]
- Gavezzotti, A.; Filippini, G. Geometry of the Intermolecular X-H…Y (X, Y = N, O) Hydrogen Bond and the Calibration of Empirical Hydrogen-Bond Potentials. J. Phys. Chem. 1994, 98, 4831–4837. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXT–Integrated space-group and crystal-structure determination. ActaCryst. 2015, A71, 3–8. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Cryst. 2015, C71, 3–8. [Google Scholar] [CrossRef]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Cryst. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Verdonk, M.L.; Cole, J.C.; Hartshorn, M.J.; Murray, C.W.; Taylor, R.D. Improved protein-ligand docking using GOLD. Proteins 2003, 52, 609–623. [Google Scholar] [CrossRef]
- Liebeschuetz, J.W.; Cole, J.C.; Korb, O. Pose prediction and virtual screening performance of GOLD scoring functions in a standardized test. J. Comput. Mol. Des. 2012, 26, 737–748. [Google Scholar] [CrossRef]
- GOLD User Guide. Available online: https://www.ccdc.cam.ac.uk/support-and-resources/ccdcresources/GOLD_User_Guide_2020_1.pdf (accessed on 12 December 2020).












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compd | X | Yield,% | m.p., °С | Rf * | Compd | t, h ** | Yield,% | m.p., °С | Rf * |
|---|---|---|---|---|---|---|---|---|---|
| 4a | p-CH3O | 62 | 230 | 0.71 | 5a | 40 | 63 | 248 | 0.80 |
| 4b | p-CH3 | 70 | 220 | 0.62 | 5b | 40 | 63 | 235 | 0.75 |
| 4c | H | 92 | 216 | 0.66 | 5c | 25 | 77 | 220 | 0.75 |
| 4d | p-Br | 83 | 224 | 0.67 | 5d | 25 | 80 | 240 | 0.77 |
| 4e | m-Cl | 56 | 190 | 0.62 | 5e | 25 | 43 | 200 | 0.70 |
| Compd | 5a | 5b | 5c | 5d | 5e | 6 | Rifampicin | |
|---|---|---|---|---|---|---|---|---|
| MIC, μg/mL | H37Rv | 50 | 10 | 20 | 100 | 100 | 1 | 1 |
| I | 50 | 50 | 50 | 100 | 100 | 2 | 2 | |
| Receptor | 1NBU | 4XT4 | 5ICJ | |||
|---|---|---|---|---|---|---|
| Ligand | Type A | Type B | Type A | Type B | Type A | Type B |
| H-bonds | −3.00 | −2.00 | −3.12 | −2.00 | −2.79 | −3.00 |
| Electrostatic | −4.37 | −1.68 | −6.34 | −5.80 | −0.51 | −2.12 |
| Nonpolar | −35.72 | −32.51 | −31.93 | −34.80 | −38.43 | −35.43 |
| Repulsive | 0.25 | 0.35 | 0.99 | 0.36 | 0.04 | 0.01 |
| Total | −42.84 | −35.84 | −40.40 | −42.24 | −41.69 | −40.54 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kayukova, L.; Vologzhanina, A.; Praliyev, K.; Dyusembaeva, G.; Baitursynova, G.; Uzakova, A.; Bismilda, V.; Chingissova, L.; Akatan, K. Boulton-Katritzky Rearrangement of 5-Substituted Phenyl-3-[2-(morpholin-1-yl)ethyl]-1,2,4-oxadiazoles as a Synthetic Path to Spiropyrazoline Benzoates and Chloride with Antitubercular Properties. Molecules 2021, 26, 967. https://doi.org/10.3390/molecules26040967
Kayukova L, Vologzhanina A, Praliyev K, Dyusembaeva G, Baitursynova G, Uzakova A, Bismilda V, Chingissova L, Akatan K. Boulton-Katritzky Rearrangement of 5-Substituted Phenyl-3-[2-(morpholin-1-yl)ethyl]-1,2,4-oxadiazoles as a Synthetic Path to Spiropyrazoline Benzoates and Chloride with Antitubercular Properties. Molecules. 2021; 26(4):967. https://doi.org/10.3390/molecules26040967
Chicago/Turabian StyleKayukova, Lyudmila, Anna Vologzhanina, Kaldybai Praliyev, Gulnur Dyusembaeva, Gulnur Baitursynova, Asem Uzakova, Venera Bismilda, Lyailya Chingissova, and Kydyrmolla Akatan. 2021. "Boulton-Katritzky Rearrangement of 5-Substituted Phenyl-3-[2-(morpholin-1-yl)ethyl]-1,2,4-oxadiazoles as a Synthetic Path to Spiropyrazoline Benzoates and Chloride with Antitubercular Properties" Molecules 26, no. 4: 967. https://doi.org/10.3390/molecules26040967
APA StyleKayukova, L., Vologzhanina, A., Praliyev, K., Dyusembaeva, G., Baitursynova, G., Uzakova, A., Bismilda, V., Chingissova, L., & Akatan, K. (2021). Boulton-Katritzky Rearrangement of 5-Substituted Phenyl-3-[2-(morpholin-1-yl)ethyl]-1,2,4-oxadiazoles as a Synthetic Path to Spiropyrazoline Benzoates and Chloride with Antitubercular Properties. Molecules, 26(4), 967. https://doi.org/10.3390/molecules26040967