Abstract
Carboxylation of bis(pyrazol-1-yl)alkanes by oxalyl chloride was studied. It was found that 4,4′-dicarboxylic derivatives of substrates with electron-donating methyl groups and short linkers (from one to three methylene groups) can be prepared using this method. Longer linkers lead to significantly lower product yields, which is probably due to instability of the intermediate acid chlorides that are initially formed in the reaction with oxalyl chloride. Thus, bis(pyrazol-1-yl)methane gave only monocarboxylic derivative even with a large excess of oxalyl chloride and prolonged reaction duration. An alternative approach involves the reaction of ethyl 4-pyrazolecarboxylates with dibromoalkanes in a superbasic medium (potassium hydroxide–dimethyl sulfoxide) and is suitable for the preparation of bis(4-carboxypyrazol-1-yl)alkanes with both short and long linkers independent of substitution in positions 3 and 5 of pyrazole rings. The obtained dicarboxylic acids are interesting as potential building blocks for metal-organic frameworks.
1. Introduction
Bis(pyrazol-1-yl)alkanes are bidentate ligands widely used for the synthesis of the coordination compounds [1], some of which were shown to exhibit catalytic [2,3,4,5,6], anticancer [7,8], antibacterial [9] and SOD-like activity [10,11], and electroluminescent properties [12,13]. The properties of the ligands can be varied by introducing the functional groups into the pyrazole rings [14,15,16,17], besides, carboxylic groups themselves can act as donor groups for the formation of coordination bond with the metal ions. Carboxy-substituted pyrazoles were used for the construction of highly porous metal-organic frameworks [18,19,20,21]. Carboxylic acids based on bis(pyrazol-1-yl)alkanes are much less explored and only a few research papers were published so far [22,23,24,25]. In addition, a series of works devoted to the synthesis of metal-organic frameworks based on structurally related bis(4-carboxyphenylpyrazol-1-yl)methane was carried out by Sumby et al., the presence of the free chelating units in the structure of the framework allowed them to prepare catalysts with single metal sites [26,27].
In this contribution we report a facile synthesis of a series of 4,4′-dicarboxy-substituted bis(pyrazol-1-yl)alkanes with varied linker length and substitution in positions 3 and 5 of the pyrazole rings.
2. Results and Discussion
To introduce carboxyl groups into bis(pyrazol-1-yl)alkanes, a reaction with oxalyl chloride was used, in which it was both a reagent and a solvent. Previously oxalyl chloride was successfully used for the carboxylation of 1-phenyl- and 1-alkylpyrazoles [28]. In this reaction, a pyrazole-containing derivative of oxalic acid chloride is initially formed, which is converted to a carboxylic acid chloride with the release of carbon monoxide. The acid chloride is hydrolyzed without isolation to form a carboxylic acid. Other methods for the introduction of carboxylic groups into pyrazole rings include oxidation of alkyl [29,30] or formyl [31] groups, substitution of halogens via intermediate organolithium derivatives [32], and hydrolysis of trichloromethyl derivatives [33].
Carboxylation of bis(3,5-dimethylpyrazol-1-yl)methane 1a with subsequent hydrolysis gave the corresponding dicarboxylic acid 1b in high yield (Scheme 1). Similarly, 1,2-bis(3,5-dimethylpyrazol-1-yl)ethane 2a and 1,3-bis(3,5-dimethylpyrazol-1-yl)propane 3a gave dicarboxylic acids 2b and 3b, albeit in lower yield (Scheme 1). When bis(3,5-dimethylpyrazol-1-yl)alkanes with longer spacers (tetramethylene, (CH2)4 and longer) were introduced into the reaction, full conversion of the starting materials was achieved (GC–MS control), but no dicarboxylic acids were isolated. Instead, mixtures of monosubstituted carboxy-derivatives (not isolated) and starting substrates were obtained, which is probably a result of instability of the intermediate acid chlorides leading to their decarboxylation during the hydrolysis.
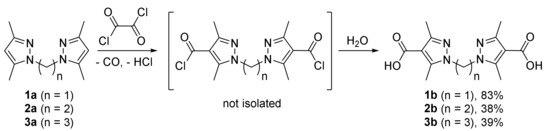
Scheme 1.
Carboxylation of bis(3,5-dimethylpyrazol-1-yl)alkanes with short linkers.
To overcome the instability of acid chlorides their methanolysis instead of hydrolysis was attempted and indeed led to the dimethyl esters 4c–6c, although in low yields (Scheme 2). Alkaline hydrolysis of the esters proceeded smoothly with the formation of dicarboxylic acids 4b–6b (Scheme 2).
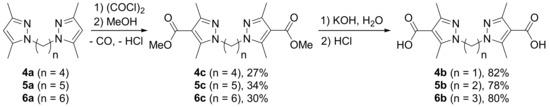
Scheme 2.
Carboxylation of bis(3,5-dimethylpyrazol-1-yl)alkanes with long linkers through the methanolysis step.
Carboxylation by oxalyl chloride proved to be poorly suitable for bis(pyrazol-1-yl)alkanes without electron-donating methyl groups. Thus, only in the case of 1,3-bis(pyrazol-1-yl)propane 3d a dicarboxylation product could be isolated, while bis(pyrazol-1-yl)methane 1d gave solely a monocarboxylated product 7 (Scheme 3). Even a large excess of oxalyl chloride and prolonged heating did not lead to the formation of the dicarboxylic acid. For derivatives with longer spacers (four to six methylene groups) only traces of carboxylation products (after conversion to methyl esters) were detected (for substrate 4c) but were not isolated (Scheme 3).
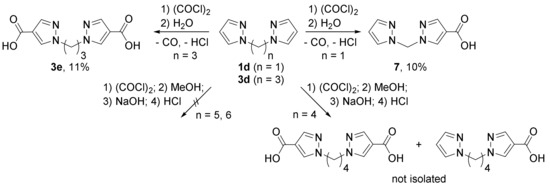
Scheme 3.
Carboxylation of bis(pyrazol-1-yl)alkanes.
In order to gain insight into the reasons for low reactivity of bis(pyrazol-1-yl)methane 1d, DFT calculations of charge distribution in the starting compound 1d and compounds 1a, 3a, and 3d for comparison and the corresponding monosubstituted acid chlorides formed during the first electrophilic substitution step were carried out. The structures were optimized at the B3LYP 6-31G(d) level of theory and absence of imaginary frequencies confirmed that the found stationary points correspond to minima on the potential energy surface. In order to get a more precise electron density distribution, MP2 6-31G(d) single point calculations were carried out for the found structures. Next, charges on carbon atoms (qC) in position 4 of the pyrazole ring (since an electrophilic attack is directed at this position) and the hydrogen atom bound to it (qH,) were calculated using the Bader’s theory of atoms in molecules [34]. The calculation results are presented in Table 1.

Table 1.
Selected atomic charges in some bis(pyrazol-1-yl)alkanes and their monoacid chloride derivatives.
The following conclusions can be drawn from the obtained charge distribution:
- (1)
- The introduction of methyl groups into pyrazole rings (in pairs of compounds 1a–1d and 3a–3d) noticeably increases the negative charge at position 4 of the heterocycle, i.e., makes it more active in the electrophilic substitution reaction, and the effect of electron-donor groups is best manifested when comparing the sum of charges on carbon and hydrogen atoms;
- (2)
- An increase in the length of the linker from one to three methylene groups also increases the excess negative charge at position 4, which is apparently associated with the negative inductive effect of the pyrazole ring;
- (3)
- The introduction of an electron-withdrawing acid chloride group into one of the pyrazole rings deactivates the other cycle in the electrophilic substitution reaction, and to a greater extent, deactivation manifests itself in pyrazole derivatives without methyl substituents, and with a short methylene linker.
Based on the charge distribution, bis(pyrazol-1-yl)methane 1d is the least active in electrophilic substitution reactions, the charge on the 4-CH group in which is close to the charge on this group in the acid chloride of the propane derivative 3d′. Experimental data show that monochloro anhydride 3d′ can undergo carboxylation with the formation of dicarboxylic acid 3e. At the same time, bis(pyrazol-1-yl)methane 1d at the first step of electrophilic substitution gives acid chloride 1d′, in which position 4 of the other pyrazole ring is so deactivated by the electron-withdrawing effect of the already substituted heterocycle that the reaction halts on monochloroanhydride 1d′, the hydrolysis of which gives monocarboxylic acid 7. Therefore, the pyrazole ring with an electron-withdrawing functional group (i.e., chlorocarbonyl) is a substituent with a strong negative inductive effect, which affects the reactivity of the neighboring pyrazole ring located even across two aliphatic bonds.
Taking into account the limitations of direct carboxylation of bis(pyrazol-1-yl)alkanes, an alternative approach involving the double alkylation of 4-pyrazolecarboxylic acid and its 3,5-dimethylderivative by α,ω-dibromoalkanes was evaluated. Ethyl esters of 4-pyrazolecarboxylic acid and 3,5-dimethyl-4-pyrazole carboxylates were smoothly alkylated by α,ω-dibromoalkanes in a superbasic KOH-DMSO system and gave the corresponding diethyl dicarboxylates in high yields (Scheme 4). Alkaline hydrolysis of the diesters and subsequent neutralization gave the target dicarboxylic acids, including the ones with longer spacers, unavailable by direct carboxylation (Scheme 4). Unfortunately, derivatives of 1,2-bis(pyrazol-1-yl)ethane cannot be prepared by this route, since, as it is known, the reaction of pyrazole and its derivatives with 1,2-dibromoethane leads to the formation of 1-vinylpyrazoles, which undergo hydrolysis to the starting materials upon isolation [35].
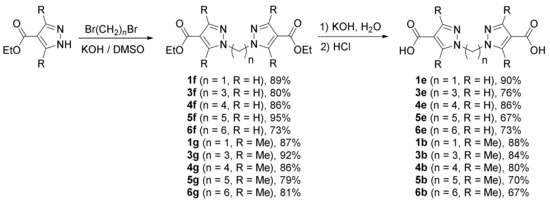
Scheme 4.
Synthesis of bis(pyrazol-1-yl)alkane-4,4′-dicarboxylic acids from ethyl 4-pyrazolecarboxylates.
Since both alkylation and ester hydrolysis take place under basic catalysis, we explored the possibility to carry out the synthesis of diacid 1e in a one-pot process. Indeed, after the alkylation was complete (TLC control), two additional equivalents of KOH and excess of water were added to the reaction mixture, which gave the hydrolysis product in several minutes in 92% overall yield.
3. Materials and Methods
Gas chromatography-mass spectrometry analysis was performed using Agilent 7890A gas chromatograph (Santa Clara, CA, USA) equipped with Agilent MSD 5975C mass-selective detector with quadrupole mass-analyzer (electron impact ionization energy 70 eV). NMR spectra were recorded on Bruker DRX400 and Bruker Advance 500 instruments (Billerica, MA, USA), solvent residual peaks were used as internal standards. Elemental analyses were carried out on a Vario Micro-Cube analyzer (Elementar Analysensysteme GmbH, Langenselbold, Germany). IR-spectra of solid samples were recorded on Agilent Cary 630 FT IR (Santa Clara, CA, USA) spectrophotometer equipped with diamond ATR accessory. Melting points of compounds 1b, 3b, and 1d (with melting points higher than 300 °C) were determined in helium atmosphere on NETZSCH TG 209 F1 thermoanalyzer (NETZSCH TAURUS Instruments GmbH, Weimar, Germany) with the heating rate of 10°/min.
The calculations were performed using the Gaussian 09 package, revision D.01 [36]. Atomic charges were calculated using the AIMAll Professional 10.05.04 package.
Ethyl 4-pyrazolecarboxylate [37], ethyl 3,5-dimethyl-4-pyrazolecarboxylate [25], bis(pyrazol-1-yl)alkanes 1a and 1d [38], 2a [35], 3d and 3a [39], 4a–6a, and 4d–6d [40] were prepared following the literature procedures. Dimethyl sulfoxide was distilled from KOH under vacuum (10 Torr) prior to use. Other commercially reagents and solvents were of reagent grade and used as received. FT-IR and NMR plots of the synthesized compounds can be found in the Supplementary Material.
Bis(3,5-dimethylpyrazol-1-yl)methane-4,4′-dicarboxylic acid (1b). Method A (carboxylation by oxalyl chloride). Oxalyl chloride (1.91 g, 1.29 mL, 15 mmol) was added dropwise to 0.51 g (2.5 mmol) of compound 1a. The mixture was refluxed during 3 h, after which the excess of oxalyl chloride was removed in vacuum of water aspiration pump. The residue was treated by 10 mL of water, the precipitate was filtered and washed with water. Yield 0.606 g, 83%, colorless power, m.p. 322–324 °C (EtOH, dec.). C13H16N4O4 (292.12): calcd. C 53.42; H 5.52; N 19.17; found C 53.4; H 5.7; N 19.0. 1H-NMR (400 MHz, D2O): δ 2.21 (s, 6H, 3-CH3), 2.51 (s, 6H, 5-CH3), 6.16 (s, 2H, CH2), ppm. 13C-NMR (100 MHz, D2O): δ 10.1, 12.2, 58.6, 113.9, 148.0, 152.6, 189.5, ppm. FT-IR (cm−1): ν = 2598 (w), 2532 (w), 1668 (m), 1554 (m), 1472 (w), 1435 (m), 1370 (w), 1316 (m), 1251 (m), 1164 (m), 1119 (m), 1040 (w), 1005 (w), 928 (w), 868 (w), 790 (m), 770 (m), 732 (w), 691 (m). Compounds 2b and 3b can be prepared similarly.
Method B (hydrolysis of diethyl diesters). Compound 1f (0.696 g, 2 mmol) was suspended in 10 mL of the aqueous solution of KOH (0.448 g, 8 mmol) and heated at 80 °C until complete dissolution of a starting diester. After cooling, the obtained solution of potassium salts was acidified by concentrated HCl solution, the resulting precipitate was filtered, washed with water and dried. Yield 0.514 g, 88%.
1,2-Bis(3,5-dimethylpyrazol-1-yl)ethane-4,4′-dicarboxylic acid (2b) was prepared similarly to compound 1b by Method A. Yield 38%, colorless crystals, m.p. 224–225 °C (H2O). C14H18N4O4 (306.32): calcd. C 54.89; H 5.92; N 18.29; found C 54.7; H 6.0; N 18.1. 1H-NMR (400 MHz, (CD3)2SO): δ 2.09 (s, 6 H, 3-CH3), 2.25 (s, 6 H, 5-CH3), 4.43 (s, 4 H, CH2), ppm. 13C-NMR (100 MHz, (CD3)2SO): δ 9.6 (5-CH3), 13.1 (3-CH3), 47.4 (CH2), 113.1 (C4(Pz)), 145.8 (C5(Pz)), 149.9 (C3(Pz)), 184.0 (COOH), ppm. FT-IR (cm−1): 3500 (br., COOH), 1664 (C=O), 1535 (Pz), 1007 (Pz breathing).
1,3-Bis(3,5-dimethylpyrazol-1-yl)propane-4,4′-dicarboxylic acid (3b) was prepared similarly to compound 1b by Methods A and B. Yield 39% (Method A) and 84% (Method B), colorless crystals, m.p. 290–292 °C (EtOH). C15H20N4O4 (320.35): calcd. C 56.24; H 6.29; N 17.49; found C 56.4, H 6.1, N 17.1. 1H-NMR (400 MHz, (CD3)2SO): δ 2.07 (p, 2 H, β-CH2), 2.24 (s, 6 H, 3-CH3), 2.42 (s, 6 H, 5-CH3), 4.07 (t, 4 H, CH2), ppm 13C-NMR (100 MHz, (CD3)2SO): δ 10.4 (5-CH3), 13.2 (3-CH3), 28.5 (PzCH2CH2), 45.3 (PzCH2CH2), 112.9 (C4(Pz)), 144.9 (C5(Pz)), 149.3 (C3(Pz)), 185.0 (COOH) ppm. FT-IR (cm−1): 3430 (br., COOH), 1651 (C=O), 1536 (Pz), 1005 (Pz breathing).
1,4-Bis(3,5-dimethylpyrazol-1-yl)butane-4,4′-dicarboxylic acid (4b) was prepared similarly to compound 1b by Method B from diesters 4c or 4g. Yield 80%, colorless crystals, m.p. 273–275 °C (reprecipitation; dec.). C16H22N4O4 (334.38): calcd. C 57.47; H 6.63; N 16.76; found C 57.1; H 6.5; N 16.9. 1H-NMR (400 MHz, (CD3)2SO): δ 1.66 (p, 4H, β-CH2), 2.25 (s, 6H, 3-CH3), 2.43 (s, 6H, 5-CH3), 3.98 (t, 4H, α-CH2), 12.12 (s, 2 H, COOH), ppm. 13C-NMR (100 MHz, (CD3)2SO): δ 11.1, 14.5, 26.8, 47.9, 109.4, 143.7, 149.4, 165.8 ppm. FT-IR (cm−1): ν = 3464 (w), 2932 (w), 2649 (w), 1674 (m), 1556 (w), 1506 (w), 1458 (w), 1431 (m), 1394 (w), 1366 (w), 1302 (m), 1264 (m), 1189 (m), 1118 (m), 1089 (m), 1038 (w), 1005 (w), 887 (w), 863 (w), 787 (m), 757 (s).
1,5-Bis(3,5-dimethylpyrazol-1-yl)pentane-4,4′-dicarboxylic acid (5b) was prepared similarly to compound 1b by Method B from diesters 5c or 5g. Yield 70%, colorless crystals, m.p. 243–244 °C (reprecipitation; dec.). C17H24N4O4 (348.40): calcd. C 58.61; H 6.94; N 16.08; found C 58.3; H 6.7; N 15.9. 1H-NMR (400 MHz, (CD3)2SO): δ 1.20 (p, 2 H, γ-CH2), 1.69 (p, 4 H, β-CH2), 2.25 (s, 6 H, 3-CH3), 2.42 (s, 6 H, 5-CH3), 3.95 (t, 4 H, α-CH2), ppm. 13C-NMR (100 MHz, (CD3)2SO): δ 11.1, 14.5, 23.6, 29.3, 48.3, 109.4, 143.6, 149.3, 165.8, ppm. FT-IR (cm−1): ν = 2933 (w), 2866 (w), 1689 (m), 1669 (m), 1545 (m), 1499 (w), 1435 (w), 1384 (w), 1301 (w), 1268 (m), 1237 (w), 1216 (m), 1165 (m), 1114 (m), 1043 (w), 1012 (w), 824 (w), 787 (w), 752 (m).
1,6-Bis(3,5-dimethylpyrazol-1-yl)hexane-4,4′-dicarboxylic acid (6b) was prepared similarly to compound 1b by Method B from diesters 6c or 6g. Yield 67%, colorless crystals, m.p. 193–195 °C (reprecipitation; dec.). 1H-NMR (400 MHz, (CD3)2SO): δ 1.23 (p, 4 H, γ-CH2), 1.66 (p, 4 H, β-CH2), 2.26 (s, 6 H, 3-CH3), 2.42 (s, 6 H, 5-CH3), 3.94 (t, 4 H, α-CH2), 12.13 (s, 2 H, COOH), ppm. 13C-NMR (100 MHz, (CD3)2SO): δ 11.1, 14.5, 26.0, 29.6, 48.3, 109.3, 143.60, 149.3, 165.8, ppm. FT-IR (cm−1): ν = 2933 (w), 2861 (w), 1697 (s), 1543 (m), 1497 (m), 1470 (m), 1430 (m), 1380 (m), 1277 (m), 1235 (m), 1178 (m), 1108 (s), 1044 (w), 1002 (m), 977 (w), 862 (m), 787 (m), 746 (s), 730 (m), 664 (w).
1,4-Bis(4-methoxycarbonyl-3,5-dimethylpyrazol-1-yl)butane (4c). Oxalyl chloride (1.91 g, 1.29 mL, 15 mmol) was added dropwise to 0.51 g (2.5 mmol) of compound 4a. The mixture was heated in a screw-cup vial for 33 h, after which the reaction mixture was transferred to 50 mL of chloroform and the solvent was evaporated in vacuum of water aspiration pump to remove the excess of oxalyl chloride. Methanol (5 mL) was added dropwise to the residue, 50 mL of water was added and the precipitate was filtered and washed with water. Yield 27%, colorless crystals, m.p. 129–132 °C (EtOAc/hexane, 1:1). C18H26N4O4 (362.42): calcd. C 59.65; H 7.23; N 15.46; found C 59.8; H 7.5; N 15.2. 1H-NMR (400 MHz, CDCl3): δ =1.80 (p, 4 H, β-CH2), 2.40 (s, 6 H, 3-CH3), 2.47 (s, 6 H, 5-CH3), 3.82 (s, 6 H, OCH3), 4.01 (t, 4 H, α-CH2), ppm. 13C-NMR (100 MHz, CDCl3): δ 11.2, 14.2, 26.9, 48.1, 50.9, 109.4, 143.7, 150.4, 164.9, ppm. FT-IR (cm−1): ν = 2988 (w), 2930 (w), 2861 (w), 1685 (s), 1544 (s), 1493 (m), 1442 (s), 1430 (s), 1303 (m), 1261 (m), 1223 (m), 1191(m), 1175 (m), 1104 (s), 1082 (s), 1041 (m), 1005 (m), 960 (m), 842 (w), 803 (m), 785 (s), 768 (m), 657 (w). m/z: 362 (35%), 347 (12%), 331 (16%), 315 (4%), 303 (4%), 209 (29%), 195 (31%), 181 (57%), 167 (100%).
1,5-Bis(4-methoxycarbonyl-3,5-dimethylpyrazol-1-yl)pentane (5c) was prepared similarly to compound 4c. Yield 34%, colorless crystals, m.p. 86–87 °C (EtOAc/hexane, 1:1). C19H28N4O4 (376.46): calcd. C 60.62; H 7.50; N 14.88; found C 60.3; H 7.4; N 14.6. 1H-NMR (400 MHz, CDCl3): δ 1.30 (p, 2 H, γ-CH2), 1.81 (p, 4 H, β-CH2), 2.40 (s, 6 H, 3-CH3), 2.48 (s, 6 H, 5-CH3), 3.82 (s, 6 H, OCH3), 3.99 (t, 4 H, α-CH2), ppm. 13C-NMR (100 MHz, CDCl3): δ 11.2, 14.2, 23.7, 29.5, 48.5, 50.9, 109.3, 143.6, 150.3, 164.9, ppm. FT-IR (cm−1): ν = 2934 (w), 2863 (w), 1690 (s), 1546 (s), 1495 (w), 1439 (s), 1377 (w), 1324 (w), 1297 (s), 1271 (m), 1236 (m), 1213 (s), 1192 (m), 1160 (m), 1102 (s), 1040 (w), 1001 (w), 959 (m), 856 (w), 825 (w), 805 (w), 785 (s), 736 (w), 713 (w). m/z: 376 (27%), 361 (11%), 345 (14%), 329 (5%), 317 (4%), 223 (24%), 209 (39%), 195 (25%), 181 (37%), 167 (100%).
1,6-Bis(4-methoxycarbonyl-3,5-dimethylpyrazol-1-yl)hexane (6c) was prepared similarly to compound 4c. Yield 30%, colorless crystals, m.p. 112–114 °C (EtOAc/hexane, 1:1). 1H-NMR (400 MHz, CDCl3): δ 1.23 (p, 2 H, γ-CH2), 1.65 (p, 4 H, β-CH2), 2.26 (s, 6 H, 3-CH3), 2.43 (s, 6 H, 5-CH3), 3.72 (s, 6 H, OCH3), 3.95 (t, 4 H, α-CH2), ppm. 13C-NMR (100 MHz, CDCl3): δ 11.2, 14.5, 25.9, 29.5, 48.4, 51.1, 108.6, 143.7, 149.1, 164.6, ppm. FT-IR (cm−1): ν = 2925 (m), 2862 (w), 1701 (s), 1540 (s), 1488 (s), 1485 (m), 1431 (s), 1375 (m), 1293 (s), 1241 (s), 1205 (s), 1191 (s), 1161 (m), 1147 (m), 1101 (s), 1053 (s), 998 (m), 965 (m), 917 (m), 876 (w), 845 (w), 802 (m), 792 (m), 780 (s), 738 (m). m/z: 390 (10%), 375 (4%), 359 (13%), 343 (4%), 331 (2%), 237 (21%), 223 (88%), 209 (17%), 195 (25%), 181 (19%), 167 (100%).
Bis(pyrazol-1-yl)methane-4,4′-dicarboxylic acid (1e) was prepared similarly to compound 1b by Method B from diester 1f. Yield 90%, colorless crystals, m.p. 335–337 °C (reprecipitation; dec.). C9H8N4O4 (236.19): calcd. C 45.77; H 3.41; N 23.72; found C 45.9; H 3.6; N 23.5. 1H-NMR (500 MHz, (CD3)2SO): δ 6.49 (s, 2 H, CH2), 7.87 (s, 2 H, 3-H), 8.53 (s, 2 H, 5-H), 12.52 (s, 2 H, COOH), ppm. 13C-NMR (125 MHz, (CD3)2SO): δ 64.6, 115.9, 134.6, 141.7, 163.4, ppm. FT-IR (cm−1): ν = 3108 (m), 1669 (s), 1560 (s), 1419 (m), 1400 (m), 1366 (m), 1345 (m), 1294 (m), 1241 (s), 1133 (m), 1003 (m), 995 (s), 944 (m), 915 (s), 863 (m), 784 (m), 769 (s), 729 (m).
One-pot procedure. A suspension of finely powdered KOH (1.68 g, 30 mmol) and ethyl 4-pyrazole carboxylate (1.40 g, 10 mmol) in 5 mL of DMSO was vigorously stirred for 30 min at 80 °C. After that, the reaction mixture was cooled to room temperature and solution of dibromomethane (0.87 g, 0.35 mL, 5 mmol) in 5 mL of DMSO was added dropwise with stirring and cooling by water over 30 min. Stirring and heating at 80 °C was continued for additional 4 h (TLC control), then 20 mL of water and 1.12 g (20 mmol) of KOH were added to the reaction flask. Heating was continued until complete dissolution of the initially formed precipitate (1 h), after that, additional 80 mL of water were added and the resulting solution was acidified by concentrated HCl. The precipitate was filtered, washed with water and dried. Yield 1.34 g, 92%.
1,3-Bis(pyrazol-1-yl)propane-4,4′-dicarboxylic acid (3e) was prepared similarly to compound 1b by Method A (yield 11%) or by Method B from diester 3f. Yield 76%, colorless crystals, m.p. 225–227 °C (EtOH). C11H12N4O4 (264.24): calcd. C 50.00; H 4.58; N 21.20; found C 50,4, H 4,8, N 21,1. 1H-NMR (400 MHz, (CD3)2SO): δ 2.35 (p, 2 H, β-CH2), 4.22 (t, 4 H, α-CH2), 8.07 (s, 2 H, 5-H), 8.60 (s, 2 H, 3-H), ppm. 13C-NMR (100 MHz, (CD3)2SO): δ 29.9, 49.0, 118.8, 137.0, 141.4, 179.4, ppm. FT-IR (cm−1): ν = 3127 (w), 3093 (w), 2948 (w), 1695 (s), 1556 (s), 1473 (w), 1369 (w), 1407 (m), 1356 (m), 1343 (m), 1298 (m), 1224 (s), 1172 (s), 1149 (m), 1083 (m), 1051 (m), 982 (s), 889 (s), 863 (s), 822 (m), 765 (s), 743 (s).
1,4-Bis(pyrazol-1-yl)butane-4,4′-dicarboxylic acid (4e) was prepared similarly to compound 1b by Method B from diester 4f. Yield 86%, colorless crystals, m.p. 300 °C (dec.). C12H14N4O4 (278.27): calcd. C 51.80; H 5.07; N 20.13; found C 51.6; H 5.1; N 20.3. 1H-NMR (500 MHz, (CD3)2SO): δ 1.71 (p, 4 H, β-CH2), 4.15 (t, 4 H, α-CH2), 7.79 (s, 2 H, 5-H), 8.25 (s, 2 H, 3-H), 12.30 (s, 2 H, COOH), ppm. 13C-NMR (125 MHz, (CD3)2SO): δ 26.5, 50.8, 114.6, 133.5, 140.4, 163.8, ppm. FT-IR (cm−1): ν = 3122 (w), 3088 (w), 2953 (w), 1685 (s), 1558 (s), 1438 (m), 1404 (m), 1379 (m), 1349 (m), 1304 (m), 1237 (s), 1221 (s), 1188 (s), 1149 (m), 1122 (m), 1018 (s), 996 (m), 986 (s), 947 (m), 896 (m), 867 (m), 805 (s), 775 (s), 748 (s).
1,5-Bis(pyrazol-1-yl)pentane-4,4′-dicarboxylic acid (5e) was prepared similarly to compound 1b by Method B from diester 5f. Yield 67%, colorless powder, m.p. 216–219 °C (reprecipitation; dec.). C13H16N4O4 (292.30): calcd. C 53.42; H 5.52; N 19.17; found C 53.6; H 5.7; N 19.0. 1H-NMR (500 MHz, (CD3)2SO): δ 1.15 (p, 2 H, γ-CH2), 1.80 (p, 4 H, β-CH2), 4.11 (t, 4 H, α-CH2), 7.78 (s, 2 H, 5-H), 8.24 (s, 2 H, 3-H), 12.27 (s, 2 H, COOH), ppm. 13C-NMR (125 MHz, (CD3)2SO): δ 22.7, 28.9, 51.2, 114.5, 133.5, 140.3, 163.8, ppm. FT-IR (cm−1): ν = 3128 (w), 2974 (w), 1696 (s), 1554 (s), 1472 (w), 1438 (w), 1403 (m), 1376 (w), 1343 (w), 1333 (w), 1298 (w), 1269 (w), 1217 (s), 1098 (m), 1041 (w), 998 (m), 987 (s), 915 (m), 885 (s), 769 (s), 745 (s), 672 (m).
1,6-Bis(pyrazol-1-yl)hexane-4,4′-dicarboxylic acid (6e) was prepared similarly to compound 1b by Method B from diester 6f. Yield 73%, colorless powder, m.p. 252–254 °C (reprecipitation; dec.). C14H18N4O4 (306.32): calcd. C 54.89; H 5.92; N 18.29; found C 55.0; H 6.1; N 18.0. 1H-NMR (500 MHz, (CD3)2SO): δ 1.22 (p, 4 H, γ-CH2), 1.75 (p, 4 H, β-CH2), 4.10 (t, 4 H, α-CH2), 7.78 (s, 2 H, 5-H), 8.24 (s, 2 H, 3-H), 12.27 (s, 2 H, COOH), ppm 13C-NMR (125 MHz, (CD3)2SO): δ 25.3, 29.3, 51.4, 114.5, 133.4, 140.3, 163.8, ppm. FT-IR (cm−1): ν = 3114 (w), 2940 (w), 2861 (w), 1652 (m), 1549 (w), 1497 (w), 1469 (w), 1438 (w), 1384 (w), 1352 (w), 1258 (w), 1228 (w), 1129 (w), 1053 (w), 995 (w), 982 (w), 931 (w), 860 (w), 778 (m), 736 (w).
Bis(4-ethoxycarbonylpyrazol-1-yl)methane (1f). A suspension of finely powdered KOH (1.68 g, 30 mmol) and ethyl 4-pyrazole carboxylate (1.40 g, 10 mmol) in 5 mL of DMSO was vigorously stirred for 30 min at 80 °C. After that, the reaction mixture was cooled to room temperature and solution of dibromomethane (0.87 g, 0.35 mL, 5 mmol) in 5 mL of DMSO was added dropwise with stirring and cooling by water over 30 min. Stirring and heating at 80 °C was continued for additional 4 h (TLC control), and the reaction mixture was poured into 100 mL of water, the precipitate was filtered and washed by water. Yield 1.30 g, 89%, colorless crystals, m.p. 144–145 °C (EtOAc/hexane, 1:1). C13H16N4O4 (292.30): calcd. C 53.42; H 5.52; N 19.17; found C 53.6; H 5.7; N 18.9. 1H-NMR (400 MHz, CDCl3): δ 1.34 (t, 6 H, CH3), 4.29 (q, 4 H, OCH2), 6.31 (s, 2 H, α-CH2), 7.96 (s, 2 H, 3-H), 8.18 (s, 2 H, 5-H), ppm. 13C-NMR (100 MHz, CDCl3): δ 14.3, 60.5, 65.7, 117.0, 133.3, 142.5, 162.3, ppm. FT-IR (cm−1): ν = 3149 (w), 3099 (w), 3069 (w), 2987 (w), 1724 (s), 1705 (s), 1558 (s), 1478 (w), 1445 (m), 1412 (w), 1383 (m), 1345 (w), 1290 (m), 1192 (s), 1230 (s), 1161 (m), 1133 (s), 1109 (m), 1029 (s), 1015 (s), 993 (s), 979 (m), 953 (m), 908 (m), 886 (m), 827 (m), 764 (s), 739 (s), 652 (m). m/z: 292 (23%), 247 (68%), 219 (13%), 153 (100%).
1,3-Bis(4-ethoxycarbonylpyrazol-1-yl)propane (3f) was prepared similarly to compound 1f. Yield 80%, colorless crystals, m.p. 92–94 °C (EtOAc/hexane, 1:1). C15H20N4O4 (320.35): calcd. C 56.24; H 6.29; N 17.49; found C 56.5; H 6.0; N 17.2. 1H-NMR (400 MHz, CDCl3): δ 1.36 (t, 6 H, CH3), 2.48 (p, 2 H, β-CH2), 4.16 (t, 4 H, α-CH2), 4.30 (q, 4 H, OCH2), 7.94 (s, 2 H, 5-H), 7.95 (s, 2 H, 3-H), ppm. 13C-NMR (100 MHz, CDCl3): δ 14.4, 30.4, 49.0, 60.3, 115.3, 133.1, 141.3, 162.8, ppm. FT-IR (cm−1): ν = 3126 (w), 3091 (w), 2984 (w), 2940 (w), 2875 (w), 1691 (s), 1556 (s), 1472 (w), 1448 (w), 1405 (m), 1373 (s), 1350 (m), 1131 (m), 1286 (m), 1238 (s), 1124 (s), 1218 (s), 1111 (m), 1023 (s), 1084 (s), 976 (s), 874 (s), 830 (m), 769 (s), 708 (w), 653 (m). m/z: 320 (3%), 291 (1%), 275 (24%), 181 (26%), 167 (100%), 153 (76%), 139 (13%).
1,4-Bis(4-ethoxycarbonylpyrazol-1-yl)butane (4f) was prepared similarly to compound 1f. Yield 86%, colorless crystals, m.p. 121 °C (EtOAc/hexane, 1:1). C16H22N4O4 (334.38): calcd. C 57.47; H 6.63; N 16.76; found C 57.5; H 6.5; N 16.9. 1H-NMR (400 MHz, CDCl3): δ 1.27 (t, 6 H, CH3), 1.81 (p, 4 H, β-CH2), 4.06 (t, 4 H, α-CH2), 4.22 (q, 4 H, OCH2), 7.78 (s, 2 H, 5-H), 7.83 (s, 2 H, 3-H), ppm. 13C-NMR (100 MHz, CDCl3): δ 14.4, 27.0, 51.8, 60.2, 115.2, 132.5, 141.2, 162.9, ppm. FT-IR (cm−1): ν = 3122 (w), 3089 (w), 2980 (w), 2948 (w), 2869 (w), 1691 (s), 1554 (s), 1464 (m), 1432 (w), 1405 (m), 1394 (m), 1373 (s), 1355 (w), 1338 (w), 1298 (w), 1276 (m), 1223 (s), 1194 (s), 1172 (s), 1113 (s), 1092 (m), 1018 (s), 984 (s), 884 (s), 832 (m), 770 (s), 740 (m), 657 (m). m/z: 334 (2%), 305 (19%), 289 (50%), 195 (44%), 181 (19%), 167 (59%), 153 (100%), 139 (10%).
1,5-Bis(4-ethoxycarbonylpyrazol-1-yl)pentane (5f) was prepared similarly to compound 1f. Yield 95%, colorless crystals, m.p. 82–83 °C (EtOAc/hexane, 1:1). C17H24N4O4 (348.40): calcd. C 58.61; H 6.94; N 16.08; found C 58.6; H 6.7; N 16.0. 1H-NMR (400 MHz, CDCl3): δ 1.21 (p, 2 H, γ-CH2, partially overlapped with 1.28), 1.28 (t, 6 H, CH3), 1.84 (p, 4 H, β-CH2), 4.05 (t, 4 H, α-CH2), 4.22 (q, 4 H, OCH2), 7.78 (s, 2 H, 5-H), 7.83 (s, 2 H, 3-H), ppm. 13C-NMR (100 MHz, CDCl3): δ 14.4, 23.4, 29.5, 52.2, 60.2, 115.0, 132.5, 141.0, 163.0, ppm. FT-IR (cm−1): ν = 3129 (w), 2970 (w), 2935 (w), 2874 (w), 1699 (s), 1554 (s), 1459 (m), 1441 (m), 1404 (s), 1372 (m), 1352 (m), 1333 (w), 1304 (m), 1263 (w), 1224 (s), 1209 (s), 1190 (s), 1113 (s), 1096 (s), 1021 (s), 980 (s), 877 (m), 826 (w), 762 (s). m/z: 348 (11%), 319 (24%), 303 (47%), 275 (4%), 209 (14%), 195 (47%), 181 (22%), 167 (33%), 153 (100%), 139 (4%).
1,6-Bis(4-ethoxycarbonylpyrazol-1-yl)hexane (6f) was prepared similarly to compound 1f. Yield 73%, colorless crystals, m.p. 85–86 °C (EtOAc/hexane, 1:1). C18H26N4O4 (362.43): calcd. C 59.65; H 7.23; N 15.46; found C 59.8; H 7.4; N 15.2. 1H-NMR (400 MHz, CDCl3): δ 1.24 (p, 4 H, γ-CH2, partially overlapped with 1.27), 1.27 (t, 6 H, CH3), 1.80 (p, 4 H, β-CH2), 4.04 (t, 4 H, α-CH2), 4.22 (q, 4 H, OCH2), 7.79 (s, 2 H, 5-H), 7.83 (s, 2 H, 3-H), ppm. 13C-NMR (100 MHz, CDCl3): δ 14.4, 25.9, 29.8, 52.4, 60.2, 115.0, 132.4, 140.9, 163.0, ppm. FT-IR (cm−1): ν = 3130 (w), 2983 (w), 2940 (w), 2866 (w), 1693 (s), 1546 (s), 1466 (w), 1442 (w), 1408 (w), 1373 (m), 1352 (w), 1256 (s), 1219 (s), 1201 (s), 1123 (s), 1110 (m), 1052 (m), 1028 (s), 995 (m), 978 (s), 899 (m), 869 (w), 830 (m), 775 (s), 739 (m), 654 (m). m/z: 362 (10%), 333 (11%), 317 (35%), 289 (4%), 223 (7%), 209 (44%), 195 (29%), 181 (19%), 167 (22%), 153 (100%), 139 (3%).
Bis(4-ethoxycarbonyl-3,5-dimethylpyrazol-1-yl)methane (1g). A suspension of finely powdered KOH (1.68 g, 30 mmol) and ethyl 3,5-dimethyl-4-pyrazolecarboxylate (1.68 g, 10 mmol) in 5 DMSO was vigorously stirred for 30 min at 80 °C. After that, the reaction mixture was cooled to room temperature and solution of dibromomethane (0.87 g, 0.35 mL, 5 mmol) in 5 mL of DMSO was added dropwise under stirring and cooling by water over 30 min. Stirring and heating at 80 °C was continued for additional 4 h (TLC control), and the reaction mixture was poured into 100 mL of water, the precipitate was filtered and washed by water. Yield 1.51 g, 87%, colorless crystals, m.p. 152–153 °C (EtOAc/hexane, 1:2). C17H24N4O4 (348.18): calcd. C 58.61; H 6.94; N 16.08; found C 58.4; H 7.1; N 16.2. 1H-NMR (400 MHz, CDCl3): δ 1.35 (t, 6 H, OCH2CH3), 2.39 (s, 6 H, 3-CH3), 2.75 (s, 6 H, 5-CH3), 4.29 (q, 4 H, OCH2), 6.12 (s, 2 H, α-CH2), ppm. 13C-NMR (100 MHz, CDCl3): δ 11.4, 14.3, 14.4, 59.8, 59.9, 110.9, 145.8, 151.4, 164.2, ppm. FT-IR (cm−1): ν = 2990 (w), 2937 (w), 1701 (s), 1557 (s), 1476 (m), 1426 (m), 1394 (m), 1352 (m), 1309 (s), 1299 (m), 1241 (s), 1212 (m), 1166 (s), 1121 (m), 1101 (s), 1042 (m), 1002 (m), 853 (m), 815 (m), 782 (s), 735 (m), 691 (s). m/z: 348 (50%), 333 (5%), 303 (33%), 275 (2%), 181 (100%).
1,3-Bis(4-ethoxycarbonyl-3,5-dimethylpyrazol-1-yl)propane (3g) was prepared similarly to compound 1g. Yield 92%, colorless crystals, m.p. 112–113 °C (EtOAc/hexane, 1:1). C19H28N4O4 (376.46): calcd. C 60.62; H 7.50; N 14.88; found C 60.3; H 7.7; N 14.9.1H-NMR (400 MHz, CDCl3): δ 1.36 (t, 6 H, OCH2CH3), 2.38 (p, 2 H, β-CH2, partially overlapped with 2.41), 2.41 (s, 6 H, 3-CH3), 2.46 (s, 6 H, 5-CH3), 4.04 (t, 4 H, α-CH2), 4.29 (q, 4 H, OCH2), ppm. 13C-NMR (100 MHz, CDCl3): δ 11.0, 14.3, 14.4, 29.3, 45.5, 59.6, 109.7, 144.0, 150.6, 164.4, ppm. FT-IR (cm−1): ν = 2980 (w), 2929 (w), 1712 (m), 1681 (m), 1545 (m), 1489 (m), 1425 (m), 1385 (m), 1370 (m), 1323 (w), 1293 (s), 1249 (w), 1231 (m), 1200 (w), 1138 (s), 1120 (m), 1100 (s), 1043 (m), 1013 (m), 1000 (m), 984 (w), 853 (w), 815 (w), 782 (s), 716 (w). m/z: 376 (11%), 331 (10%), 209 (18%), 195 (100%), 181 (51%), 167 (6%).
1,4-Bis(4-ethoxycarbonyl-3,5-dimethylpyrazol-1-yl)butane (4g) was prepared similarly to compound 1g. Yield 86%, colorless crystals, m.p. 122–123 °C (EtOAc/hexane, 1:1). C20H30N4O4 (390.48): calcd. C 61.52; H 7.74; N 14.35; found C 61.7; H 8.0; 14.4. 1H-NMR (400 MHz, CDCl3): δ 1.36 (t, 6 H, OCH2CH3), 1.82 (p, 4 H, β-CH2), 2.42 (s, 6 H, 3-CH3), 2.49 (s, 6 H, 5-CH3), 4.03 (t, 4 H, α-CH2), 4.29 (q, 4 H, OCH2), ppm. 13C-NMR (100 MHz, CDCl3): δ 11.1, 14.3, 14.4, 26.9, 48.0, 59.6, 109.7, 143.7, 150.4, 164.4, ppm. FT-IR (cm−1): ν = 2985 (w), 2933 (w), 2875 (w), 1695 (s), 1547 (s), 1491 (m), 1472 (m), 1436 (m), 1400 (m), 1383 (m), 1364 (m), 1298 (s), 1248 (s), 1188 (s), 1122 (m), 1102 (s), 1076 (m), 1044 (m), 1015 (m), 993 (m), 910 (w), 888 (w), 849 (m), 813 (w), 784 (s), 742 (w), 711 (m). m/z: 390 (36%), 375 (12%), 361 (12%), 345 (29%), 329 (1%), 317 (4%), 223 (25%), 209 (29%), 195 (68%), 181 (100%).
1,5-Bis(4-ethoxycarbonyl-3,5-dimethylpyrazol-1-yl)pentane (5g) was prepared similarly to compound 1g. Yield 79%, colorless crystals, m.p. 100–101 °C (EtOAc/hexane, 1:1). C21H32N4O4 (404.51): calcd. C 62.35; H 7.97; N 13.85; found C 62.0; H 8.1; N 13.9. 1H-NMR (400 MHz, CDCl3): δ 1.32 (p, 2 H, γ-CH2, partially overlapped with 1.36), 1.36 (t, 6 H, OCH2CH3), 1.81 (p, 4 H, β-CH2), 2.42 (s, 6 H, 3-CH3), 2.49 (s, 6 H, 5-CH3), 3.99 (t, 4 H, α-CH2), 4.29 (q, 4 H, OCH2), ppm. 13C-NMR (100 MHz, CDCl3): δ 11.2, 14.3, 14.4, 23.7, 29.5, 48.5, 59.6, 109.5, 143.5, 150.3, 164.5, ppm. FT-IR (cm−1): ν = 2981 (w), 2930 (w), 2866 (w), 1686 (s), 1548 (s), 1496 (m), 1473 (m), 1436 (m), 1399 (w), 1371 (m), 1296 (s), 1281 (m), 1246 (s), 1219 (m), 1190 (s), 1166 (m), 1122 (s), 1101 (s), 1042 (m), 997 (m), 889 (w), 852 (m), 812 (w), 783 (s), 746 (w), 718 (w), 698 (w). m/z: 404 (29%), 389 (8%), 375 (10%), 359 (30%), 343 (6%), 331 (4%), 237 (28%), 223 (47%), 209 (35%), 195 (47%), 181 (100%).
1,6-Bis(4-ethoxycarbonyl-3,5-dimethylpyrazol-1-yl)hexane (6g) was prepared similarly to compound 1g. Yield 81%, colorless crystals, m.p. 107–108 °C (EtOAc/hexane, 1:1). C22H34N4O4 (418.54): calcd. C 63.13; H 8.19; N 13.39; found C 63.2; H 8.3; N 13.4. 1H-NMR (400 MHz, CDCl3): δ 1.33 (p, 4 H, γ-CH2, partially overlapped with 1.36), 1.36 (t, 6 H, OCH2CH3), 1.79 (p, 4 H, β-CH2), 2.42 (s, 6 H, 3-CH3), 2.49 (s, 6 H, 5-CH3), 3.98 (t, 4 H, α-CH2), 4.29 (q, 4 H, OCH2), ppm. 13C-NMR (100 MHz, CDCl3): δ 11.2, 14.3, 14.4, 26.2, 29.8, 48.6, 59.6, 109.4, 143.5, 150.2, 164.6, ppm. FT-IR (cm−1): ν = 2983 (w), 2929 (w), 2863 (w), 1686 (s), 1548 (s), 1481 (m), 1469 (m), 1436 (m), 1374 (m), 1361 (m), 1320 (m), 1311 (m), 1291 (s), 1238 (s), 1185 (s), 1121 (s), 1102 (s), 1041 (m), 1018 (m), 997 (m), 851 (m), 819 (w), 783 (s), 744 (w), 716 (m). m/z: 418 (21%), 403 (2%), 389 (6%), 373 (29%), 357 (8%), 345 (4%), 251 (25%), 237 (100%), 223 (22%), 209 (26%), 195 (20%), 181 (87%).
Bis(pyrazol-1-yl)methane-4-carboxylic acid (7) was prepared following the procedure for compound 1b (Method A). Yield 10%, colorless crystals, m.p. 205–207 °C (EtOH). C8H8N4O2 (192.18): calcd. C 50.00; H 4.20; N 29.15; found C 50.4; H 4.5; N 28.8. 1H-NMR (400 MHz, (CD3)2SO): δ 6.32 (t, 1 H, 4-H-Pz), 6.50 (s, 2 H, CH2), 7.53 (d, 1 H, 3-H-Pz), 8.01 (d, 1 H, 5-H-Pz), 8.08 (s, 1 H, 3-H-PzCOOH), 8.12 (s, 1 H, 5-H-PzCOOH), ppm. 13C-NMR (100 MHz, (CD3)2SO): δ 64.5, 106.7, 119.7, 131.4, 136.8, 140.9, 142.2, 179.6, ppm. FT-IR (cm−1): ν = 3430 (br., COOH), 1682 (C=O), 1544 (νPz), 1002 (Pz breathing).
4. Conclusions
In summary, approaches to the synthesis of 4,4′-dicarboxy-substituted bis(pyrazol-1-yl)alkanes were evaluated. It was found that direct carboxylation by oxalyl chloride is feasible only for the preparation of bis(3,5-dimethylpyrazol-1-yl)methane derivates due to electron-donating methyl groups and short methylene linker. Longer linkers lead to significantly lower product yields, which is probably due to the instability of the intermediate acid chlorides that are formed in the reaction with oxalyl chloride. A more universal method is based on the reaction of ethyl 4-pyrazolecarboxylates with dibromoalkanes in a superbasic medium and is applicable for the preparation of bis(4-carboxypyrazol-1-yl)alkanes with both short and long linkers. The obtained dicarboxylic acids are interesting as potential building blocks for metal-organic frameworks.
Supplementary Materials
The following are available online: FT-IR and NMR plots of the synthesized compounds.
Author Contributions
Conceptualization, A.S.P.; methodology, N.P.B.; investigation, N.P.B.; writing—original draft preparation, N.P.B.; writing—review and editing, A.S.P.; supervision, A.S.P.; funding acquisition, A.S.P. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Ministry of Science and Higher Education of the Russian Federation.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data presented in this study are available on request from the corresponding author.
Acknowledgments
The Siberian Branch of the Russian Academy of Sciences (SB RAS) Siberian Supercomputer Center is gratefully acknowledged for providing supercomputer facilities. The authors thank the Analytical laboratory and thermal analysis facilities of NIIC SB RAS for carrying out the corresponding analyses.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
Sample Availability
Samples of the dicarboxylic acids reported in this article are available from the authors.
References
- Pettinari, C.; Pettinari, R. Metal Derivatives of Poly(pyrazolyl)alkanes: II. Bis(pyrazolyl)alkanes and Related Systems. Coord. Chem. Rev. 2005, 249, 663–691. [Google Scholar] [CrossRef]
- Dehury, N.; Tripathy, S.K.; Sahoo, A.; Maity, N.; Patra, S. Facile Tandem Suzuki Coupling/Transfer Hydrogenation Reaction with a Bis-Heteroscorpionate Pd-Ru Complex. Dalton Trans. 2014, 43, 16597–16600. [Google Scholar] [CrossRef] [PubMed]
- Martínez, J.; Otero, A.; Lara-Sánchez, A.; Castro-Osma, J.A.; Fernández-Baeza, J.; Sánchez-Barba, L.F.; Rodríguez, A.M. Heteroscorpionate Rare-Earth Catalysts for the Hydroalkoxylation/Cyclization of Alkynyl Alcohols. Organometallics 2016, 35, 1802–1812. [Google Scholar] [CrossRef]
- Otero, A.; Fernández-Baeza, J.; Lara-Sánchez, A.; Sánchez-Barba, L.F. Metal Complexes with Heteroscorpionate Ligands Based on the Bis(pyrazol-1-yl)methane Moiety: Catalytic Chemistry. Coord. Chem. Rev. 2013, 257, 1806–1868. [Google Scholar] [CrossRef]
- Zubkevich, S.V.; Tuskaev, V.A.; Gagieva, S.C.; Pavlov, A.A.; Khrustalev, V.N.; Polyakova, O.V.; Zarubin, D.N.; Kurmaev, D.A.; Kolosov, N.A.; Bulychev, B.M. Catalytic Systems Based on Nickel(ii) Complexes with Bis(3,5-dimethylpyrazol-1-yl)methane—Impact of PPh3 on the Formation of Precatalysts and Selective Dimerization of Ethylene. New J. Chem. 2020, 44, 981–993. [Google Scholar] [CrossRef]
- Moegling, J.; Hoffmann, A.; Herres-Pawlis, S. Insights into Copper-Poly(pyrazolyl)methane-Catalyzed Reactions for Organic Transformations. Synthesis 2017, 49, 225–236. [Google Scholar]
- Montani, M.; Pazmay, G.V.B.; Hysi, A.; Lupidi, G.; Pettinari, R.; Gambini, V.; Tilio, M.; Marchetti, F.; Pettinari, C.; Ferraro, S.; et al. The Water Soluble Ruthenium(II) Organometallic Compound [Ru(p-cymene)(bis(3,5 dimethylpyrazol-1-yl)methane)Cl]Cl Suppresses Triple Negative Breast Cancer Growth by Inhibiting Tumor Infiltration of Regulatory T Cells. Pharmacol. Res. 2016, 107, 282–290. [Google Scholar] [CrossRef] [PubMed]
- Silvestri, S.; Cirilli, I.; Marcheggiani, F.; Dludla, P.; Lupidi, G.; Pettinari, R.; Marchetti, F.; Di Nicola, C.; Falcioni, G.; Marchini, C.; et al. Evaluation of Anticancer Role of a Novel Ruthenium(II)-Based Compound Compared with NAMI-A and Cisplatin in Impairing Mitochondrial Functionality and Promoting Oxidative Stress in Triple Negative Breast Cancer Models. Mitochondrion 2021, 56, 25–34. [Google Scholar] [CrossRef]
- Fonseca, D.; Páez, C.; Ibarra, L.; García-Huertas, P.; Macías, M.A.; Triana-Chávez, O.; Hurtado, J.J. Metal Complex Derivatives of Bis(pyrazol-1-yl)methane Ligands: Synthesis, Characterization and Anti-Trypanosoma Cruzi Activity. Transit. Met. Chem. 2019, 44, 135–144. [Google Scholar] [CrossRef]
- Schepetkin, I.; Potapov, A.; Khlebnikov, A.; Korotkova, E.; Lukina, A.; Malovichko, G.; Kirpotina, L.; Quinn, M.T. Decomposition of Reactive Oxygen Species by Copper(II) bis(1-pyrazolyl) Methane Complexes. J. Biol. Inorg. Chem. 2006, 11, 499–513. [Google Scholar] [CrossRef]
- Potapov, A.S.; Nudnova, E.A.; Domina, G.A.; Kirpotina, L.N.; Quinn, M.T.; Khlebnikov, A.I.; Schepetkin, I.A. Synthesis, Characterization and Potent Superoxide Dismutase-Like Activity of Novel Bis(pyrazole)-2,2′-bipyridyl Mixed Ligand Copper(II) Complexes. Dalton Trans. 2009, 4488–4498. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Li, W.; Wei, D.; Li, C.; Pan, C.; Dong, X.; Li, Z.; Li, S.; Wei, B.; Zhang, F.; et al. Synthesis, Characterization, Photo- and Electro-Luminescent Properties of Blue Cationic Iridium Complexes with Nonconjugated Bis(pyrazole-1-yl)methane as the Ancillary Ligand. Dye. Pigment. 2016, 134, 19–26. [Google Scholar] [CrossRef]
- Meng, S.; Jung, I.; Feng, J.; Scopelliti, R.; Di Censo, D.; Grätzel, M.; Nazeeruddin, M.K.; Baranoff, E. Bis(pyrazol-1-yl)methane as Non-Chromophoric Ancillary Ligand for Charged Bis-Cyclometalated Iridium(III) Complexes. Eur. J. Inorg. Chem. 2012, 2012, 3209–3215. [Google Scholar] [CrossRef]
- Alkorta, I.; Claramunt, R.M.; Díez-Barra, E.; Elguero, J.; de la Hoz, A.; López, C. The Organic Chemistry of Poly(1H-pyrazol-1-yl)methanes. Coord. Chem. Rev. 2017, 339, 153–182. [Google Scholar] [CrossRef]
- Potapov, A.S.; Khlebnikov, A.I.; Vasilevskii, S.F. Synthesis of Monomeric and Oligomeric 1,1′-methylenebis-(1H-pyrazoles) Contaning Ethynyl Fragments. Russ. J. Org. Chem. 2006, 42, 1368–1373. [Google Scholar] [CrossRef]
- Zhang, S.; Gao, Z.; Lan, D.; Jia, Q.; Liu, N.; Zhang, J.; Kou, K. Recent Advances in Synthesis and Properties of Nitrated-Pyrazoles Based Energetic Compounds. Molecules 2020, 25, 3475. [Google Scholar] [CrossRef] [PubMed]
- Usami, Y.; Tatsui, Y.; Yoneyama, H.; Harusawa, S. C4-Alkylamination of C4-Halo-1H-1-tritylpyrazoles Using Pd(dba)2 or CuI. Molecules 2020, 25, 4634. [Google Scholar] [CrossRef]
- Liu, Q.; Song, Y.; Ma, Y.; Zhou, Y.; Cong, H.; Wang, C.; Wu, J.; Hu, G.; O’Keeffe, M.; Deng, H. Mesoporous Cages in Chemically Robust MOFs Created by a Large Number of Vertices with Reduced Connectivity. J. Am. Chem. Soc. 2019, 141, 488–496. [Google Scholar] [CrossRef]
- Jia, Y.-Y.; Ren, G.-J.; Li, A.-L.; Zhang, L.-Z.; Feng, R.; Zhang, Y.-H.; Bu, X.-H. Temperature-Related Synthesis of Two Anionic Metal–Organic Frameworks with Distinct Performance in Organic Dye Adsorption. Cryst. Growth Des. 2016, 16, 5593–5597. [Google Scholar] [CrossRef]
- Wang, L.-D.; Tao, F.; Cheng, M.-L.; Liu, Q.; Han, W.; Wu, Y.-J.; Yang, D.-D.; Wang, L.-J. Syntheses, Crystal Structures, and Luminescence of Two Main-Group Metal Complexes Based on 3,4-pyrazoledicarboxylic Acid. J. Coord. Chem. 2012, 65, 923–933. [Google Scholar] [CrossRef]
- Chen, Y.; Liu, C.-B.; Gong, Y.-N.; Zhong, J.-M.; Wen, H.-L. Syntheses, Crystal Structures and Antibacterial Activities of Six Cobalt(II) Pyrazole Carboxylate Complexes with Helical Character. Polyhedron 2012, 36, 6–14. [Google Scholar] [CrossRef]
- Li, F.-L.; Chen, Q.; Song, H.-B.; Dai, B.; Tang, L.-F. Synthesis of Organotin bis(pyrazol-1-yl)methane-tetracarboxylates and Tris(pyrazol-1-yl)methane-hexacarboxylates. Polyhedron 2014, 83, 102–107. [Google Scholar] [CrossRef]
- Cheng, M.; Wang, Q.; Bao, J.; Wu, Y.; Sun, L.; Yang, B.; Liu, Q. Synthesis and Structural Diversity of d10 Metal Coordination Polymers Constructed from New Semi-Rigid Bis(3-methyl-1H-pyrazole-4-carboxylic acid)alkane Ligands. New J. Chem. 2017, 41, 5151–5160. [Google Scholar] [CrossRef]
- Radi, S.; El-Massaoudi, M.; Benaissa, H.; Adarsh, N.N.; Ferbinteanu, M.; Devlin, E.; Sanakis, Y.; Garcia, Y. Crystal Engineering of a Series of Complexes and Coordination Polymers Based on Pyrazole-carboxylic Acid Ligands. New J. Chem. 2017, 41, 8232–8241. [Google Scholar] [CrossRef]
- Kivi, C.E.; Gelfand, B.S.; Dureckova, H.; Ho, H.T.K.; Ma, C.; Shimizu, G.K.H.; Woo, T.K.; Song, D. 3D Porous Metal-Organic Framework for Selective Adsorption of Methane over Dinitrogen under Ambient Pressure. Chem. Commun. 2018, 54, 14104–14107. [Google Scholar] [CrossRef]
- Bloch, W.M.; Burgun, A.; Coghlan, C.J.; Lee, R.; Coote, M.L.; Doonan, C.J.; Sumby, C.J. Capturing Snapshots of Post-Synthetic Metallation Chemistry in Metal-Organic Frameworks. Nat. Chem. 2014, 6, 906. [Google Scholar] [CrossRef]
- Burgun, A.; Coghlan, C.J.; Huang, D.M.; Chen, W.; Horike, S.; Kitagawa, S.; Alvino, J.F.; Metha, G.F.; Sumby, C.J.; Doonan, C.J. Mapping-Out Catalytic Processes in a Metal-Organic Framework with Single-Crystal X-ray Crystallography. Angew. Chem. Int. Ed. 2017, 56, 8412–8416. [Google Scholar] [CrossRef]
- Chiriac, C.I. The Direct Carboxylation of Pyrazoles. Synthesis 1986, 1986, 753–755. [Google Scholar] [CrossRef]
- Padial, N.M.; Quartapelle Procopio, E.; Montoro, C.; López, E.; Oltra, J.E.; Colombo, V.; Maspero, A.; Masciocchi, N.; Galli, S.; Senkovska, I.; et al. Highly Hydrophobic Isoreticular Porous Metal-Organic Frameworks for the Capture of Harmful Volatile Organic Compounds. Angew. Chem. Int. Ed. 2013, 52, 8290–8294. [Google Scholar] [CrossRef]
- Zhao, B.; Liang, Q.; Ren, H.; Zhang, X.; Wu, Y.; Zhang, K.; Ma, L.-Y.; Zheng, Y.-C.; Liu, H.-M. Discovery of Pyrazole Derivatives as Cellular Active Inhibitors of Histone Lysine Specific Demethylase 5B (KDM5B/JARID1B). Eur. J. Med. Chem. 2020, 192, 112161. [Google Scholar] [CrossRef]
- Batista, D.C.; Silva, D.P.B.; Florentino, I.F.; Cardoso, C.S.; Gonçalves, M.P.; Valadares, M.C.; Lião, L.M.; Sanz, G.; Vaz, B.G.; Costa, E.A.; et al. Anti-Inflammatory Effect of a New Piperazine Derivative: (4-methylpiperazin-1-yl)(1-phenyl-1H-pyrazol-4-yl)methanone. Inflammopharmacology 2018, 26, 217–226. [Google Scholar] [CrossRef] [PubMed]
- Hüttel, R.; Schön, M.E. Über Pyrazolyl-lithium-Verbindungen. Justus Liebigs Ann. Chem. 1959, 625, 55–65. [Google Scholar] [CrossRef]
- Janin, Y.L. Synthetic Accesses to 3/5-pyrazole Carboxylic Acids. Mini Rev. Org. Chem. 2010, 7, 314–323. [Google Scholar] [CrossRef]
- Bader, R.F.W. Atoms in Molecules. A Quantum Theory; Clarendon Press: Oxford, UK, 1994. [Google Scholar]
- Torres, J.; Lavandera, J.L.; Cabildo, P.; Claramunt, R.M.; Elguero, J. Synthesis and Physicochemical Studies on 1,2-bisazolylethanes. J. Heterocycl. Chem. 1988, 25, 771–782. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision D.01; Gaussian, Inc.: Wallingford, UK, 2013. [Google Scholar]
- Skidmore, J.; Heer, J.; Johnson, C.N.; Norton, D.; Redshaw, S.; Sweeting, J.; Hurst, D.; Cridland, A.; Vesey, D.; Wall, I.; et al. Optimization of Sphingosine-1-phosphate-1 Receptor Agonists: Effects of Acidic, Basic, and Zwitterionic Chemotypes on Pharmacokinetic and Pharmacodynamic Profiles. J. Med. Chem. 2014, 57, 10424–10442. [Google Scholar] [CrossRef]
- Potapov, A.S.; Khlebnikov, A.I. Synthesis of Mixed-Ligand Copper(II) Complexes Containing Bis(pyrazol-1-yl)methane Ligands. Polyhedron 2006, 25, 2683–2690. [Google Scholar] [CrossRef]
- Potapov, A.S.; Domina, G.A.; Khlebnikov, A.I.; Ogorodnikov, V.D. Facile Synthesis of Flexible bis(pyrazol-1-yl)alkane and Related Ligands in a Superbasic Medium. Eur. J. Org. Chem. 2007, 5112–5116. [Google Scholar] [CrossRef]
- Zatonskaya, L.V.; Schepetkin, I.A.; Petrenko, T.V.; Ogorodnikov, V.D.; Khlebnikov, A.I.; Potapov, A.S. Synthesis and Cytotoxicity of Bis(pyrazol-1-yl)-Alkane Derivatives with Polymethylene Linkers and Related Mono- and Dipyrazolium Salts. Chem. Heterocycl. Compd. 2016, 52, 388–401. [Google Scholar] [CrossRef]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).