
From Angiotensin II to Cyclic Peptides and Angiotensin Receptor Blockers (ARBs): Perspectives of ARBs in COVID-19 Therapy


 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
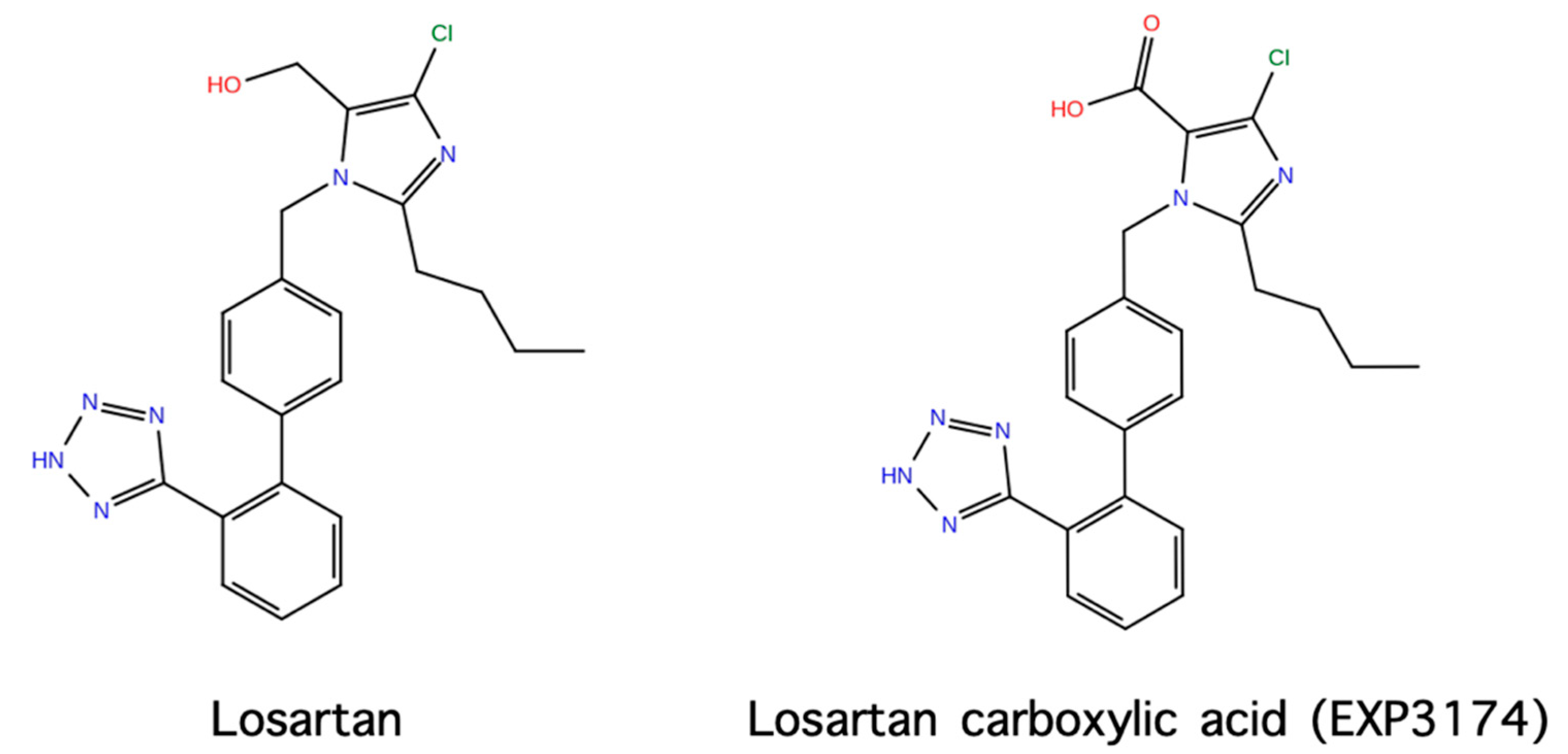
1.1. From Angiotensin II to Losartan and ARBS
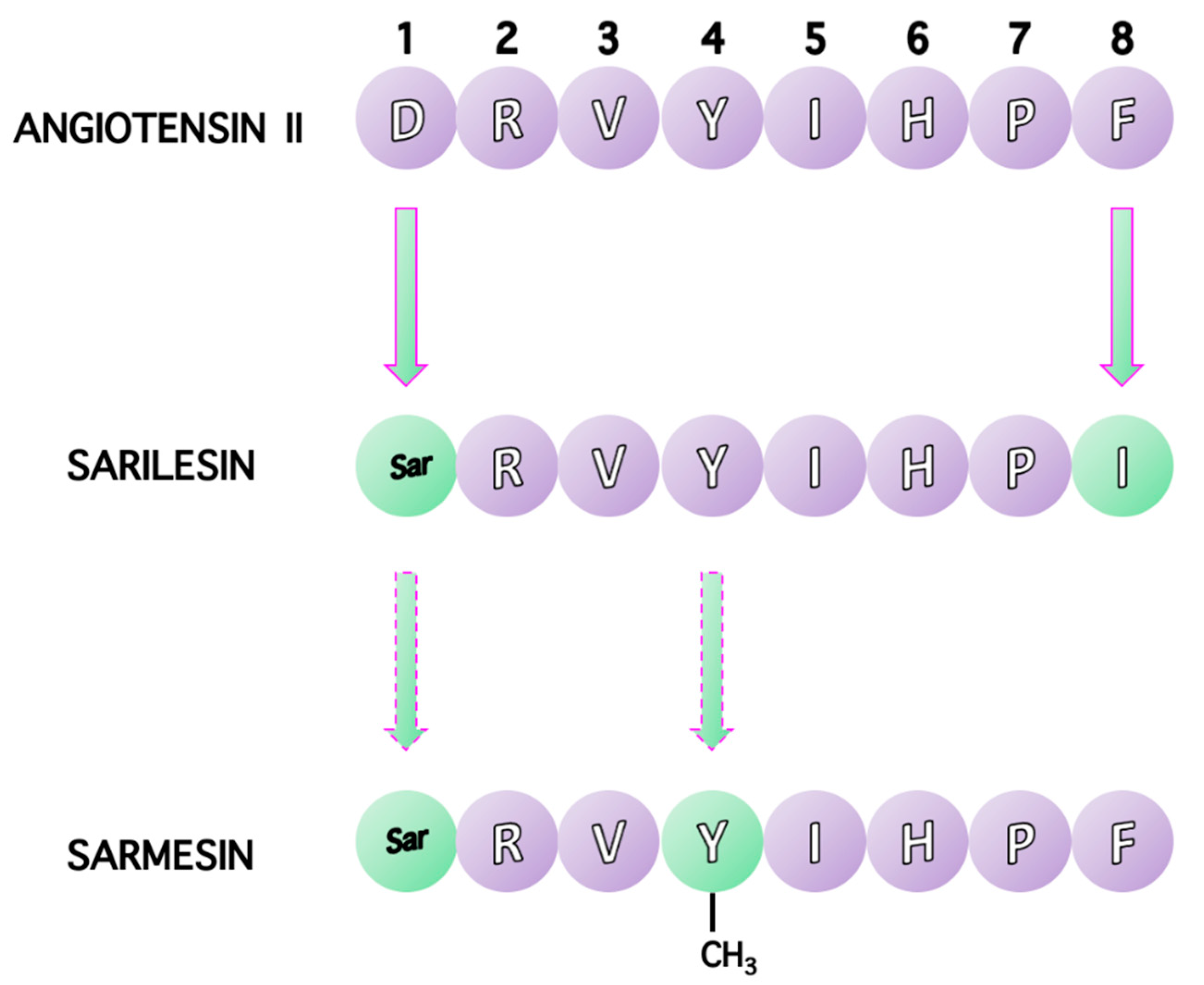
1.2. From Linear to Cyclic Peptides
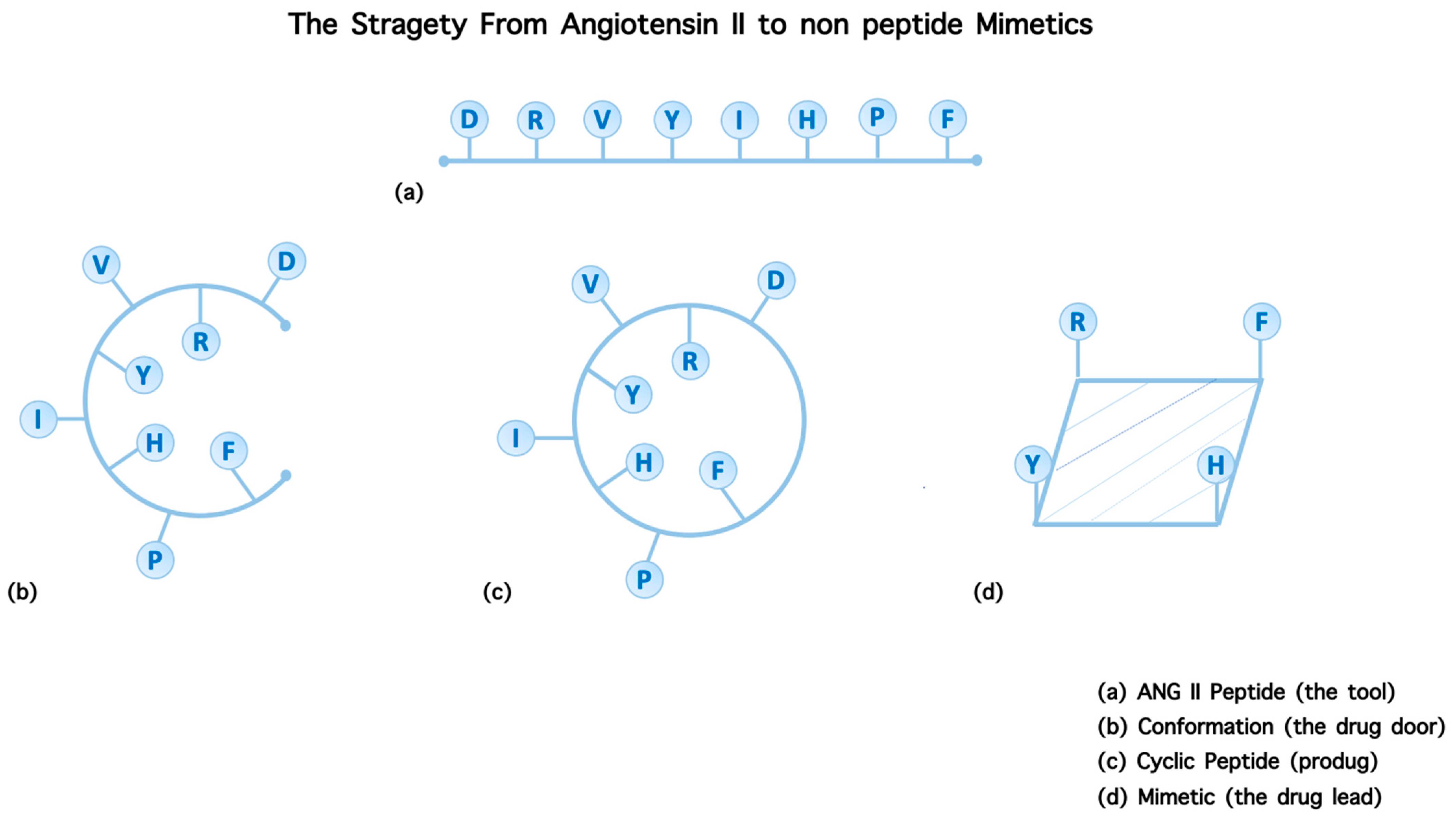
2. The Strategy to Build Non-Peptide Mimetics
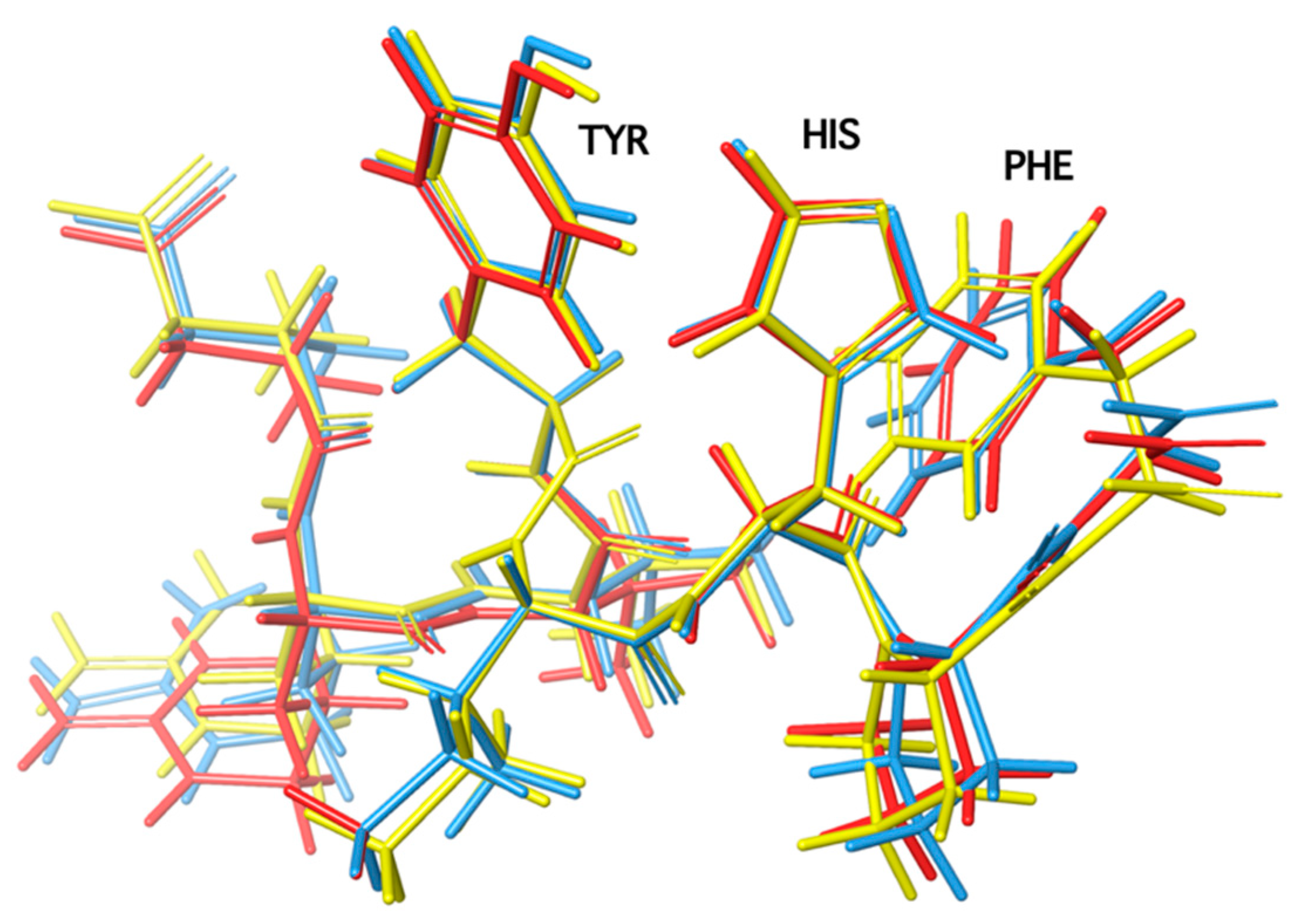
3. Rational Design of Potent Cyclic Peptides Based on SAR (Structure–Activity Relationship) and NMR Studies
4. The Renin–Angiotensin System (RAS) Regulates Vasodilation
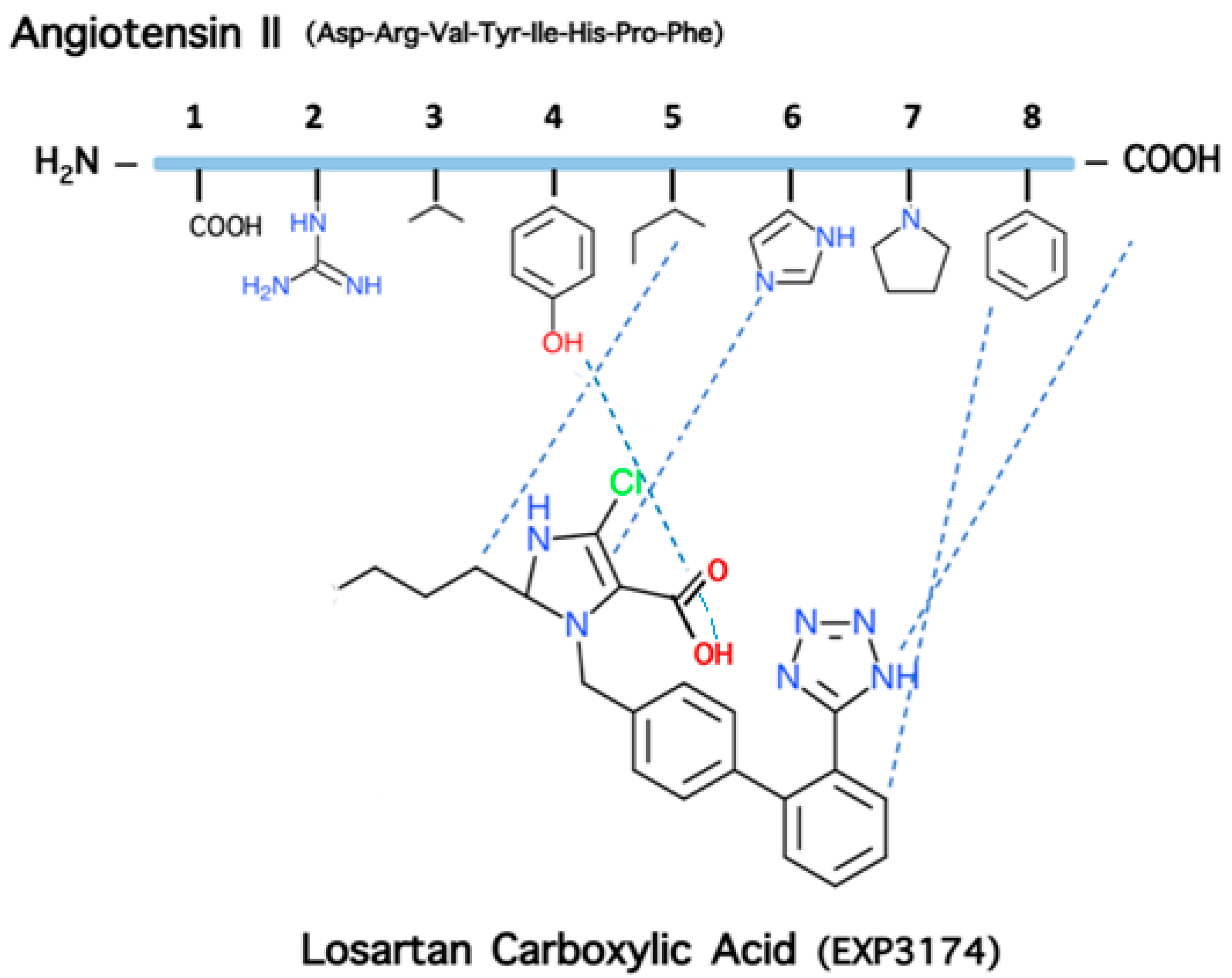
5. The Role of Phenylalanine for Hypertensive Activity of Angiotensin
6. Charge Relay System (CRS) and the Role of Tyrosine Hydroxyl in Triggering Activity of Angiotensin II
7. Cyclic Angiotensin II Analogues Confirm the Ring Cluster Conformation of ANG II
8. From Potent Cyclic (3,5) Angiotensin II to Losartan and ARBS—A Simulation Study
9. Losartan Carboxylic Acid (EXP 3174) is a Stronger Binder Compared to Losartan
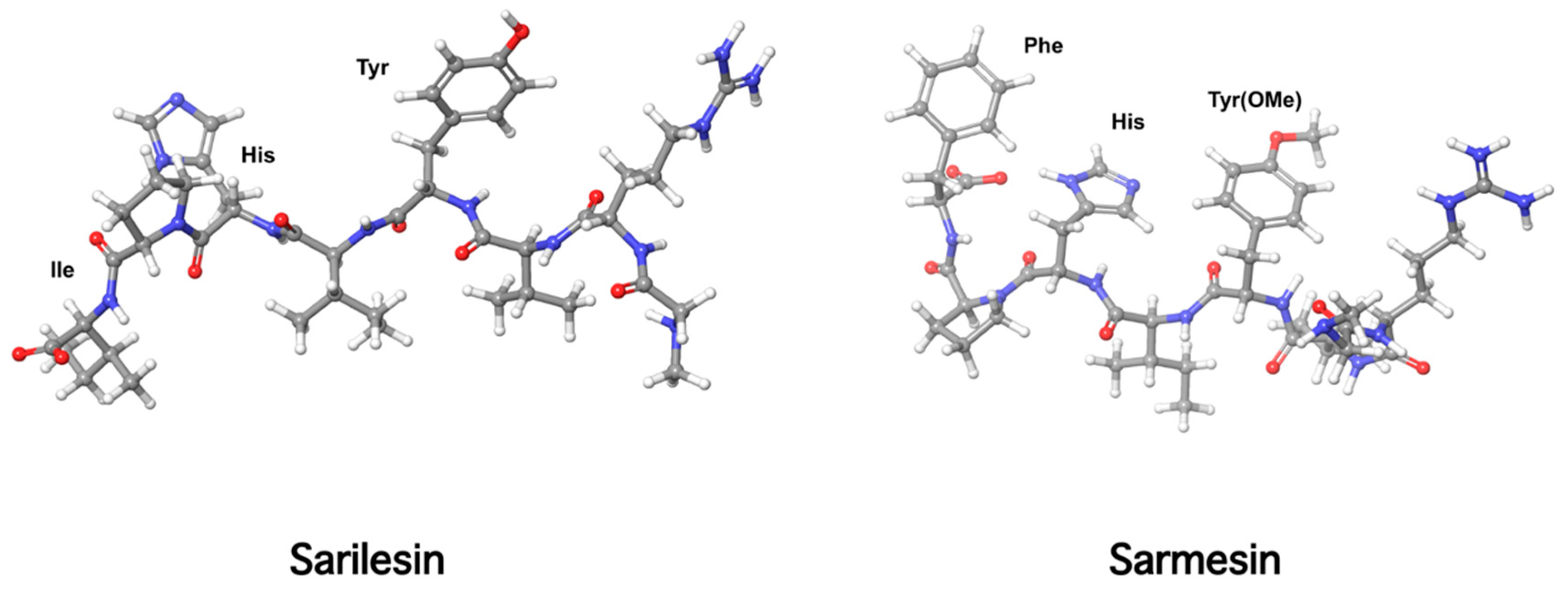
10. Receptor Desensitization by Angiotensin II Antagonist Sarilesin
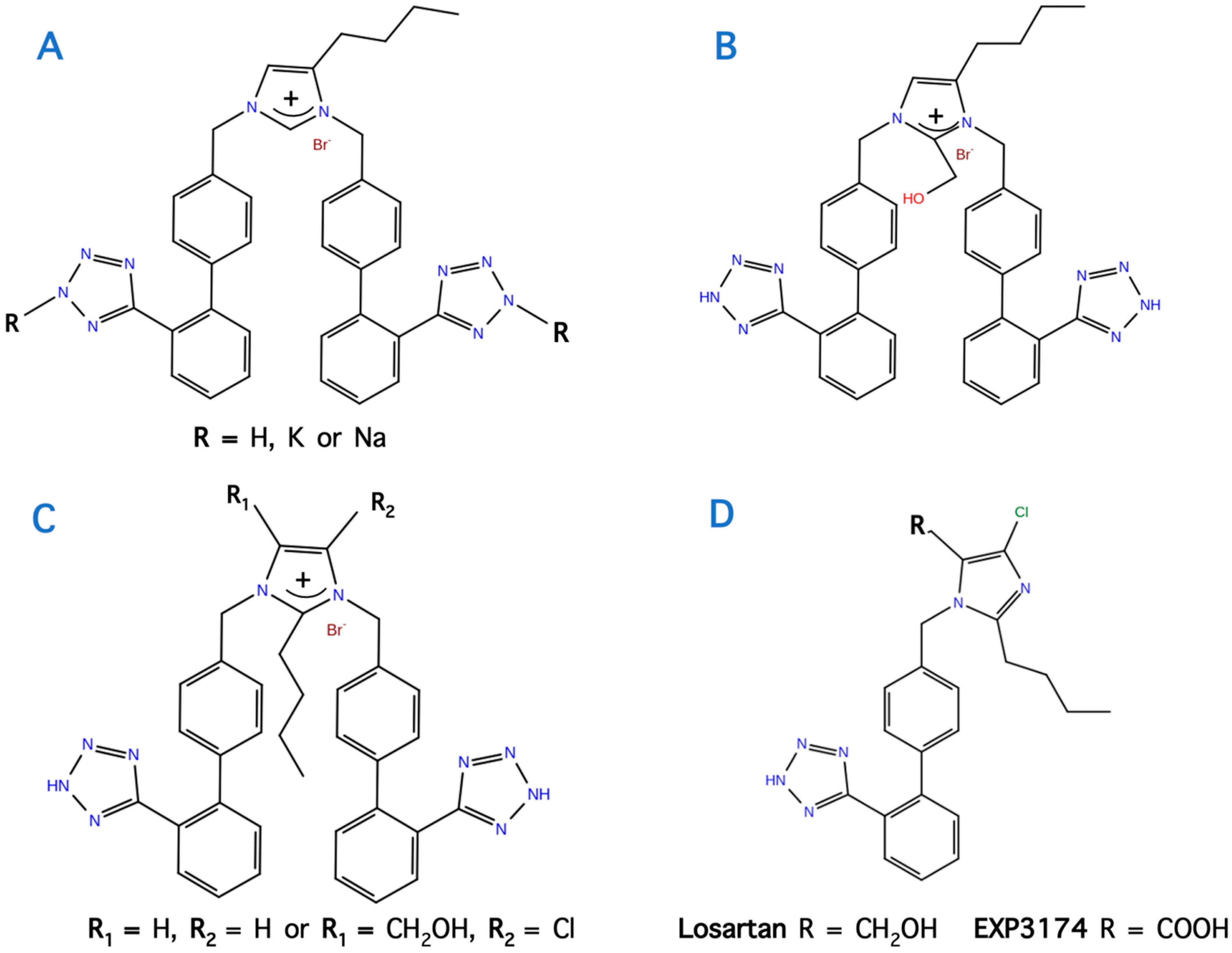
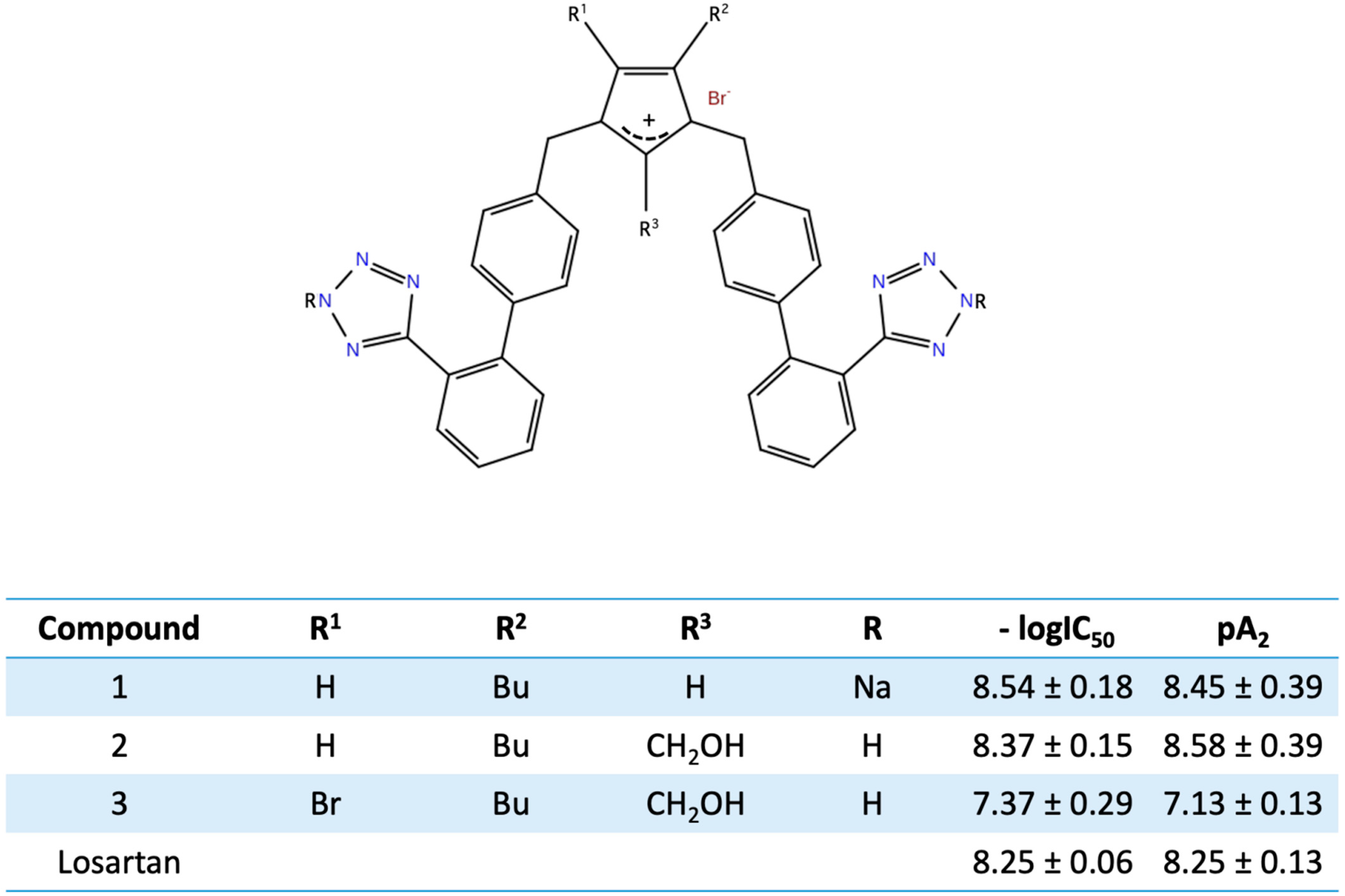
11. Bis Alkylated Arbs Display Strong Affinity and Antagonist Activity
12. Perspectives of Arbs in Transdermal Treatment of Hypertension
13. Perspectives of ARBs in COVID-19 Therapy
14. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Timmermans, P.B.; Duncia, J.V.; Carini, D.J.; Chiu, A.Τ.; Wong, P.C.; Wexler, R.R.; Smith, R.D. Discovery of Losartan, the first angiotensin II receptor antagonist. J. Hum. Hypertens. 1995, 9, 3–18. [Google Scholar]
- Timmermans, P.B. Angiotensin II receptor antagonistsL an emerging new class of cardiovascular therapeutic. Hypertens. Res. 1999, 22, 147–153. [Google Scholar] [CrossRef]
- Khairnar, A.K.; Baviskar, D.T.; Jain, D.K. Angiotensin II Receptor Blockers: An Overview. Int. J. Pharm. Sci. 2012, 4, 50–56. [Google Scholar]
- Moore, G.J.; Smith, J.; Baylis, B.; Matsoukas, J. Design and pharmacology of peptide mimetics. Adv. Pharmacol. 1995, 33, 91–141. [Google Scholar]
- Mavromoustakos, T.; Apostolopoulos, V.; Matsoukas, J. Antihypertensive drugs that act on Renin-Angiotensin system with emphasis in AT1 antagonists. Mini Rev. Med. Chem. 2001, 1, 207–217. [Google Scholar] [CrossRef] [PubMed]
- Neitzel, J.J. Enzyme Catalysis: The Serine Proteases. Nature 2010, 3, 21. [Google Scholar]
- Blow, D.M.; Birktoft, J.J.; Hartley, B.S. Role of a Buried Acid Group in the Mechanism of Action of Chymotrypsin. Nature 1969, 221, 337–338. [Google Scholar] [CrossRef] [PubMed]
- Katsara, M.; Tselios, T.; Deraos, S.; Deraos, G.; Matsoukas, M.; Lazoura, E.; Matsoukas, J.; Apostolopoulos, V. Round and round we go: Cyclic peptides in disease. Curr. Med. 2006, 13, 2221–2232. [Google Scholar]
- Katsara, M.; Deraos, G.; Tselios, T.; Matsoukas, M.T.; Friligou, I.; Matsoukas, J.; Apostolopoulos, V. Design and Synthesis of a Cyclic Double Mutant Peptide (cyclo(87-99)[A91,A96]MBP87-99) Induces Altered Responses in Mice after Conjugation to Mannan: Implications in the Immunotherapy of Multiple Sclerosis. J. Med. Chem. 2009, 52, 214–218. [Google Scholar] [CrossRef]
- Mavromoustakos, T.; Kolocouris, A.; Zervou, M.; Roumelioti, P.; Matsoukas, J.; Weisemann, R. An Effort to Understand the Molecular Basis of Hypertension through the Study of Conformational Analysis of Losartan and Sarmesin Using a Combination of Nuclear Magnetic Resonance Spectroscopy and Theoretical Calculations. J. Med. Chem. 1999, 42, 1714–1722. [Google Scholar] [CrossRef] [PubMed]
- Matsoukas, J.; Agelis, G.; Wahhab, A.; Hondrelis, J.; Panagiotopoulos, D.; Yamdagni, R.; Wu, Q.; Mavromoustakos, T.; Maia, H. Differences in backbone structure between angiotensin II agonists and type I antagonists. J. Med. Chem. 1995, 38, 4660–4669. [Google Scholar] [CrossRef]
- Matsoukas, J.; Hondrelis, J.; Keramida, M.; Mavromoustakos, T.; Makriyannis, A.; Yamdagni, R.; Wu, Q.; Moore, G.J. Role of the NH2-terminal domain of angiotensin II (ANG II) and [Sar1]angiotensin II on conformation and activity. NMR evidence for aromatic ring clustering and peptide backbone folding compared with [des-1,2,3] angiotensin II. J. Biol. Chem. 1994, 269, 5303–5312. [Google Scholar] [CrossRef]
- Matsoukas, J.; Bigam, G.; Zhou, N.; Moore, G.J. 1H-NMR studies of [Sar1]angiotensin II conformation by nuclear Overhauser effect spectroscopy in the rotating frame (ROESY): Clustering of the aromatic rings in dimethylsulfoxide. Peptides 1990, 11, 359–366. [Google Scholar] [CrossRef]
- Qaradakhi, T.; Gadanec, L.; Matsoukas, J.; Apostolopoulos, V.; Zulli, A. Could DIZE be the answer to Covid 19? Maturitas 2020, 140, 83–84. [Google Scholar] [CrossRef] [PubMed]
- Qaradakhi, T.; Matsoukas, M.T.; Hayes, A.; Rybalka, E.; Caprnda, M.; Rimarova, K.; Sepsi, M.; Busselber, D.; Kruzliak, P.; Matsoukas, J.; et al. Alamandine reverses hyperhomocysteinemia-induced vascular dysfunction via PKA dependent mechanisms. Cardiovasc. Ther. 2017, 35, 1–27. [Google Scholar] [CrossRef] [PubMed]
- Qaradakhi, T.; Gadanek, L.K.; McSweeney, K.R.; Tacey, A.; Apostolopoulos, V.; Rimarova, K.; Egom, E.E.; Rodrigo, L.; Kubatka, P.; Zulli, A.; et al. The potential actions of angiotensin-converting enzyme II (ACE2) activator diminazene aceturate (DIZE) in various diseases. Clin. Exp. Pharmacol. Physiol. 2020, 47, 751–758. [Google Scholar] [CrossRef]
- Scanlon, M.; Matsoukas, J.; Franklin, K.; Moore, G.J. A new approach to angiotensin antagonists: Methylation of the tyrosine hydroxyl in angiotensin II. Life Sci. 1984, 34, 317–321. [Google Scholar] [CrossRef]
- Matsoukas, J.; Goghari, M.; Scanlon, M.; Franklin, K.; Moore, G.J. Synthesis and biological activities of analogs of angiotensins II and III containing O-methyltyrosine and D-tryptophan. J. Med. Chem. 1985, 28, 780–783. [Google Scholar] [CrossRef]
- Moore, G.J.; Genter, R.C.; Matsoukas, J.M.; Hondrelis, J.; Agelis, G.; Barlos, K.; Wilkinson, S.; Sandall, J.; Fowler, P. Receptor interactions of the position 4 side chains of angiotensin II analogues: Importance of aromatic ring quadrupole. J. Mol. Recognit. 1994, 7, 251–256. [Google Scholar] [CrossRef]
- Matsoukas, J.; Agelis, G.; Hondrelis, J.; Yamdagni, R.; Wu, Q.; Ganter, R.; Moore, D.; Moore, G.J.; Smith, J. Synthesis and biological activities of angiotensin II, sarilesin, and sarmesin analogs containing Aze or Pip at position 7. J. Med. Chem. 1993, 36, 904–911. [Google Scholar] [CrossRef]
- Turner, R.; Matsoukas, J.; Moore, G.J. Tyrosinate fluorescence lifetimes for oxytocin and vasopressin in receptor-simulating environments: Relationship to biological activity and 1H-NMR data. Biochem. Biopsys. Res. Commun. 1990, 171, 996–1001. [Google Scholar] [CrossRef]
- Turner, R.; Matsoukas, J.M.; Moore, G.J. Fluorescence properties of angiotensin II analogues in receptor-simulating environments: Relationship between tyrosinate fluorescence lifetime and biological activity. Biochim. Biophys. Acta 1991, 1065, 21–28. [Google Scholar] [CrossRef]
- Matsoukas, J.M.; Hondrelis, J.; Aggelis, G.; Barlos, K.; Gatos, D.; Ganter, R.; Moore, D.; Moore, G.J. Novel Synthesis of Cyclic Amide-Linked Analogues of Angiotensins II and III. J. Med. Chem. 1994, 37, 2958–2969. [Google Scholar] [CrossRef] [PubMed]
- Polevaya, L.; Mavromoustakos, T.; Zoumboulakis, P.; Grdadolnik, S.G.; Roumelioti, P.; Giatas, N.; Mutule, I.; Keivish, T.; Vlahakos, D.V.; Iliodromitis, E.K.; et al. Synthesis and Study of a Cyclic Angiotensin II Antagonist Analogue Revelas the Role of π*-π* Interactions in the C-terminal Aromatic Residue for Agonist Activity and Its Structure Resemblance with At1 Non-peptide Antagonists. Bioorg. Med. Chem. 2001, 9, 1639–1647. [Google Scholar] [CrossRef]
- Matsoukas, J.M.; Polevaya, L.; Ancans, J.; Mavromoustakos, T.; Kolocouris, A.; Roumelioti, P.; Vlahakos, D.V.; Yamdagni, R.; Wu, Q.; Moore, G.J. The Design and Synthesis of a Potent Angiotensin II Cyclic Analogue Confirms the Ring Cluster Receptor Conformation of the Hormone Angiotensin II. Bioorg. Med. Chem. 2000, 8, 1–10. [Google Scholar] [CrossRef]
- Agelis, G.; Roumelioti, P.; Resvani, A.; Durdagi, S.; Androutsou, M.-E.; Kelaidonis, K.; Vlahakos, D.; Mavromoustakos, T.; Matsoukas, J. An efficient synthesis of a rationally designed 1,5 disubstituted imidazole AT1 Angiotensin II receptor antagonist: Reorientation of imidazole pharmacophore groups in losartan reserves high receptor affinity and confirms docking studies. J. Comput. Aided Mol. Des. 2010, 24, 749–758. [Google Scholar] [CrossRef]
- Agelis, G.; Resvani, A.; Durdagi, S.; Spyridaki, K.; Tumova, T.; Slaninova, J.; Giannopoulos, P.; Vlahakos, D.; Liapakis, G.; Mavromoustakos, T.; et al. The discovery of new potent non-peptide Angiotensin II AT1 receptor blockers: A concise synthesis, molecular docking studies and biological evaluation of N-substituted 5-butylimidazole derivatives. Eur. J. Med. Chem. 2012, 55, 358–374. [Google Scholar] [CrossRef]
- Agelis, G.; Kelaidonis, K.; Resvani, A.; Kalavrizioti, D.; Androutsou, M.; Plotas, P.; Vlahakos, D.; Koukoulitsa, C.; Tselios, T.; Mavromoustakos, T.; et al. Facile and efficient syntheses of a series of N-benzyl and N-biphenylmethyl substituted imidazole derivatives based on (E)-urocanic acid, as angiotensin II AT1 receptor blockers. Molecules 2013, 18, 7510–7532. [Google Scholar] [CrossRef]
- Wahhab, A.; Smith, J.R.; Ganter, R.C.; Moore, D.M.; Hondrelis, J.; Matsoukas, J.; Moore, G.J. Imidazole based non-peptide angiotensin II receptor antagonists. Investigation of the effect of the orientation of the imidazole ring on biological activity. Arzneim. Forsch. 1993, 43, 1157–1168. [Google Scholar]
- Wexler, R.R.; Greenlee, W.J.; Irvin, J.D.; Goldberg, M.R.; Prendergast, K.; Smith, R.D.; Timmermans, P.B.M.W.M. Nonpeptide Angiotensin II receptor antagonists: The next generation in antihypertensive therapy. J. Med. Chem. 1996, 39, 625. [Google Scholar] [CrossRef]
- Moore, G.; Matsoukas, J. Angiotensin as a model for hormone–receptor interactions. Biosci. Rep. 1985, 5, 407–416. [Google Scholar] [CrossRef]
- Moore, G.J.; Scanlon, M.N. Methods for analyzing and interpreting co-operativity in dose-response curves. Gen. Pharmacol. 1989, 20, 124–129. [Google Scholar]
- Moore, G.J. Designing peptide mimetics. Trends Pharmacol. Sci. 1994, 15, 124–129. [Google Scholar] [CrossRef]
- Zhang, H.; Unal, H.; Denoyer, R.; Han, G.W.; Patel, N.; Katrina, Y.; Karnik, S.; Cheriton, V.; Steven’s, R. Structural basis for ligand recognition and functional selectivity are angiotensin receptor. J. Biol. Chem. 2015, 290, 29127–29139. [Google Scholar] [CrossRef] [PubMed]
- Matsoukas, J.; Cordopatis, P.; Belte, U.; Goghari, M.H.; Ganter, R.C.; Franklin, K.J.; Moore, G.J. Importance of the N-terminal domain of the type II angiotensin antagonist sarmesin for receptor blockade. J. Med. Chem. 1988, 31, 1418–1421. [Google Scholar] [CrossRef] [PubMed]
- Tchekalarova, J.; Pechlivanova, D.; Kampourova, T.; Matsoukas, J.; Georgiev, V. The effects of sarmesin, an angiotensin II analogue on seizure susceptibility, memory retention and nociception. Regul. Pept. 2003, 1, 191–197. [Google Scholar] [CrossRef]
- Mavromoustakos, T.; Zevou, M.; Zoumpoulakis, P.; Kyrikou, I.; Polevaya, L.; Roumelioti, P.; Giatas, N.; Zoga, A.; Minakakis, P.M.; Kolocouris, A.; et al. Conformation and Bioactivity. Design and Discovery of Novel Antihypertensive Drugs. Curr. Top. Med. Chem. 2004, 4, 385–401. [Google Scholar] [CrossRef]
- Matsoukas, M.T.; Potamitis, C.; Plotas, P.; Androutsou, M.E.; Agelis, G.; Matsoukas, J.; Zoumboulakis, P. Insights into AT1 Receptor Activation through AngII Binding Studies. J. Chem. Inf. Mod. 2013, 53, 2798–2811. [Google Scholar] [CrossRef]
- Agelis, G.; Resvani, A.; Koukoulitsa, C.; Tumova, T.; Slaninova, J.; Kalavrizioti, D.; Spyridaki, K.; Afanitis, A.; Melagraki, G.; Siafaka, A.; et al. Rational design, efficient synthesis and biological evaluation of N,N-symmetrically bis-substituted butylimidazole analogs as a new class potent Angiotensin II receptor blockers. Eur. J. Med. Chem. 2013, 62, 352–370. [Google Scholar] [CrossRef]
- Agelis, G.; Resvani, A.; Ntountaniotis, D.; Chatzigeorgiou, P.; Koukoulitsa, C.; Androutsou, M.E.; Plotas, P.; Matsoukas, J.; Mavromoustakos, T.; Cendak, T.; et al. Interactions of the potent synthetic AT1 antagonist analog BV6 with membrane bilayers and mesoporous silicate matrices. Biochim. Biophys. Acta 2013, 1828, 1846–1855. [Google Scholar] [CrossRef]
- Michalatou, M.; Androutsou, M.E.; Antonopoulos, M.; Vlahakos, D.V.; Agelis, G.; Zulli, A.; Qaradakhi, T.; Mikkelsen, K.; Apostolopoulos, V.; Matsoukas, J. Transdermal Delivery of AT1 Receptor Antagonists Reduce Blood Pres-sure and Reveal a Vasodilatory Effect on Kidney Blood Vessels. Curr. Mol. Pharmacol. 2018, 11, 226–236. [Google Scholar] [CrossRef] [PubMed]
- Ahad, A.; Al-Mohizea, A.M.; Al-Jenoobi, F.I.; Aqil, M. Transdermal delivery of angiotensin II receptor blockers (ARBS), angiotensin-converting enzyme inhibitors (ACEIS) and others for management of hypertension. Drug Deliv. 2014, 32, 569–590. [Google Scholar] [CrossRef] [PubMed]
- Rizwan, M.; Aqil, M.; Ahad, A.; Sultana, Y.; Ali, M. Transdermal Delivery of Valsartan: I. Effect of various terpenes. Drug Dev. Ind. Pharm. 2008, 34, 618–626. [Google Scholar] [CrossRef] [PubMed]
- Martin del Valle, E.; Galan, M.; Carbonell, R. Drug Delivery Technologies: The way forward in the new decade. Ind. Eng. Chem. Res. 2009, 48, 2475–2486. [Google Scholar] [CrossRef]
- Paudel, K.S.; Milewski, M.; Swadley, C.L.; Brogden, N.K.; Ghosh, P.; Stinchcomb, A.L. Challenges and opportunities in dermal/transdermal delivery. Ther. Deliv. 2010, 1, 109–131. [Google Scholar] [CrossRef]
- Walczak, A.; Siger, M.; Ciach, A.; Szczepanik, M.; Selmaj, K. Transdermal application of myelin peptides in multiple sclerosis treatment. JAMA Neurol. 2013, 70, 1105–1109. [Google Scholar] [CrossRef]
- Amit, A.; Shubhangi, D.; Ajazuddin, D.; Tapan, K.; Swarnlata, S.; Shailendra, S.; Dulal, K. Approaches for breaking the barriers of drug permeation through transdermal drug delivery. J. Control Release 2012, 164, 26–40. [Google Scholar]
- Sheth, N.; Mistry, R. Formulation and evaluation of transdermal patches and to study permeation enhancement of eugenol. Int. J. Appl. Pharm. Sci. 2011, 3, 96–101. [Google Scholar]
- Keleb, E.; Sharma, R.; Mosa, E.; Aljahwi, A. Transdermal Drug Delivery System—Design and Evaluation. Int. J. Adv. Pharm Sci. 2010, 1, 201–211. [Google Scholar]
- Nishida, N.; Taniyama, K.; Sawabe, T.; Manome, Y. Development and evaluation of a monolithic drug-in-adhesive patch for valsartan. Int. J. Pharm. 2010, 402, 103–109. [Google Scholar] [CrossRef]
- Vijayan, V.; Sumanth, M.; Suman, L.; Vinay, T.; Srinivasrao, D.; Kumar, K. Development and Physiochemical, in vitro Evaluation of Antihypertensive Transdermal Patches. J. Pharm. Sci. Res. 2010, 2, 171–177. [Google Scholar]
- Sinha, V.; Kaur, M. Permeation Enhancers for Transdermal Drug Delivery. Drug Dev. Ind. Pharm. 2000, 26, 1131–1140. [Google Scholar] [CrossRef]
- Abassi, Z.A.; Skorecki, K.; Heyman, S.N.; Kinaneh, S.; Armaly, Z. Covid-19 infection and mortality: A physiologist’s perspective enlightening clinical features and plausible interventional strategies. Am. J. Physiol. Lung Cell Mol. 2020, 318, 1020–1022. [Google Scholar] [CrossRef] [PubMed]
- Walther, T.; Kuebler, W.M. Don’t judge too RAShly: The multifaceted role of the renin-angiotensin system and its therapeutic potential in COVID-19. Am. J. Physiol. Lung Cell Mol. 2020, 318, 1023–1024. [Google Scholar] [CrossRef] [PubMed]
- Abassi, Z.A.; Skorecki, K.; Heyman, S.N.; Kinaneh, S.; Armaly, Z. Reply to Letter to the Editor: “Don’t judge too RAShly: The multifaceted role of the renin-angiotensin system and its therapeutic potential in COVID-19. Am. J. Physiol. Lung Cell Mol. 2020, 318, 1029–1030. [Google Scholar] [CrossRef] [PubMed]
- Gurwitz, D. Angiotensin Receptor blockers (ARBs) as tentative SARS-Cov-2 therapeutics. Drug Dev. Res. 2020, 81, 537–540. [Google Scholar] [CrossRef] [PubMed]
- Dambha-Miller, H.; Albasri, A.; Hodgson, S.; Wilcox, C.R.; Khan, S.; Islam, N.; Little, P.; Griffin, S.J. Currently prescribed drugs in UK that could upregulate or downregulate ACE2 in Covid-19 disease: A systematic review. Br. Med. J. 2020, 10, 1–10. [Google Scholar] [CrossRef]
- Rico-Mesa, J.S.; White, A.; Anderson, A.S. Outcomes in Patients with COVID-19 Infection Taking ACEI/ARB. Curr. Cardiol. Rep. 2020, 22, 31. [Google Scholar] [CrossRef]
- Nejat, R.; Sadr, A.S. Are losartan and Imatinib Effective against SARS-CoV2 Pathogenesis? A Pathophysiologic-Based In Silico Study. In Silico Pharmacol. 2020, 9, 1–39. [Google Scholar] [CrossRef]
- Shete, A. Urgent need for evaluating agonists of angiotensin-(1-7)/Mas receptor axis for treating patients with COVID-19. Int. J. Infect. Dis. 2020, 96, 348–351. [Google Scholar] [CrossRef]
- Marin, G.H. Facts and reflections on COVID-19 and anti-hypertensives drugs. Drug Discov Ther 2020, 14, 105–106. [Google Scholar] [CrossRef]
- Meng, J.; Xiao, G.; Zhang, J.; He, X.; Ou, M.; Bi, J.; Yang, R.; Di, W.; Wang, Z.; Li, Z.; et al. Renin-angiotensin system inhibitors improve clinical outcomes of Covid 19 patients with hypertension. Emerg. Microbes Infect. 2020, 9, 757–760. [Google Scholar] [CrossRef] [PubMed]
- Sriram, K.; Rohit, L.; Paul, A.I. Targeting the renin-angiotensin signaling pathway in COVID-19: Unanswered questions, opportunities, and challenges. PNAs 2020, 117, 29274–29282. [Google Scholar] [CrossRef] [PubMed]
- Warner, F.J.; Rajapakhsa, H.; Shackel, N.; Herath, N. ACE2: From protection of liver disease to propagation of COVID-19. Clin. Sci. 2020, 134, 3137–3158. [Google Scholar] [CrossRef] [PubMed]
- Ni, W.; Yang, X.; Yang, D.; Bao, J.; Li, R.; Xiao, Y.; Hou, C.; Wang, H.; Liu, J.; Yang, D.; et al. Role of angiotensin-converting enzyme 2 (ACE2) in COVID-19. Crit. Care 2020, 24, 422. [Google Scholar] [CrossRef]
- Li, H.; Zhang, P.; Zhu, L.; Cai, J.; Lei, F.; Qin, J.; Xie, J.; Liu, Y.M.; Zhao, Y.C.; Huang, X.; et al. Association of inpatient use of angiotensin converting enzyme inhibitors and angiotensin II receptor blockers with mortality among patients with hypertension hospitalized with Covid-19. Circ. Res. 2020, 126, 1671–1681. [Google Scholar]
- Amanat, A.; Ranjit, V. Dynamics of the ACE-2-SARS-CoV-2/SARS-CoV spike protein interface reveal unique mechanisms. Nature 2020, 10, 214. [Google Scholar]
- Vaduganathan, M.; Vardeny, O.; Michel, T.; McMurray, J.J.V.; Pfefer, M.A.; Solomon, S.D. Renin-Angiotensin-Aldosterone System Inhibitors in Patients with Covid-19. N. Engl. J. Med. 2020, 382, 1653–1659. [Google Scholar] [CrossRef]
- Lopes, R.D.; Macedo, A.V.; Silva, P.; Moll-Bernardes, R.J.; Feldman, A.; Saba Arruda, G.D.; Silvestre de Souza, A.; Campos de Albuquerque, D.; Mazza, L.; Fraga Santos, M.; et al. Continuing versus suspending angiotensin-converting enzyme inhibitors and angiotensin receptor blockers: Impact on adverse outcomes in hospitalized patients with severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2)—The BRACE CORONA trial. Am. Heart J. 2020, 226, 49–59. [Google Scholar] [CrossRef]
- Phua, J.; Weng, L.; Ling, L.; Egi, M.; Lim, C.M.; Divatia, J.; Shrestha, B.R.; Arabi, Y.M.; Ng, J.; Gomersall, C.D.; et al. Intensive care management of coronavirus disease 2019 (COVID-19): Challenges and recommendations. Lancet Respir. Med. 2020, 8, 506–517. [Google Scholar] [CrossRef]












Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Matsoukas, J.; Apostolopoulos, V.; Zulli, A.; Moore, G.; Kelaidonis, K.; Moschovou, K.; Mavromoustakos, T. From Angiotensin II to Cyclic Peptides and Angiotensin Receptor Blockers (ARBs): Perspectives of ARBs in COVID-19 Therapy. Molecules 2021, 26, 618. https://doi.org/10.3390/molecules26030618
Matsoukas J, Apostolopoulos V, Zulli A, Moore G, Kelaidonis K, Moschovou K, Mavromoustakos T. From Angiotensin II to Cyclic Peptides and Angiotensin Receptor Blockers (ARBs): Perspectives of ARBs in COVID-19 Therapy. Molecules. 2021; 26(3):618. https://doi.org/10.3390/molecules26030618
Chicago/Turabian StyleMatsoukas, John, Vasso Apostolopoulos, Anthony Zulli, Graham Moore, Konstantinos Kelaidonis, Kalliopi Moschovou, and Thomas Mavromoustakos. 2021. "From Angiotensin II to Cyclic Peptides and Angiotensin Receptor Blockers (ARBs): Perspectives of ARBs in COVID-19 Therapy" Molecules 26, no. 3: 618. https://doi.org/10.3390/molecules26030618
APA StyleMatsoukas, J., Apostolopoulos, V., Zulli, A., Moore, G., Kelaidonis, K., Moschovou, K., & Mavromoustakos, T. (2021). From Angiotensin II to Cyclic Peptides and Angiotensin Receptor Blockers (ARBs): Perspectives of ARBs in COVID-19 Therapy. Molecules, 26(3), 618. https://doi.org/10.3390/molecules26030618