
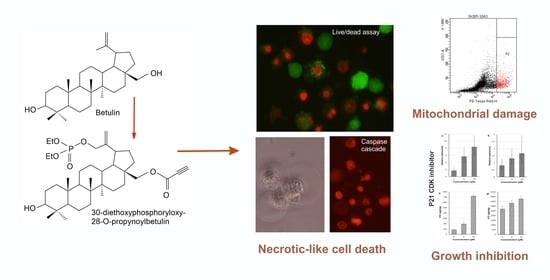
Anticancer Activity of the Acetylenic Derivative of Betulin Phosphate Involves Induction of Necrotic-Like Death in Breast Cancer Cells In Vitro

 , , , , ,
, , , , , 
Abstract
1. Introduction
2. Results
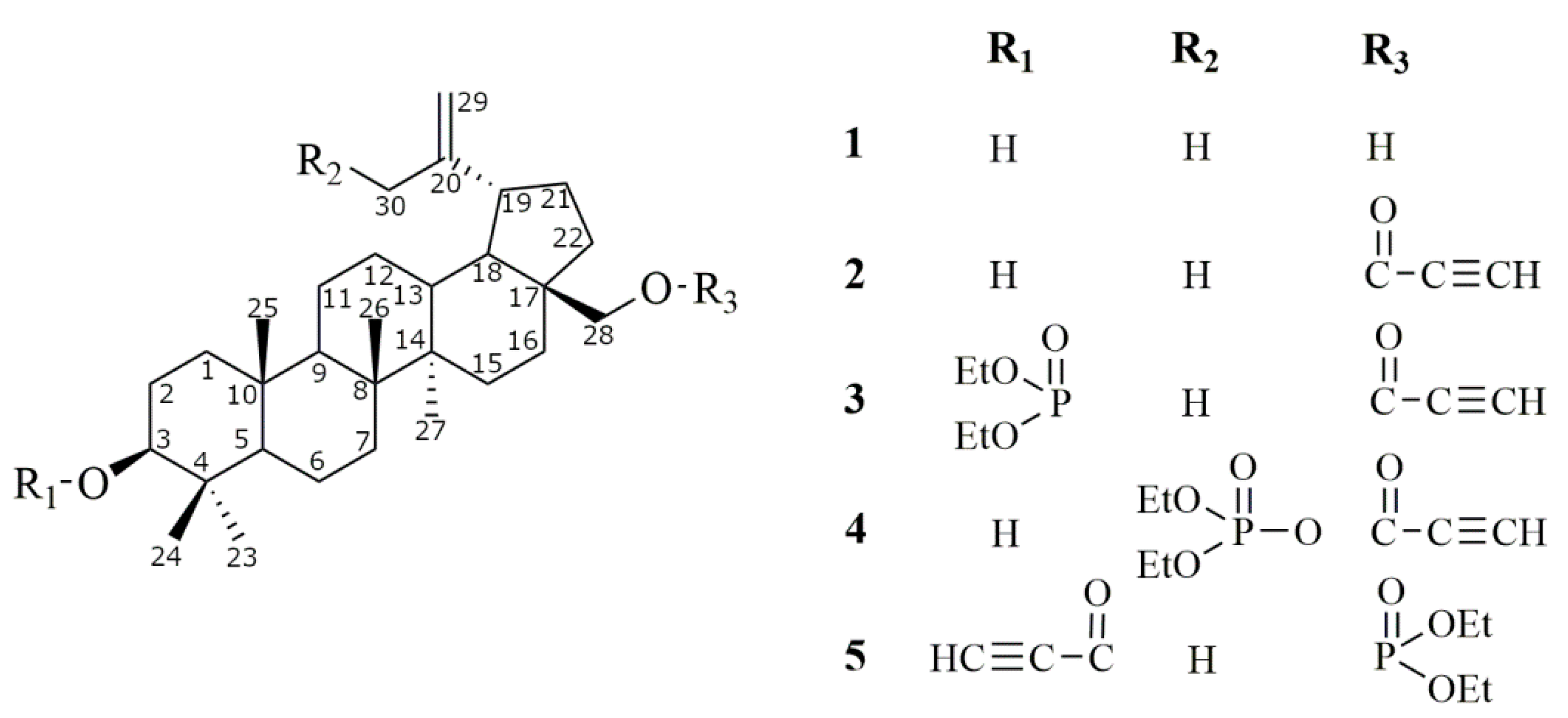
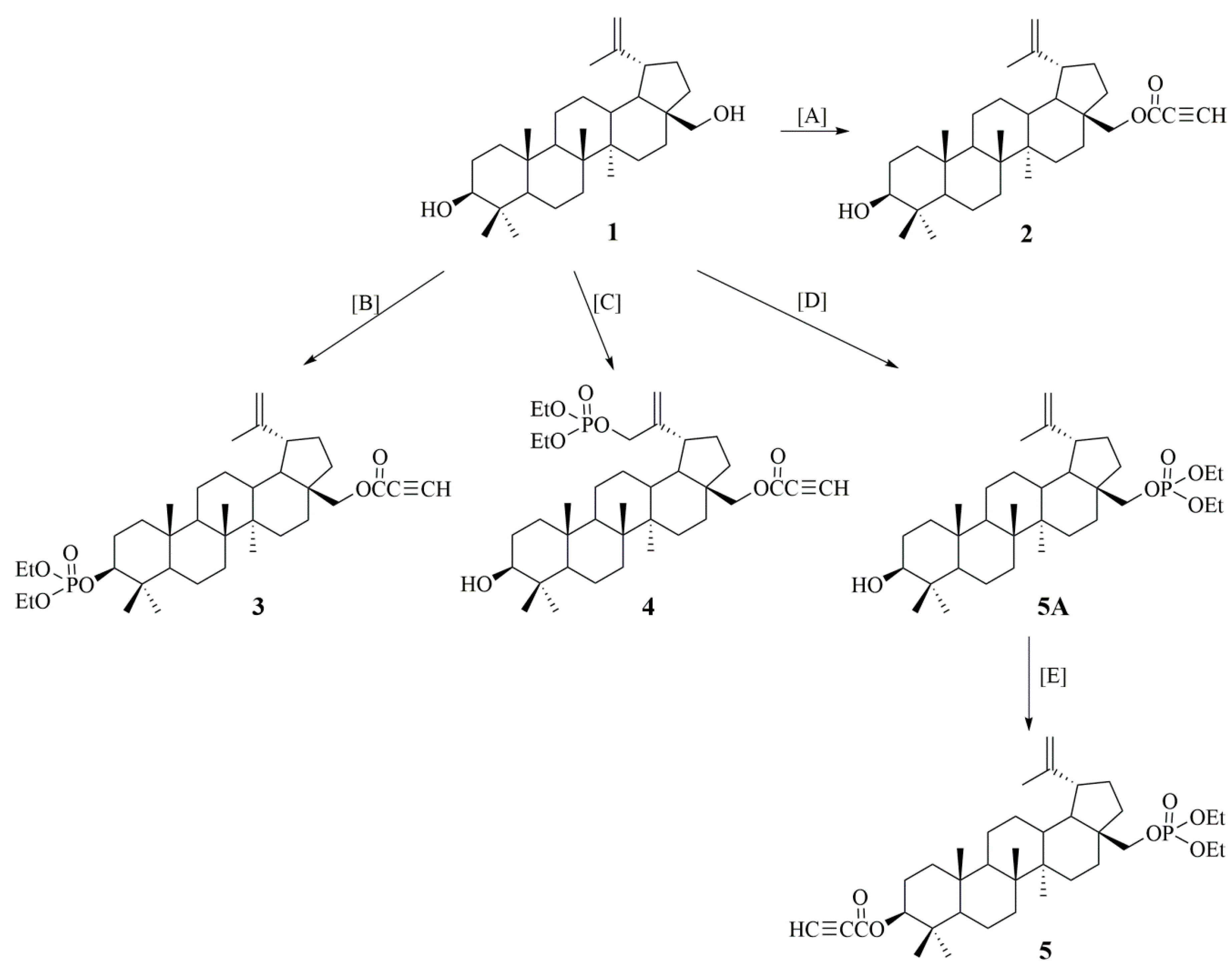
2.1. Synthesis of Betulin Derivatives (2–5)
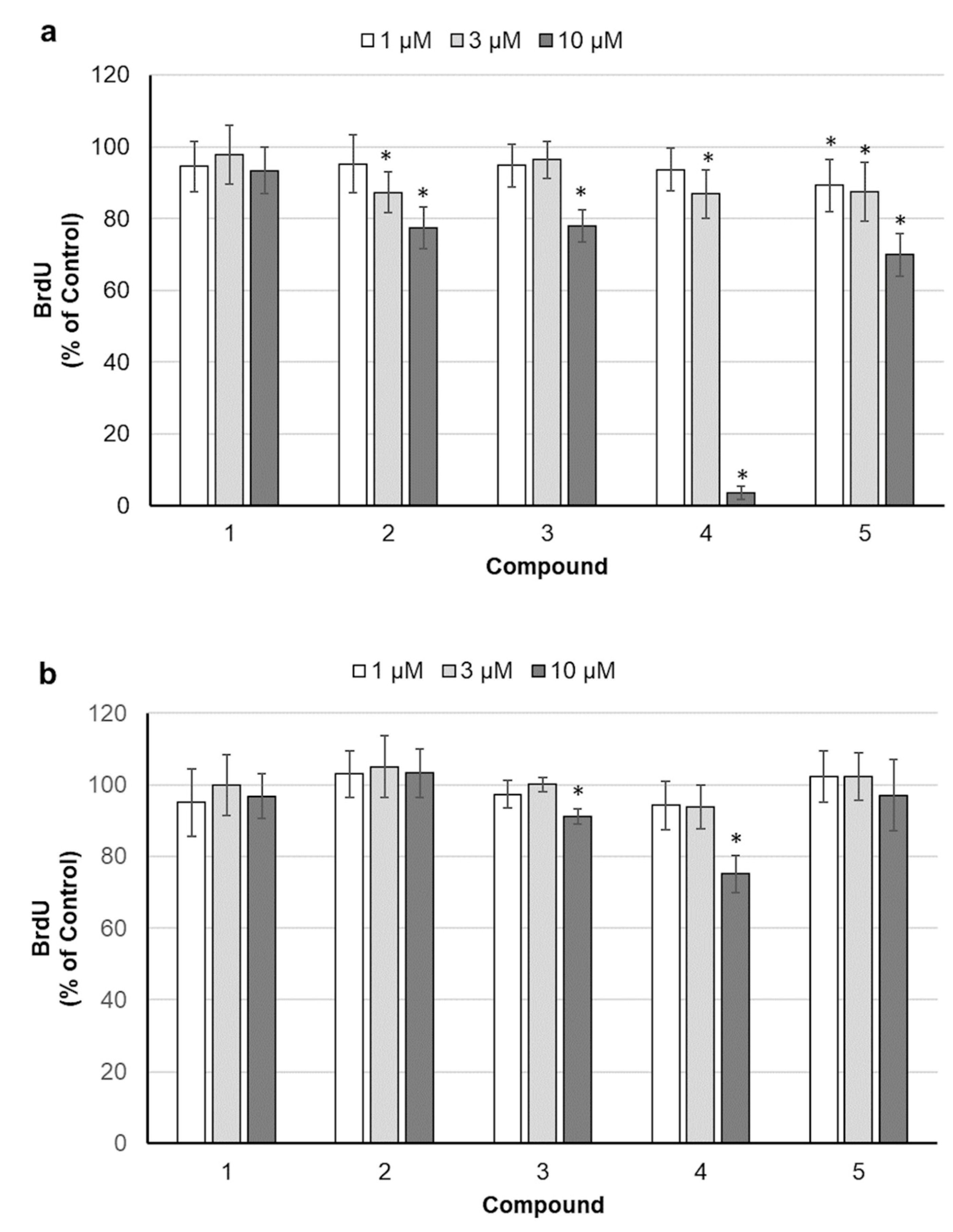
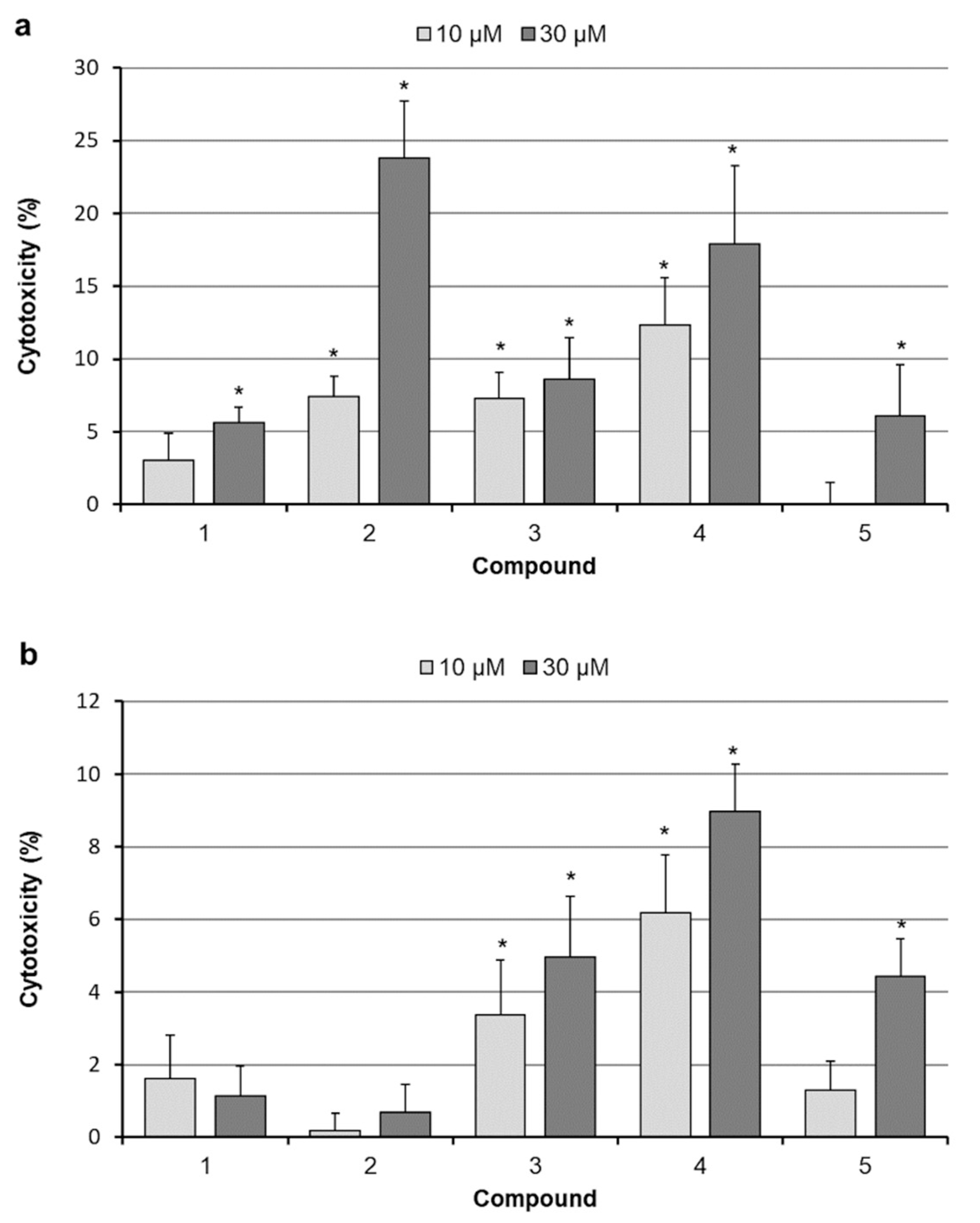
2.2. Assessment of Cell Proliferation
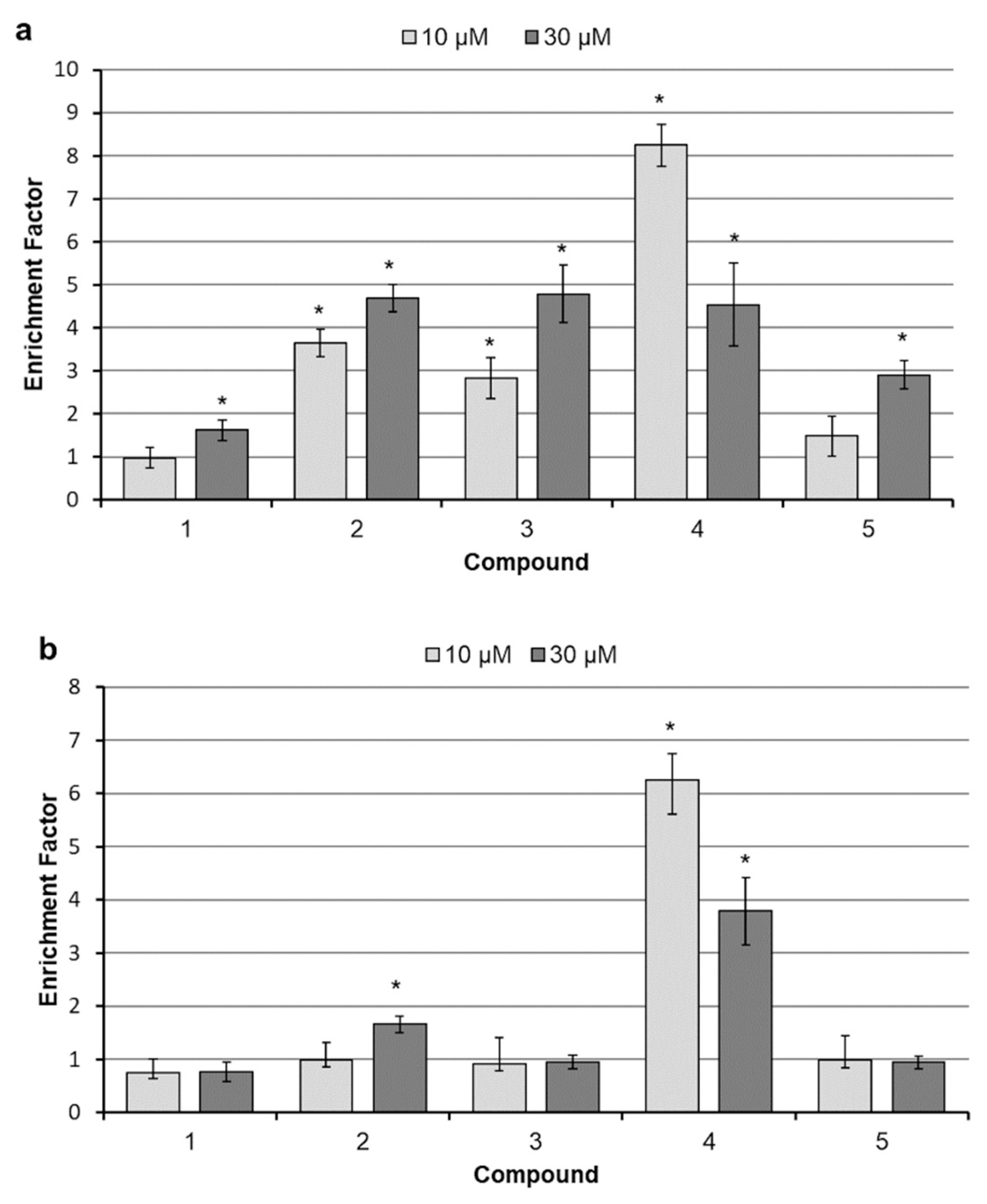
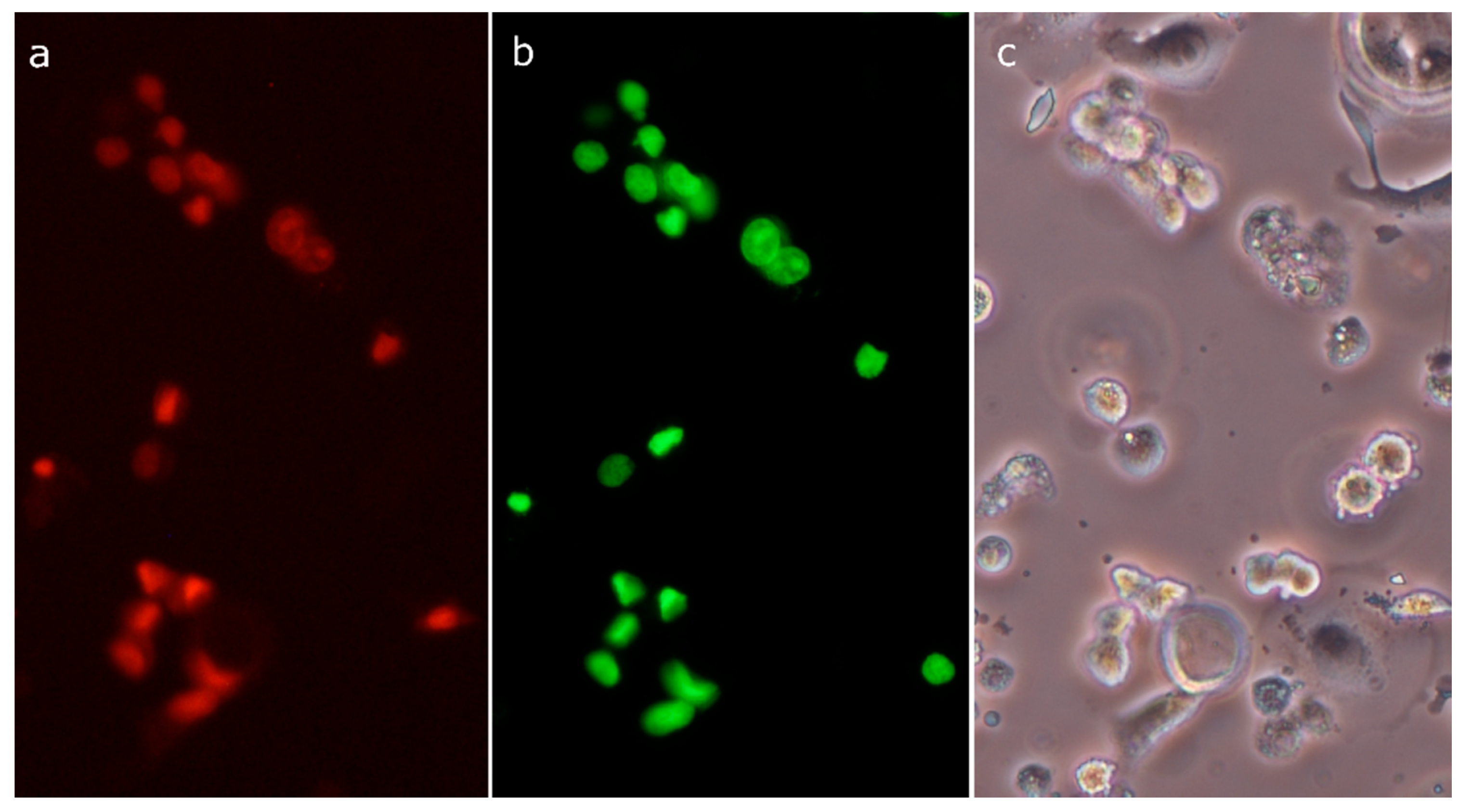
2.3. Analysis of Cell Death
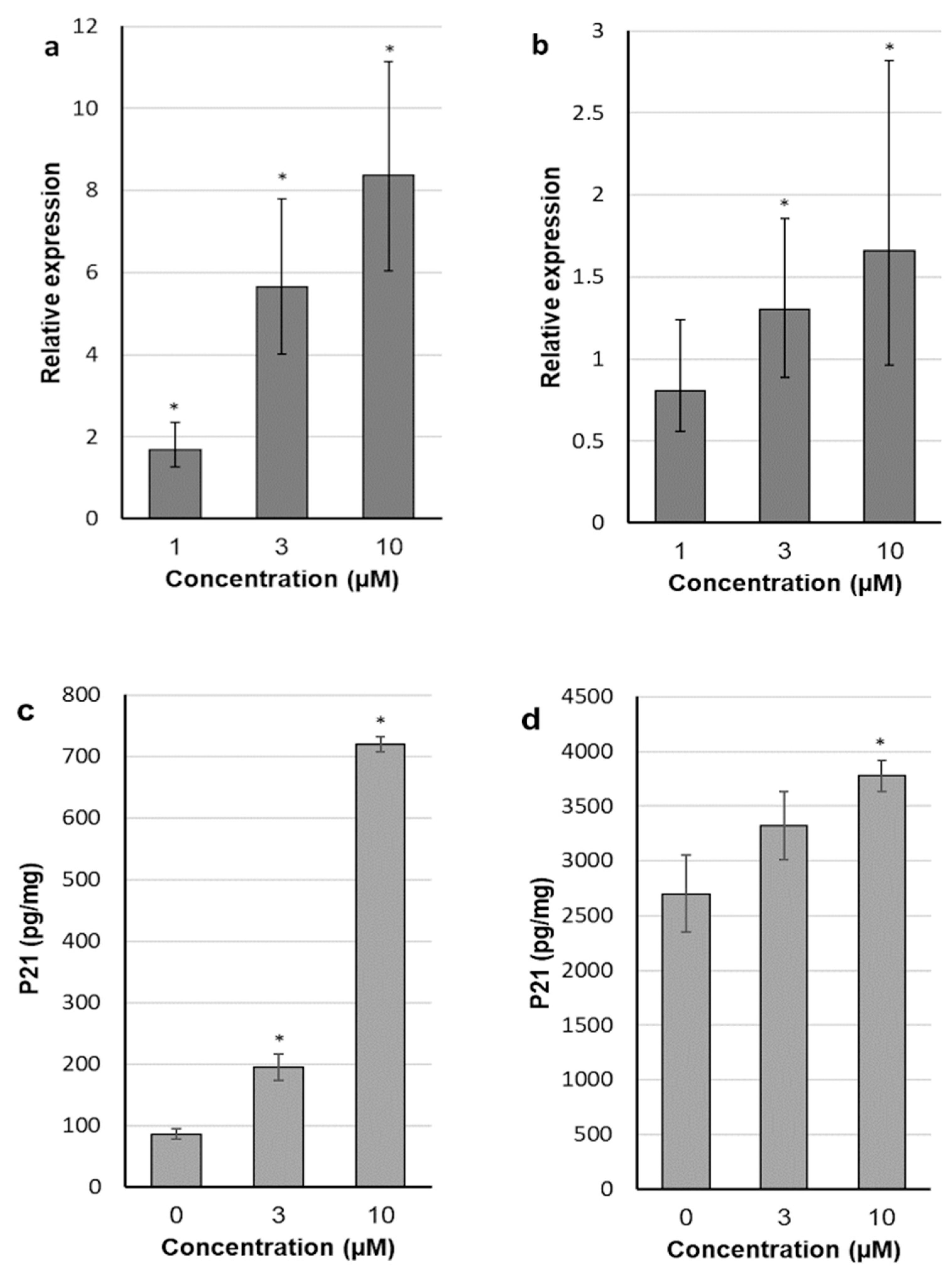
2.4. 30-diethoxyphosphoryl-28-propynoylbetulin Induces P21WAF1/Cip1 mRNA and Protein Expression
2.5. 30-diethoxyphosphoryl-28-propynoylbetulin Causes an Early Damage of Mitochondrial Function
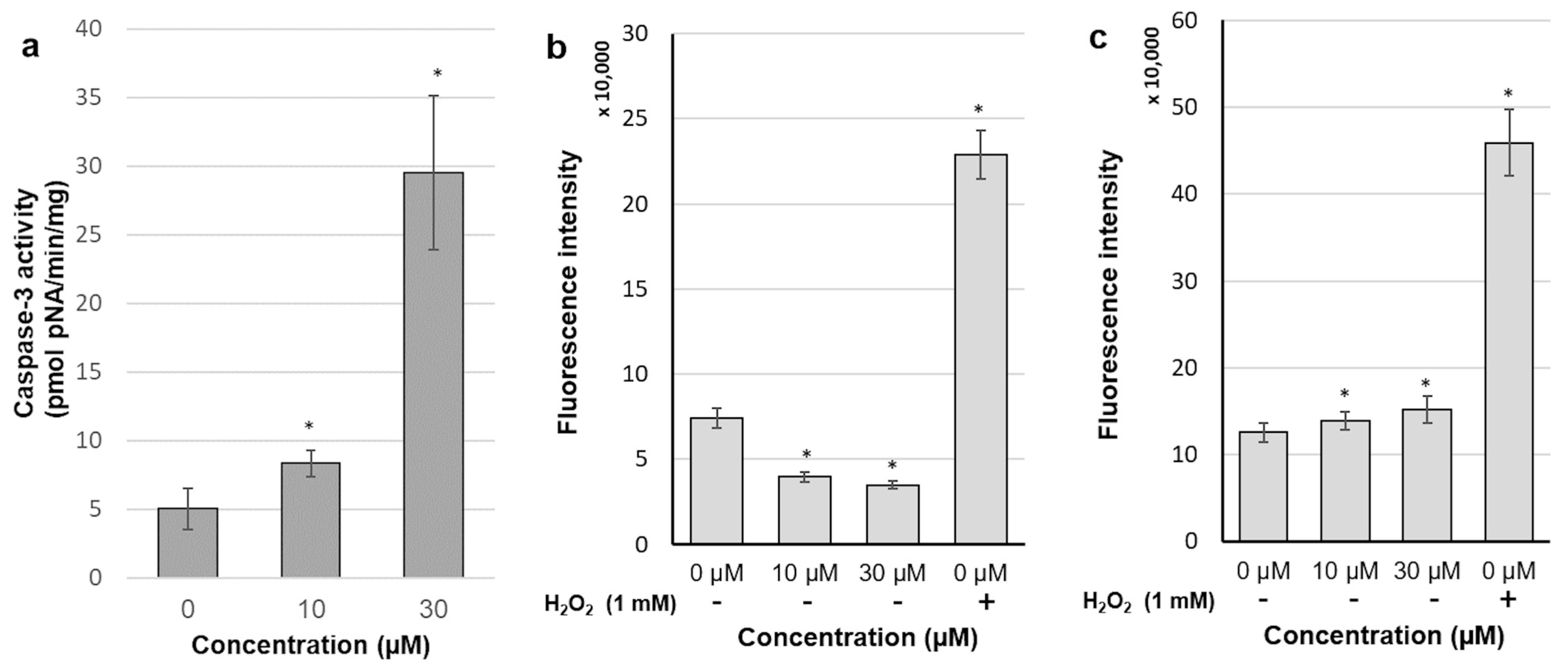
2.6. 30-diethoxyphosphoryl-28-propynoylbetulin Increases Caspase-3 Activity
2.7. Intracellular Production of Reactive Oxidative Species
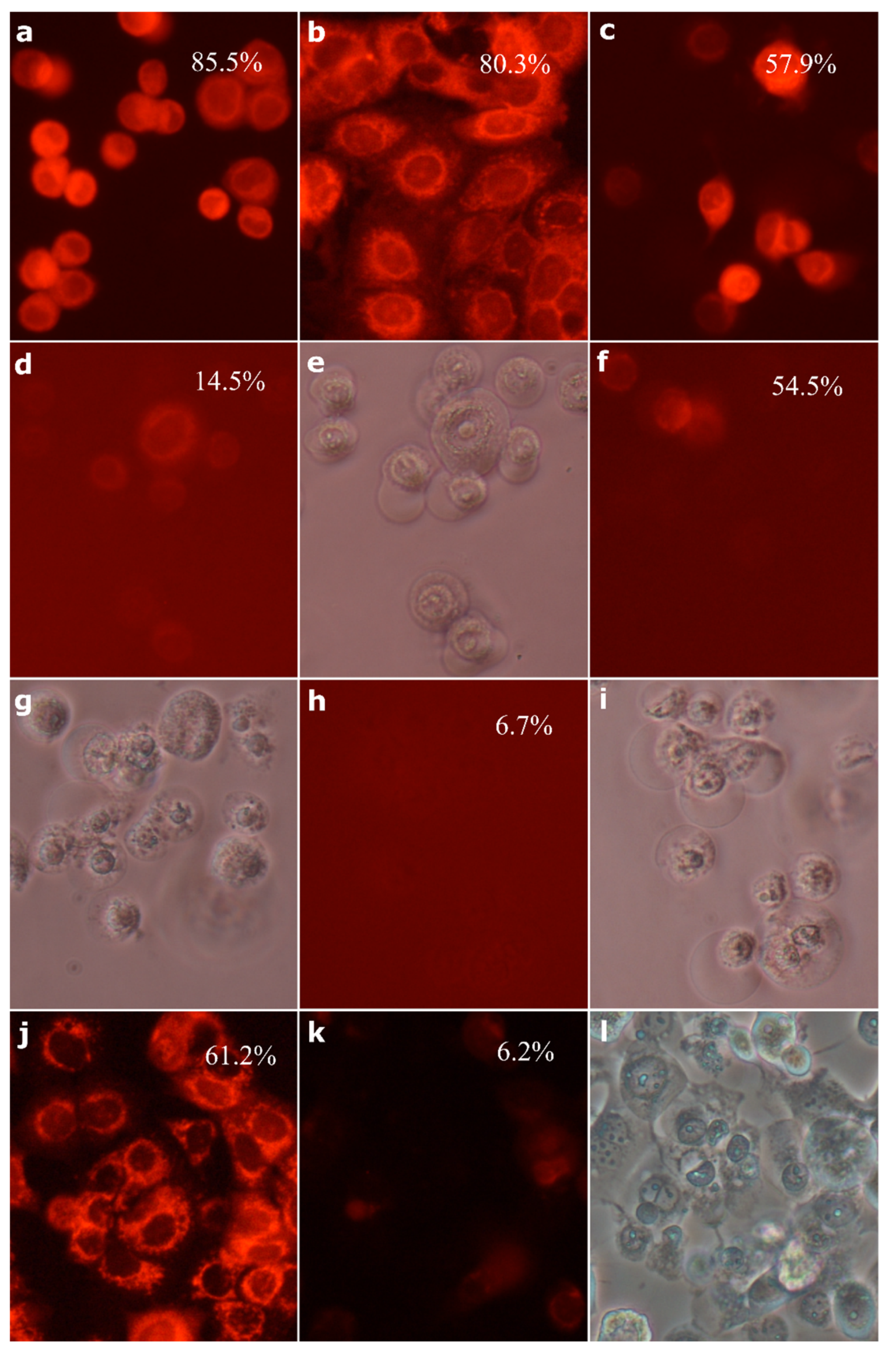
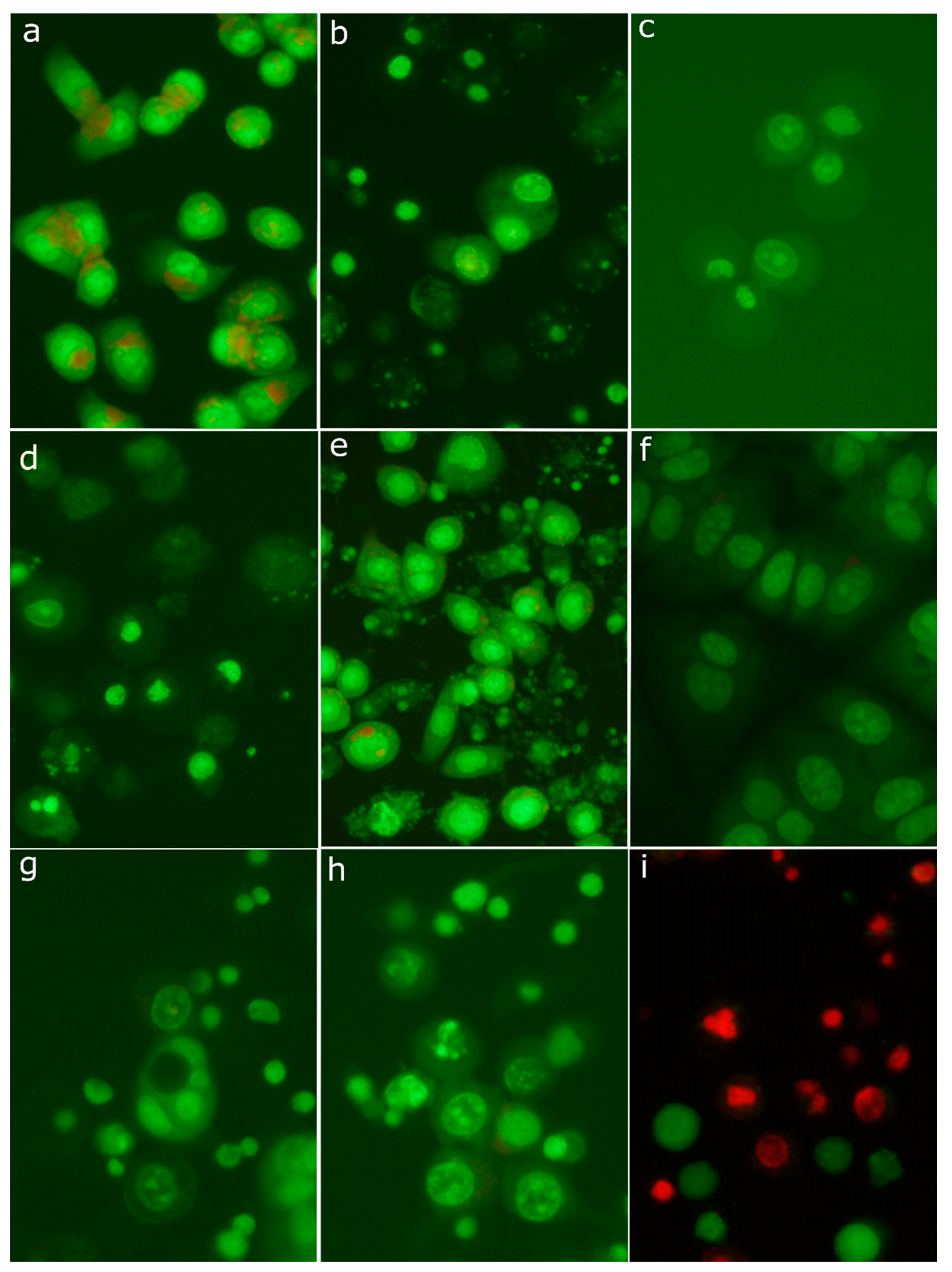
2.8. Morphological Analysis of Cells Treated with 30-diethoxyphosphoryl-28-propynoylbetulin
3. Discussion
4. Materials and Methods
4.1. Materials
Synthesis of 28-diethoxyphosphoryl-3-propynoylbetulin (5)
4.2. Cell Cultures
4.3. Sulforhodamine B (SRB) Assay
4.4. Determination of DNA Synthesis
4.5. Cytotoxicity Assays
4.6. DNA Fragmentation Assay
4.7. Gene and Protein Expression
4.8. Assessment of Mitochondrial Membrane Potential
4.9. Caspase-3 Activity Assay
4.10. Measurement of the Production of Reactive Oxygen Species
4.11. Acridine Orange Staining
4.12. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Rzeski, W.; Stepulak, A.; Szymanski, M.; Juszczak, M.; Grabarska, A.; Sifringer, M.; Kaczor, J.; Kandefer-Szerszen, M. Betulin elicits anti-cancer effects in tumour primary cultures and cell lines in vitro. Basic Clin. Pharmacol. Toxicol. 2009, 105, 425–432. [Google Scholar] [CrossRef] [PubMed]
- Alakurtti, S.; Makela, T.; Koskimies, S.; Yli-Kauhaluoma, J. Pharmacological properties of the ubiquitous natural product betulin. Eur. J. Pharm. Sci. 2006, 29, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; He, K.; Huang, Y.; Zheng, D.; Gao, C.; Cui, L.; Jin, Y.H. Betulin induces mitochondrial cytochrome c release associated apoptosis in human cancer cells. Mol. Carcinog. 2010, 49, 630–640. [Google Scholar] [CrossRef] [PubMed]
- Orchel, A.; Kulczycka, A.; Chodurek, E.; Bebenek, E.; Borkowska, P.; Boryczka, S.; Kowalski, J.; Dzierzewicz, Z. Influence of betulin and 28-O-propynoylbetulin on proliferation and apoptosis of human melanoma cells (G-361). Postepy Hig Med. Dosw (Online) 2014, 68, 191–197. [Google Scholar] [CrossRef]
- Pyo, J.S.; Roh, S.H.; Kim, D.K.; Lee, J.G.; Lee, Y.Y.; Hong, S.S.; Kwon, S.W.; Park, J.H. Anti-cancer effect of Betulin on a human lung cancer cell line: A pharmacoproteomic approach using 2 D SDS PAGE coupled with nano-HPLC tandem Mass Spectrometry. Planta Med. 2009, 75, 127–131. [Google Scholar] [CrossRef]
- Zhou, Z.; Zhu, C.; Cai, Z.; Zhao, F.; He, L.; Lou, X.; Qi, X. Betulin induces cytochrome c release and apoptosis in colon cancer cells via NOXA. Oncol. Lett. 2018, 15, 7319–7327. [Google Scholar] [CrossRef]
- Fulda, S.; Kroemer, G. Targeting mitochondrial apoptosis by betulinic acid in human cancers. Drug Discov Today 2009, 14, 885–890. [Google Scholar] [CrossRef]
- Zeng, A.Q.; Yu, Y.; Yao, Y.Q.; Yang, F.F.; Liao, M.; Song, L.J.; Li, Y.L.; Yu, Y.; Li, Y.J.; Deng, Y.L.; et al. Betulinic acid impairs metastasis and reduces immunosuppressive cells in breast cancer models. Oncotarget 2018, 9, 3794–3804. [Google Scholar] [CrossRef]
- Fulda, S.; Friesen, C.; Los, M.; Scaffidi, C.; Mier, W.; Benedict, M.; Nunez, G.; Krammer, P.H.; Peter, M.E.; Debatin, K.M. Betulinic acid triggers CD95 (APO-1/Fas)- and p53-independent apoptosis via activation of caspases in neuroectodermal tumors. Cancer Res. 1997, 57, 4956–4964. [Google Scholar] [PubMed]
- Mullauer, F.B.; Kessler, J.H.; Medema, J.P. Betulin is a potent anti-tumor agent that is enhanced by cholesterol. PLoS ONE 2009, 4, e1. [Google Scholar] [CrossRef] [PubMed]
- Fulda, S. Tumor resistance to apoptosis. Int. J. Cancer 2009, 124, 511–515. [Google Scholar] [CrossRef] [PubMed]
- Elmore, S. Apoptosis: A review of programmed cell death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef] [PubMed]
- Zeiss, C.J. The apoptosis-necrosis continuum: Insights from genetically altered mice. Vet. Pathol. 2003, 40, 481–495. [Google Scholar] [CrossRef] [PubMed]
- Slee, E.A.; Harte, M.T.; Kluck, R.M.; Wolf, B.B.; Casiano, C.A.; Newmeyer, D.D.; Wang, H.G.; Reed, J.C.; Nicholson, D.W.; Alnemri, E.S.; et al. Ordering the cytochrome c-initiated caspase cascade: Hierarchical activation of caspases-2, -3, -6, -7, -8, and -10 in a caspase-9-dependent manner. J. Cell Biol. 1999, 144, 281–292. [Google Scholar] [CrossRef]
- Wang, J.; Guo, W.; Zhou, H.; Luo, N.; Nie, C.; Zhao, X.; Yuan, Z.; Liu, X.; Wei, Y. Mitochondrial p53 phosphorylation induces Bak-mediated and caspase-independent cell death. Oncotarget 2015, 6, 17192–17205. [Google Scholar] [CrossRef]
- Vanden Berghe, T.; Linkermann, A.; Jouan-Lanhouet, S.; Walczak, H.; Vandenabeele, P. Regulated necrosis: The expanding network of non-apoptotic cell death pathways. Nat. Rev. Mol. Cell Biol. 2014, 15, 135–147. [Google Scholar] [CrossRef]
- Leist, M.; Single, B.; Castoldi, A.F.; Kuhnle, S.; Nicotera, P. Intracellular adenosine triphosphate (ATP) concentration: A switch in the decision between apoptosis and necrosis. J. Exp. Med. 1997, 185, 1481–1486. [Google Scholar] [CrossRef]
- Garcia-Belinchon, M.; Sanchez-Osuna, M.; Martinez-Escardo, L.; Granados-Colomina, C.; Pascual-Guiral, S.; Iglesias-Guimarais, V.; Casanelles, E.; Ribas, J.; Yuste, V.J. An Early and Robust Activation of Caspases Heads Cells for a Regulated Form of Necrotic-like Cell Death. J. Biol. Chem. 2015, 290, 20841–20855. [Google Scholar] [CrossRef]
- Mullauer, F.B.; Kessler, J.H.; Medema, J.P. Betulinic acid induces cytochrome c release and apoptosis in a Bax/Bak-independent, permeability transition pore dependent fashion. Apoptosis 2009, 14, 191–202. [Google Scholar] [CrossRef]
- Potze, L.; Mullauer, F.B.; Colak, S.; Kessler, J.H.; Medema, J.P. Betulinic acid-induced mitochondria-dependent cell death is counterbalanced by an autophagic salvage response. Cell Death Dis. 2014, 5, e1169. [Google Scholar] [CrossRef]
- Kessler, J.H.; Mullauer, F.B.; de Roo, G.M.; Medema, J.P. Broad in vitro efficacy of plant-derived betulinic acid against cell lines derived from the most prevalent human cancer types. Cancer Lett. 2007, 251, 132–145. [Google Scholar] [CrossRef] [PubMed]
- Gauthier, C.; Legault, J.; Lavoie, S.; Rondeau, S.; Tremblay, S.; Pichette, A. Synthesis and cytotoxicity of bidesmosidic betulin and betulinic acid saponins. J. Nat. Prod. 2009, 72, 72–81. [Google Scholar] [CrossRef] [PubMed]
- Rajendran, P.; Jaggi, M.; Singh, M.K.; Mukherjee, R.; Burman, A.C. Pharmacological evaluation of C-3 modified Betulinic acid derivatives with potent anticancer activity. Invest. New Drugs 2008, 26, 25–34. [Google Scholar] [CrossRef] [PubMed]
- Drag-Zalesinska, M.; Drag, M.; Poreba, M.; Borska, S.; Kulbacka, J.; Saczko, J. Anticancer properties of ester derivatives of betulin in human metastatic melanoma cells (Me-45). Cancer Cell Int. 2017, 17, 4. [Google Scholar] [CrossRef] [PubMed]
- Boryczka, S.; Bebenek, E.; Wietrzyk, J.; Kempinska, K.; Jastrzebska, M.; Kusz, J.; Nowak, M. Synthesis, structure and cytotoxic activity of new acetylenic derivatives of betulin. Molecules 2013, 18, 4526–4543. [Google Scholar] [CrossRef]
- Chrobak, E.; Bebenek, E.; Kadela-Tomanek, M.; Latocha, M.; Jelsch, C.; Wenger, E.; Boryczka, S. Betulin Phosphonates; Synthesis, Structure, and Cytotoxic Activity. Molecules 2016, 21, 1123. [Google Scholar] [CrossRef]
- Yan, X.; Yang, L.; Feng, G.; Yu, Z.; Xiao, M.; Cai, W.; Xing, Y.; Bai, S.; Guo, J.; Wang, Z.; et al. Lup-20(29)-en-3beta,28-di-yl-nitrooxy acetate affects MCF-7 proliferation through the crosstalk between apoptosis and autophagy in mitochondria. Cell Death Dis. 2018, 9, 241. [Google Scholar] [CrossRef]
- Chakraborty, B.; Dutta, D.; Mukherjee, S.; Das, S.; Maiti, N.C.; Das, P.; Chowdhury, C. Synthesis and biological evaluation of a novel betulinic acid derivative as an inducer of apoptosis in human colon carcinoma cells (HT-29). Eur. J. Med. Chem. 2015, 102, 93–105. [Google Scholar] [CrossRef]
- Szoka, L.; Karna, E.; Hlebowicz-Sarat, K.; Karaszewski, J.; Boryczka, S.; Palka, J.A. Acetylenic derivative of betulin induces apoptosis in endometrial adenocarcinoma cell line. Biomed. Pharmacother. 2017, 95, 429–436. [Google Scholar] [CrossRef]
- Bebenek, E.; Kadela-Tomanek, M.; Chrobak, E.; Wietrzyk, J.; Sadowska, J.; Boryczka, S. New acetylenic derivatives of betulin and betulone, synthesis and cytotoxic activity. Med. Chem. Res. 2017, 26, 1–8. [Google Scholar] [CrossRef]
- Zaklos-Szyda, M.; Pawlik, N.; Polka, D.; Nowak, A.; Koziolkiewicz, M.; Podsedek, A. Viburnum opulus Fruit Phenolic Compounds as Cytoprotective Agents Able to Decrease Free Fatty Acids and Glucose Uptake by Caco-2 Cells. Antioxidants (Basel) 2019, 8, 262. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Mackenzie, G.G.; Sun, Y.; Ouyang, N.; Xie, G.; Vrankova, K.; Komninou, D.; Rigas, B. Chemotherapeutic properties of phospho-nonsteroidal anti-inflammatory drugs, a new class of anticancer compounds. Cancer Res. 2011, 71, 7617–7627. [Google Scholar] [CrossRef] [PubMed]
- Rosłon, M.; Jastrzębska, A.; Sitarz, K.; Książek, I.; Koronkiewicz, M.; Anuszewska, E.; Jaworska, M.; Dudkiewicz-Wilczyńska, J.; Ziemkowska, W.; Basiak, D.; et al. The toxicity in vitro of titanium dioxide nanoparticles modified with noble metals on mammalian cells. Int. J. Appl. Ceram. Technol. 2019, 16, 481–493. [Google Scholar] [CrossRef]
- Boryczka, S.; Chrobak, E.; Szymura, A.; Latocha, M.; Kadela, M.; Bębenek, E. Acetylenowe pochodne 30-fosforanu betuliny o działaniu przeciwnowotworowym, sposób ich wytwarzania i zastosowanie (Antitumor acetylenic derivatives of betulin 30-phosphate, method of their preparation and application). RP Patent PL230002, 27 February 2017. [Google Scholar]
- Chrobak, E.; Kadela-Tomanek, M.; Bebenek, E.; Marciniec, K.; Wietrzyk, J.; Trynda, J.; Pawelczak, B.; Kusz, J.; Kasperczyk, J.; Chodurek, E.; et al. New phosphate derivatives of betulin as anticancer agents: Synthesis, crystal structure, and molecular docking study. Bioorg. Chem. 2019, 87, 613–628. [Google Scholar] [CrossRef] [PubMed]
- Sachet, M.; Liang, Y.Y.; Oehler, R. The immune response to secondary necrotic cells. Apoptosis 2017, 22, 1189–1204. [Google Scholar] [CrossRef]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef]
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Mathers, C.; Parkin, D.M.; Pineros, M.; Znaor, A.; Bray, F. Estimating the global cancer incidence and mortality in 2018: GLOBOCAN sources and methods. Int. J. Cancer 2018. [Google Scholar] [CrossRef]
- Barnard, M.E.; Boeke, C.E.; Tamimi, R.M. Established breast cancer risk factors and risk of intrinsic tumor subtypes. Biochim. Biophys. Acta 2015, 1856, 73–85. [Google Scholar] [CrossRef]
- Nelson, H.D.; Zakher, B.; Cantor, A.; Fu, R.; Griffin, J.; O’Meara, E.S.; Buist, D.S.; Kerlikowske, K.; van Ravesteyn, N.T.; Trentham-Dietz, A.; et al. Risk factors for breast cancer for women aged 40 to 49 years: A systematic review and meta-analysis. Ann. Intern. Med. 2012, 156, 635–648. [Google Scholar] [CrossRef]
- ACS. Breast Cancer Facts & Figures 2017–2018; American Cancer Society, Inc.: Atlanta, GA, USA, 2017. [Google Scholar]
- ACS. Cancer Facts & Figures 2018; American Cancer Society, Inc.: Atlanta, GA, USA, 2018. [Google Scholar]
- Tamimi, R.M.; Colditz, G.A.; Hazra, A.; Baer, H.J.; Hankinson, S.E.; Rosner, B.; Marotti, J.; Connolly, J.L.; Schnitt, S.J.; Collins, L.C. Traditional breast cancer risk factors in relation to molecular subtypes of breast cancer. Breast Cancer Res. Treat. 2012, 131, 159–167. [Google Scholar] [CrossRef]
- Oakes, S.R.; Vaillant, F.; Lim, E.; Lee, L.; Breslin, K.; Feleppa, F.; Deb, S.; Ritchie, M.E.; Takano, E.; Ward, T.; et al. Sensitization of BCL-2-expressing breast tumors to chemotherapy by the BH3 mimetic ABT-737. Proc. Natl. Acad. Sci. USA 2012, 109, 2766–2771. [Google Scholar] [CrossRef] [PubMed]
- Booy, E.P.; Henson, E.S.; Gibson, S.B. Epidermal growth factor regulates Mcl-1 expression through the MAPK-Elk-1 signalling pathway contributing to cell survival in breast cancer. Oncogene 2011, 30, 2367–2378. [Google Scholar] [CrossRef] [PubMed]
- Zhuo, Z.J.; Xiao, M.J.; Lin, H.R.; Luo, J.; Wang, T. Novel betulin derivative induces anti-proliferative activity by G2/M phase cell cycle arrest and apoptosis in Huh7 cells. Oncol. Lett. 2018, 15, 2097–2104. [Google Scholar] [CrossRef] [PubMed]
- Drag-Zalesinska, M.; Wysocka, T.; Borska, S.; Drag, M.; Poreba, M.; Choromanska, A.; Kulbacka, J.; Saczko, J. The new esters derivatives of betulin and betulinic acid in epidermoid squamous carcinoma treatment—In vitro studies. Biomed. Pharmacother. 2015, 72, 91–97. [Google Scholar] [CrossRef]
- Majeed, R.; Hamid, A.; Sangwan, P.L.; Chinthakindi, P.K.; Koul, S.; Rayees, S.; Singh, G.; Mondhe, D.M.; Mintoo, M.J.; Singh, S.K.; et al. Inhibition of phosphotidylinositol-3 kinase pathway by a novel naphthol derivative of betulinic acid induces cell cycle arrest and apoptosis in cancer cells of different origin. Cell Death Dis. 2014, 5, e1459. [Google Scholar] [CrossRef] [PubMed]
- Bebenek, E.; Jastrzebska, M.; Kadela-Tomanek, M.; Chrobak, E.; Orzechowska, B.; Zwolinska, K.; Latocha, M.; Mertas, A.; Czuba, Z.; Boryczka, S. Novel Triazole Hybrids of Betulin: Synthesis and Biological Activity Profile. Molecules 2017, 22, 1876. [Google Scholar] [CrossRef]
- Rello, S.; Stockert, J.C.; Moreno, V.; Gamez, A.; Pacheco, M.; Juarranz, A.; Canete, M.; Villanueva, A. Morphological criteria to distinguish cell death induced by apoptotic and necrotic treatments. Apoptosis 2005, 10, 201–208. [Google Scholar] [CrossRef]
- Soriano, J.; Mora-Espi, I.; Alea-Reyes, M.E.; Perez-Garcia, L.; Barrios, L.; Ibanez, E.; Nogues, C. Cell Death Mechanisms in Tumoral and Non-Tumoral Human Cell Lines Triggered by Photodynamic Treatments: Apoptosis, Necrosis and Parthanatos. Sci. Rep. 2017, 7, 41340. [Google Scholar] [CrossRef]
- Ormerod, M.G.; Sun, X.M.; Brown, D.; Snowden, R.T.; Cohen, G.M. Quantification of apoptosis and necrosis by flow cytometry. Acta Oncol. 1993, 32, 417–424. [Google Scholar] [CrossRef]
- Gukovskaya, A.S.; Perkins, P.; Zaninovic, V.; Sandoval, D.; Rutherford, R.; Fitzsimmons, T.; Pandol, S.J.; Poucell-Hatton, S. Mechanisms of cell death after pancreatic duct obstruction in the opossum and the rat. Gastroenterology 1996, 110, 875–884. [Google Scholar] [CrossRef]
- Ankarcrona, M.; Dypbukt, J.M.; Bonfoco, E.; Zhivotovsky, B.; Orrenius, S.; Lipton, S.A.; Nicotera, P. Glutamate-induced neuronal death: A succession of necrosis or apoptosis depending on mitochondrial function. Neuron 1995, 15, 961–973. [Google Scholar] [CrossRef]
- Dacheux, D.; Toussaint, B.; Richard, M.; Brochier, G.; Croize, J.; Attree, I. Pseudomonas aeruginosa cystic fibrosis isolates induce rapid, type III secretion-dependent, but ExoU-independent, oncosis of macrophages and polymorphonuclear neutrophils. Infect. Immun. 2000, 68, 2916–2924. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, T.; Marchetti, P.; Susin, S.A.; Dallaporta, B.; Zamzami, N.; Marzo, I.; Geuskens, M.; Kroemer, G. The apoptosis-necrosis paradox. Apoptogenic proteases activated after mitochondrial permeability transition determine the mode of cell death. Oncogene 1997, 15, 1573–1581. [Google Scholar] [CrossRef]
- Lemasters, J.J.; Qian, T.; Bradham, C.A.; Brenner, D.A.; Cascio, W.E.; Trost, L.C.; Nishimura, Y.; Nieminen, A.L.; Herman, B. Mitochondrial dysfunction in the pathogenesis of necrotic and apoptotic cell death. J. Bioenerg. Biomembr. 1999, 31, 305–319. [Google Scholar] [CrossRef] [PubMed]
- Eguchi, Y.; Shimizu, S.; Tsujimoto, Y. Intracellular ATP levels determine cell death fate by apoptosis or necrosis. Cancer Res. 1997, 57, 1835–1840. [Google Scholar]
- Eguchi, Y.; Srinivasan, A.; Tomaselli, K.J.; Shimizu, S.; Tsujimoto, Y. ATP-dependent steps in apoptotic signal transduction. Cancer Res. 1999, 59, 2174–2181. [Google Scholar]
- Kushnareva, Y.; Newmeyer, D.D. Bioenergetics and cell death. Ann. N. Y. Acad. Sci. 2010, 1201, 50–57. [Google Scholar] [CrossRef]
- Robertson, J.D.; Orrenius, S.; Zhivotovsky, B. Review: Nuclear events in apoptosis. J. Struct. Biol. 2000, 129, 346–358. [Google Scholar] [CrossRef]
- Oppenheim, R.W.; Flavell, R.A.; Vinsant, S.; Prevette, D.; Kuan, C.Y.; Rakic, P. Programmed cell death of developing mammalian neurons after genetic deletion of caspases. J. Neurosci. 2001, 21, 4752–4760. [Google Scholar] [CrossRef]
- Zheng, T.S.; Flavell, R.A. Divinations and surprises: Genetic analysis of caspase function in mice. Exp. Cell Res. 2000, 256, 67–73. [Google Scholar] [CrossRef]
- Wen, Y.; Chen, Z.; Lu, J.; Ables, E.; Scemama, J.L.; Yang, L.V.; Lu, J.Q.; Hu, X.H. Quantitative analysis and comparison of 3D morphology between viable and apoptotic MCF-7 breast cancer cells and characterization of nuclear fragmentation. PLoS ONE 2017, 12, e0184726. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Bi, Y.; Yang, C.; Yang, J.; Jiang, Y.; Meng, F.; Yu, B.; Khan, M.; Ma, T.; Yang, H. Magnolol induces apoptosis in MCF-7 human breast cancer cells through G2/M phase arrest and caspase-independent pathway. Pharmazie 2013, 68, 755–762. [Google Scholar] [PubMed]
- Mooney, L.M.; Al-Sakkaf, K.A.; Brown, B.L.; Dobson, P.R. Apoptotic mechanisms in T47D and MCF-7 human breast cancer cells. Br. J. Cancer 2002, 87, 909–917. [Google Scholar] [CrossRef] [PubMed]
- Feinstein-Rotkopf, Y.; Arama, E. Can’t live without them, can live with them: Roles of caspases during vital cellular processes. Apoptosis 2009, 14, 980–995. [Google Scholar] [CrossRef]
- Miossec, C.; Dutilleul, V.; Fassy, F.; Diu-Hercend, A. Evidence for CPP32 activation in the absence of apoptosis during T lymphocyte stimulation. J. Biol. Chem. 1997, 272, 13459–13462. [Google Scholar] [CrossRef]
- Redza-Dutordoir, M.; Averill-Bates, D.A. Activation of apoptosis signalling pathways by reactive oxygen species. Biochim. Biophys. Acta 2016, 1863, 2977–2992. [Google Scholar] [CrossRef]
- Burkitt, M.J.; Wardman, P. Cytochrome C is a potent catalyst of dichlorofluorescin oxidation: Implications for the role of reactive oxygen species in apoptosis. Biochem. Biophys. Res. Commun. 2001, 282, 329–333. [Google Scholar] [CrossRef]
- Gupta, M.K.; Neelakantan, T.V.; Sanghamitra, M.; Tyagi, R.K.; Dinda, A.; Maulik, S.; Mukhopadhyay, C.K.; Goswami, S.K. An assessment of the role of reactive oxygen species and redox signaling in norepinephrine-induced apoptosis and hypertrophy of H9c2 cardiac myoblasts. Antioxid Redox Signal. 2006, 8, 1081–1093. [Google Scholar] [CrossRef]
- Kalota, A.; Selak, M.A.; Garcia-Cid, L.A.; Carroll, M. Eltrombopag modulates reactive oxygen species and decreases acute myeloid leukemia cell survival. PLoS ONE 2015, 10, e0126691. [Google Scholar] [CrossRef]
- Ma, E.; Jeong, S.J.; Choi, J.S.; Nguyen, T.H.; Jeong, C.H.; Joo, S.H. MS-5, a Naphthalene Derivative, Induces the Apoptosis of an Ovarian Cancer Cell CAOV-3 by Interfering with the Reactive Oxygen Species Generation. Biomol. Ther. (Seoul) 2019, 27, 48–53. [Google Scholar] [CrossRef]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Halliwell, B.; Whiteman, M. Measuring reactive species and oxidative damage in vivo and in cell culture: How should you do it and what do the results mean? Br. J. Pharmacol. 2004, 142, 231–255. [Google Scholar] [CrossRef] [PubMed]
- Ruijter, J.M.; Ramakers, C.; Hoogaars, W.M.; Karlen, Y.; Bakker, O.; van den Hoff, M.J.; Moorman, A.F. Amplification efficiency: Linking baseline and bias in the analysis of quantitative PCR data. Nucleic Acids Res. 2009, 37, e45. [Google Scholar] [CrossRef]
- Pfaffl, M.W.; Horgan, G.W.; Dempfle, L. Relative expression software tool (REST) for group-wise comparison and statistical analysis of relative expression results in real-time PCR. Nucleic Acids Res. 2002, 30, e36. [Google Scholar] [CrossRef] [PubMed]
- Melcher, A.; Gough, M.; Todryk, S.; Vile, R. Apoptosis or necrosis for tumor immunotherapy: What’s in a name? J. Mol. Med. (Berl.) 1999, 77, 824–833. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | IC50 (µM) | |
|---|---|---|
| SK-BR-3 | MCF7 | |
| 1 | 7.93 ± 1.05 | 13.26 ± 0.75 |
| 2 | 3.83 ± 0.34 | n.d. |
| 3 | 2.89 ± 0.56 | n.d. |
| 4 | 2.12 ± 0.59 | 5.34 ± 0.03 |
| 5 | 3.49 ± 0.32 | 12.88 ± 0.96 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Orchel, A.; Chodurek, E.; Jaworska-Kik, M.; Paduszyński, P.; Kaps, A.; Chrobak, E.; Bębenek, E.; Boryczka, S.; Borkowska, P.; Kasperczyk, J. Anticancer Activity of the Acetylenic Derivative of Betulin Phosphate Involves Induction of Necrotic-Like Death in Breast Cancer Cells In Vitro. Molecules 2021, 26, 615. https://doi.org/10.3390/molecules26030615
Orchel A, Chodurek E, Jaworska-Kik M, Paduszyński P, Kaps A, Chrobak E, Bębenek E, Boryczka S, Borkowska P, Kasperczyk J. Anticancer Activity of the Acetylenic Derivative of Betulin Phosphate Involves Induction of Necrotic-Like Death in Breast Cancer Cells In Vitro. Molecules. 2021; 26(3):615. https://doi.org/10.3390/molecules26030615
Chicago/Turabian StyleOrchel, Arkadiusz, Ewa Chodurek, Marzena Jaworska-Kik, Piotr Paduszyński, Anna Kaps, Elwira Chrobak, Ewa Bębenek, Stanisław Boryczka, Paulina Borkowska, and Janusz Kasperczyk. 2021. "Anticancer Activity of the Acetylenic Derivative of Betulin Phosphate Involves Induction of Necrotic-Like Death in Breast Cancer Cells In Vitro" Molecules 26, no. 3: 615. https://doi.org/10.3390/molecules26030615
APA StyleOrchel, A., Chodurek, E., Jaworska-Kik, M., Paduszyński, P., Kaps, A., Chrobak, E., Bębenek, E., Boryczka, S., Borkowska, P., & Kasperczyk, J. (2021). Anticancer Activity of the Acetylenic Derivative of Betulin Phosphate Involves Induction of Necrotic-Like Death in Breast Cancer Cells In Vitro. Molecules, 26(3), 615. https://doi.org/10.3390/molecules26030615